Études d’association pangénomique pour
l’identification des régions génomiques influençant la
qualité nutritionnelle chez le soya canadien.
Thèse
Sidiki Malle
Doctorat en biologie végétale
Philosophiæ doctor (Ph. D.)
Études d’association pangénomique pour
l’identification des régions génomiques influençant
la qualité nutritionnelle chez le soya canadien
Thèse
Sidiki MALLE
Sous la direction de :
François BELZILE
Résumé
Le soya est une source importante de protéines, d'huile, de glucides, ainsi que d'autres nutriments bénéfiques, tels que des éléments minéraux. Une fonction majeure des protéines dans la nutrition est de fournir des quantités adéquates d'acides aminés. Bien qu’ils soient essentiels pour la santé humaine et la nutrition animale, les acides aminés soufrés que sont la cystéine (Cys) et la méthionine (Met) sont souvent en concentration limitée et le déterminisme génétique de leur teneur dans la graine de soya est mal caractérisé. Un autre facteur non moins important pour la qualité nutritionnelle du soya est sa teneur en éléments minéraux, laquelle affecte les caractéristiques d'utilisation finale des fractions d'huile et de protéines ainsi que la qualité des semences (germination, vigueur des jeunes plantules). Malheureusement, très peu d’attention a été portée sur les variétés canadiennes de soya en ce qui a trait à leur teneur en acides aminés soufrés ainsi qu’en éléments minéraux.
L’amélioration génétique est l’une des voies les plus efficientes et économiques pour contribuer à une alimentation saine et nutritive, laquelle apporte au consommateur la quantité de nutriments nécessaires à une bonne santé. Pour que puisse se pratiquer une sélection en faveur d’une qualité nutritionnelle accrue, il faut idéalement identifier les déterminants génétiques de la teneur en divers nutriments et développer des marqueurs facilitant cette sélection.
Présentement, les analyses d’association pangénomique (GWAS en anglais) constituent l’approche la plus puissante pour déterminer l’assise génétique d’un caractère. Dans les cas les plus favorables, non seulement ces analyses permettent-elles d’identifier des régions génomiques qui contrôlent en tout ou en partie les caractères étudiés, mais elles peuvent même permettre d’identifier des gènes candidats qui jouent un rôle direct dans ce déterminisme.
Dans le cadre de cette thèse, nous avons souhaité déterminer l’assise génétique de composantes clés de la valeur nutritive du soya, soit la teneur en acides aminés soufrés (Cys/Met) et en quatre éléments minéraux majeurs (Ca, K, P et S). Dans les deux cas, des analyses d’association pangénomique ont été réalisées sur une collection de 137 lignées
représentatives de la diversité génétique rencontrée au sein du soya canadien à maturité hâtive.
Dans le premier volet, les teneurs en Cys et en Met ont été mesurées par spectroscopie proche infrarouge (NIR) sur des graines produites dans cinq environnements au total. Des données génotypiques pour 2,2 M de polymorphismes mononucléotidiques (SNP) ont été exploitées pour réaliser une analyse d’association. Dans une première phase de découverte, nous avons réussi à identifier un total de dix régions génomiques (QTL) en nous appuyons sur les phénotypes rencontrés dans deux environnements. Afin d’assurer la fiabilité ainsi que la reproductibilité de ces QTL, nous avons validé une forte majorité de ces QTL dans trois environnements additionnels. Ces QTL, dont la plupart sont inédits, nous ont permis d’identifier deux gènes candidats fort prometteurs codant pour des protéines impliquées dans la synthèse de la cystéine.
Dans le deuxième volet, les teneurs en éléments minéraux ont été mesurées chez les 137 lignées à l’aide d’un spectromètre de fluorescence à rayons X (XRF) sur des grains récoltés dans cinq environnements. Les analyses d’association ont été réalisées avec le même jeu de données génotypiques (2,2 M de SNP) que dans le volet précédant. Huit QTL significativement associés à la teneur en Ca, K, P et S ont été identifiés de manière robuste à l’aide d’au moins deux modèles statistiques différents (sur les trois employés). La reproductibilité de ces QTL dans trois environnements additionnels a fait l’objet d’une seconde analyse pour des fins de validation. Une forte reproductibilité de l’effet de ces régions génomiques sur ces phénotypes a été observée par la validation de sept QTL sur huit. Trois gènes candidats ont été identifié impliqués soit dans le transport ou l’assimilation de ces éléments minéraux.
Par rapport aux études précédentes, la forte densité de marqueurs utilisés dans cette étude a contribué à la détection reproductible de plusieurs nouveaux loci associés à la teneur en acides aminés soufrés ou en éléments minéraux. De plus, cela a rendu possible l’identification de gènes candidats prometteurs. Les marqueurs et les gènes identifiés dans cette étude seront utiles pour l'amélioration génétique du soya à travers la sélection génétique assistée par marqueur.
Abstract
Soybean is an important source of protein, oil, carbohydrates, and other beneficial nutrients, such as minerals. A major function of protein in nutrition is to provide adequate amounts of amino acids. Although essential for human health and animal nutrition, the sulfur amino acids cysteine (Cys) and methionine (Met) are often limiting and the genetic basis underlying their accumulation in soybeans seeds is poorly characterized. Another factor no less important for the nutritional quality of soybeans is its mineral content, which affects the end-use traits of both the oil and protein fractions as well as the quality of seed (germination rate, vigor of seedlings). Unfortunately, very little attention has been paid to Canadian soybean varieties in terms of their content in sulfur amino acids and important minerals in seeds.
The enhancement of seed nutrient content via genetic improvement is considered as the most promising and cost-effective approach to contribute to a healthy and nutritious diet, which provides the consumer with the necessary quantity of nutrients for good health. To facilitate breeding for increased nutritional quality, it is necessary to identify the genetic determinants underlying various nutrients and to develop markers allowing this selection.
Currently, genome-wide association analysis (GWAS) is the most powerful approach for determining the genetic basis of a trait. In the most favorable cases, not only do these analyses make it possible to identify genomic regions which control all or part of the trait of interest, but they can even make it possible to identify candidate genes which play a direct role in the trait of interest.
The goals of this thesis were to determine the genetic basis of key components of the nutritional value of soybeans, namely the seed content in sulfur amino acids (Cys / Met) and four major mineral elements (Ca, K, P and S). In both cases, a GWAS was performed on a collection of 137 lines representative of the genetic diversity encountered in early-maturing Canadian soybeans.
In part 1, Cys and Met content were measured using near infrared spectroscopy (NIR) on seed from five environments in total. Genotypic data for 2.2 M single nucleotide polymorphisms (SNPs) were used to perform an association analysis. In an initial discovery
phase based on the data from two environments, we were able to identify a total of ten genomic regions (QTL), most of which were identified for the first time. To ensure the reliability and reproducibility of these QTLs, we validated a large majority of these in three additional environments. These QTLs allowed us to identify two candidate genes, both of which code for proteins involved in cysteine synthesis.
In part 2, mineral content was measured in seed of the same 137 lines using an X-ray fluorescence spectrometer (XRF) harvested from five environments in total. The association analyses were carried out with the same genotypic data set (2.2 M SNP) as in part 1. Eight QTLs significantly associated with the Ca, K, P and S content were identified by at least two of the three statistical models used. These QTLs were found to be highly reproducible as they influenced the studied traits in three additional environments. Indeed, seven of the eight QTLs were validated in this fashion. For these QTLs regions, we were able to identify thee candidate gene annotated as being involved in the transport or the assimilation of these mineral elements.
Compared to previous studies, the high density of markers used in this study has contributed to the reproducible detection of several new loci associated with the content of sulfur amino acids or mineral elements. In addition, it has made it possible to identify promising candidate genes. The markers and genes identified in this study will be useful for the genetic improvement of soybeans through marker-assisted selection.
Table des matières
Résumé ... ii
Abstract ... iv
Table des matières ... vi
Liste des tableaux ... ix
Liste des abréviations ... xi
Remerciements ... xii
Avant-propos ... xiii
Introduction générale ... 1
Chapitre 1 : Revue bibliographique ... 6
Brève description du soya ... 6
Les acides aminés ... 7
1.2.1 La méthionine ... 7
1.2.2 La cystéine ... 8
Les éléments minéraux ... 8
1.3.1 Le calcium ... 9
1.3.2 Potassium ... 9
1.3.3 Phosphore ... 10
1.3.4 Soufre ... 10
Amélioration génétique ... 11
1.4.1 Les marqueurs moléculaires ... 12
1.4.2 La cartographie génétique ... 18
1.4.3. Validation des QTL identifiés et analyse post-GWAS ... 24
1.5. Hypothèses et objectifs ... 26
Chapitre 2: GENOME-WIDE ASSOCIATION IDENTIFIES SEVERAL QTLS CONTROLLING CYSTEINE AND METHIONINE CONTENT IN SOYBEAN SEED INCLUDING A CYSTEINE SYNTHASE GENE ... 27
2.1. Résumé ... 28
2.2. Abstract ... 29
2.3. Introduction ... 30
2.4. Results ... 33
2.4.1. Phenotypic variation and correlations among traits ... 33
2.4.2. Genotyping and SNP calling ... 35
2.4.3. Population structure ... 35
2.4.5. QTL validation ... 39
2.4.6. Refinement of the GWA scan ... 41
2.4.7. Prediction of candidate genes within the QTL regions ... 41
2.4.8. Structural and nucleotide variation within candidate genes and their predicted functional impact ... 43
2.5. Discussion ... 44
2.5.1. Phenotypic variation and correlations among traits ... 44
2.5.2. Genome-wide association analysis ... 44
2.5.3. Candidates genes and their functions for sulfur amino acid accumulation ... 46
2.6. Materials and methods ... 47
2.6.1. Plants material and field trials ... 47
2.6.2. Phenotyping and Statistical Analysis ... 47
2.6.3. Genotyping and quality control ... 48
2.6.4. Population structure and kinship ... 49
2.6.5. Genome-wide association analysis ... 49
2.6.6. Validation of the allelic effect of the reported QTLs in three environments ... 50
2.6.7. Candidate genes and their functional analysis ... 50
2.7. Supplementary Materials ... 51
2.8. Conflict of interest disclosure ... 51
2.9. Acknowledgments ... 51
2.10. Author contributions ... 51
2.11. References ... 52
Chapitre 3: IDENTIFICATION OF LOCI CONTROLLING MINERAL ELEMENT CONCENTRATION IN SOYBEAN SEEDS ... 57
3.1. Résumé ... 58
3.2. Abstract ... 59
3.3. Introduction ... 60
3.4. Results ... 62
3.4.1. Correlation between wet chemistry and energy-dispersive X-ray fluorescence method 62 3.4.2. Phenotypic variation and correlations among traits ... 63
3.4.3. Genotyping and SNP calling ... 65
3.4.4. Population structure ... 65 3.4.5. Genome-wide association scan for mineral elements content in soybean seeds
3.4.6. Validation of the eight co-identified QTL across three environments ... 70
3.4.7. Refinement of the GWA scan for co-identified QTL ... 71
3.4.8. Prediction of candidate genes within the robust QTL regions ... 71
3.4.9. Structural and nucleotide variation within candidate genes and their predicted functional impact ... 74
3.5. Discussion ... 74
3.5.1. Phenotypic variation and correlations among traits ... 74
3.5.2. Genome-wide association scan for mineral elements content in soybean seeds 75 3.5.3. Candidates genes and their functions for mineral elements accumulation ... 76
3.6. Materials and methods ... 77
3.6.1. Plant material and experimental design ... 77
3.6.2. Calibration and validation ... 78
3.6.3. Phenotyping and statistical analysis ... 78
3.6.4. Genotyping and SNPs imputation ... 79
3.6.5. Population structure and kinship analyses ... 79
3.6.6. Genome-wide association analysis ... 80
3.6.7. Validation of the allelic effect of the priority QTLs in three environments ... 80
3.6.8. Candidate genes and their functional analysis ... 81
3.7. Supplementary Materials ... 81
3.8. Conflict of Interest Disclosure ... 81
3.9. Acknowledgments ... 82
3.10. Author contributions ... 82
3.11. References ... 82
Conclusion générale ... 87
Bibliographie ... 89
Annexe A : Matériels supplémentaires chapitre 2 ... 94
Liste des tableaux
Table II. 1: Descriptive statistics for cysteine, methionine and the sulfur amino acid content across two sites (two replicates per site). ... 34 Table II. 2: The most highly associated SNP markers within each QTL and their effects on phenotypic variation for sulfur amino acid content in a Canadian soybean collection. ... 38 Table III. 1: Descriptive statistics for Ca, K, P and S content across two sites (two replicates per site) in the seed of 137 Canadian soybean lines ... 64 Table III. 2: ANOVA results for Ca, K, P and S content across two sites (two replicates per site) in seed of 137 Canadian soybean lines. ... 64 Table III. 3: List of QTLs for mineral element content identified by at least two statistical models in 137 Canadian soybean lines. ... 69 Table III. 4: Identification of candidate genes for seven QTLs associated with mineral element content in a core set of 137 Canadian soybean lines. ... 73
Liste des Figures
Figure I. 1: Vue d'ensemble d'une plante de soya (source
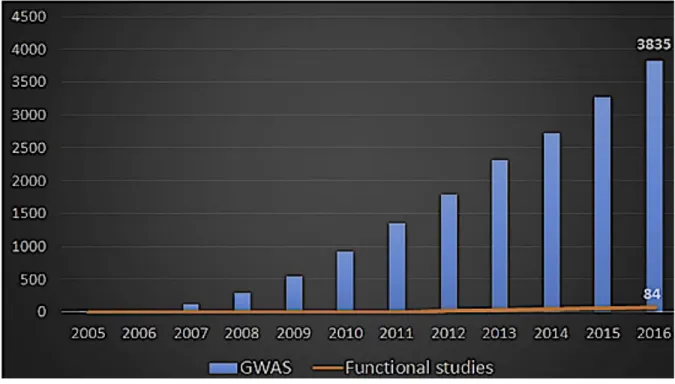
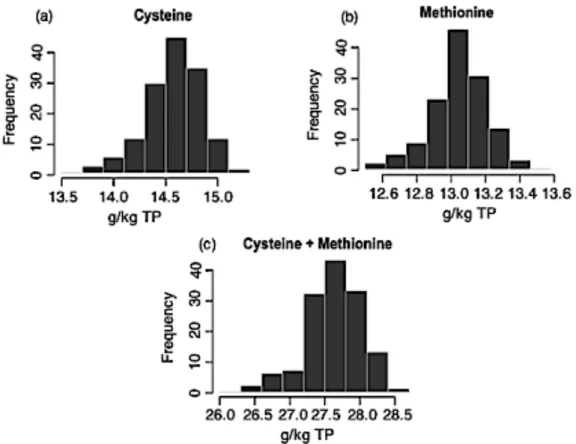
http://www.naturemania.com/bioproduits/soja.html) ... 6 Figure I. 2: Différentes plateformes de nouvelle technologie de séquençage à haut débit .. 15 Figure I. 3: Coût du séquençage (https://www.biostars.org/p/3367/) ... 15 Figure I. 4: Procédure de détection des marqueurs SNP par une approche GBS. ... 17 Figure I. 5: Illustration de deux méthodes d'association. Réadapté selon (Zhu, et al., 2008). ... 20 Figure I. 6: Les étapes les plus importantes pour réussir une GWAS ... 24 Figure I. 7: visualisation du résultat d’un GWAS. Diagramme Quantile-Quantile (a) et de Manhattan (b) ... 24 Figure I. 8: Études cumulatives de GWAS et nombre d’études de suivi fonctionnel par année (Gallagher, et al., 2018). ... 25 Figure II. 1 : Distribution of Cys (a), Met (b) and (c) Cys+ Met content (g/kg of total protein) in the seed of 137 Canadian soybean lines. 34
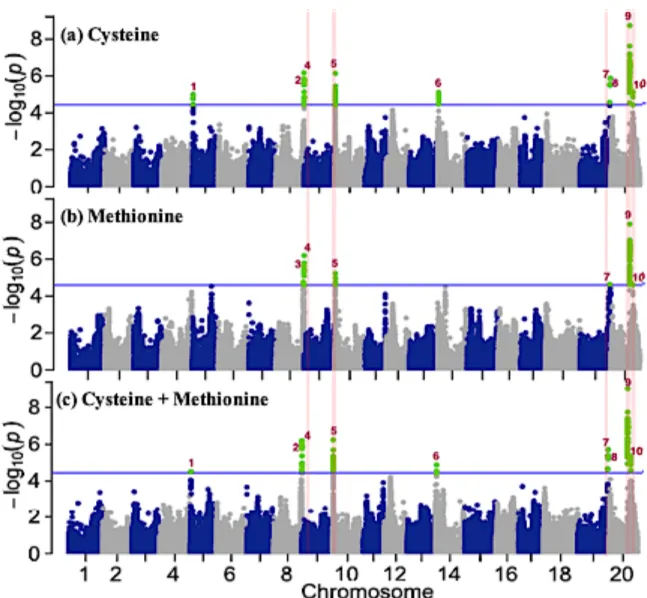
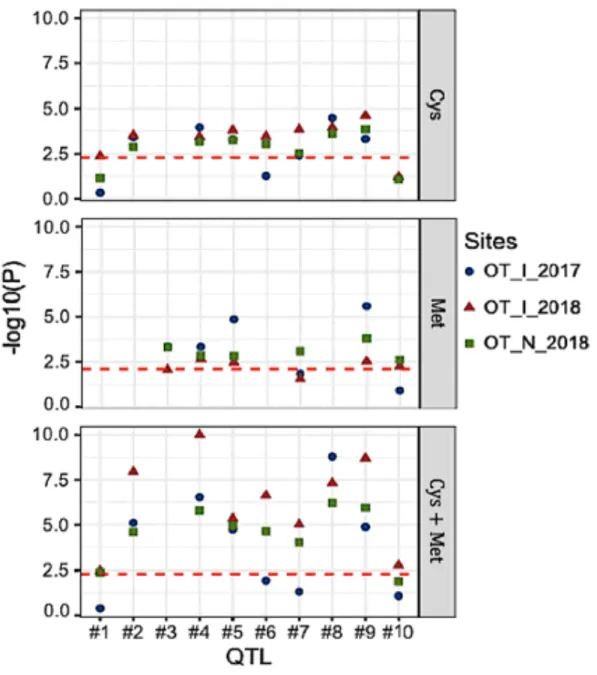
Figure II. 2: Manhattan plots for (a) Cys, (b) Met, and (c) Cys + Met content in a core set of 137 Canadian soybean accessions using a mixed linear model. ... 37 Figure II. 3: Degree of significance (p-value) of the contrast between phenotypic means for lines carrying different alleles at candidate QTLs for sulfur-containing amino acid content in whole soybean seeds. ... 40 Figure II. 4: Identification of candidate genes within the haplotype block corresponding to QTL 7 on chromosome 19. ... 42 Figure II. 5: eFP browser view (www.bar.utoronto.ca) of Glyma.19g242300 expression in soybeans. Expression strength coded by color: yellow = low, red = high. ... 43 Figure III. 1: Pearson correlation between wet chemistry and ED-XRF for Ca, K, P and S content on a dry-weight basis among 30 soybean seed samples. 62
Figure III. 2: Distribution of Ca, K, P and S content in the seed of 137 Canadian soybean lines. ... 63 Figure III. 3:Models-based population structure in a core set of 137 Canadian soybean lines. ... 66 Figure III. 4: Manhattan plots for (a) calcium (b) potassium, (c) phosphorus and (d) sulfur content in a core set of 137 Canadian soybean accessions using three models for measuring marker-trait associations. Each dot indicates the degree of association between a single marker and a trait (y-axis) while the x-axis shows the physical position of each marker. A blue horizontal bar indicates the significance threshold (FDR £ 0.05) and significantly associated markers are coloured in red for FarmCPU, (+) for CMLM and (*) for MLMM. ... Erreur ! Signet non défini.68 Figure III. 5: Venn diagram for the 32 identified QTLs through three analytical approaches. ... 69 Figure III. 6: Stability of the eight QTLs detected by at least two models for Ca, K, P and S content. ... 71 Figure III. 7: Identification of a candidate gene underlying QTL S_#10 within the haplotype block on chromosome 20. ... 73
Liste des abréviations
ADN : …Acide désoxyribonucléique DNA : … deoxyribonucleic acid
AFLP : Amplified fragment length polymorphism GBS: Genotyping by sequencing
GWAS: Genome-wide association study LD: Linkage disequilibrium
NGS : Next-generation sequencing NIL: Near isogenic line
PCA: Principal component analysis QTL : Quantitative trait locus
RAPD: Random amplified polymorphic DNA RFLP: Restriction fragment length polymorphism RILs: Recombinant inbred lines
SNP: Single nucleotide polymorphism SSR: Simple sequence repeat
USDA: United States Department of Agriculture WGS: Whole-genome sequencing
Remerciements
Au terme de ces années d’études et de collaboration, je ne pourrai jamais assez remercier certaines personnes qui ont su positivement marquer ma vie à jamais.
Je tiens à remercier du fond du cœur Dr François Belzile un brillant et rigoureux chercheur, d’avoir bien voulu m’accepter dans son labo tout en sachant mes connaissances limitées dans le domaine de la génomique ainsi que les outils bio-informatiques. Cher Pr Belzile, en tant que mon directeur de thèse, vous avez été une source d’inspiration grâce à votre souci du travail bien fait, d’ouverture d’esprit, de connaissance très étendue et approfondie, mais surtout de votre patience d’écoute. Vous avez été d’une source de motivation grâce à votre disponibilité malgré un planning chargé car vous avez cru en moi. Vous m’avez été un support aussi bien matériel que financier. Vous m’avez été aussi un ami grâce à nos échanges autour du feu dans votre domicile après avoir bien mangé un bon steak ou dans des restaurants et PUB universitaire.
Je tiens aussi à remercier le projet FASAM d’avoir financé la réalisation de la présente thèse ainsi que la bourse qui m’était offerte mensuellement. Je remercie également le projet d’avoir mis en place une équipe efficace, qui était là à m’aider dans l’accomplissement de ma thèse. Je souhaite également à remercier Dre Martine Jean pour sa disponibilité, son sens critique, mais surtout son souci du détail. Merci aussi pour tes super bonbons d’Halloween et ta surveillance inoubliable pour les dates et achats des gâteaux d’anniversaire.
J’aimerais aussi profiter de cette occasion pour remercier Dr Steve Boahen, Dre Nathalie Isabel et Dr Pierre-Mathieu Charest pour l’honneur qu’ils m’accordent en tant que membres du jury de ma thèse.
Un remerciement particulier à Dre Amina Abed pour son support intellectuel, son encouragement qui m’a été d’une aide inestimable.
Mes vifs remerciements à ma fiancée, Djelika Toure, et à toute ma famille à qui j’ai beaucoup manqué durant ces années passées loin d’eux. Je les remercie aussi pour leur support moral. Pour finir, je remercie mes collègues du labo présents ou absents: Suzanne, Davoud, Manel, Marc-André́ dit Mar-en-ciel, Vincent-thomas Boucher St-Amour dit Tom, Sébastien, Honoré Tekeu, Chiheb et Patricio pour nos échanges fructueux, leur disponibilité et pour toutes les connaissances qu’ils m’ont enseignées à travers nos discussions quotidiennes.
Avant-propos
La présente thèse comporte une introduction générale, trois chapitres et une conclusion générale. L’introduction générale est une présentation du cadre général faisant ressortir l’état des lieux au sujet de l’insécurité alimentaire et nutritionnelle dans le monde et en particulier en Afrique ainsi que les solutions envisagées. Elle sert également de préambule pour les chapitres 1 à 3. Le chapitre 1 est présenté sous forme d’une revue bibliographique qui dresse l’importance particulière du soya à travers sa composition en acides aminés soufrés et en éléments minéraux dans l’alimentation humaine/animale et chez les plantes. Dans ce chapitre, nous donnons également des brefs aperçus sur les marqueurs moléculaires, les technologies de séquençage et génotypage à haut débit ainsi que les approches pour cartographier des loci de caractère quantitatif associés à des caractères d’intérêt. Les hypothèses et objectifs spécifiques sont définis à la fin de ce chapitre.
Les chapitres deux et trois de cette thèse sont sous la forme d’articles scientifiques. Le chapitre 2 a été soumis pour publication en décembre 2019 dans le journal « Scientific Reports » sous : Malle S., Eskandari M., Morrison M. and Belzile F. (2019). Genome-wide association identifies several QTLs controlling cysteine and methionine content in soybean seed including a cysteine synthase gene. S.M. et F.B. ont conçu le projet, interprété les données et rédigé le manuscrit. S.M. a effectué la plupart du phénotypage et toutes les analyses de données. M.M. a effectué les essais (trois sites) utilisés pour valider les QTL. M.E. a effectué le phénotypage (NIR) sur les graines des essais utilisés pour valider les QTL. Tous les auteurs ont lu et approuvé le manuscrit final.
Le chapitre 3 a été soumis pour publication dans le journal « BMC Plant Biology » en avril 2020 sous : Malle S., Morrison M. and Belzile F. (2020). Identification of loci controlling mineral element concentration in soybean seeds. S.M. et F.B. ont conçu le projet, interprété les données et rédigé le manuscrit. S.M. a effectué les phénotypages et toutes les analyses de données. M.M. a effectué les essais (trois sites) utilisés pour valider les QTL.
Introduction générale
La population mondiale est en perpétuelle croissance. Estimée à 7,7 milliards de personnes en 2020 (Worldometer 2020) dont la majorité vit dans les régions urbaines, cette population atteindra 9,7 milliards en 2050 (ONU 2020). Tout comme la population mondiale, le climat évolue à un rythme sans précédent (Notre-planete, 2020). Les changements climatiques ont des effets défavorables sur la productivité́ agricole, la production alimentaire et les ressources naturelles. Cela a des répercussions néfastes sur l’existence des populations rurales, sur les systèmes de production, de distribution et de consommation des aliments partout dans le monde occasionnant un problème d’insécurité et de malnutrition sévère (OMS 2019). Actuellement, 1 personne sur 9 souffre de faim et de malnutrition dans le monde dont 515 millions en Asie, 257 millions en Afrique et 39 millions en Amérique latine et dans les Caraïbes (OMS 2019).
Aussi, il faut noter que notre système agricole est conçu dans le but de promouvoir l'augmentation du rendement et de la productivité au détriment de la qualité nutritionnelle (Prat 2017) augmentant ainsi la malnutrition chez les consommateurs (Garg, et al. 2018). On estime que les carences en nutriments (vitamines et minéraux), connues sous le nom de « faim invisible », touchent plus de deux milliards de personnes à travers le monde (OMS 2019). En Afrique, il a été estimé que 30 % des enfants souffrent de malnutrition et d’un retard de croissance, 42 % des femmes âgées de 15 à 49 ans souffrent de carence en vitamine A (Bafan 2014) et 1 femme sur 2 en état de grossesse souffre d’anémie (CTA 2015).
Que faire face à ce défi majeur? Afin d’assurer une sécurité nutritionnelle durable, des solutions alternatives ont été adoptées, notamment l’ajout des suppléments alimentaires dans les rations alimentaires. Hélas, ces ajouts de suppléments ont été très vite délaissés, surtout dans les pays pauvres en raison de leur faible pouvoir d'achat, leur accès limité aux marchés et aux systèmes de soins de santé (Garg, et al. 2018).
En tenant compte des contraintes évoquées, un renforcement des interventions à court et long terme est nécessaire pour améliorer l’état de la sécurité alimentaire et nutritionnelle de la population par la création des variétés qui seront plus productives et riches en nutriment. C’est ainsi que l’approche de « biofortification » ou de « bioenrichissement » (OMS 2019) a
été proposée. Elle consiste à améliorer les cultures vivrières (céréales, légumineuses et à tubercules, etc..) sur le plan nutritionnel avec une biodisponibilité accrue des nutriments à l'aide des techniques biotechnologiques modernes, de sélection variétale (amélioration génétique) et de pratiques agronomiques (Garg, et al. 2018).
Dans cette optique, plusieurs instituts de recherche agricole, projets et programmes de recherche ont été fondés ou initiés. En 2012, le centre de recherche « HarvestPlus » et l’agence des États-Unis pour le développement international (« USAID ») ont lancé une initiative « Feed the Future » aboutissant à l’introduction d’une nouvelle variété de patates douces enrichis en micronutriments et en vitamine A, laquelle a été adoptée par 60 % des producteurs agricoles ougandais (Bafan 2014). De même grâce à des collaborations entre des centres de recherche comme le groupe consultatif pour la recherche agricole internationale (« CGIAR »), le Centre international d’agriculture tropicale (« CIAT »), l’institut international d’agriculture tropicale (« IITA »), et le Centre international de recherche sur le maïs et le blé (« CIMMYT ») financées par la fondation Bill & Melinda Gates et la Banque Mondiale, différentes espèces cultivées ont été biofortifiées. Ce fut le cas du maïs biofortifié en Zambie, du manioc au Nigeria et au Congo ainsi que du riz au Bangladesh et en Inde (Prat 2017). Certains de ces pays ont d’ores et déjà bénéficié de l’introduction de ces nouvelles variétés afin de diversifier leur régime alimentaire. C’est le cas du Rwanda, où environ un demi-million d’agriculteurs cultivent de nouvelles variétés de fèves riches en fer (Bafan 2014).
Ainsi, tout comme certains de ces pays, le Mali est un pays africain dont la population connaît des difficultés alimentaires plus ou moins sévères environ une année sur trois (Martin et Niaméogo, 2005) du fait de la forte sensibilité de l’agriculture aux variations des précipitations qui rend le recours à l’aide alimentaire d’urgence indispensable dans certaines années (FAO 2010). Mais aussi son régime alimentaire reste très peu diversifié et pauvre en certains nutriments essentiels, causant une carence tant en certains acides aminés qu’en éléments minéraux tels que le fer et le calcium, etc. (OMS 2019). De plus, on note une prévalence de la malnutrition chronique élevée (>20 %, dans certaines parties du pays) et une malnutrition aiguë globale au-dessus du seuil d’alerte (>10 %) qui perdure depuis 2013 (Nkunzimana et al., 2019).
Ainsi, c’est dans le but de contribuer à la sécurité alimentaire et nutritionnelle au Mali par la diversification du régime alimentaire et la biofortification des cultures vivrières que la présente thèse a été entreprise. Elle a été financée grâce au projet dénommé Formation Agricole pour la Sécurité Alimentaire au Mali (« FASAM »). Le projet FASAM est le fruit d’un partenariat entre l’Institut polytechnique rural de formation et de recherche appliquée (« IPR/IFRA ») au Mali et l’Université Laval à Québec. Ce partenariat est financé par le gouvernement canadien via Affaires Mondiales Canada.
Pour faire face au défi majeur de cette insécurité alimentaire et nutritionnelle au Mali, l’une des solutions qui s’offrent à nous est la diversification du régime alimentaire par l’introduction de nouvelles variétés ayant une teneur élevée en certains nutriments, mais aussi par l’utilisation de variétés améliorées « biofortifiées » qui seraient dotées d’une teneur accrue en certains éléments nutritifs. A cet effet, l’une des espèces cultivées qui répondent mieux aux besoins alimentaires et nutritionnels du Mali tant en protéines que d’éléments minéraux est le soya.
En effet, le poids sec d’une graine du soya est composé en moyenne 42 % de protéine (Bellaloui, et al. 2011), 19,5 % d’huile (Wilson 2004), 28 % de glucide, mais aussi 4 % par des éléments minéraux (Stowe 2017). Grâce à cette composition, le soya entre dans la composition d'une variété de produits alimentaires comme le tofu, la sauce soya, et divers substituts de viande. Son tourteau constitue un supplément alimentaire très riche en protéines pour le bétail. Dans le secteur industriel, les usages du soya sont divers, allant de la production de levures à la fabrication de savons et de désinfectants (Dorff 2007). À l’échelle mondiale, sa production était estimée à plus de 350 millions de tonnes métriques en 2018-2019 (SOPA 2018-2019) dont 3 millions de tonnes métriques étaient produites en Afrique (World Agricultural Production 2019)
En dépit de ses nombreuses qualités en tant qu’aliment, le soya n’est pas parfait. De nombreuses études ont montré que le soya présente une concentration limitée quant à sa teneur en acides aminés soufrés. En effet, la cystéine (Cys) et la méthionine (Met) ne sont pas présentes à des niveaux adéquats pour une croissance et un développement optimal des animaux monogastriques (<3 % de tous les acides aminés des grains) (Panthee, et al., 2006; Ramamurthy, et al., 2014; Ziegler, et al., 2017). Une autre composante de la graine de soya
non moins importante est sa composition en éléments minéraux importants en tant que nutriments tant pour la santé humaine qu’animale (Ning et al., 2015). De plus, une carence en éléments minéraux essentiels entraîne chez la plante elle-même une perte de rendement, une plus grande sensibilité aux maladies, une altération du métabolisme, une interruption du développement normal et une mauvaise qualité des semences (Glass 1989).
De ce fait, afin de faciliter son introduction dans le régime alimentaire des Maliens, il serait important d’améliorer la teneur du soya en acides aminés soufrés et en certains éléments minéraux par les techniques de biofortification.
Malheureusement, les mécanismes moléculaires contrôlant l'accumulation d’acides aminés soufrés et en éléments minéraux sont des caractères génétiquement complexes faisant intervenir un certain nombre de loci (QTL) et sont souvent peu caractérisés (Ning et al. 2015). Cependant, l’amélioration de la teneur en acides aminés soufrés et en certains éléments minéraux essentiels via la sélection variétale est considérée comme l'approche la plus prometteuse et la plus rentable. Elle consiste donc à faire l'identification des cultivars présentant une variabilité génétique utile pour les caractères agronomiques d’intérêt et, par la suite, à identifier les déterminants génétiques qui sont en partie responsables de la variation pour la concentration de ces caractères dans la graine (Panthee et al., 2004 et Ramamurthy et al., 2014).
L’identification de variétés ayant une teneur accrue nécessite des méthodes et outils permettant de mesurer quantitativement ces différentes composantes au sein de la graine comme la spectrométrie proche infra-rouge (NIR) (pour mesurer la teneur en acides aminés) ou la spectrométrie à fluorescence des rayons X (pour mesurer la teneur en éléments minéraux) (Warrington, et al., 2015 et Chavez, et al., 2019).
Quant à l’identification des déterminants génétiques, elle est basée sur la recherche de marqueurs qui sont étroitement associés à des gènes contrôlant des caractères d’intérêt (Panthee et al., 2006). Il existe quelques méthodes de cartographie génétique permettant l’identification de QTL associés à un caractère agronomique d’intérêt. Il y a d’abord la méthode de cartographie biparentale, laquelle repose sur l’étude de plusieurs descendants issus du croisement des deux parents. Une autre méthode est la méthode d’association
pangénomique (GWAS); celle-ci repose plutôt sur l’étude d’une large collection de lignées non-apparentées (Korte et Farlow 2013). Le GWAS fournit une résolution relativement élevée en termes de définition de la position du QTL au sein du génome (Scherer et Christensen 2016) et c’est cette dernière approche qui sera utilisée dans le cadre de cette thèse.
Dans le cadre de cette thèse, nous avons souhaité déterminer l’assise génétique de composantes clés de la valeur nutritive du soya, soit la teneur en acides aminés soufrés (Cys/Met) et en quatre éléments minéraux majeurs (Ca, K, P et S). Dans les deux cas, des analyses d’association pangénomique ont été réalisées sur une collection de 137 lignées représentatives de la diversité génétique rencontrée au sein du soya canadien à maturité hâtive.
Chapitre 1 : Revue bibliographique Brève description du soya
Le soya cultivé, Glycine max (L.) Merrill, aurait été domestiqué à partir de son parent sauvage
Glycine soja en Chine 1100 ans avant J.C et il se serait répandu dans les pays voisins vers le
1er siècle de notre ère (Kim, et al. 2012). C’est une plante herbacée appartenant à la famille des Fabacées et au genre Glycine, d’où son nom scientifique Glycine max. Le soya mesure de 0,50 à 1,50 m de hauteur, selon les variétés et les conditions culturales, et elle est entièrement hérissée de poils roux. Elle présente de grandes feuilles trilobées et de petites fleurs, de couleur violette ou blanche, groupées en grappes. Les fruits sont des gousses velues, longues de 3 à 8 cm, qui contiennent des graines comestibles (2 à 4) de la taille et forme d'un pois dont le plus couramment cultivé est de couleur jaune (SIA 2017). Le système racinaire, à nodosités, est formé d'une racine pivotante où prend naissance un système de racines latérales.
Les variétés de soya sont classées en groupes de maturité (GM) en fonction de leur réponse à la photopériode. Aujourd'hui, il existe 13 grands groupes allant de GM 000 à GM 10 repartis du nord au sud en Amérique du Nord (Stowe et Dumphy, 2017).
Figure I. 1: Vue d'ensemble d'une plante de soya (source http://www.naturemania.com/bioproduits/soja.html)
Les graines du soya ont une teneur élevée en protéines, ce qui en fait une excellente source de protéines. Il y a majoritairement deux types de protéines de stockage au niveau de la graine de soya, les albumines et globulines (Warrington, et al. 2015). La principale protéine de stockage est la fraction de globulines, laquelle représente environ 70 % de la fraction
protéique au sein des graines (Krishnan et Jez 2018) et, par conséquent, celle-ci fournit la majorité des acides aminés.
Les acides aminés
Les acides aminés sont les unités de base des protéines. Ils sont caractérisés par la présence d’un atome de carbone comme structure de base autour duquel sont reliés une fonction acide (-COOH), une fonction basique amine (-NH2), un atome d'hydrogène (H) et un radical (R). Ce dernier est l’élément variable entre les différents acides aminés qui forment naturellement une protéine (Kamoun, 2020). Les acides aminés sont à l’origine de la synthèse d’une large gamme de composés métaboliques jouant un rôle clé dans les fonctions biologiques (Wu 2009). Parmi ces substances bioactives, nous avons des neurotransmetteurs, des hormones, des gaz de signalisation, des antioxydants et régulateurs du métabolisme, de la croissance, du développement, de la réponse immunitaire et de la santé (Brosnan et Brosnan 2010). Ainsi, sur le plan nutritionnel et selon la capacité de l’organisme (humain ou animal) de les synthétiser, les acides aminés sont classés en deux groupes (MedlinePlus 2020). Nous avons les acides aminés essentiels que l’organisme est incapable de synthétiser (histidine, isoleucine, leucine, lysine, méthionine, phénylalanine, thréonine, tryptophane et valine) et les acides aminés non-essentiels que l’organisme est capable de synthétiser (alanine, arginine, asparagine, acide aspartique, cystéine, acide glutamique, glutamine, glycine, proline, sérine et tyrosine). Parmi ces derniers, certains sont synthétisés en faible quantité surtout en cas de maladie et de stress et nécessite un apport supplémentaire à travers l’alimentation (ex. la cystéine) (MedlinePlus 2020).
Parmi les 20 acides aminés cités ci-dessus, la méthionine et la cystéine contiennent chacune une molécule du soufre et de ce fait sont classés en tant qu’acides aminés soufrés.
1.2.1 La méthionine
La méthionine (Met) est un acide aminé essentiel pour l'humain nécessitant un apport suffisant au travers de l'alimentation ou de compléments alimentaires. Elle est codée sur les ARN messagers par le codon AUG et est caractérisée par la présence d'un atome de soufre engagé dans une fonction thioéther (Wikipédia 2019). La méthionine joue un rôle
particulièrement important dans la biosynthèse des protéines, puisque toutes les chaînes polypeptidiques démarrent par l'incorporation d'une méthionine en position N-terminale. Elle est rencontrée sous deux formes : la forme L- ou S-méthionine et la forme R- ou D-méthionine fabriquée de façon synthétique (Ambrogelly 2007). Sa formule chimique est C5H11NO2S avec une masse molaire de 149,21g/mol. En plus de son rôle clé dans l’initiation de la traduction de l’ARN messager en protéine, la méthionine intervient dans le développement et la différenciation cellulaire, la synthèse des neurotransmetteurs, l’équilibre nerveux et se veut essentielle pour le maintien de la santé des phanères (peau, cheveux, ongles et poils, etc…). La méthionine est également impliquée dans la lutte contre le vieillissement, la neutralisation des métaux lourds comme le mercure et le plomb (Sante Science 2019) ainsi que dans la prévention des dépôts de matière grasse au niveau du foie.
1.2.2 La cystéine
La cystéine (Cys) est un acide aminé considéré comme non-essentiel pour l'homme mais elle peut être produite en quantité insuffisante par l'organisme selon l'état de santé des individus notamment dans le cas de certaines maladies métaboliques et de syndromes de malabsorption ainsi que chez les enfants et les personnes âgées (Wikipédia 2019). Elle est codée par les codons UGU et UGC. De formule chimique C3H7NO2S, la cystéine intervient dans la synthèse de la mélanine, le pigment naturel de la peau et des cheveux. Elle intervient aussi dans la synthèse du coenzyme A qui joue un grand rôle dans le métabolisme chez l’humain notamment dans le cycle de Krebs aboutissant à la production d’énergie sous forme d’ATP. Elle est également impliquée dans certains processus de détoxication naturelle de l'organisme par la chélation de métaux lourds (Moenne 2001). La cystéine jouerait un rôle bénéfique sur l’hypertension artérielle en favorisant une baisse de la pression sanguine (Sante Science 2019).
En plus de leur richesse en composés protéiques, les graines du soya contiennent également des éléments minéraux importants dans la santé humaine et animale.
Les éléments minéraux
Les éléments minéraux jouent un rôle fondamental dans les fonctions biochimiques et physiologiques des systèmes biologiques. Les humains et les autres animaux dépendent des
espèces végétales pour leur fournir des minéraux alimentaires. De ce fait, les plantes peuvent contenir une large gamme d'éléments minéraux, mais les concentrations dans une même plante varient en fonction des espèces, du génotype et des contraintes environnementales (Grusak et Cakmak 2004). Il y a 16 éléments minéraux jugés essentiels pour l'homme. Il s'agit notamment des macronutriments N, S, K, Ca, P, Cl, Na et Mg, ainsi que des micronutriments Fe, Zn, Mn, Cu, Mo, Cr, I et Se (Board 1989). Parmi les macronutriments, seuls Ca, K, P et S sont concernés par les travaux réalisés dans cette thèse. Ces quatre éléments minéraux essentiels sont aussi essentiels pour la plante qui les obtient à partir du sol à travers des mécanismes de transport complexe.
1.3.1 Le calcium
Le calcium est un nutriment essentiel nécessaire à de nombreuses fonctions en santé humaine comme la contraction vasculaire, la vasodilatation, les fonctions musculaires, la transmission nerveuse, la signalisation intracellulaire et la sécrétion hormonale (Beto 2015). Il constitue le minéral le plus abondant dans le corps et 99 % du Ca est présent dans les dents et les os. Chez la plante, le calcium est impliqué dans la division et l'élongation cellulaires et joue un rôle majeur dans le maintien de l'intégrité de la membrane cellulaire (Fageria 2009). De plus, le calcium joue un rôle important dans le maintien de l'équilibre nutritif dans les tissus végétaux et intervient dans la prévention contre la toxicité des métaux lourds.
1.3.2 Potassium
Le potassium est le troisième minéral le plus abondant dans le corps. Il aide le corps à réguler le fluide, à envoyer des signaux nerveux et à réguler les contractions musculaires. Environ 80 % du potassium dans le corps se trouve dans les cellules musculaires (Cheng, et al. 2014). Le potassium joue un rôle important dans la croissance et le développement des plantes, y compris le système racinaire, l’activation d’enzymes et la synthèse des protéines (Blevins 1994). Une carence en K entraîne une diminution du taux net de photosynthèse (Zhao, et al. 2001) et une baisse spectaculaire du rendement des cultures (Ding, et al.2006).
1.3.3 Phosphore
Le phosphore est un composant important parmi les minéraux osseux, il représente le deuxième minéral le plus abondant dans le corps humain. Environ 85 % du phosphore dans le corps d’un adulte se trouve dans les os. Le phosphore intervient dans le stockage et transfert d’énergie nécessaire aux processus métaboliques (Marcus, et al. 2013). Une carence en phosphore peut entraîner des douleurs osseuses et une faiblesse musculaire. Pris principalement sous forme de phosphate par la plante, le phosphore est impliqué dans la photosynthèse, le transfert d'énergie, la division cellulaire, la croissance des racines. De plus, il améliore la qualité des fruits et légumes, est essentiel à la formation des graines et aide à accélérer la maturité (Nutri-Facts, 2020).
1.3.4 Soufre
Le soufre est considéré comme le quatrième minéral le plus abondant dans le corps humain (Wikipédia 2019) après le calcium, le phosphore et le potassium, constituant près de 0,25 % du poids du corps. Le soufre est un macronutriment essentiel nécessaire à la croissance et au développement de tous les organismes vivants. Il est présent dans une grande variété de métabolites importants pour le maintien de la structure cellulaire et des activités biologiques. Le soufre inorganique dans l'environnement, l'ion sulfate dans le sol et le dioxyde de soufre gazeux dans l'air, sont fixés et incorporés dans la cystéine par la voie d'assimilation du soufre chez les plantes. Par la suite, la cystéine est convertie en méthionine. Cependant, les animaux n'ont pas de mécanisme d'assimilation du soufre inorganique; ils l’utilisent sous forme de méthionine via leur alimentation (Takahashi, et al. 2011).
Comme précédemment mis en évidence, les humains ainsi que les animaux ont besoin d’un apport adéquat de ces nutriments essentiels pour assurer leur développement. Par conséquent, un apport pauvre en nutriments peut occasionner une mauvaise santé, des maladies, une morbidité une invalidité accrue et un développement altéré (Chizuru, et al. 2003). Diverses approches ont été développées afin de favoriser la biofortification des produits agricoles qui constituent la base de notre alimentation. La biofortification des nutriments essentiels dans les plantes cultivées par amélioration génétique est considérée comme l’approche la plus
rentable et stable (Garg, et al. 2018) permettant d’améliorer la teneur nutritionnelle des produits agricoles.
Amélioration génétique
Durant la dernière décennie, près de 50 % de l'augmentation du rendement en grains ont été attribués à l'amélioration génétique (Singh et al. 2016). Cette approche nécessite l’acquisition d’une connaissance intime des assises génétiques et moléculaires des caractères qu’on veut améliorer. Elle nécessite l’utilisation des outils moléculaires permettant d’optimiser la justesse de la sélection. Chez le soya, la majeure partie des caractères agronomiques sont considérés complexes en raison du contrôle d’un certain nombre de gènes ou des loci de caractère quantitatif (« en anglais : Quantitatif trait loci (QTL)») mais aussi parce qu’ils sont fortement influencés par l'environnement rendant difficile leur amélioration (Tripathi et Khare 2016).
Parmi les outils moléculaires, nous avons la sélection assistée par marqueurs moléculaires (SAM) (Tripathi et Khare 2016). La sélection assistée par marqueurs moléculaires est une méthode qui est basée en premier temps sur la recherche de marqueurs qui sont étroitement associés à des QTL contrôlant les caractères d’intérêt. Individuellement, ces QTL sont généralement responsables d’une faible partie de la variation phénotypique observée pour les caractères agronomiques recherchés (teneur en acides aminés soufrés, en éléments minéraux, en protéine, en huile, rendement, etc.…). Une fois identifiés, les marqueurs génétiques associés à ces QTL permettront de réaliser une sélection en faveur du caractère souhaité.
Cependant, il existe quelques méthodes principales permettant l’identification des QTL associés à un caractère agronomique recherché. Nous avons d’abord la méthode de cartographie biparentale qui repose sur l’étude de plusieurs descendants issus du croisement de deux parents (Xiaopeng, et al. 2016). Une autre méthode, très répandue et offrant une très grande résolution quant à l’identification des QTL, est la méthode d’association pangénomique (« Genome-Wide Association Study (GWAS) »), laquelle repose plutôt sur l’étude d’une large collection de lignées non apparentées (Scherer et Christensen 2016) Qu’un QTL soit identifié par la méthode de cartographie biparentale ou par GWAS, l’intérêt qu’on lui donne est corrélé avec la proportion de la variation phénotypique qu’il contrôle
mais surtout avec sa stabilité (reproductibilité à travers différents environnements). D’une façon générale, pour pouvoir être utilisé dans un programme de sélection assisté par marqueur moléculaire, un QTL à besoin d’être validé travers différents environnements (Orazaly, et al. 2018).
1.4.1 Les marqueurs moléculaires
Les marqueurs moléculaires sont des marqueurs génétiques composés de fragments d'ADN qui servent de repères pour suivre la transmission d'un segment de chromosome d'une génération à l'autre. En fait, de nouvelles mutations sont introduites dans le génome à chaque génération qui passe (Scherer et Christensen 2016). Au fil du temps, ces menues différences s’accumulent et font en sorte qu’on arrive à distinguer les différents individus/variétés au sein d’une espèce et à suivre la transmission de segments chromosomiques hérités d’un ancêtre commun. Ces mutations courantes sont généralement appelées variants ou polymorphismes (Scherer et Christensen 2016) et peuvent survenir suite à un changement d’une paire de bases, du réarrangement d’un segment d’ADN (translocation/inversion), d’une insertion/délétion ou simplement d’une variation du nombre de copies d’une région particulière. Ces polymorphismes sont donc des indicateurs de la variabilité génétique au sein d’une famille, genre, espèce, variété, population et même individu (Wikipédia 2019). Depuis leur première utilisation chez l’homme par Botstein et al. (1980), des progrès substantiels ont été réalisés dans le développement et l'amélioration des techniques moléculaires permettant la découverte de marqueurs d'intérêt à grande échelle, ainsi que leur utilisation répandue en génétique humaine, animale et végétale (Jiang 2013). Parmi les techniques qui ont été largement utilisées dans les deux dernières décennies, surtout en matière de sélection végétale, figurent le polymorphisme de longueur des fragments de restriction (abrégé RFLP en anglais), le polymorphisme de longueur des fragments amplifiés (AFLP), l'ADN polymorphe amplifié au hasard (RAPD), les microsatellites ou les séquences simples répétées (SSR) et le polymorphisme mononucléotidique (SNP).
De nos jours, grâce aux avancées des nouvelles technologies de génotypage à haut débit, les polymorphismes RFLP, AFLP, RAPD et SSR ont progressivement été abandonnés au profit des SNP (Boudhrioua 2019). Les SNP constituent la plus simple forme des marqueurs moléculaires, car ils résultent du changement d’une seule base nucléotidique dans la séquence
d’ADN. Ils sont relativement faciles à découvrir, très répandus sur le génome et bi-alléliques. De plus, les SNP peuvent se présenter à l'intérieur de séquences codantes des gènes, dans des régions non codantes des gènes ou dans les régions inter-géniques entre gènes à différentes fréquences dans différentes régions chromosomiques (Jiang 2013).
1.4.1.1 Technologies de séquençage de nouvelle génération
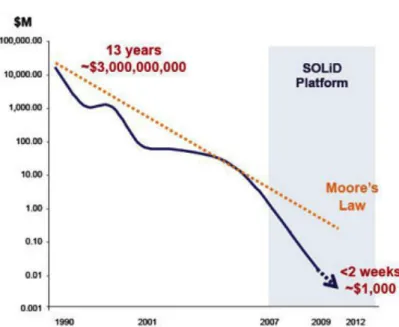
Le séquençage de l'ADN consiste à déterminer l'ordre d'enchaînement des nucléotides dans un fragment d’ADN donné (Wikipédia 2019). Les technologies de séquençage de nouvelle génération (« next-generation sequencing (NGS) ») ont révolutionné les sciences biologiques grâce à leur débit, leur évolutivité et leur vitesse ultra-élevée permettant aux chercheurs d'effectuer une grande variété d'applications et d'étudier des systèmes biologiques à un niveau jamais atteint auparavant (Illumina 2020). De nos jours, grâce aux avancées technologiques et de la biologie moléculaire, plusieurs plateformes de séquençage ont été développées (Figure I.2). Cela a joué un rôle considérable dans la diminution des coûts de séquençage de génomes entiers (« whole-genome sequencing (WGS) »), lequel a été réalisé chez un grand nombre d’eucaryotes, procaryotes et de virus. À titre de comparaison, le premier séquençage complet d’un génome humain s’est achevé en 2003 et a nécessité une dizaine d'années de travail, avec un investissement total de 2,7 milliards de dollars (Léchenet 2014). Par contre en 2012, il était déjà possible de le faire au bout de deux semaines pour moins de 1000 dollars (Figure I.3). Selon la prédiction, le coût de séquençage pourrait descendre jusqu’a jusqu’à 100 dollars d’ici 2023 (LaFrançaise 2018).
La possibilité de pouvoir séquencer à l’échelle du génome et à faible coût facilite non seulement la détermination de l’enchainement des nucléotides avec précision (assemblage de novo) mais aussi elle permet l’obtention d’une forte densité de marqueur couvrant l’ensemble du génome et, par conséquent, d’augmenter la chance d’identifier des marqueurs qui sont étroitement liés à un gène causal lors d’une analyse d’association. De plus, les données moléculaires issues du WGS permettent de capturer les grands et petits variants, très utiles pour des études de suivi de l'expression des gènes (études fonctionnelles) (Torkamaneh et al. 2015) et des mécanismes de régulation.
Chez le soya, le premier génome séquencé au complet a été celui de la variété Williams 82 (Wm82), une variété américaine (Schmutz, et al. 2010). Ce génome constituait le premier génome de référence chez le soya. Le génome du soya a ainsi été estimé à 1,1 gigabases et compte plus de 50 000 gènes codant pour des protéines dont plus de 75 % sont présents en plus d’une copie.
Quoique très utile, le WGS demeure cher pour une utilisation à grande échelle (p. ex. : séquençage de milliers d’individus). En revanche, pour contourner cet obstacle, une approche très avantageuse a été proposée par Torkamaneh, et al. (2018) basée en deux étapes principales. La première consiste à génotyper une plus grande population à l’aide des technologies de génotypage moins coûteuses (p. ex. le génotypage-par-séquençage « GBS ») et générer des milliers de SNP. Dans la seconde étape, un sous ensemble représentatif de cette grande population est choisi et séquencé par l’approche du WGS afin d’identifier des millions de SNP. Ces derniers individus complètement séquencés serviront comme panel de référence pour imputer le catalogue de SNP issus du GBS. Dans la présente thèse, cette approche combinant le WGS et GBS a été utilisée pour générer des millions de marqueurs SNP répartis sur l’ensemble du génome.
Tenant compte du coût pour séquencer une large population, différentes approches de génotypage à haut débit pour détecter des milliers de marqueurs moléculaires de type SNP ont vu le jour.
Figure I. 2: Différentes plateformes de nouvelle technologie de séquençage à haut débit
1.4.1.2 Technologies de génotypage à haut débit des SNP
Le génotypage est « la discipline qui vise à déterminer l'identité d'une variation génétique, à une position spécifique sur tout ou partie du génome, pour un individu ou un groupe d'individus donné » (Wikipédia 2019). Grâce aux progrès en biologie moléculaire, en matière de technologies de séquençage et en bio-informatique, le génotypage a beaucoup gagné en vitesse, tout en devenant moins coûteux. Cette diminution progressive du coût de séquençage a favorisé le développement des méthodes et outils de génotypage pour la détection des polymorphismes SNP. Ces méthodes vont de la détection des SNP déjà connus (la puce d’ADN) au séquençage partiel du génome par réduction de complexité à l’aide des enzymes de restriction. Bien qu’il existe actuellement un certain nombre de techniques qui sont utilisées selon les besoins de l’étude, nous pouvons citer la puce d’ADN « SoySNP50K » pour identifier les SNP déjà connus chez le soya (Song, et al. 2013). Pour les méthodes qui favorisent l’échantillonnage du génome, nous avons la réduction de la complexité des séquences polymorphes « CRoPS », le séquençage d'ADN lié aux sites de restriction « RAD-Seq » et l’approche GBS. Cette dernière est la plus prometteuse et la plus utilisée surtout dans le domaine de la recherche chez les plantes (Kumar, et al. 2012).
§ Le génotypage-par-séquençage
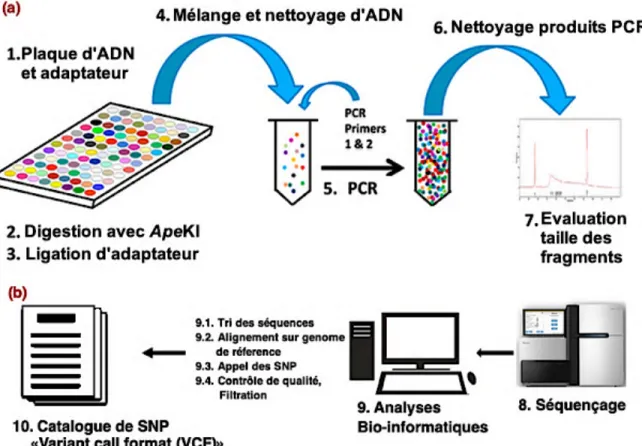
Le GBS est une procédure robuste, simple et peu coûteuse pour la découverte et le génotypage simultané des SNP. Développée par Elshire et al. (2011), cette approche réduit la complexité du génome en capturant de manière reproductible une fraction du génome à l’aide d’enzymes de restriction (ER).
Le principe du GBS peut être résumé comme suit (Figure I.4): (i) l’ADN de haut poids moléculaire est extrait et digéré en utilisant une ER spécifique préalablement définie ; (ii) des adaptateurs dotés de codes à barres sont ensuite ligaturés aux extrémités collantes de chaque individu ; et (iii) les différents échantillons sont alors combinés en une seule librairie, (iv) une amplification PCR est effectuée. Par la suite, (v) ces librairies sont envoyées à séquencer pour obtenir des lectures (« reads ») de 100 à 200 paires de base (pb), selon l’appareil employé.
Cependant, après le séquençage de la librairie GBS, la réalisation de l’analyse bio-informatique des séquences constitue un véritable défi dont l’aboutissement permet d’obtenir des SNP de bonne qualité dépendamment de la nature de l’étude. A cet effet, le laboratoire de Dr François Belzile à l’Université Laval s’est spécialisé dans les analyses bio-informatiques à travers la mise en place de pipelines d’analyse bio-informatique comme « Fast-GBS » (Torkamaneh, et al. 2017). Ainsi, il est possible d'appeler rapidement des SNP connus dans des échantillons nouvellement séquencés.
Figure I. 4: Procédure de détection des marqueurs SNP par une approche GBS.
(a) préparation de la librairie GBS (1,2,3,4,5,6, et 7 (Elshire, et al. 2011)) et analyses bio-informatiques (8, 9 et 10).
Grâce à l'avènement du séquençage de nouvelle génération, des technologies de génotypage, des outils d’analyse bio-informatique ainsi que des bases de données génétiques accessibles au grand public, d’énormes progrès ont été effectués dans l’amélioration génétique des plantes tant sur le plan du rendement, de la résistance aux maladies, de la qualité nutritionnelle (biofortification) permettant d’améliorer autant la santé humaine qu’animale. Parmi ces méthodes, nous avons la cartographie génétique.
1.4.2 La cartographie génétique
Globalement, la cartographie génétique est la détermination de la position d’un locus (gène ou marqueur génétique) sur un chromosome. Ces gènes sont très souvent en partie responsables de la variation phénotypique pour un caractère d’intérêt. En effet, dépendamment de la nature génétique du caractère d’intérêt, il pourrait être contrôlé un seul gène (monogénique) dans le cas de caractères qualitatifs comme la couleur des fleurs ou des graines. Par contre, la plupart des caractères agronomiques d’intérêt économique sont contrôlés par un grand nombre de gènes répartis sur plusieurs chromosomes du génome et sont donc appelés des caractères quantitatifs (« quantitative trait locus » ou QTL). Ce n'est pas une tâche facile d’identifier ces derniers et, par conséquent, cela nécessite des approches de cartographie fiables. Les approches de cartographie génétique peuvent être résumées en deux types selon le matériel génétique utilisé. (i) la cartographie biparentale nécessitant des d'individus issus du croisement de deux parents et (ii) la méthode d’association pangénomique faisant intervenir des individus non apparentés par un croisement récent.
1.4.2.1 Cartographie biparentale
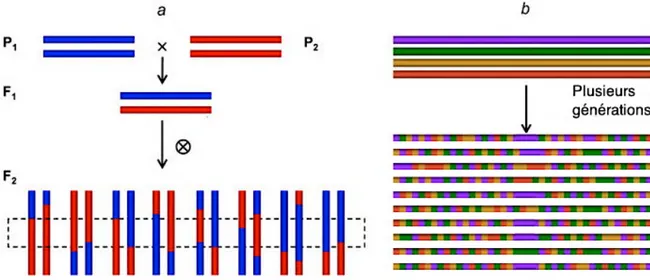
La cartographie biparentale aussi connue sous le nom de cartographie de liaison génétique nécessite la production d’une population en ségrégation, laquelle est généralement obtenue en croisant deux parents présentant des différences phénotypiques significatives pour au moins un caractère d'intérêt. À cet égard, différentes populations de cartographie peuvent être utilisées comme la seconde génération filiale (F2), des lignées haploïdes doublées « double haploide (DH) », des lignées consanguines recombinantes (« recombinant inbred line » ou RIL), des lignées isogéniques « near isogenic lines » ou NIL) ainsi que des lignées rétrocroisées (« backcross » ou BC) (Meksem et Kahl 2005).
L’approche de cartographie biparentale nécessite la création d'une carte génétique rassemblant les informations génétiques de deux parents différents (Iquira 2014) Chez le soya, la première carte de liaison génétique moléculaire a été rapportée en 1990 (Keimp, et al. 1990) utilisant une population F2 avec 60 descendants issus d'un croisement de A81-356022 (G. max) × PI468916 (G. soja) (Song, et al. 2016). Le fondement de la cartographie QTL est basée sur le principe selon lequel si un gène particulier est très proche d'un marqueur moléculaire, le gène et le marqueur resteront probablement ensemble pendant le processus
de recombinaison (méiose) et ils seront probablement transmis ensemble du parent aux descendants. Ainsi, si un gène est en partie responsable de la variation d’un caractère phénotypique, l’identification de ce marqueur permettra d’affirmer la présence potentielle du gène chez l’individu donné (Mammadov, et al., 2012).
Dans les deux dernières decennie, la méthode de cartographie biparentale à été utilisée pour identifier des QTL associés à une très large gamme de caractères d’intérêt. Parmi les caractères qui sont en lien avec le sujet de cette thèse, nous pouvons souligner que de telles études ont été réalisées sur la teneur en acides aminés soufrés (cystéine et méthionine) chez des lignées de soya de diverses origines (Panthee, et al., 2006; Ramamurthy, et al., 2014; Warrington, et al., 2015) ou pour identifier des QTL associés à la teneur en éléments minéraux (Zhang, et al., 2009 ; Bellaloui, et al., 2011 ; King, et al., 2013 et Hacisalihoglu et al., 2018).
Malgré ses nombreuses contributions à l’acquisition de connaisssances sur l’architecture génétique des caractères d’intérêt, la cartographie biparentale présente toujours certains défis et lacunes. Elle necessite plusieurs années de travail pour obtenir une population de cartographie (Figure I.5a). Les QTL identifiés à l'issue de ces méthodes ne sont valables que pour cette population, car la diversité allélique (une différence dans la chaîne d’acides nucléiques constituant le gène) observée se limite à celle des deux parents. Finalement, elle offre une faible résolution pour identifier d'éventuels gènes candidats, car les régions génomiques dans lesquelles se trouvent les QTL sont généralement très grandes (Figure I.5a). En revanche, la méthode d’association pangénomique « Genome wide association study (GWAS) » représente une approche passionnante et prometteuse pour surmonter aux problèmes cités ci-dessus au sein d’une population d’individu non apparentée (Figure I.5b).
Figure I. 5: Illustration de deux méthodes d'association. Réadapté selon (Zhu, et al. 2008).
1.4.2.2. Méthode d’association pangénomique
Initialement développée pour étudier le génome humain (Scherer et Christensen 2016), les études GWAS peuvent être conçues pour évaluer les déterminants génétiques de presque tous les caractères qualitatifs ou quantitatifs (Gurgul, et al. 2014). Alors que de plus en plus de chercheurs se lancent dans ce domaine, nous aimerions donner un bref aperçu des concepts clés qui sous-tendent le GWAS.
Le GWAS consiste à évaluer statistiquement l'association entre chaque marqueur génotypé (le plus souvent des SNP) et la variation d’un phénotype d'intérêt (Korte et Farlow 2013). Contrairement à la méthode biparentale où la diversité allélique se limitait à celle présente au sein des deux parents, le GWAS bénéficie d’une diversité allélique plus large grâce à la diversité génétique naturelle au sein de la population d’étude. Il ne nécessite pas un croisement et offre également une plus grande résolution pour la détection des QTL grâce aux évènements historiques de recombinaison entre les individus de la population d’étude (Rafalski 2010). Le succès d’une analyse GWAS repose sur un certain nombre de facteurs décrits comme suit.
1.4.2.2.1. Déséquilibre de liaison
Le déséquilibre de liaison « linkage disequilibrium (LD) » est une propriété des SNP qui décrit le degré avec lequel un allèle d'un SNP est hérité ou corrélé avec un allèle d'un autre
SNP au sein d'une population (Alqudah, et al. 2020). La puissance et la précision du GWAS dans la détection des QTL reposent sur ce concept de LD. En effet, les évènements historiques de recombinaison occasionnent une diminution rapide du LD au sein de la population. En contrepartie, dans une population de GWAS, la décroissance rapide du LD nécessite l'utilisation d'un grand nombre de marqueurs pour pouvoir couvrir le génome entier à une densité suffisante (Myles, et al. 2009). Le LD s’étend sur des longues distances chez les individus issus d’un croisement biparental par rapport aux individus non apparentés (Zhu, et al. 2008). De plus, le LD varie selon les régions génomiques surtout dans les régions péricentromériques qui sont pauvres en gènes et en évènements de recombinaison (Lee, et al. 2013). Également, le LD a tendance à être plus élevé entre les allèles des loci qui sont situés à proximité les uns des autres et inversement si les loci ont éloignés à cause des échanges de fragments chromosomiques lors de la méiose.
Ainsi, dépendamment de la population à l’étude, le calcul de LD au début de l'analyse d'association est essentiel. Le r2 et le D’sont des métriques utilisées comme coefficients du LD pour mesurer la corrélation entre les allèles entre deux loci suite aux évènements historiques de mutation et de la recombinaison (Flint-Garcia, et al. 2003).
Ce LD peut être conservé à travers les différents évènements de recombinaison entre les allèles d’un sous-ensemble de marqueurs sur le chromosome créant ce qu’on appelle un haplotype. La conservation du LD est un facteur très important pour un GWAS puisqu’il nous renseigne sur le degré de liaison entre les marqueurs d’une région génomique sur un chromosome. Le variant génétique (SNP ou variant structural) qui cause la variation d’un caractère particulier peut ne pas être directement testé, car non génotypé dans le GWAS, mais du fait de sa liaison conservée avec un autre marqueur, sa signature peut toujours être évidente par l'association à ce SNP (Scherer et Christensen 2016) ; on parle alors d’association indirecte.
1.4.2.2.2. La variation phénotypique
La réussite d’une étude de GWAS dépend aussi d’un phénotypage aussi précis que possible du caractère d'intérêt ainsi que l’intensité du phénotypage (taille de la population) (Würschum 2012). D'autre part, une variation phénotypique significative et une héritabilité
au sens large modérée à élevée est cruciale dans une étude d’association. En effet, le niveau de l’héritabilité au sens large estimé à partir de la variance phénotypique nous renseigne sur la part de contribution de la variance génétique au phénotype.
Un autre facteur limitant la puissance de détection du GWAS est l’erreur issue des répétitions des essais et l’effet de l’environnement (lorsque plusieurs sites sont inclus). Différentes méthodes ont été proposées pour réduire ces effets comme la méthode de meilleur prédicteur linéaire sans biais « best linear unbiased predictor (BLUP) » ou le meilleur estimateur linéaire sans biais « best linear unbiased estimator (BLEU) » (Alqudah, et al. 2020).
1.4.2.2.3. Structure de population et relations familiales
Bien que les études GWAS aient le potentiel de détecter les polymorphismes génétiques sous-jacents aux caractères importants, les faux positifs sont une préoccupation majeure et peuvent être partiellement attribués à de fausses associations causées par la structure de la population ou relations familiales (« kinship ») entre les individus d'une cohorte donnée (Zhang, et al. 2010). La structure de la population est due à la présence de deux ou plusieurs sous-populations principales, tandis que la structure familiale fait référence à différents niveaux de parenté entre les individus (Würschum 2012). Actuellement, différents outils ont été développés comme SPAGeDi (Hardy et Vekemans 2002) pour l’analyse du kinship et FastSTRUCTURE (Raj et al. 2014) pour l’analyse de la structure de population. Ces outils permettent de générer une matrice de parenté (matrice K) et de structure (matrice Q). Par la suite, ces matrices sont incorporées dans les modèles statistiques d’association pour ainsi réduire le risque de déclarer des fausses associations.
1.4.2.2.4. Méthodes d’analyse statistique
Dans une situation idéale, des analyses statistiques « classiques » (régression linéaire, l'analyse de variance (ANOVA), le test de t ou le test du chi carré) suffiraient à mesurer le degré d’association entre chaque marqueur et le phénotype. Cependant, comme la structure de la population ainsi que le kinship peuvent occasionner des fausses associations génotype-phénotype, différentes approches statistiques ont été conçues pour traiter ces facteurs limitants dans une étude d’association pangénomique (Zhu, et al. 2008). Ces approches peuvent être groupées en deux grandes catégories selon la méthode analytique. Ces approches