HAL Id: pastel-00002160
https://pastel.archives-ouvertes.fr/pastel-00002160
Submitted on 27 Feb 2007
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Utilisation de l’électrophorèse capillaire (EC) pour la
caractérisation des liquides ioniques (LI) et intérêt des
LI comme nouveaux milieux de séparation en EC
Yannis Francois
To cite this version:
Yannis Francois. Utilisation de l’électrophorèse capillaire (EC) pour la caractérisation des liquides ioniques (LI) et intérêt des LI comme nouveaux milieux de séparation en EC. Chemical Sciences. Chimie ParisTech, 2006. English. �pastel-00002160�
Thèse de doctorat
de l’université Pierre et Marie Curie (Paris VI)
École doctorale de Chimie Physique et Chimie Analytique de Paris-Centre
Spécialité : Chimie Analytique
Présentée par Yannis FRANCOIS
Pour obtenir le grade de docteur de l’université Paris VI
Utilisation de l’électrophorèse capillaire (EC)
pour la caractérisation des liquides ioniques
(LI) et intérêt des LI comme nouveaux milieux
de séparation en EC
Soutenue le 17 novembre 2006,
devant le jury composé de
Mme Valérie CABUIL, Présidente
Mr Paul-Louis DESBENE, Rapporteur
Mr Jacques CROMMEN, Rapporteur
Mr Didier VILLEMIN
Mlle Anne VARENNE Codirectrice de thèse
Mr Pierre GAREIL, Directeur de thèse
Aucune expérience humaine n’est dénuée de sens ou indigne d’analyse
Primo Levi
Extrait de Si c’est un homme
Remerciements
Ce travail a été réalisé au Laboratoire d’Electrochimie et Chimie Analytique (CNRS-UMR 7575) dirigé par le Docteur Daniel LINCOT. Je lui suis très reconnaissant pour son accueil au sein de ce laboratoire.
Je tiens particulièrement à remercier le Professeur Pierre GAREIL pour m’avoir accueilli au sein de son équipe. Je le remercie pour sa patience, sa compréhension, sa grande disponibilité et son aide indispensable qui ont rendu ce travail de thèse très enrichissant sur le plan scientifique mais aussi sur le plan humain. Je lui exprime également toute ma gratitude pour m’avoir fait confiance en tant que moniteur dans l’enseignement de la formation continue, en tant que conférencier dans des événements scientifiques et en tant qu’encadrant de stage.
Ce paragraphe pourrait être copié-collé et adressé au Docteur Anne VARENNE tant tous ces remerciements sont partagés entre mes deux co-encadrants. Je remercie donc dans les mêmes termes le Docteur Anne VARENNE pour qui j’ai une grande amitié. Sa bonne humeur constante, sa grande disponibilité, sa façon bien à elle de faire avancer rapidement les choses en douceur et sa très grande (voir immense…) pédagogie m’ont permis de prendre confiance en moi et donc de faire de ce travail de thèse un vrai apprentissage du monde de la recherche et de l’enseignement.
Je tiens à remercier le Professeur Valérie CABUIL pour m’avoir fait l’honneur de présider mon jury de thèse, le Professeur Paul-Louis DESBENE et Jacques CROMMEN pour avoir accepté de juger ce travail et d’en être les rapporteurs auprès de l’Université Paris 6. Merci pour l’intérêt que vous avez porté à ce manuscrit et pour le temps que vous y avez consacré. Je remercie également le Professeur Didier VILLEMIN pour avoir fait partie de mon jury de thèse en tant qu’examinateur et surtout pour la collaboration très importante que nous avons eue.
Au niveau des collaborations, je tiens tout d’abord à remercier les Docteurs Patrice CHIAP et Anne-Catherine SERVAIS de l’Université de Liège pour m’avoir permis de mieux appréhender le monde de la chimiométrie. Je tiens aussi à remercier les docteurs Laurent GAILLON et Juliette SIRIEIX pour les synthèses des différents liquides à longue chaîne.
Enfin, un grand merci aux Docteurs Christian GIRARD, Marie-Noëlle RAGER et Béatrice ALLARD pour l’aide matérielle indispensable qu’ils ont mis à ma disposition durant ces trois années de thèse.
Au niveau de l’UMR 7575, je remercie l’ensemble des personnels du laboratoire avec qui je me suis très bien entendu, et plus particulièrement les « gens du dessous », c'est-à-dire Elisabeth BROCHET, Agnès PAILLOUX, Armelle RINGUEDE, Michel CASSIR, Virginie LAIR, Cyrine BRAHIM, Valérie ALBIN et Sophie GRIVEAU (pour moi tu es toujours au LECA…).
Changeons de LECA et d’école, direction l’ESPCI. Un grand merci aux différents membres du LECA de PC avec qui les discussions scientifiques et personnelles ont toujours été très agréables. Et puis bien sûr, un grand merci à Stéphanie DESCROIX qui m’a beaucoup aidé grâce à sa gentillesse et sa bonne humeur lorsque je suis arrivé à l’ENSCP.
Un grand merci à l’ensemble des personnels de l’école avec qui j’ai eu des rapports cordiaux et amicaux, plus particulièrement Gilles le Squasheur fou, Ali l’homme aux clés et Yougo le beau gosse du réseau. J’ai aussi une très forte pensée pour Alain BOYER qui est parti trop tôt et qui aurait dû être là le soir de ma soutenance. Sans lui, il manque quelque chose à l’ENSCP.
Passons maintenant aux collègues les plus proches, c'est-à-dire les thèsards du laboratoire. Sachez que sans vous, cette thèse n’aurait pas eu le même goût. D’abord le marseillais, la roussette, électrode man… Définitivement, plein de surnoms vont à Jean ROUSSET. Nazaré la Portugaise du Japon, celle qui a des cheveux en paille (toi-même tu sais…) et Denis GUIMARD, l’homme qui sait toujours applaudir au bon moment (respect…). Thomas le SAUX, l’homme que l’on nomme le chercheur, t’es mon maître en électrophorèse capillaire. Fanny d’ORLYE, madame « j’suis jamais contente de ce que je fais » mais qui au fond va tout déchirer parce qu’elle le vaut bien. Laurent BENZERARA, alias « papa », la force tranquille. Et bien sûr, celle venue de cette belle province du Maroc qu’on appelle la Tunisie et surtout celle qui a subi les pires moqueries de ma part tout au long de ces trois années sans jamais le prendre mal, évidemment Meriem MOKKADEM. Pour finir, de nombreux stagiaires (Pierre, Wen, Estelle, Julie entre autres) ont aussi fait partie de cette belle aventure et chacun a eu une grande importance pour moi.
Avant de passer à la famille et aux amis, je tiens à remercier trois professeurs qui ont marqué ma scolarité. Tout d’abord Mme France HARMAND, institutrice de CE2, pour qui j’ai toujours eu une petite pensée affectueuse et qui m’a certainement donné goût à l’école. Mme
COHEN TANNOUDJII, professeur de chimie de 3ème, qui m’a définitivement donné le goût
des sciences et plus particulièrement de la chimie. M. Jan DUSSOL, professeur de violon, qui m’a énormément appris sur la musique, l’art, mais aussi la rigueur et la patience.
Une page de remerciement ne serait pas complète sans les remerciements à la famille. Evidemment je remercie mes parents pour tout ce qu’ils ont fait pour moi, pour m’avoir aidé et soutenu dans tous les moments de doutes de ma scolarité. Ma sœur et mon beauf qui m’ont donné les deux plus beaux neveux du monde. Mes grands-parents qui ont toujours été très important pour moi et bien sûr tous les oncles et tantes, cousins et cousines. Une petite spéciale pour Judith et Thibaut qui m’ont bien suivi pendant ma thèse, pour l’une parce que je suis un exemple pour elle, et pour l’autre, parce qu’il vit la même chose mais en physique théorique (courage…).
Enfin, pour finir cette page de remerciements, il me reste à remercier tous mes amis. Tout d’abord Jean-Marc BUSNEL et son doudou. Je pense que c’est toi la personne la plus importante de tout ce travail (après ma femme, bien sûr…) parce que sans toutes les phö que tu m’as fait manger, j’aurais jamais eu assez d’énergie pour finir… Hormis ce coté gastronomique que nous avons en commun, maîtrise, DEA et thèse, y’a plus qu’à monter notre labo…
Je tiens à remercier tous mes amis du lycée même si pendant ce travail de thèse j’ai presque totalement coupé les ponts. Particulièrement Aline PICARD, Laetitia BROT et Pierre LOUVEAU.
Un gros bisou à ma future témoin et à son mari, Amaëlle et Vincent. Je me rappelle encore l’attente en bas de chez toi avant d’aller au collège, et ce match France – Angleterre au Frog… Mémorable. Définitivement vous faite partie de ma vie.
Un grand merci à Lauriane pour tous ces coca light que nous avons bu dans le hall de l’école. Merci pour ta bonne humeur.
Reste maintenant à remercier ma bande de potes. Ceux qui sont indissociables, pour qui j’ai la plus grande amitié. Tout d’abord, celui qui aurait pu être mon frère si j’en avais eu un, le
Celle qui restera toujours la plus belle morue que je connaisse (après ma femme bien sûr…) et son saltimbanque de copain, Céline PARIS et Dave FOQUIN. La plus gentille du monde et de l’univers et son beau, Amande et Mat CHARTER (comme les avions…). Le plus jeune d’entre nous qui va prendre un sacré coup de vieux dans quelques mois, Aurel CANE. Sa compagne (et oui, je peux pas encore dire sa femme….) la belle Julie GUEDON. Et pis les deux derniers célibataires Magali SCHEMBRI et le plus beau des hidalgos, François « Paco » NEGRO.
Pour finir cette page, il me reste juste une personne à remercier. Evidemment la plus importante de toutes à mes yeux, puisque c’est la femme qui partage ma vie. C’est elle qui a subi ma thèse de l’intérieur, les moments de doutes et de joies, les WE au labo, les répétitions et j’en passe…
Bref, tu es la femme de ma vie.
Sommaire
ABREVIATIONS
10
INTRODUCTION GENERALE
13
Chapitre I
:Etude bibliographique « les liquides ioniques »
1. Les liquides ioniques 17
1.1. Historique des liquides ioniques 17
1.2. Structure des liquides ioniques 18
2. Synthèse des liquides ioniques 20
2.1. Réaction de quaternarisation 21
2.2. Réaction d’échange d’anions 22
2.2.1. Traitement avec un acide de Lewis 22
2.2.2. Métathèse anionique 22
3. Impuretés des liquides ioniques 24
3.1. Les sels organiques de départ et les composés volatils 24
3.2. Les cations alcalins 25
3.3. L’eau et les ions halogénure 26
4. Propriétés physico – chimiques des liquides ioniques 27
4.1. Le point de fusion 27
4.2. Stabilité thermique 28
4.3. Densité 29
4.4. Viscosité 29
4.5. Conductivité 30
4.6. Solubilité dans les autres solvants 30
4.6.1. Solubilité dans l’eau 30
4.6.2. Solubilité dans les solvants organiques 31
5. Applications des liquides ioniques 37
5.1. Applications en électrochimie 37
5.2. Applications en synthèse organique et en catalyse 37
5.2.1. Réaction de Diels – Alder 38
5.2.2. Réaction d’hydrogénation 38
5.3. Applications dans le domaine des procédés de séparation et de l’analyse 39
5.3.1. Extraction liquide – liquide 39
5.3.1.1 Extraction liquide – liquide 39
5.3.1.2 Microextraction en phase liquide (LPME) 40
5.3.2. Chromatographie en phase gazeuse (GC) 40
5.3.3. Chromatographie en phase liquide (LC) 43
5.3.4. Electrophorèse capillaire (CE) 45
5.3.4.1 Electrophorèse capillaire en phase aqueuse (CE) 46
5.3.4.2 Electrophorèse capillaire en phase non aqueuse (NACE) 48
5.3.5. Spectrométrie de masse (MALDI) 49
6. Les liquides ioniques chiraux 53
6.1. Les liquides ioniques chiraux existant 53
6.2. Applications des liquides ioniques chiraux 63
6.2.1. Synthèse organique 63
6.2.2. Chimie analytique 63
Chapitre II
:Développement d’un nouveau protocole de mesure mettant en jeu
l’électrophorèse capillaire pour la détermination de la viscosité, la conductivité
et l’absorbance d’un liquide ionique pur ou en mélange avec un solvant
moléculaire
A Introduction 70
B Article 72
C Conclusion 89
D Annexe 90
Chapitre III
:Détermination des paramètres d’inclusion (constante et
stœchiométrie)
de
liquides
ioniques
à
base
de
cation
alkyl(méthyl)méthylimidazolium et de cyclodextrines neutres par électrophorèse
capillaire d’affinité
A Introduction 91
B Article 93
C Conclusion 113
D Référence 111
Chapitre IV
:Détermination du seuil d’agrégation de liquides ioniques à base
de cation alkylmétthylimidazolium par conductométrie et analyse frontale
électrocinétique capillaire continue : étude préliminaire
1. Introduction 115
2. Généralités sur les tensioactifs 116
2.1. Les tensioactifs 116
2.2. Méthode de détermination de la concentration micellaire critique (CMC) 117
3. Réactifs et produits 124
4. Etude du liquide ionique [C12MIM,Br] 124
4.1. Détermination du seuil d’agrégation du [C12MIM,Br] par la méthode
conductométrique 125
4.2. Détermination du seuil d’agrégation du [C12MIM,Br] par analyse frontale
électrocinétique capillaire continue 129
4.2.1. Etude du Palier 1 (P1) 133
4.2.2. Etude du Palier 2 (P2) 136
4.2.2. Etude du Palier 3 (P3) 139
5. Conclusion 140
Chapitre V
:Etude en électrophorèse capillaire en phase non aqueuse, du
comportement électrophorétique d’acides 2-arylpropioniques en présence d’un
liquide ionique achiral : mise en place d’un plan d’expérience
A Introduction 144
B Article 145
C Conclusion 162
Chapitre VI
:Evaluation de liquides ioniques chiraux comme nouveaux
sélecteurs pour des séparations électrocinétiques chirales en veine liquide
capillaire
A Introduction 163 B Article 165 C Conclusion 184CONCLUSION GENERALE
185
PERSPECTIVES
188
Abréviations
Techniques
ACE : électrophorèse capillaire d’affinité CE : électrophorèse capillaire
ESI : électrospray ionisation
GC : chromatographie en phase gazeuse LC : chromatographie en phase liquide MS : spectropétrie de masse
MALDI : Matrix Assisted Laser Desorption Ionisation MEKC : électrochromatographie en phase micellaire NACE : non-aqueuse électophorèse capillaire
UV : ultra violet
Grandeurs
µeo : mobilité électroosmotique
µep : mobilité électrophorétique
Rs : résolution
αeff : sélectivité effective
CMC : concentration micellaire critique
Solvants moléculaires ACN : acétonitrile DMF : diméthylformamide EtOH : éthanol MeOH : méthanol TFE : trifluoroéthanol Cyclodextrine
CD : cyclodextrine α−CD : α-cyclodextrine β−CD : β-cyclodextrine γ−CD : γ-cyclodextrine HP−α−CD : hydroxypropyl-α-CD HP−β−CD : hydroxypropyl-β-CD (HP−β−CD) HP−γ−CD : hydroxypropyl-γ-CD DM-β-CD : heptakis-(2,6-di-O-méthyl)-β-CD TM-β-CD heptakis-(2,3,6-tri-O-méthyl)-β-CD Liquide ionique LI : liquide ionique cations IM : imidazolium MIM : méthylimidazolium MMIM : 1,3-diméthyimidazolium EMIM : 1-éthyl-3-méthylimidazolium EMMIM : 1-éthyl-2,3-diméthylimidazolium PMIM : 1-propyl-3-méthylimidazolium PMMIM : 1-propyl-2,3-diméthylimidazolium BMIM : 1-butyl-3-méthylimidazolium BMMIM : 1-butyl-2,3-diméthylimidazolium HMIM : 1-hexa-3-méthylimidazolium
OMIM ou C8MIM : 1-octyl-3-méthylimidazolium
C10MIM : 1-décyl-3-méthylimidazolium C12MIM : 1-dodécyl-3-méthylimidazolium EtChol : éthylcholine PhChol : phénylcholine anions Cl- : chlorure
Br- : bromure I- : iodure NO3- : nitrate SO42- : sulfate BF4- : tétrafluoroborate PF6- : hexafluorophosphate CH3CO2- : acétate CF3CO2- : trifluoroacétate CF3SO3- ou Tf : trifluorométylsulfonate N(CF3SO2-)2 ou NTf2 : bis(trifluorométhylsulfonyl)imide N(CN)2 : dicyanamide
Introduction Générale
L'utilisation continue de grandes quantités de solvants organiques en tant que milieu réactionnel est une préoccupation majeure dans l'industrie chimique d’aujourd’hui. Les effets délétères remarquables de ces solvants sur la santé humaine, la sécurité et l'environnement combinés avec leur volatilité et leur inflammabilité ont mené à une pression croissante pour réduire au minimum leur utilisation.
Bien que les approches alternatives telles que la catalyse hétérogène et les milieux aqueux de réaction existent, elles peuvent ne pas représenter les solutions génériques à ces problèmes. Alors que la chimie de base a trouvé une solution qui consiste en la conception de procédés de fabrication alternatifs avec des risques réduits et une génération d’effluents minimale, la chimie verte cherche des solutions plus efficaces qui consistent à substituer les solvants organiques par une nouvelle classe de solvants dits « solvants verts ». Une catégorie qui inclut le CO2 supercritique, les systèmes aqueux biphasés et les liquides ioniques .Dans cette
approche le rejet d’effluent devrait être nul et la réutilisation de ces solvants serait possible. Ces dernières années, l'attention prêtée par la communauté scientifique vers l'utilisation des liquides ioniques (LI) en tant que solvants innovants est très importante.
Les LI sont des composés totalement ioniques qui possèdent un point de fusion inférieur à 100°C. Dans le cas idéal, les liquides ioniques possèdent une tension de vapeur très faible voire non mesurable, cette nature non volatile offre un avantage certain pour la séparation des produits par distillation et évite l’exposition aux vapeurs non contrôlées. Ils peuvent être facilement régénérés et recyclés, ont une stabilité chimique et thermique élevée et sont relativement peu coûteux et faciles à synthétiser. Ils offrent, de plus, une forte solvatation pour un grand nombre de solutés organiques et inorganiques, et ont une conductivité élevée. Le principal désavantage des LI est leur haute viscosité qui implique des coefficients de diffusion faibles. Les principaux domaines d’applications des LI sont la synthèse organique, la catalyse et l’électrochimie. Plus récemment, dans le domaine de l’analyse et des procédés, les LI ont été évalués en extraction liquide-liquide. En effet, certains d’entre eux ont la particularité de former des systèmes biphasiques avec l’eau ou certains solvants moléculaires. Ces dernières années, les LI ont suscité un très grand intérêt dans le domaine des sciences séparatives. Depuis 1985, les LI ont été utilisés en tant que nouvelle phase stationnaire en
chromatographie en phase gazeuse (GC) car ils peuvent se comporter soit comme une phase polaire, soit comme une phase apolaire. Concernant la chromatographie en phase liquide (LC) les LI ont été utilisés en tant qu’additif dans la phase mobile. Le fait que l’anion et le cation interagissent avec les groupements silanol fait que les LI ont été utilisés en LC pour augmenter la résolution des séparations. Depuis l’année 2000, une vingtaine de publications est sorties dans la littérature sur des applications en électrophorèse capillaire (CE) mettant en jeu des LI. Ces applications qui ont été réalisées en phase aqueuse et non-aqueuse, ont permis d’observer de nouveaux types d’interaction.
L’évaluation des LI en CE en temps que nouveaux milieux de séparation a donc été le sujet principal de ce travail de thèse. Cependant, il est apparu très rapidement que le principal frein au développement des applications des LI en CE était le manque de données sur les propriétés physico-chimiques des LI, particulièrement la viscosité et la conductivité. La première partie de ce travail a donc consisté à évaluer l’apport de la CE pour la caractérisation des LI, en développant des méthodes mettant en jeu l’instrumentation de la CE et des techniques
électrophorétiques pour la détermination de paramètres physico-chimiques et
thermodynamiques des LI.
Préalablement à l’exposé des résultats obtenus dans ce travail, une étude bibliographique sur les liquides ioniques a été décrite dans le chapitre I.
La première étude, décrite dans le chapitre II, a été consacrée au développement d’un protocole de mesure mettant en jeu l’électrophorèse capillaire pour la détermination de l’absorbance, la viscosité et la conductivité d’un liquide ionique pur ou en mélange avec un solvant organique. La grande complémentarité de l’instrumentation de la CE, tels que le système de pression, le générateur de tension, le détecteur à barrette de diodes et le thermorégulateur, a permis la mise en place d’une mesure en ligne, rapide, automatique et miniaturisée de ces trois propriétés physico-chimiques. Ce protocole a été validé sur quatre LI différant par la nature de leur cation (1-éthyl-3-méthylimidazolium, EMIM, et 1-butyl-3-méthylimidazolium, BMIM) et par la nature de leur anion (trifluorométhanesulfonate, Tf, et bis(trifluorméthylsulfonyl)imide, NTf2). Quatre solvants moléculaires de choix pour les
applications en CE ont été sélectionnés (acétonitrile, méthanol, trifluoroéthanol et diméthylformamide). Les résultats de l’évolution de la viscosité et de la conductivité en fonction de la proportion en solvant moléculaire ont permis d’obtenir des informations sur le comportement de ces liquides ioniques en milieu solvant et plus particulièrement sur la force
des interactions anion – cation des liquides ioniques étudiés. La deuxième étude sur la détermination de paramètres physico-chimiques des LI (décrite dans le chapitre III) a consisté à déterminer les paramètres d’inclusion (constante et stoechiométrie) entre le cation d’un LI à base d’alkyl(méthyl)méthylimidazolium et une cyclodextrine neutre par une méthode d’électrophorèse capillaire d’affinité (ACE). Cette méthode a été appliquée à sept LI différant par la longueur de la chaîne alkyle portée par le cation imidazolium en position C1, par la présence (ou non) d’un groupement méthyle en position C2 et par la nature de l’anion, et huit CDs neutres différant par la taille et la forme de la cavité, et par la nature du groupement dérivatif externe. Les résultats obtenus sur l’étude de ces 56 systèmes a permis de dégager quelques règles sur le comportement de ces LI en présence de CD et plus particulièrement sur les facteurs influençant la force de l’inclusion du cation du LI dans une CD. Enfin, une troisième étude préliminaire (décrite dans le chapitre IV) a été menée sur la détermination du seuil d’agrégation de LI à base de cation alkylméthylimidazolium par conductométrie et analyse frontale électrophorétique capillaire continue. Ce travail a eu un double objectif, tout d’abord un objectif appliqué, avec la détermination du seuil d’agrégation de différents LI, puis un but plus fondamental, avec l’évaluation de la méthode d’analyse frontale électrocinétique capillaire continue pour cette détermination. Cependant, cette étude n’a pas pu être menée à terme et laisse encore ouverte de nombreuses perspectives.
La deuxième partie de ce travail de thèse a été dédiée à l’étude d’une application des LI en électrophorèse capillaire. Comme nous l’avons vu au début de cette introduction, depuis l’an 2000, une vingtaine d’articles a été publié sur des applications des LI en CE. La grande viscosité et conductivité des LI fait que dans la totalité de ces applications, les LI sont utilisés comme additifs à l’électrolyte support. De nouvelles interactions ont été observées, mais dans la plupart des cas, les phénomènes réactionnels restent inexpliqués.
Notre étude a porté sur l’évaluation de liquides ioniques chiraux en temps que nouveaux sélecteurs pour des séparations énantiomériques en électrophorèse capillaire. Cette étude a été réalisée en deux parties, permettant de mieux comprendre les phénomènes d’interactions mis en jeu.
Dans la première partie de ce travail (décrite dans le chapitre V), l’étude par électrophorèse capillaire en phase non-aqueuse (NACE) du comportement électrophorétique de composés modèles chiraux, les acides 2-arylpropioniques (profènes), en présence d’un liquide ionique achiral ([BMIM,NTf2]) a été réalisée. Afin d’estimer l’influence des nombreux paramètres
entrant en jeu dans le système interactif, un plan d’expérience a été développé puis appliqué. Ce plan d’expérience a permis d’identifier les interactions entre paramètres, et plus particulièrement d’évaluer la possibilité de formation de paires d’ions entre le cation du LI et les analytes anioniques. La deuxième partie de ce travail (décrite dans le chapitre VI) a été centrée sur l’évaluation des performances énantiosélectives de deux LI chiraux (éthyl- (EtChol) et phénylcholine (PhChol) de bis(trifluorométhylsulfonyl)imide) sur des profènes, par électrophorèse capillaire. Dans un premier temps, les conditions de la NACE ont été appliquées, mais n’ont pas permis d’observer un pouvoir énantiosélectif de ces composés. Ce travail a donc été réorienté vers l’étude en milieu aqueux et hydroorganique du comportement électrophorétique des profènes mettant en jeu un LI chiral et un deuxième sélecteur chiral classique (di- ou triméthyl-β-cyclodextrine).
CHAPITRE I : Etude Bibliographique « Les liquides
ioniques »
1. Les liquides ioniques
Les liquides ioniques sont des composés totalement ioniques qui possèdent un point de fusion inférieur à 100°C. Dans le cas idéal, les liquides ioniques possèdent une tension de vapeur très faible voire non mesurable, cette nature non volatile offre un avantage certain pour la séparation des produits par distillation et évite l’exposition aux vapeurs non contrôlées. Ils ont une stabilité thermique élevée et sont relativement peu coûteux et faciles à synthétiser. Ils offrent une forte solvatation car c’est un milieu non coordinant dans lequel un certain nombre de solutés organiques et inorganiques peuvent être dissous. L’ensemble de ces propriétés en font des milieux de choix pour le développement de la chimie verte.
1.1. Historique des liquides ioniques
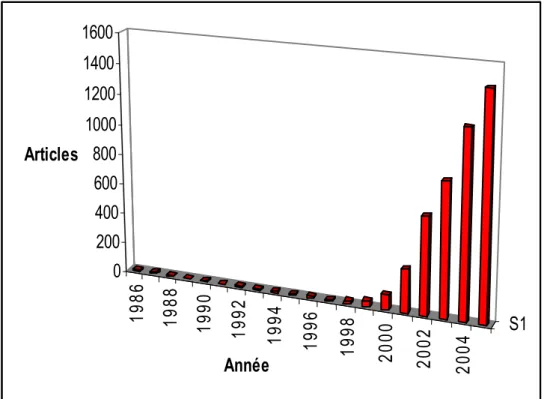
Le premier liquide ionique, le nitrate d’éthylammonium, fut synthétisé par Walden et coll. en 1914 durant la première guerre mondiale, en cherchant de nouveaux explosifs [1]. En 1951, Hurley et coll. ont mis au point la première synthèse des liquides ioniques à base d’anions choloraluminate dans le développement d’électrodépôt de l’aluminium à basse température [2]. Durant les années 1970 et 1980, les liquides ioniques ont été utilisés principalement dans le domaine de l’électrochimie. Au milieu des années 1980, ce nouveau type de composés a été mis en œuvre comme solvant pour des synthèses organiques. Depuis les années 1990 et jusqu’à maintenant, les liquides ioniques ont connu un réel engouement de la part de nombreux chercheurs, dans les domaines de l’électrochimie, de la synthèse organique et de la catalyse et plus récemment, dans le domaine des sciences séparatives : l’extraction liquide – liquide, la chromatographie en phase gazeuse et en phase liquide et l’électrophorèse capillaire. La Figure 1 montre le nombre de publications portant sur ce nouveau thème de recherche depuis 1986 [3].
Figure 1 : Nombre d’articles publiés par année sur le sujet « room temperature ionic liquids »
(Base de données : SciFinder)
A ce jour, il existe à peu près cinq cent liquides ioniques, mais Seddon et coll. ont estimé que le nombre total de ces nouveaux solvants pourrait atteindre un million [4].
1.2. Structure des liquides ioniques
La plus simple définition d’un liquide ionique est un liquide composé exclusivement d’ions contrôlés principalement par des forces coulombiennes. Un liquide ionique est le plus généralement constitué d’un cation organique de faible symétrie. Ils font partie le plus souvent de la famille des ammonium, sulfonium, phosphonium, imidazolium, pyridinium différemment substitués (Figure 2). Les liquides ioniques les plus utilisés ces dernières années sont les composés à base de cations asymétriques de N,N’-dialkylimidazolium.
1
9
8
6
1
9
8
8
1
9
9
0
1
9
9
2
1
9
9
4
1
9
9
6
1
9
9
8
2
0
0
0
2
0
0
2
2
0
0
4
S1
0
200
400
600
800
1000
1200
1400
1600
Articles
Année
N N R' R N R R R R R' R R R R' R R R S Ion sulfonium + + N + P +
Ion imidazolium Ion pyridinium
Ion ammonium Ion phosphonium
+
Figure 2 : Exemple de classes de cations des liquides ioniques
La nature des anions qui composent les liquides ioniques peut être divisée en trois groupes : tout d’abord, les anions polynucléaires (le plus souvent Cl-/AlCl3) qui constituent la
classe de liquides ioniques dite de « première génération », puis, les anions mononucléaires inorganiques comme les anions halogénures (Cl-, Br-…), l’anion nitrate (NO3-), l’anion
hexafluorophosphate (PF6-) et l’anion tétrafluoroborate (BF4-) ; ou enfin, plus récemment, les
anions mononucléaires organiques tels que l’anion trifluorométhanesulfonate (Tf-) (ou triflate), l’anion bis(trifluorométhylsulfonyl)imide (NTf2-) et l’anion dicyanamide (N(CN)2-)
(Figure 3).
Figure 3 : Quelques exemples d’anions Cl- , Br- , I- NO3- , SO42- BF4- PF6- , SbF6- Organique Polynucléaire Cl-/[AlCl3] CF3CO2-, CH3CO2- CF3SO3- (Tf-) N(CF3SO2)2- (NTf2-) N(CN)2- CF3(CF2)3CO2 -Inorganique
Nature de l’anion
Mononucléaire2. Synthèse des liquides ioniques [8]
Ce chapitre se concentrera sur la préparation des liquides ioniques à base de cation 1,3-dialkylimidazolium, qui sont mis en jeu dans la majorité des études de ces vingt dernières années. Les techniques discutées dans ce chapitre, sont généralement applicables pour les autres classes de cations (Figure 2).
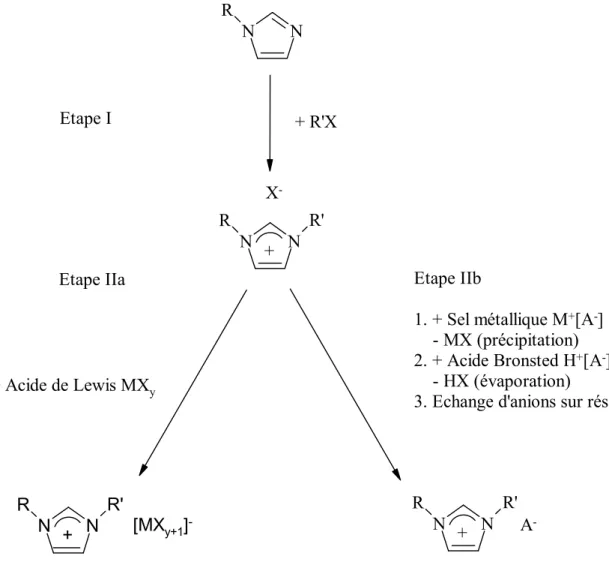
La synthèse des liquides ioniques est généralement réalisée en deux étapes (Figure 4). La première est une réaction de quaternarisation afin d’obtenir le cation souhaité, et la seconde, une réaction d’échange d’anions. Dans la plupart des cas, il est possible d’obtenir commercialement, et à faible coup, le cation souhaité sous forme d’halogénure. De ce fait, la synthèse des liquides ioniques se résume à la réaction d’échange d’anions.
N N R N N R R' N N R R' N N R R' + X -+ A -Etape IIb 1. + Sel métallique M+[A-] - MX (précipitation) 2. + Acide Bronsted H+[A-] - HX (évaporation)
3. Echange d'anions sur résine Etape IIa
+ Acide de Lewis MXy
Etape I + R'X
+ [MXy+1]
2.1. Réaction de quaternarisation
La formation du cation du liquide ionique peut être menée soit par protonation avec un acide libre, soit par quaternarisation d’une amine ou d’une phosphine, le plus communément par un halogénoalcane. Dans le cas de la protonation avec un acide libre, Evans et coll. ont réalisé la synthèse d’un sel de nitrate d’éthylammonium par l’addition d’acide nitrique 3 M dans une solution aqueuse d’éthylamine [5]. Ce procédé, généralisé, peut être employé pour la préparation de tous les sels de ce type. Par contre, il existe de grands risques de contamination des produits résiduels aminés pour la synthèse de sels d’amine de plus hauts poids moléculaires. Le procédé d’alkylation présente tout d’abord l’avantage d’avoir une large gamme d’halogénoalcanes disponible commercialement et peu onéreuse, mais aussi le fait que la réaction est douce et a lieu généralement à des températures raisonnables. De plus, les sels d’halogénures peuvent être facilement convertis en sels avec d’autres anions. La voie de synthèse décrite sur la Figure 4, qui met en jeu un sel de 1,3-dialkylimidazolium, peut être généralisée pour les sels de pyridine [6], 1-methylpyrrolidine [7 et phosphine entre autres. La température et la durée de la réaction sont des paramètres qui dépendent en grande partie du type d’halogénoalcanes mis en jeu et de la longueur de la chaîne alkyle. Les chloroalcanes sont les moins réactifs et les iodoalcanes les plus réactifs. De plus, la réactivité des halogénoalcanes décroît avec l’augmentation de la longueur de la chaîne alkyle [8]. Une technique alternative pour la réaction de quaternarisation des amines et des phosphines avec les halogénoalcanes, a été récemment rapportée dans la littérature [9]. Elle met en jeu l’utilisation d’une irradiation aux micro-ondes. Cette technique permet d’obtenir de très bons rendements en un temps très rapide (quelques minutes comparées à quelques heures). Malheureusement, cette technique ne s’applique que pour de très faibles quantités de réactif.
Il est à noter que la quaternarisation des amines et des phosphines n’est pas une technique réservée uniquement aux liquides ioniques à base d’anions halogénure. Cette voie de synthèse peut en effet être mise en œuvre pour d’autres types d’anions comme le tosylate et le triflate [10,11]. Cette méthode présente l’avantage de réaliser une synthèse en une seule étape avec la possibilité de s’affranchir des impuretés d’ions halogénure. Le principal désavantage de cette méthode est le fait que les réactifs d’alkyltosylate ou triflate sont extrêmement sensibles à l’eau. Il faut donc travailler sous une atmosphère inerte.
2.2. Réaction d’échange d’anions
Les réactions d’échange d’anions des liquides ioniques peuvent être divisées en deux voies de synthèse distinctes : le traitement direct des sels d’halogénures par les acides de Lewis, et la formation d’anion par métathèse anionique. Ces deux approches seront traitées séparément car elles nécessitent des méthodes expérimentales différentes.
2.2.1. Traitement avec un acide de Lewis
La synthèse de liquides ioniques par traitement des sels d’halogénures avec des acides de Lewis (le plus communément AlCl3) a été la méthode précurseur dans ce domaine de la
chimie. Une grande avancée est venue en 1951 avec Hurley et coll. qui réalisèrent la formation d’un sel liquide à température ambiante basée sur la combinaison du
1-butylpyridinium avec AlCl3 dans des proportions molaires 1:2 [2]. Plus récemment, Robinson
et coll. et Wilkes et coll. ont développé des techniques pour synthétiser des liquides ioniques chloroaluminates à base de cation 1-alkylpyridinium [12] et 1,3-dialkylimidazolium [13]. Le paramètre principal de cette voie de synthèse est la proportion relative de sel d’halogénure et d’acide de Lewis. En effet, il est possible que l’excès d’acide de Lewis puisse impliquer la formation d’autres espèces anioniques comme le montrent les équations suivantes :
[EMIM]+Cl- + AlCl3 ⇔ [EMIM]+[AlCl4]
-[EMIM]+[AlCl4-] + AlCl3 ⇔ [EMIM]+[Al2Cl7]
-[EMIM]+ [Al2Cl7]- + AlCl3 ⇔ [EMIM]+[Al3Cl10]
-Ce schéma réactionnel met en jeu les anions chloroaluminates, mais d’autres acides de Lewis tels que AlEtCl2 [14], CuCl [15], SnCl2 [16], permettent, par la même méthode, la
synthèse de liquides ioniques.
2.2.2. Métathèse anionique
La première synthèse de liquides ioniques stables à l’air et l’humidité à base de cations 1,3-dialkylméthylimidazolium (désignés parfois comme liquides ioniques de deuxième génération) a été rapportée par Wilkes et coll. en 1992 [17]. Cette préparation comprenait une
réaction métathétique entre [EMIM,I] et une série de sels d’argent (AgNO3, AgNO2, AgBF4,
Ag2SO4 et Ag [CO2CH3]) dans le méthanol ou dans des mélanges eau/méthanol. La très faible
solubilité de l’iodure d’argent dans ces solvants permet sa séparation par simple filtration, et l’élimination des solvants permet d’isoler des liquides ioniques de très haute pureté avec des rendements élevés. Cette méthode reste la plus efficace pour la synthèse des liquides ioniques miscibles à l’eau mais elle est évidemment limitée par le coût relativement élevé des sels d’argent et les grandes quantités de sous produits solides.
Les méthodes de préparation employées suivent généralement les mêmes lignes directrices. L’objectif de ces réactions d’échange d’anions est la formation du liquide ionique souhaité non contaminé par les cations et les anions non désirés. Un objectif qui est plus facile à atteindre pour les liquides ioniques non miscibles à l’eau.
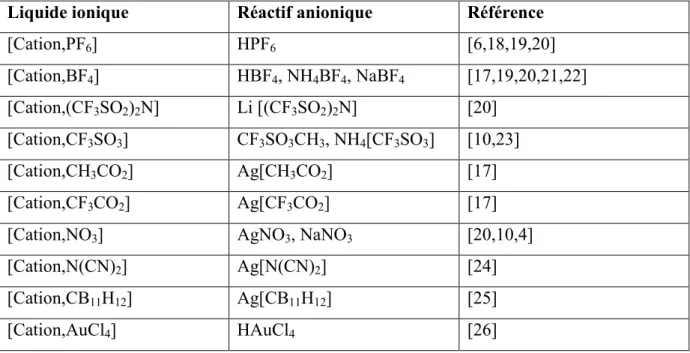
Ces dernières années, une grande variété de réactions d’échange d’anions a été investiguée dans la préparation des liquides ioniques (Tableau 1).
Tableau 1 : Exemple de liquides ioniques préparés par métathèse anionique
Liquide ionique Réactif anionique Référence
[Cation,PF6] HPF6 [6,18,19,20] [Cation,BF4] HBF4, NH4BF4, NaBF4 [17,19,20,21,22] [Cation,(CF3SO2)2N] Li [(CF3SO2)2N] [20] [Cation,CF3SO3] CF3SO3CH3, NH4[CF3SO3] [10,23] [Cation,CH3CO2] Ag[CH3CO2] [17] [Cation,CF3CO2] Ag[CF3CO2] [17]
[Cation,NO3] AgNO3, NaNO3 [20,10,4]
[Cation,N(CN)2] Ag[N(CN)2] [24]
[Cation,CB11H12] Ag[CB11H12] [25]
[Cation,AuCl4] HAuCl4 [26]
Il est à noter que les réactions d’échange d’anions peuvent être réalisées par des résines échangeuses d’anions. Wasserscheid et coll. suggèrent que cette méthode est la meilleure pour la synthèse de liquides ioniques de pureté très élevée [27]. Cette alternative, bien qu’elle soit intéressante, n’est pas très répandue et rares sont les travaux qui mentionnent les échangeurs d’anions pour les préparations à grande échelle.
3. Impuretés des liquides ioniques
La pureté d’un liquide ionique est l’un des paramètres les plus importants qui conditionne son utilisation. Le schéma réactionnel général qui régit la synthèse des liquides ioniques indique la présence possible de plusieurs sortes d’impuretés : les sels organiques de départ, les composés volatils, l’eau, les cations alcalins et les ions halogénure. La présence de ces impuretés a, à différents niveaux, des effets dramatiques sur les propriétés physico-chimiques des liquides ioniques. Diverses méthodes analytiques ont été mises au point afin de déceler la présence de ces impuretés et de déterminer leurs teneurs. Les méthodes d’analyses souffrent d’un manque de matrice de référence, les méthodes séparatives telles que la chromatographie ionique trouvent une limitation en la nature de la matrice.
3.1. Les sels organiques de départ et les composés volatils
Les impuretés volatiles dans les liquides ioniques peuvent avoir plusieurs origines. Elles peuvent résulter des solvants utilisés lors des étapes d’extraction durant la synthèse, du réactif qui n’a pas été consommé par la réaction d’alkylation ou de tout autre composé organique volatil précédemment dissous dans le liquide ionique.
Théoriquement, ces impuretés peuvent être facilement éliminées du liquide ionique, non volatil, par évaporation, mais ce procédé peut être lent. Les facteurs qui influencent la durée nécessaire pour l’élimination de toutes les substances volatiles d’un liquide ionique (à une température et une pression données) sont : la quantité de ces substances volatiles, leurs points d’ébullition, leurs interactions avec le liquide ionique, la viscosité du liquide ionique, et la surface libre du liquide ionique.
Les impuretés volatiles potentielles qui peuvent exister dans les liquides ioniques 1,3-dialkylimidazolium proviennent du 1-alkylimidazole de départ. Ces composés sont difficiles à éliminer, même à haute température et pression réduite, en raison de leur température d’ébullition élevée (par exemple, méthylimidazole tébullition = 198 °C) et des fortes interactions
avec le liquide ionique.
Ces impuretés affectent drastiquement les performances des liquides ioniques. Lorsque ces derniers sont utilisés en tant qu’électrolyte pour les batteries, de larges quantités d’impuretés affectent le nombre de transport des espèces inclues dans le système. Des études
préalables suggèrent que différents substituants alkyle des cations imidazolium, y compris les isomères, engendrent diverses propriétés des liquides ioniques. Des traces de bases telles que le méthylimidazole dans le liquide ionique final peuvent jouer un rôle défavorable dans certaines applications connues (telles que les catalyses bi-phasiques). Plusieurs catalyseurs électrophiles vont se coordiner avec la base d’une manière irréversible et seront alors désactivés.
De nombreuses méthodes analytiques ont été développées pour détecter la présence de ces impuretés dans le liquide ionique final. La spectroscopie RMN est utilisée par la plupart des groupes académiques, mais elle a une limite de détection d’environ 1 % en mole. La spectroscopie IR, la spectrophotométrie UV et la spectrométrie de masse ont été aussi utilisées. Ces méthodes, bien qu’elles soient très sensibles, sont soit coûteuses, soit peu convenables pour les analyses de routine.
Holbrey et coll. ont développé une méthode colorimétrique pour évaluer la teneur en imidazoles dans les liquides ioniques à base des cations 1-éthyl-3-méthylimidazolium (EMIM) [28]. Elle est fondée sur la complexation du 1-méthylimidazole par le chlorure cuivrique dans l’éthanol. Ainsi l’ion [Cu(MI)4]2+, qui est d’une couleur bleue intense, se
forme. La limite de détection de cette méthode est de 0,2 % en mole dans le chlorure de 1-éthyl-3-méthylimidazolium ([EMIM,Cl]). Bien qu’elle soit relativement rapide et convenable pour le contrôle de routine, cette méthode présente l’inconvénient de ne pas disposer de matrice de référence. En outre, d’autres imidazoles tels que l’imidazole et les 1,2-di-méthylimidazoles peuvent interférer du moment où ils peuvent être complexés par l’ion cuivrique.
Qin et coll. ont développé une méthode d’analyse par électrophorèse capillaire de zone pour la détermination des dérivés imidazole présents dans le chlorure d’éthylméthylimidazole ([EMIM,Cl]) [29]. La limite de détection de cette méthode est de 0,27 % en mole. Bien que cette valeur soit supérieure à celle obtenue lors du précédent travail d’Holbrey et coll., elle est plus précise et plus exacte puisque l’électrophorèse capillaire permet la séparation des différents imidazoles.
3.2. Les cations alcalins
Les liquides ioniques peuvent être contaminés par d’autres impuretés issues de la métathèse d’anions, notamment par les sels alcalins qui présentent une solubilité significative dans le liquide ionique formé. Bien que la présence des cations alcalins puisse ne pas altérer le
rendement de certaines réactions catalytiques, elle est d’une grande influence sur les propriétés physico-chimiques du produit. Dans cette optique, il est important de noter que la détection de ces cations dans les liquides ioniques n’est pas aisée en utilisant des techniques traditionnelles telles que la spectroscopie RMN.
Seule une étude parue en 2002 mentionne la détermination des ions sodium par spectrométrie d’émission atomique [30]. Mais, comme l’objectif du travail était l’investigation de l’effet des impuretés sur les performances des systèmes catalytiques dans les liquides ioniques, la méthode d’analyse n’a pas été décrite.
L’analyse de ces cations nécessite des techniques plus spécialisées telles que la chromatographie ionique et l’électrophorèse capillaire.
3.3. L’eau et les ions halogénure
L’eau et les ions chlorure altèrent significativement les propriétés physiques telles que le point de fusion, la viscosité, la densité, la conductivité et la stabilité thermique [4,18]. La présence d’une contamination par les ions chlorure augmente la viscosité des liquides ioniques alors que la présence de l’eau ou d’autres co-solvants la diminue. La présence d’eau dans les liquides ioniques contenant l’anion hexafluorophosphate favorise la dégradation par formation du fluorure d’hydrogène à température élevée (> 100 °C).
Comme la plupart des sels organiques sont hygroscopiques, l’eau est un contaminant commun. La teneur en eau est déterminée par la méthode de Karl-Fisher. Si les liquides ioniques sont laissés en contact avec l’air libre, ils peuvent adsorber facilement de l’eau à des quantités de 0,2 à 2 M en fonction du type du liquide ionique, l’humidité relative et la température [4].
Les ions halogénure sont aussi présents dans plusieurs liquides ioniques préparés par des réactions de métathèse et peuvent être déterminés par potentiométrie au moyen d’une électrode d’argent. Les ions halogénure sont connus par leur coordination aux catalyseurs des métaux de transitions et ceci influence (généralement négativement) le rendement des réactions chimiques.
La détermination des impuretés chlorure par chromatographie ionique a été décrite par Villagrán et coll. Pour les liquides ioniques analysés (famille des alkylimidazoliums avec Cl-, PF6-, BF4-, Tf-, NTf2-) la limite de quantification des ions chlorure est de l’ordre de 8 ppm
[31]. Villagrán et coll. ont aussi développé une méthode électroanalytique pour quantifier des traces de chlorure avec électrode à disque d’argent par voltampérométrie à tension carrée
[32]. Cette méthode est la plus sensible, avec une limite de détection de l’ordre de la centaine de ppb dans les liquides ioniques analysés.
Un autre travail intéressant a été mené par Berthier et coll. sur la mise au point d’une méthode de détermination des impuretés halogénure dans les liquides ioniques ([BMIM,NTf2]) par électrophorèse capillaire. Le seuil de détection de la méthode pour les
ions chlorure est de l’ordre de 2 ppm [33].
4. Propriétés physico – chimiques des liquides ioniques
La recherche, menée sur les propriétés des liquides ioniques purs, a porté dans un premier temps sur la compréhension de la relation entre la structure du cation et de l’anion, et leurs propriétés physico-chimiques. Très rapidement, il est apparu que la pureté des liquides ioniques était un paramètre influençant de façon très importante les mesures de leurs propriétés physico-chimiques. Comme il a été rappelé dans le paragraphe 3, la présence de contaminants tels que l’eau ou/et les ions halogénure, a un effet significatif sur les propriétés physico-chimiques telles que la densité, la viscosité, le point de fusion et la conductivité [4,18]. Malheureusement, les données concernant la pureté des liquides ioniques ne sont pas systématiquement mentionnées dans la littérature car les méthodes de détermination des impuretés sont encore en cours de recherche. Ce manque de données peut, en grande partie expliquer la grande variabilité des valeurs des propriétés physico-chimiques des liquides ioniques purs issues de la littérature [8]. Le Tableau 3, repris des travaux de Wasserscheid et coll. [8], est un récapitulatif exhaustif des principales propriétés physico chimiques des liquides ioniques existants suivant la nature du cation.
4.1. Le point de fusion
Le critère clé pour l’évaluation d’un liquide ionique est, par définition, son point de fusion. Un sel fondu est défini liquide ionique lorsque son point de fusion est inférieur à 100°C. Le point de fusion est difficile à corréler avec la composition chimique. Les principaux facteurs qui influencent le point de fusion des liquides ioniques sont : la distribution de charge sur les ions, la possibilité de liaisons hydrogène, la symétrie des ions et les interactions de Van der Waals. Un succès modeste a été obtenu en utilisant des descripteurs moléculaires modélisés par ordinateur pour prévoir le point de fusion des bromures d’imidazolium [34].
Il existe une grande incertitude sur la valeur de nombreux points de fusion de liquides ioniques tirée de la littérature, car certains liquides ioniques présentent la propriété d’être surfondus, c'est-à-dire, qu’ils possèdent une plage de température dans laquelle ils passent par une phase cristalline vitreuse. Ngo et coll. ont montré que le point de fusion de certains liquides ioniques pouvait fortement varier selon qu’ils sont chauffés ou qu’ils sont refroidis [35]. L’état surfondu est une caractéristique associée à de nombreux liquides ioniques à base de cations imidazolium [10, 35-37]. Pour exemple, la littérature [10] indique que les cations
imidazolium combinés avec les anions NTf2- sont généralement liquides au dessus de 30° à
-50°, mais ils deviennent très visqueux jusqu’à l’état vitreux sans que l’on puisse observer le point de fusion.
Ngo et coll. ont aussi travaillé sur l’influence de la symétrie du cation (plus particulièrement sur les cations N,N’-alkylméthylimidazolium) sur le point de fusion des liquides ioniques [35]. Les résultats de leurs études indiquent une diminution notable du point de fusion des liquides ioniques lorsqu’il y a une forte asymétrie des substituants du cation imidazolium.
La longueur de la chaîne alkyle substituée sur les cations des liquides ioniques a une grande influence sur leur point de fusion. Holbrey et coll. [28], Visser et coll. [63] et Chun et coll. [65] ont systématiquement étudié l’influence de ce paramètre. Il apparaît, en règle générale, une diminution de la valeur du point de fusion lorsque l’on augmente la chaîne alkyle du méthyle au butyle, puis un palier pour les chaînes butyle à hexyle, et enfin une augmentation pour les chaînes alkyle supérieure à l’hexyle. Il est à noter que certaines conclusions peuvent être critiquées du fait de l’état surfondu de certains liquides ioniques.
L’effet de l’anion sur le point de fusion est plus difficile à expliquer. Dans le cas des liquides ioniques à base de cations imidazolium combinés à des anions tels que le trifluorométhanesulfonate ou le bis(trifluorométhylsulfonyl)imide, les faibles valeurs de points de fusion sont attribuées à une importante délocalisation de la charge sur l’anion, et à une faible interaction de liaison hydrogène [38].
4.2. Stabilité thermique
Un grand nombre de ces sels, ayant un anion faiblement nucléophile, montre une stabilité thermique exceptionnelle permettant leur utilisation pour des applications à des températures supérieures à 250 °C et dans certains cas supérieures à 400 °C dans l’air ou dans une atmosphère inerte [10]. Pour des températures supérieures à 400 °C, par analyse calorimétrique différentielle, une tension de vapeur significative ou une décomposition
thermique est observée. La nature des anions a une influence significative sur la stabilité thermique des liquides ioniques. Il apparaît que la température de décomposition diminue (PF6- > NTf2- ≈ BF4- > ions halogénure) [35] quand le caractère hygroscopique de l’anion
augmente.
La présence des impuretés peut influencer considérablement la fidélité de ces mesures en agissant, par exemple, en tant que catalyseurs pour les réactions de décomposition.
4.3. Densité
La densité à température ambiante des liquides ioniques de la classe 1,3-dialkylimidazolium est en général plus grande que celle de l’eau (0,9 à 1,6 g.cm-3). Aussi bien pour les liquides ioniques hydrophiles qu’hydrophobes, elle diminue presque linéairement avec la longueur de la chaîne alkyle du cation [39]. De plus, il est à noter que l’augmentation de la teneur en eau provoque une diminution de la densité [37].
4.4. Viscosité
La viscosité de la plupart des liquides ioniques du type 1,3-dialkylimidazolium reste considérablement plus élevée que celle des solvants moléculaires conventionnels. Les valeurs de viscosité des sels 1,3-dialkylimidazolium à température ambiante vont de 40 cP à 1000 cP. Par comparaison, les viscosités de l’eau, de l’éthylène glycol et du glycérol à 25°C sont respectivement : 0,89, 16,1 et 934 cP (1 cP = 1 mPa.s).
La viscosité des liquides ioniques est déterminée essentiellement par leur tendance à former des liaisons hydrogènes et par la force des interactions de Van der Waals [40,10]. La délocalisation de la charge sur l’anion semble favoriser une viscosité faible par l’affaiblissement de la liaison hydrogène avec le cation (exemple de l’anion NTf2-) [41,42]. La
longue de la chaîne alkyle portée par le cation est un paramètre influent sur la viscosité. L’augmentation de la longueur de la chaîne alkyle a pour conséquence l’augmentation de la viscosité due à des interactions de van der Waals plus fortes [10,18,39,43].
Pour le même cation, la viscosité diminue, selon l’anion, dans l’ordre : Cl- > PF6- > BF4- > NO3- > NTf2- [43].
L’importance de l’influence des impuretés sur la viscosité des liquides ioniques est prépondérante. Seddon et coll. ont étudié les effets de l’eau, des ions chlorure et des solvants organiques sur le liquide ionique [BMIM,BF4] [4]. Pour l’exemple, le liquide ionique
[BMIM,BF4] a une viscosité de 154 mPa.s-1 avec 0,01 mol.kg-1 de Cl- et augmente à 201
peuvent absorber facilement de l’eau à des quantités de 0,2 à 2 M en fonction du type du liquide ionique, l’humidité relative et la température [4]. L’augmentation de la teneur en eau a pour effet la diminution très importante de la viscosité.
4.5. Conductivité
Les liquides ioniques présentent une grande conductivité ionique, généralement de l’ordre de 10-1.S.m-1 [44]. Bonhôte et coll. rapportent la relation entre la conductivité et différentes propriétés [10]. )] ) ( ) ) )[( 6 ( 1 1 2 − − + = c c a a W AM r r N d yF
ζ
ζ
η
π
σ
Notation : viscosité (η), masse molaire (MW), nombre d’Avogadro (NA), nombre de faraday
(F), densité (d), degré de dissociation (0<y<1), rayon de l’anion et du cation (ra, rc
respectivement), facteur de microviscosité corrigée de l’anion et du cation (ζa, ζc
respectivement).
Il apparaît que la viscosité n’est pas le seul paramètre ayant une influence sur la valeur de la conductivité : il faut aussi prendre en considération la taille et la masse moléculaire des ions, qui ont également un effet important.
Le modèle de Bonhôte et coll. [10] est donc incompatible avec le modèle plus simple de Walden, qui exprime que le produit de la conductivité par la viscosité (produit de Walden) est une constante à une température donnée. Les valeurs de produits de Walden rapportées dans la littérature (Tableau 3) varient en fait selon le LI dans un rapport allant de 1 à 2.
Ce phénomène a souvent été évoqué, mais n’a jamais été confirmé [41]. Ce modèle prend en compte, en outre, un phénomène d’appariement d’ions.
4.6. Solubilité dans les autres solvants
4.6.1. Solubilité dans l’eau
Le caractère hydrophile ou hydrophobe des liquides ioniques est un paramètre important dans l’étude de leurs propriétés de solvatation. La majorité des résultats relatifs à la miscibilité des liquides ioniques dans l’eau concerne les sels à base de N,N’-dialkylimidazolium. La solubilité de ces liquides ioniques dans l’eau dépend de la nature de l’anion et de la longueur de la chaîne alkyle portée par le cation imidazolium. Le Tableau 2 donne qualitativement la solubilité de quelques liquides ioniques dans l’eau et certains solvants organiques.
Tableau 2 : Miscibilité dans les solvants de certains liquides ioniques [59] Solvanta
Liquide ionique
Eau MeOH ACN THF EtOAc Acet
EMIM PF6- M M M CH3SO3- M M M CF3CO2- M PM CF3SO3- I M M (CF3SO2)2N- I M M BMIM Cl- M PF6- I M M I- M CH3SO3- M M M BF4- M M M I CF3CO2- M M CF3SO3- I M M (CF3SO2)2N- I M M
M = Miscible ; PM = Partiellement miscible ; I = Immiscible
a
MeOH = Méthanol ; ACN = Acétonitrile ; THF = Tétrahydrofurane ; EtOAc = Acétate d’éthyle ; Acet = Acétone
Les liquides ioniques ont la propriété d’être hygroscopiques, c'est-à-dire qu’ils peuvent absorber de l’eau contenue dans l’atmosphère [4]. Cammarata et coll. ont établi que les molécules d’eau absorbées dans les liquides ioniques sont à l’état « libre » en interaction par des liaisons hydrogènes avec PF6-, BF4-, SbF6-, ClO4-, Tf- et NTf2- avec des concentrations
allant de 0,2 à 1.10-3 mol.L-1 [45]. La force des interactions hydrogène entre l’anion du liquide ionique et l’eau augmente suivant l’ordre PF6- < SbF6- < BF4- < NTf2- < ClO4- < Tf- < NO3-.
4.6.2. Solubilité dans les solvants organiques
Les liquides ioniques sont miscibles avec tous les solvants dont la constante diélectrique est supérieure à 7 [10] (les alcools, les cétones, le THF, le dichlorométane…). Ils sont non-miscibles avec les alcanes, le dioxane, le toluène. L’acétate d’éthyle (εr = 6,0)
5. Applications des liquides ioniques
Les liquides ioniques ont été développés il y a plus d’une vingtaine d’années dans le domaine de l’électrochimie pour la recherche de nouveaux systèmes d’énergie. Ces nouveaux milieux ont ensuite connu un grand intérêt dans les domaines de la synthèse organique et de la catalyse. Plus récemment, de nombreux chercheurs ont tenté de mettre en évidence l’intérêt des liquides ioniques dans le domaine des procédés de l’analyse, et plus particulièrement dans les sciences séparatives : l’extraction liquide – liquide, la chromatographie en phase liquide et gazeuse, et l’électrophorèse capillaire.
Quelques exemples d’applications des liquides ioniques dans les domaines de l’électrochimie, la synthèse organique et la catalyse vont être développées, mais une plus grande attention sera donnée aux applications analytiques.
5.1. Applications en électrochimie [43,46]
Les propriétés des liquides ioniques telles que leur très large domaine d’électroactivité, leur forte conductivité, et leur grande stabilité thermique ont fait de ces nouveaux milieux des candidats de choix dans la recherche de nouveaux systèmes d’énergie (cellule photovoltaïque, batterie…).
Pour exemple, Caja et coll. ont montré, dans la recherche des batteries aux ions Li+ qui constitue un axe très important des batteries rechargeables, que les liquides ioniques, utilisés comme électrolyte, permettaient d’avoir une conductivité cinq fois supérieure aux électrolytes à base de solvant organique et de sel [47,48]. De plus, leurs domaines d’électroactivité peuvent atteindre 4,5 V comparé à 1,2 V pour les électrolytes aqueux et ils offrent une très bonne stabilité thermique et une meilleure solubilité que les composés usuels à base d’ammonium quaternaire.
5.2. Applications en synthèse organique et en catalyse
La synthèse organique et la catalyse sont certainement les deux domaines en expansion dans l’utilisation des liquides ioniques. Il existe de nombreuses applications des liquides ioniques dans ces domaines [43,49]. D’un point de vue chimique, le principal
potentiel des liquides ioniques est d’augmenter le rendement et la cinétique de la réaction et d’améliorer la chimio- et la régiosélectivité par rapport aux solvants organiques. D’un point de vue pratique et économique, la grande variété de liquides ioniques permet d’améliorer les réactions selon les propriétés propres à chaque liquide. De plus, il est possible de séparer plus facilement le produit de la réaction et le catalyseur utilisé permettant ainsi un possible recyclage des liquides ioniques (aspect environnemental).
5.2.1. Réaction de Diels – Alder
La réaction de Diels – Alder est l’une des réactions de condensation carbone – carbone les plus utilisées en chimie organique. Récemment, les liquides ioniques ont été évalués en tant que nouveaux solvants pour ce type de réaction. Chiappe et coll. rapportent dans leur revue que les phénomènes dus aux liquides ioniques qui influencent la réaction sont encore controversés [41]. Un effet solvophobe capable de générer une « pression interne » qui va dans le sens d’une association des réactifs dans une cavité du solvant, a été évoqué par Early et coll. et Debreul et coll. pour expliquer l’aspect cinétique et stéréochimique de cette réaction [50,51]. La réaction réalisée dans les liquides ioniques est un peu plus rapide que dans l’eau et beaucoup plus rapide que dans les solvants organiques, mais l’ajout d’un acide de Lewis permet une amélioration de la rapidité et de la sélectivité de la réaction. Avec cette propriété, Lee et coll. ont mis en évidence l’intérêt des liquides ioniques chloroaluminate dans ce type de réactions [52]. Plus récemment, Aggarwal et coll. ont montré l’importance de la force de la liaison hydrogène anion – cation du liquide ionique sur la cinétique et la régiosélectivité en évaluant plusieurs liquides ioniques [53].
5.2.2. Réaction d’hydrogénation
La réaction catalytique d’hydrogénation d’une double liaison carbone – carbone par transition d’un complexe métallique, est l’une des réactions les plus étudiées en catalyse homogène. Cependant, la séparation des produits et des réactifs reste problématique. Chauvin (Prix Nobel 2005) et coll. ont évalué avec succès le potentiel de certains liquides ioniques à isoler le catalyseur [54]. De nombreuses études ont montré que l’utilisation des liquides ioniques améliorait les rendements et la sélectivité de ce type de réactions [55-57].
5.3. Applications dans le domaine des procédés de séparation et de
l’analyse [3,58-61]
Les différentes propriétés originales des liquides ioniques, décrites dans le paragraphe 4, présentent un très grand intérêt dans le domaine des procédés de séparation et de l’analyse chimique. Leurs capacités à dissoudre des composés organiques apolaires aussi bien que des composés inorganiques ioniques en ont fait des milieux de choix pour les sciences séparatives.
L’extraction liquide – liquide est certainement le domaine dans lequel l’évaluation des liquides ioniques est la plus avancée. L’étude des liquides ioniques dans le domaine des techniques chromatographiques et électrocinétiques est encore peu investiguée, toutefois un grand intérêt lui est porté ces dernières années. Enfin, une application des liquides ioniques comme matrice en MALDI – TOF a été récemment étudiée.
5.3.1. Extraction liquide – liquide
Les propriétés physico – chimiques des liquides ioniques telles que la pression de vapeur négligeable, la non-miscibilité avec d’autres solvants, et la bonne solubilité des composés organiques et inorganiques, en font un milieu de choix pour les techniques d’extraction liquide – liquide et de microextraction en phase liquide (LPME).
5.3.1.1 Extraction liquide – liquide
L’évaluation des liquides ioniques comme alternative aux solvants organiques classiques, a porté dans un premier temps sur l’étude de l’extraction de composés inorganiques ioniques. Dai et coll. ont été les premiers à rapporter la très grande efficacité de l’utilisation du [BMIM,PF6] et [BMIM,NTf2] dans l’extraction du Sr2+ avec l’utilisation de
l’éther-couronne dicyclohexano-18-6 comme agent extractant [62]. De nombreuses études similaires ont ensuite été réalisées selon la nature du liquide ionique (longueur de la chaîne alkyle, nature de l’anion…) [63,65]. Wei et coll. ont réalisé l’extraction des composés Ag+, Hg2+, Cu2+, Pb2+, Cd2+ et Zn2+ dans du [BMIM,PF6] avec de la dithizone comme agent
dans le chloroforme. De plus, il est possible de recycler facilement le liquide ionique par désextraction des ions métalliques en faisant varier le pH.
Les liquides ioniques ont aussi été appliqués à de nombreuses extractions de composés organiques comme les dérivés de benzène substitués [67] ou les hydrocarbures polyaromatiques (HAP) [68]. Carda-Broch et coll. ont évalué en détail les propriétés du
[BMIM,PF6] et ont déterminé le coefficient de distribution [BMIM,PF6]/eau et
[BMIM,PF6]/heptane de quarante composés à différents pH [69]. Une comparaison faite avec
le coefficient de partage octanol/eau a montré une meilleure affinité des composés aromatiques basiques (amines aromatiques) pour la phase liquide ionique et une moins bonne affinité des composés acides (dérivés d’acides aromatiques et phénols) pour la phase liquide ionique.
5.3.1.2 Microextraction en phase liquide (LPME)
La non-volatilité et les propriétés de miscibilité des liquides ioniques ont permis de s’intéresser à ces nouveaux milieux pour réaliser des microextractions sur goutte pendante. Liu et coll. ont réalisé une étude de trois liquides ioniques [CnMIM,PF6] (avec n = 4,6,8)
comparé avec l’octanol en LPME sur des hydrocarbures polyaromatiques (HAP) [70]. Les résultats montrent une amélioration de l’extraction avec les liquides ioniques comparés à l’octanol, et une augmentation de l’efficacité de l’extraction avec les liquides ioniques à chaînes alkyles plus grandes.
Cette technique a aussi été mise en œuvre pour la quantification de formaldéhyde dans les champignons shiitake [71] et dans l’extraction de 45 polluants environnementaux (benzène, toluène, HAP, phénols…) [72].
5.3.2. Chromatographie en phase gazeuse (GC)
Les propriétés des liquides ioniques telles que leur non-volatilité, leur bonne solubilité, leur polarité, leur grande viscosité et le fait qu’ils soient ininflammables, ont fait de ces composés, des candidats uniques dans la recherche de nouvelles phases stationnaires en chromatographie en phase gazeuse. La première application des liquides ioniques comme phase stationnaire en GC a été réalisée par Barber et coll. [73], mais c’est au cours des années 1980 que cet axe de recherche a connu un réel essor par les travaux de l’équipe de Poole
![Figure 3 : Quelques exemples d’anions Cl- , Br- , I- NO3- , SO42- BF4- PF6- , SbF6- Organique Polynucléaire Cl-/[AlCl3] CF3CO2-, CH3 CO 2 - CF3SO3- (Tf-) N(CF3SO2)2- (NTf2- ) N(CN)2- CF3(CF2)3CO2-Inorganique Nature de l’anion Mononucléaire](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/20.892.128.795.807.1097/figure-exemples-anions-organique-polynucléaire-inorganique-nature-mononucléaire.webp)
![Figure 6 : Proposition d’un schéma d’interaction entre le [BMIM,BF 4 ] et la surface](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/45.892.100.783.611.996/figure-proposition-schéma-interaction-bmim-bf-surface.webp)
![Figure 7 : Mécanismes de séparation de composés polyphénoliques mettant en jeu des liquides ioniques à base de cation 1-alkyl-3-méthylimidazolium [99]](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/47.892.123.760.864.1079/figure-mécanismes-séparation-composés-polyphénoliques-liquides-ioniques-méthylimidazolium.webp)
![Figure 8 : Schéma réactionnel de la procédure du greffage covalent des liquides ioniques [112]](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/49.892.134.788.195.403/figure-schéma-réactionnel-procédure-greffage-covalent-liquides-ioniques.webp)
![Figure 11 : Spectre de masse MALDI de l’insuline dans des matrices (A) CHCA et (B) IL [61]](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/52.892.130.791.111.367/figure-spectre-masse-maldi-l-insuline-matrices-chca.webp)
![Tableau 4 : Liquides ioniques à base de cation imidazolium possédant un anion chiral [131] Structure Réf](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/55.892.139.806.102.1105/tableau-liquides-ioniques-cation-imidazolium-possédant-chiral-structure.webp)
![Tableau 5 (Part. 1): Liquides ioniques à base de cation ammonium chiral [131] Réf. 135 136 137 137 137 137 138 138](https://thumb-eu.123doks.com/thumbv2/123doknet/2723364.64578/56.892.136.806.102.1082/tableau-part-liquides-ioniques-cation-ammonium-chiral-réf.webp)