Sénescence et cancer - Double jeu
Texte intégral
Figure


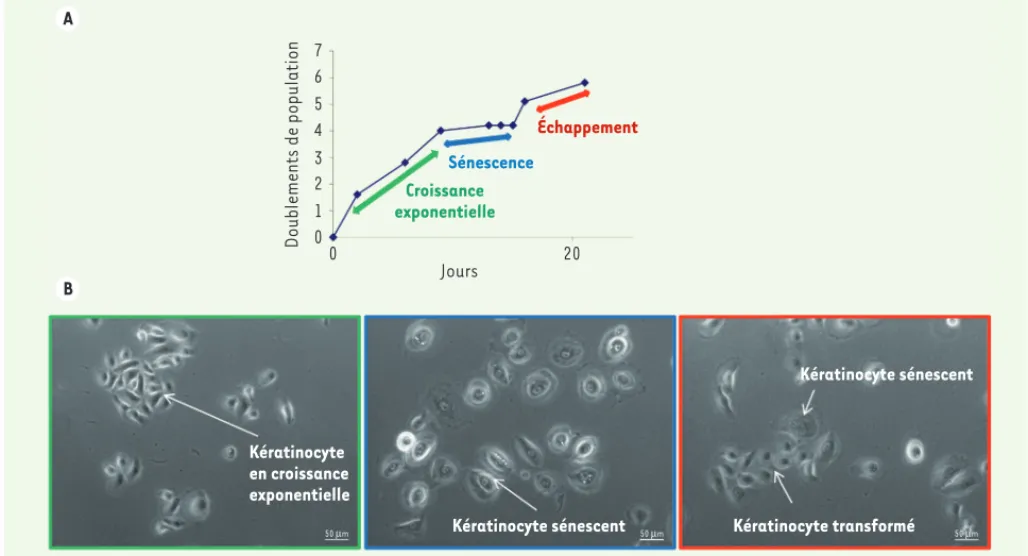
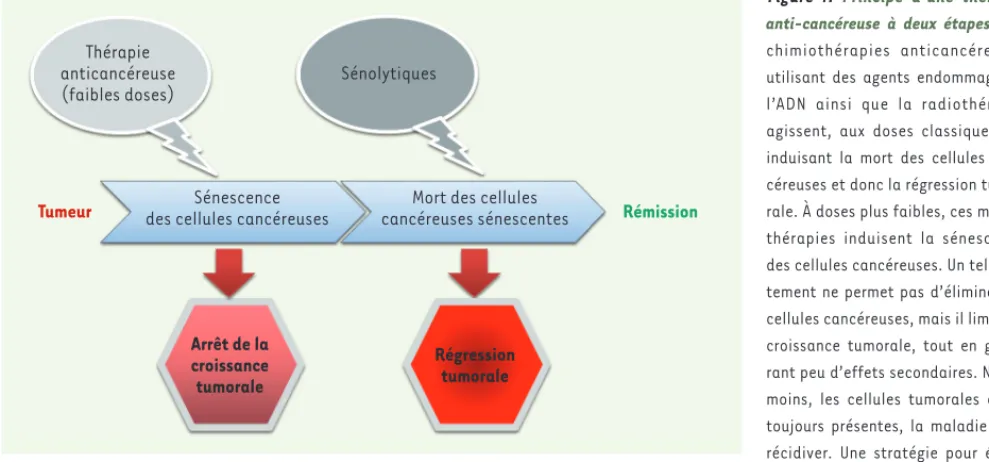
Documents relatifs
Meyerson précise qu’il ne vise pas le pan-algébrisme de l’école de Marburg, pourtant présenté comme la « forme la plus définie » d’idéalisme
Cette localisa- tion semble un élément essentiel au contrôle de l’apoptose assuré par Bcl- 2 car, dans un système cellulaire dont la survie dépend d’un facteur de croissance,
Cela concernerait les cellules sénescentes dont l’arrêt dans le cycle cel- lulaire dépend de p16 et n’implique pas la voie DDR, comme cela semble être le plus souvent le cas pour
Dans les premiers mois de l'après- guerre, où la CEL repartait à zéro, adhérer à la coopérative signifiait sa- crifier un mois de son traitement, sans compter
L’objectif de cette étude est ainsi de déterminer l’effet de l’addition de Sc sur la production du lait chez la vache laitière tout au long de la lactation et dans les conditions
Vous comprendrez pourquoi, plutôt que de nommer tout de suite quel est ce a en fonction à ce niveau qui dépasse celui de l'occultation de l'angoisse dans le désir, s'il est lié à
[r]
3 ère méthode : Avec un logiciel de pointage et d’acquisition de données AVISTEP Ouvrir avistep.exe et suivre la notice + la vidéo intitulée « tutoriel avistep ».
