T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Chimie Biologique
JURY
Mme le Professeur L. El Kihel Rapporteur Mr le Docteur G. Lizard Rapporteur Mr le Professeur H. Gornitzka Examinateur
Mr le Professeur A. Milon Président
Ecole doctorale : Biologie Santé Biotechnologies Unité de recherche : INSERM U563 / AFFICHEM Directeur(s) de Thèse : Dr M. Poirot et Dr S. Silvente-Poirot
Rapporteurs :
Présentée et soutenue par PAILLASSE Michaël Le 25 Septembre 2009
UNIVERSITE TOULOUSE III – PAUL SABATIER U.F.R Sciences de la Vie et de la Terre
T H E S E
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III Discipline :Chimie Biologique
présentée et soutenue par Michaël PAILLASSE ………. le………25 Septembre 2009 ……… Titre :
Métabolisme du cholestérol et cancer
Directeurs de thèse :
__________
Marc Poirot et Sandrine Silvente-Poirot
__________
JURY
Mme le professeur Laïla El Kihel , Rapporteur
Mr le docteur Gérard Lizard , Rapporteur
Mr le professeur Heinz Gornitzka , Examinateur
Qu’il me soit permis de remercier,
Madame le Professeur Laïla El Kihel et Monsieur le Docteur Gérard Lizard qui ont accepté de bien vouloir lire et critiquer ce travail. Qu’ils soient assurés de ma profonde reconnaissance,
Messieurs les Professeurs Heinz Gornitzka et Alain Milon d’avoir accepté d’examiner ce travail,
Madame et Monsieur les Docteurs Sandrine et Marc Poirot, de m’avoir accueilli au sein de l’équipe Métabolisme, Oncogénèse et Différenciation cellulaire de l’U563 avant même qu’elle ne soit baptisée, d’avoir dirigé ces travaux et de m’avoir guidé sur le chemin de la connaissance
Monsieur Stéphane Silvente, pour avoir cru en moi au moment de lancer ce projet Cifre et m’avoir soutenu tout au long de son avancée.
Remerciements
Comme chacun le sait, l’union fait la force. Vous êtes nombreux ceux sans qui je ne serais rien, et j’espère avoir pu, ne serait-ce que dans une infime mesure, avoir pu vous apporter à chacun un petit peu.
Maman, papa, Sophie et Marie,
Arnaud, Christelle, Emilie, Fabien, Lara, Nicolas, Pierrot, Romain, Sylvie, mes amis de toujours,
Dominique,
Les oies sauvages,
Marc, Philippe, Sandrine, Frédéric, Stéphane,
Audrey, Caroline, Grégory, Guillaume, Julia, Loubna, Michel, Et tous ceux que j’oublie de citer mais qui pour moi ont compté,
Merci du fond du cœur.
ABBREVIATIONS
11β-HSD : 11β-hydroxystéroïde deshydrogénase ABC : ATP-binding cassette
ACAT : Acyl-coenzymeA cholestérol acyl transférase ADN : Acide 2-déoxyribonucléique
AEBS : anti-estrogen binding site (site de liaison des anti-oestrogènes) AhR : Aryl hydrocarbon receptor (récepteur à la dioxine)
Akt : Protéine kinase B ARN : Acide ribonucléique C5DS : 3β-hydrxystérol-Δ5 CCK : Cholécystokinine
-désaturase CE : Cholestéryl ester
CERT : Ceramide Transfer Protein
ChEH : Cholestérol 5,6-époxyde hydrolase
COPII : Vésicule de transport antérograde caractérisée par les COat Proteins CT : Cholestane-3β,5α,6β-triol CYP : cytochrome P450 D8D7I : 3β-hydroxystérol-Δ8,Δ7 DCM : Dichlorométhane -isomérase DDA : Dendrogénine A DDB : Dendrogénine B DHCR7 : 3β-hydroxystérol- Δ7 DHCR24 : 3β-hydroxystérol- Δ -réductase 24 DMF : Diméthylformamide -réductase DMSO : Diméthylsulfoxyde DPPE : N,N-Diéthyl-2-(4-benzylphénoxyl)éthanamine α-EC : 5,6α-époxycholestan-3β-ol β-EC : 5,6β-époxycholestan-3β-ol DPPE : Tesmilifène
ECL : Boucle extracellulaire EGF : Endothelial growth factor
Erk : Extracellular signal regulated kinase FXR : Farnesoid X receptor
GDP : Guanosine diphosphate
GST B : Glutathione S-Transférase type B GTP : Guanosine triphosphate
HDL : Lipoprotéine de haure densité Hic : Histamine intracellular receptor
HMGCR : 3-Hydroxyméthyl-glutaryl coenzyme A réductase I-BABP : Ileal bile acid binding protein
I-BAT : Ileal bile acid transporter ICL : Boucle intracellulaire
IGF-1 : facteur de croissance relié à l’insuline de type 1 IL : interleukine
Insig : Insulin-induced gene
LCAT : Lecithine-cholestérol acyl transférase LDL : Lipoprotéine de faible densité
LDLR : Récepteur aux LDL LXR : Liver X receptor
MAPK : Mitogen activated protein kinase mEH : Epoxyde hydrolase microsomale ORP : protéine reliée aux OSBP
OSBP : Protéine de liaison aux oxystérols OSC : 2,3-Oxydosqualène cyclase
P21 : Cyclin-dependent kinase inhibitor 1A
PBPE : N-Pyrrolidino-2-(4-benzyl-phénoxyl)éthanamine PLC : Phospholipase C
PP2A : Protéine phosphatase 2A RCCK : Récepteur à la cholécystokinine RCCK2-WT : Récepteur CCK type 2 sauvage RCPG : Récepteur couplé aux protéines G RE : Récepteur des oestrogènes S1P : Protéase du site 1 de SREBP S2P : Protéase du site 2 de SREBP Scap : SREBP-Clivage activating protein SERM : Selective estrogen receptor modulator SF-1 : Sterol factor 1
SLO : Syndrome Smith-Lemli-Opitz SN1/2
SOD : Superoxyde dismutase
: Substitution nucléophile de type 1 ou de type 2 SREBP : Sterol regulatory element binding protein Sult 2B1 : Sulfo-transférase de type 2B1
TC : cholestérol total (libre + estérifié) Tesmilifène : Cf DPPE
TG : Triglycérides
TPBP : Tumor promoter binding protein TM : Domaine transmembranaire UV : Ultra violet
SOMMAIRE INTRODUCTION ... 3 ETUDE BIBLIOGRAPHIQUE ... 7 I . Le cholestérol ... 7 1. Sources de cholestérol ... 8 2. Homéostasie du cholestérol ... 10
2.1 Modulation de la 3-hydroxy-3-méthylglutaryl-CoA réductase (HMGCR) ... 11
2.2 Stockage sous forme d’esters ou efflux ... 13
2.3 Formation des dérivés du cholestérol ... 14
3. Fonctions physiologiques ... 18
4. Cholestérol, précurseurs et dérivés, implication dans le cancer ... 19
4.1 Les hormones stéroïdes... 19
4.2 La perte du rétrocontrôle du cholestérol sur sa biosynthèse ... 20
4.3 Le cholestérol comme biomarqueur de l’état cancéreux ... 22
4.4 Corrélation entre taux de HDL-C et métabolisme du cholestérol dans le tissu tumoral ... 24
4.5 Rôle de l’estérification du cholestérol ... 25
II. Les oxystérols ... 29
1. origine... 29 2. formation ... 32 2.1 Auto-oxydation ... 32 2.2 Formation enzymatique ... 37 3. effets biologiques ... 39 3.1 Oxystérols et cancer ... 40
3.2 Modulation de la biosynthèse du cholestérol ... 43
3.3 Les récepteurs nucléaires ... 45
3.4 Oxysterol Binding Proteins et protéines associées ... 50
3.5 Le site de liaison des anti-oestrogènes ... 52
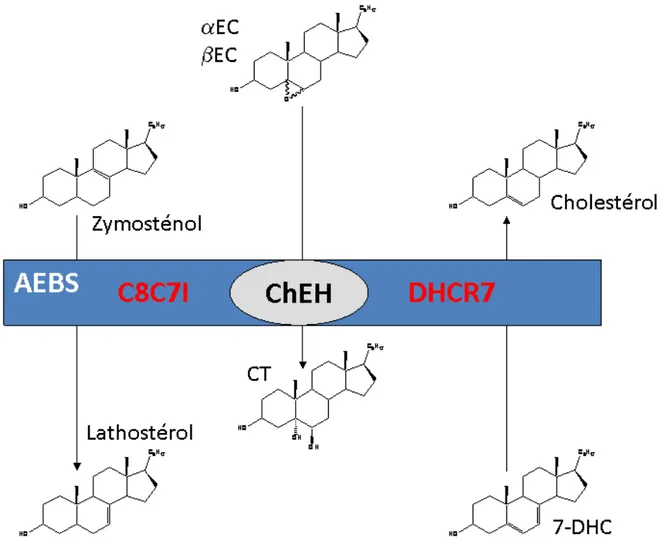
III. La cholestérol époxyde hydrolase ... 55
1. Caractérisation moléculaire et pharmacologique de la ChEH ... 55
2. Activité ChEH et cancer ... 61
3 ChEH et développement ... 62
IV Métabolisme des époxycholestanols : caractérisation des alkylamino-oxystérols ... 63
1. Structures et activités d’aminostéroïdes connus ... 65
2. Etude de la réactivité des 5,6-époxycholestanols ... 68
2.1 Régiosélectivité de l’ouverture des époxydes ... 69
2.2 Influences conformationelles sur la régiosélectivité ... 71
3. Les Dendrogénines ... 77
TRAVAUX PERSONNELS ... 79
Article 1 ... 85
Conclusion ... 87 Article 2 ... 88 Introduction ... 88 Conclusion ... 89 Article 3 ... 90 Introduction ... 90 Conclusion ... 91 Article 4 ... 92 Introduction ... 92 Conclusion ... 93 CONCLUSION ... 94 REFERENCES BIBLIOGRAPHIQUES ... 100 ANNEXES ... 113 Article 5 ... 113 Introduction ...113 Conclusion ...115 45ème RICT... 115
INTRODUCTION
Le tamoxifène (Nolvadex®) est le médicament le plus utilisé au monde pour le traitement par hormonothérapie du cancer du sein exprimant le récepteur des oestrogènes (RE). Il est approuvé par la FDA – équivalent américain de l’AFSSAPS - pour être utilisé en chémo-prévention de ce même cancer chez les sujets à risque (Jordan, 2003). Jusqu’à très récemment, l’action du tamoxifène via le RE était la seule à être prise en compte pour expliquer ses effets biologiques. Cependant, son mécanisme d’action est complexe et ses cibles multiples (Jordan, 2003). Outre ses effets concernant le potentiel anticancéreux du tamoxifène, des effets athéroprotecteurs ont également été mis en évidence. La complexité des mécanismes d’action du tamoxifène a été à l’origine des travaux menés dans l’équipe du docteur Marc Poirot, qui ont permis de mettre en évidence deux nouvelles cibles qui participent aux effets du tamoxifène. Ainsi ses effets sont également médiées par sa liaison au site de liaison des anti-oestrogènes (AEBS), pour lequel il a une affinité semblable à celle qu’il a pour le RE (Delarue et al., 1999; Kedjouar et al., 1999; Sutherland et al., 1980) ainsi que par l’Acyl-CoA Cholestérol Acyl Transférase (ACAT) (de Medina et al., 2004), pour lequel il a une affinité mille fois plus faible. La caractérisation moléculaire du site AEBS, constitué de deux enzymes cholestérogéniques, la 3β -hydroxystérol-Δ8,Δ7-isomérase et la 3β -hydroystérol-Δ7-réductase, a permis de découvrir que le tamoxifène et les autres ligands du site AEBS modulent le métabolisme du cholestérol et provoquent l’accumulation de précurseurs du cholestérol (Kedjouar et al., 2004). Ces données ont amené les membres de l’équipe à étudier le métabolisme du cholestérol en relation avec les processus cancéreux. Ceci comprend l’étude des effets médiés par les ligands du site AEBS et de leur mécanisme d’action ainsi que la caractérisation des dérégulations du métabolisme du cholestérol dans les cellules tumorales.
Le traitement de cellules tumorales cancéreuses par des ligands d’AEBS a mis en évidence une capacité anti-proliférative et différenciante de ces molécules qui a été mise en parallèle de leur capacité à accumuler des précurseurs du cholestérol. La caractérisation des dérivés de cholestérol accumulés sous l’effet des ligands d’AEBS a permis de mettre en avant
un rôle prépondérant des espèces oxygénées réactives dans l’effet anti-prolifératif des ces ligands (de Medina et al., 2009; Payre et al., 2008).
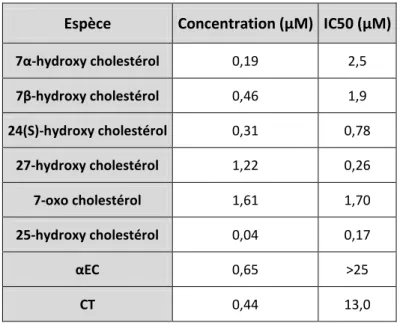
Il a été rapporté d’une part que certains oxystérols sont capables d’inhiber l’hydrolyse des 5,6-époxycholestanols en cholestestane-3β,5α,6β-triol (Hwang, 1990; Hwang and Matin, 1989) catalysée par la Cholestérol époxyde hydrolase (ChEH). Il est également décrit dans la littérature que ces oxystérols sont capables de déplacer le tamoxifène sur AEBS (Sevanian and McLeod, 1986). Nous avons fait le lien entre ces deux études indépendantes et avons cherché à étudier la relation qui pourrait exister entre le site AEBS et la ChEH. Nous avons établi que les deux enzymes formant le site AEBS sont responsables de l’activité ChEH, et que la capacité d’inhibition de cette activité d’hydrolyse par les ligands d’AEBS est proportionnelle à leur affinité pour le site AEBS, laissant à penser que tout ou partie des effets des ligands d’AEBS pourraient être dus à l’inhibition de la ChEH et à l’accumulation des époxycholestanols (travaux personnels - article 2). Le complexe protéique comprenant le site AEBS avait été décrit comme un site de fixation intracellulaire de l’histamine (HIC) et à d’autres amines biogéniques (LaBella and Brandes, 2000). De ce fait nous avons envisagé la possibilité de formation d’adduits obtenus par l’aminolyse des 5,6-époxycholestanols par les différentes amines biogéniques et l’existence d’une voie métabolique générant ces aminoalkyl-oxystérols. Dans ce but nous avons synthétisé ces différents produits et testé leurs activités sur la différenciation et la prolifération cellulaire. Deux molécules leaders ont été identifiées. La Dendrogénine A induit la différenciation cellulaire des cellules tumorales et inhibe la prolifération tumorale in vitro et in vivo. La Dendrogénine B induit la différenciation de précurseurs en cellules nerveuses et favorise la survie des neurones (travaux personnels - article 3). Ces travaux ont permis de mettre le métabolisme du cholestérol, et notamment les 5,6-époxycholestanols, au centre d’une voie d’action de molécules anticancéreuses et ont également mis au jour une nouvelle voie métabolique potentielle de production de molécules ayant des activités bénéfiques pour l’organisme par une action différenciante. Une étude a ensuite été menée afin de comprendre comment et pourquoi les Dendrogénines ne sont issues que du 5,6-époxycholestanol, faisant la relation entre caractéristiques chimiques et réalité biologique (travaux personnels - article 4).
J’ai également pu montrer un autre niveau d’implication du métabolisme du cholestérol dans les processus cancéreux. En effet, il existe une surproduction d’esters et d’époxycholestanols dans les cellules tumorales, qui participent à la prolifération et à l’invasivité cellulaires, suggérant un rôle central du cholestérol et des époxydes dans les processus de maintien d’un état différencié des cellules (travaux personnels - article 1).
Durant mon doctorat, j’ai participé à la mise en évidence du fait que le site AEBS portait l’activité ChEH, et que l’inhibition de cette activité par les ligands d’AEBS rend compte au moins en partie de leurs effets (travaux personnels - article 2), notamment pour l’étude de l’effet des oxystérols sur l’activité ChEH et pour les travaux sur cellules entières. J’ai ensuite été impliqué dans la préparation et l’étude des effets de dérivés de la famille des Dendrogénines A et B afin de réaliser une étude structure-fonction (travaux personnels - article 3), avec un travail exclusivement de synthèse chimique. J’ai également étudié la réactivité des 5,6-époxycholestanols vis-à-vis de nucléophiles afin de déterminer la nature des produits obtenus (travaux personnels - article 4).
Enfin, j’ai étudié le différentiel de métabolisme d’esters de cholestérol entre état tumoral et non tumoral et pu mettre en évidence l’implication directe de ces esters de cholestérol dans la promotion de la prolifération et de la l’invasivité des cellules (Paillasse et al., 2009).
La partie bibliographique du manuscrit présente le métabolisme du cholestérol et les dérégulations qui apparaissent lors des processus cancéreux, la genèse et des différents rôles connus à ce jour des oxystérols, ainsi qu’une présentation du site AEBS et de son implication dans l’effet de ses ligands et enfin les études portant sur les Dendrogénines. La seconde partie présente sous forme de publications parues, soumises ou en préparation les travaux menés durant mon doctorat.
HO O HO OH OH HO HO OH HN N H N HO OH S N H NH2 OH O O H N HO O O O O O O O O O Produit d'oxydation ChEH Mono-oxygénase ACAT ACAT GST B AminoT CT Adduit EC-GSH DA EC ester Cholestérol ester Cholestérol S O O OH Sult2B1 αEC EC sulfate
Résumé graphique de la thèse de doctorat
Schéma global du métabolisme actif du 5,6α-ou 5,6α-époxycholestan-3β-ol et activités biologiques des composés
Les molécules en rouge ont un effet de promotion de la prolifération et/ou de l’invasivité des cellules, celles en gris ont des effets cytotoxiques directs ou indirects à forte dose, celle en bleu induit la re-différenciation de cellules tumorales et contrôle leur prolifération, le rôle biologique de l’adduit EC-GSH n’est pas connu. ACAT : AcylCoA Cholestérol Acyl Transférase ; ChEH : Cholestérol Epoxyde Hydrolase ; GST B : Glutathione S-Transférase type B ; AminoT : Amino-Transférase non connue; Sult 2B1 : Sulfo-transférase type 2B1; EC : 5,6α-époxycholestan-3β-ol ; EC ester : ester gras de EC; CT : Cholestane-3,5,6-triol ; Adduit EC-GSH : glutathion conjugué au EC. DA : Dendrogénine A (5α-hydroxy-6β-[2-(1H-imidazol-4-yl)-ethylamino]-cholestan-3β-ol) ; EC sulfate : ester sulfurique de EC.
ETUDE BIBLIOGRAPHIQUE
I . Le cholestérol
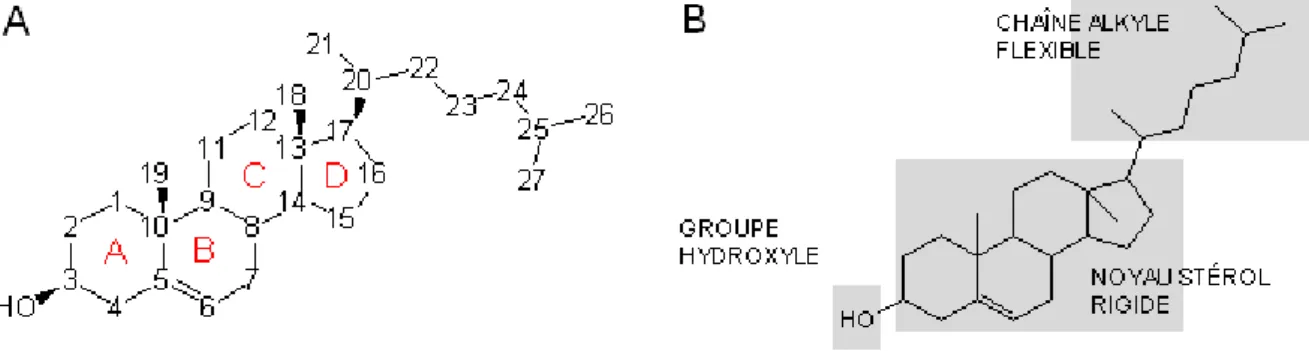
Le cholestérol est une molécule hydrophobe (logP = 7.6), à l’exception de son groupe hydroxyle en 3β qui lui donne son caractère amphiphile (fig. 1A). C’est un dérivé de triterpène comportant 27 carbones. Il est formé d’un noyau tétracyclique stérol, substitué par une chaîne iso-octyle flexible en position 17 et une fonction alcool en position 3 (fig. 1B). Les jonctions de cycles sont en trans ce qui lui confère une structure plane et rigide (fig. 2) caractérisant le cholestérol et la plupart de ses dérivés (Rog et al., 2009). Les deux groupements méthyle 18 et 19, attachés aux carbones 10 et 13 respectivement, sont du même côté, conférant une asymétrie à la molécule (fig 2B). La face plane, sans substituant, est appelée face α et la face comprenant les deux méthyles est appelée face β, et les
substituants qui seront sur une face ou l’autre seront respectivement dits en α ou en β (comme l’hydroxyle en 3).
Figure 1. Structure du cholestérol. A : Numérotation des atomes de carbone. B : Eléments structuraux
Figure 2. Structure tridimensionnelle du cholestérol. A : représentation en bâtons. B : représentation CPK avec coloration des méthyles 18 et 19 en violet (d’après Rog, BBA2009).
Sa structure tridimensionnelle plane lui permet une interaction hydrophobe très favorable avec les phospholipides au sein de la bicouche lipidique de la membrane (Fig 3). Cette caractéristique confère au cholestérol la majorité de ses fonctions (Tabas, 2002).
O H O O O O O HC H2C P RO O O PHOSPHOLIPIDE CHOLESTEROL
Figure 3. Interaction entre le cholestérol et un phospholipide.
1. Sources de cholestérol
Le cholestérol présent dans l’organisme peut avoir deux sources, soit l’alimentation qui constitue l’apport exogène, soit la biosynthèse du cholestérol, le foie étant capable d’en produire à lui seul la moitié (Repa and Mangelsdorf, 2000). On considère que chez l’humain, l’apport de cholestérol par la nourriture est compris entre 300 et 500mg par jour alors que la production endogène est comprise entre 600 et 900mg par jour. Sur ces 1200mg de cholestérol acquis en moyenne, entre 400 et 600mg sont dégradés en acides biliaires, 600mg sont sécrétés dans la bile, 85mg sont utilisés pour le renouvellement des membranes des cellules et 50mg sont utilisés pour la synthèse d’hormones stéroïdes. Dans des conditions physiologiques normales, la balance entre apport et utilisation du cholestérol par l’organisme est équilibrée (Repa and Mangelsdorf, 2000).
Le cholestérol issu de l’alimentation est capté au niveau de l’intestin, exporté vers le foie puis distribué aux autres tissus par les lipoprotéines de faible densité (LDL). L’élimination se fait par le chargement sur les lipoprotéines de haute densité (HDL) qui suivent le cheminement inverse vers le foie (transport inverse du cholestérol) schématisé à la figure 4 (Chang et al., 2006).
Figure 4. schéma simplifiée de la circulation du cholestérol dans l’organisme
partir de l’acétate, par une succession de 19 réactions enzymatiques dont l’étape limitante est la synthèse de mévalonate par l’3-Hydroxy-3-MéthylGlutaryl-CoenzymeA réductase (HMGCR) (Brown and Goldstein, 1980). La synthèse est globalement scindée en deux, les étapes pré- et post-lanostérol, le lanostérol étant le premier intermédiaire cyclique du cholestérol et de fait le premier stérol. La partie pré-lanostérol, après formation du mévalonate consiste en une série de condensations pour former le farnésyl-pyrophosphate. C’est un composé clé car il ouvre à la fois la voie du cholestérol et celle des dolichols (Rip et al., 1985). La condensation de deux farnésylpyrophosphates par la squalène synthase donne le squalène, qui est ensuite époxydé par la squalène oxydase puis cyclisé en lanostérol par la 2,3-oxydosqualène cyclase (OSC) (fig. 5A). Le lanostérol conduit ensuite au cholestérol et à ses dérivés par deux voies parallèles dites de Bloch et de Kandusch-Russel (fig. 5B).
B : étapes post lanostérol, D’après (Kedjouar et al., 2004). DHCR24 : 3β-hydroxystérol-Δ24-réductase; D8D7I : 3β-hydroxystérol-Δ8,Δ7-isomérase; C5DS : 3β-hydrxystérol-Δ5-désaturase; DHCR7 : 3β-hydroxystérol- Δ7
2. Homéostasie du cholestérol
-réductase.
Le taux intracellulaire de cholestérol est régulé par plusieurs voies dans les tissus périphériques. D’une part par la balance entre l’apport extérieur par les LDL et son efflux par les HDL, d’autre part la régulation de la synthèse endogène, principalement au niveau de l’HMGCR, et le stockage sous forme d’esters.
2.1 Modulation de la 3-hydroxy-3-méthylglutaryl-CoA réductase (HMGCR)
Les protéines de liaison aux éléments de réponse aux stérols (SREBP) sont les contrôleurs majeurs de la transcription de nombreux gènes impliqués dans le métabolisme du cholestérol, dont ceux impliqués dans sa biosynthèse (HMGCR) et son import dans les cellules (LDL-récepteur). Les SREBP sont présentes dans le réticulum endoplasmique. SREBP-1c est impliquée dans la stimulation de la synthèse des acides gras et SREBP-2 dans la stimulation de celle du cholestérol et activent donc en partie les mêmes enzymes précoces communes aux deux voies de synthèse (Brown and Goldstein, 1997). L’activation de SREBP se fait par clivage protéolytique au niveau du Golgi par les protéases S1P et S2P (protéases du site 1 et 2) lorsque la synthèse de cholestérol est nécessaire (fig. 6A). La protéine activée est alors transportée dans le noyau pour activer la transcription des gênes. Le transport et donc l’activation des SREBP est régulé par la protéine Scap (protéine activant le clivage des SREBP) qui contient un domaine de détection des stérols par lequel se fait la modulation du métabolisme. En absence de stérols, Scap se lie à SREBP, et favorise son export vers le golgi en recrutant les protéines du complexe COPII (fig. 6B). Lorsque le taux de cholestérol est suffisant, il s’immisce dans la membrane du RE, interagit avec le domaine sensible aux stérols de Scap de manière spécifique et saturable et bloque l’export vers le Golgi du complexe Scap-SREBP en favorisant l’interaction entre Scap et Insig (INSulin-Induced Gene), protéine d’encrage du RE (Sun et al., 2005).
Lorsque le taux de cholestérol est faible, Insig n’est pas liée au complexe SREBP/Scap, elle subit alors une ubiquitinylation et est transportée vers le protéasome pour être dégradée et ainsi ne pas pouvoir bloquer la biosynthèse de cholestérol. L’interaction entre Scap et le cholestérol est faible, et la liaison à Insig stabilise ce complexe, et de manière analogue, Insig est dégradée par le protéasome après ubiquitination lorsqu’elle n’est pas au sein du complexe Insig/Scap-cholestérol/SREBP. De plus, Insig est régulé par SREBP, le taux d’ARN messager d’Insig est dépendant du taux de cholestérol. Dans les cellules riches en cholestérol, le complexe est stable, et donc le taux de SREBP nucléaire est bas. Lorsque le niveau de cholestérol diminue, le complexe Insig/Scap/SREBP se dissocie, et le taux de SREBP nucléaire redevient suffisant pour induire la transcription des gênes de la cholestérogénèse et également d’Insig, mais la protéine est rapidement dégradée tant que le taux de
cholestérol ne permet pas l’interaction avec Scap. Le blocage de la synthèse de cholestérol est donc effectif seulement lorsqu’il y a à la fois suffisamment de cholestérol et d’Insig (Gong et al., 2006). Ce double contrôle serait mis en place afin de ne pas inhiber la voie du mévalonate de manière trop précoce si l’accumulation de cholestérol se fait préférentiellement à celle des autres isoprénoïdes (Goldstein et al., 2006).
Figure 6 : activation de la transcription dépendante de SREBP. A : Export et maturation de SREBP. B : mécanisme de contrôle de l’export de SREBP par le taux de stérols
HMGCR possède également un domaine de détection des stérols semblable à celui de Scap. Elle est donc de ce fait régulée de façon à délivrer un taux constant de substrats pour la synthèse des isoprénoïdes tout en évitant l’accumulation de cholestérol ou de ses précurseurs potentiellement toxiques (Goldstein and Brown, 1990). Au niveau transcriptionnel, le gène HMGCR est sous contrôle de SREBP, alors qu’au niveau post-transcriptionnel, la réductase est régulée par une dégradation stimulée par les stérols, et ces deux phénomènes impliquent Insig (fig. 7). Les précurseurs du cholestérol et notamment le lanostérol sont plus efficaces que le cholestérol lui-même pour induire la dégradation. Lorsque le taux de lanostérol dans le RE est suffisant, il va provoquer l’interaction entre HMGCR et Insig. Insig est liée de manière constitutive à un complexe gp78/ubc-7/VCP responsable de l’ubiquitination de l’HMGCR préalable à sa dégradation par le protéasome (Goldstein et al., 2006).
Figure 7. Contrôles transcriptionnel et post-traductionnel de la voie du mévalonate par le cholestérol et ses précurseurs, d’après Sun et al., 2005.
2.2 Stockage sous forme d’esters ou efflux
Outre l’inhibition de la synthèse, l’homéostasie du cholestérol peut être régulée par son efflux et son stockage. L’augmentation de l’une de ces deux voies influençant négativement l’autre (Yamauchi et al., 2004).
L’efflux du cholestérol est sous contrôle du sous-type α des récepteurs LXR (Liver X receptor), un récepteur nucléaire qui est un facteur de transcription ligand-dépendant qui module entre autres le taux de protéines de transport de la famille des ATP-binding cassettes (ABC) et plus particulièrement ABC-A1 et G1 impliquées dans le transport du cholestérol vers la membrane plasmique pour son appariement aux lipoprotéines formant les HDL et son export vers le foie (Gelissen et al., 2006).
La formation intracellulaire d’esters de cholestérol a deux buts physiologiques, la limitation du taux de cholestérol et d’oxystérols libres dans les cellules et également la constitution dans les cellules de stocks de cholestérol, au sein de gouttelettes lipidiques, rapidement utilisables pour la synthèse de membranes. De ce fait, l’hydrolyse des esters de cholestérol et la ré-estérification du cholestérol libéré ont lieu en continu (fig. 8).
Réticulum endoplasmique Cytosol CE CE Chol Chol nCEH ACAT1 LDLR LDL Endosome/lysosome Efflux via HDL Synthèse de membranes Réticulum endoplasmique Cytosol CE CE Chol Chol nCEH ACAT1 LDLR LDL Endosome/lysosome Efflux via HDL Synthèse de membranes
Figure 8. Schéma de la dynamique de stockage par estérification du cholestérol dans les tissus périphériques.
Le stockage du cholestérol se fait sous forme d’esters d’acides gras. Dans les cellules, deux enzymes sont responsable de l’estérification, ACAT 1 et ACAT-2 (Acyl-CoA Cholestérol Acyl tranférase 1 et 2) (Chang et al., 2000). Les enzymes ACAT estérifient le cholestérol ainsi que certains de ses dérivés à l’aide d’acyl-CoA (Cases et al., 1998). ACAT-2 est présente dans l’intestin et le foie où elle est impliquée dans l’absorption des stérols alimentaires. ACAT-1 est à peu près ubiquitaire chez l’homme et particulièrement présente dans le hépathocytes, les macrophages, les cellules de la peau, les neurones et les entérocytes intestinaux (Chang et al., 2000; Lee et al., 1998; Sakashita et al., 2000). Elle est impliquée dans le stockage du cholestérol estérifié sous forme de gouttelettes lipidiques, dans l’absorption du cholestérol exogène et à la production d’esters de cholestérol pour la formation des lipoprotéines (Buhman et al., 2000). L’hydrolyse des esters de cholestérol est effectuée par la cholestéryl ester hydrolase neutre (nCEH).
2.3 Formation des dérivés du cholestérol
Le cholestérol est également utilisé pour la formation de dérivés tels que les oxystérols, dont la formation, le métabolisme et les rôles biologiques seront présentés dans la suite du manuscrit, et les hormones stéroïdes. Dans le foie, le cholestérol provenant des HDL émis par les tissus périphériques est dégradé en acides biliaires et excrété vers l’intestin.
Les hormones stéroïdes (androgènes, glucocorticoïdes, minéralocorticoïdes, œstrogènes et progestagènes) sont formées à partir de cholestérol au sein des tissus stéroïdogéniques primaires (gonades et les glandes surrénales). L’étape initiale est commune à toutes les hormones et consiste à la formation de la prégnénolone à partir du cholestérol par coupure oxydative de la liaison entre les carbones 20 et 22 du cholestérol (fig. 9). Cette réaction est activée par le cytochrome P450scc (cholesterol Side-Chain Cleavage) également appelé CYP11A1 (Hu et al., 2004).
Le cholestérol est éliminé de l’organisme au niveau du foie, après avoir été transformé en acides biliaires. Les acides biliaires entrent alors dans l’iléum pour être excrétés. Les acides biliaires servent à dégrader le cholestérol mais également les oxystérols et les hormones stéroïdes. Ainsi les acides biliaires proviennent de plusieurs voies, la voie classique à partir du cholestérol qui a lieu dans le foie, et la voie acide qui permet de dégrader dans le foie certains oxystérols et les hormones stéroïdes formées dans les tissus stéroïdogènes comme décrit dans le figure 10 (Lefebvre et al., 2009). Il existe en plus deux voies ultraminoritaires
HO O HO OH O OH HO O O O O HO O O HO OH Cholestérol Prégnénolone Progestérone Aldostérone Cortisol Oestradiol Testostérone P450SCC Progestagènes Androgènes Oestrogènes Glucocorticoïdes Minéralocorticoïdes OH OH O
HO HO CYP7A1 (RE) CYP27A1 (mitochondrie) HO OH OH OH
Voie neutre ou classique (75%)
Voie acide (25%)
CYP7B1 (RE)
3β-hydroxy-∆5-C27 stéroïde oxydoréductase (HSD3B7) (RE)
O OH 7α-Hydroxycholest-4-èn-3−οne CYP8B1 (RE) Dérivés 12-hydroxylés HO OH OH O HO OH OH O H H OH
Acide chénodéoxycholique Acide cholique
HO OH O H HO OH O H OH Sels biliaires chénodéoxycholiques Sels biliaires choliques
Acide litocholique Acide déoxycholique
7α,27-Dihydroxycholest-4-èn-3−οne
O OH
OH HO
OH
Figure 10. Voies métaboliques des acides biliaires. D’après Lefebvre, 2009. Les acides biliaires sont formés par deux voies principales, dites classique et acide. Il existe deux voies minoritaires ayant pour substrat initial le 24-hydroxycholestérol ou le 25-24-hydroxycholestérol. Les acides litocholique et déoxycholique sont dits acides biliaires secondaires. RE : réticulum endoplasmique.
3. Fonctions physiologiques
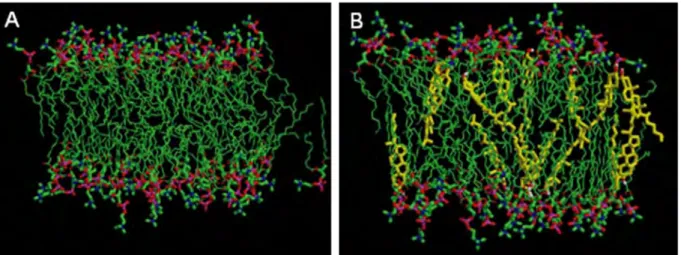
Le cholestérol est le stérol le plus abondant chez les animaux, et se retrouve principalement au sein des membranes biologiques (ref). Il est distribué de manière variable et non aléatoire dans ces membranes, au sein desquelles il interagit avec les phospholipides tels que les sphingomyélines et les phosphatidylcholines. Ces interactions dépendent de la nature de ces phospholipides (Rog et al., 2009), mais globalement le cholestérol permet d’ordonner les membranes (fig 11), ce qui a pour effet de les rigidifier dans un état fluide-pauvre en cholestérol et au contraire de les fluidifier dans l’état de gel-riche en cholestérol (Vist and Davis, 1990).
Figure 11. Effet structurant du cholestérol. A : modélisation de l’organisation d’une bicouche de dipalmitoylphosphatidylcholine (DPPC). B : modélisation de la bicouche de BPPC après ajout de cholestérol. Le cholestérol en jaune, la DPPC colorée selon la norme CPK ; d’après (Smondyrev and Berkowitz, 1999)
Cette capacité d’organisation permet également au cholestérol d’augmenter la résistance mécanique, de diminuer la perméabilité passive vis-à-vis de l’eau ou d’autre petites molécules et des gaz (Rog et al., 2009). Des zones de la membrane riches en cholestérol comprennent des structures spéciales, les cavéoles et les rafts lipidiques, impliqués dans des processus semblables à l’endocytose et dans la présentation de protéines, le trafic membranaire et la transduction du signal (Simons and Toomre, 2000).
Le cholestérol a par ailleurs une influence sur les nombreuses protéines présentes dans les membranes. Il module l’activité de ces protéines soit de manière non spécifique en influençant la dynamique membranaire (Lee, 2004; Ohvo-Rekila et al., 2002; Yeagle, 1985), soit de manière spécifique en interagissant directement avec les protéines et/ou les ligands
de ces protéines pour modifier leur structure tridimensionelle (Addona et al., 1998; Hua et al., 1996; Nunez and Glass, 1982).
Ces propriétés sont spécifiques du cholestérol et de sa structure tridimensionnelle puisque son remplacement par le desmostérol, différencié par une seule double liaison en 24,25, modifie de manière drastique la structure membranaire et l’activation des protéines (Aittoniemi et al., 2006; Vainio et al., 2006).
En plus des activités au niveau de la membrane, le cholestérol est également impliqué dans l’activation de protéines, notamment celle de la Sonic Hedgehog, protoprotéine impliquée dans le développement de l’embryon (Porter et al., 1996), et plus récemment impliquée dans différents processus cancéreux (Jacob and Lum, 2007; Rubin and de Sauvage, 2006). Le cholestérol réalise un clivage protéolytique de la protoprotéine, libérant la partie N-terminale, qui est impliquée dans la signalisation, couplée de manière covalente au cholestérol (Porter et al., 1996).
4. Cholestérol, précurseurs et dérivés, implication dans le cancer
4.1 Les hormones stéroïdes
Des études récentes ont mis en avant le rôle probable des glucocorticoïdes dans la progression tumorale. En provoquant une diminution de l’activité de défense immunitaire contre les cellules cancéreuses, les glucocorticoïdes pourraient favoriser l’échappement des cellules cancéreuses et donc la progression plus rapide des tumeurs puis des métastases (Zhang et al., 2007).
L’implication des oestrogènes dans le cancer chez la femme est très largement décrite dans la littérature. L’oestradiol, via l’activation des récepteurs des oestrogènes (ERα et β), est impliqué dans la progression tumorale pour les cancers du sein et de l’endomètre, en modulant l’activation de facteurs de croissance comme le récepteur à l’EGF (endothelial growth factor) ou celle de proto-oncogènes comme c-myc ou c-fos (Pasqualini, 2004).
De manière semblable chez l’homme, les androgènes sont impliqués dans la survenue de cancer de la prostate. Ces effets impliquent l’activation du facteur Hif (hypoxia inducible factor), et de l’angiogénèse par stimulation de la formation de VEGF (vascular endothelial growth factor) (Li and Cozzi, 2009). D’autre part, il a été montré un lien étroit entre cholestérogénèse et survenue de cancer de la prostate (Schaffner, 1981; Swinnen et al., 1996). Cette cholestérogénèse accrue dans les cancers de la prostate a été corrélée à une activité accrue des SREBP-1c et 2 (Ettinger et al., 2004) qui augmente à mesure que le cancer développe son indépendance aux androgènes.
4.2 La perte du rétrocontrôle du cholestérol sur sa biosynthèse
Dès le début du XXème siècle, des médecins ont suggéré que le cancer pourrait être dû à la cristallisation du cholestérol dans les cellules vivantes, et montré que le cholestérol s’accumulait effectivement dans certaines tumeurs. Ils proposaient alors que cet évènement, avec d’autres, pourrait être associé au contrôle de la prolifération. Cependant ces études n’ont pas été généralisées, et la découverte du rôle physiologique du cholestérol dans les membranes a prévalu sur ces études. Lorsque les recherches ont mis à jour d’autres rôles pour le cholestérol, notamment dans l’athérosclérose, de nouvelles études peu connues ont proposé de nouvelles pistes. Ainsi des études statistiques menées dans le cadre de l’athérosclérose ont montré une corrélation positive entre ces problèmes vasculaires et la survenue de certains cancers, notamment pulmonaires, colo-rectaux et des leucémies (Alcantara and Speckmann, 1976), avec comme facteur commun le cholestérol. Une explication à cette apparente accumulation de cholestérol dans les processus cancéreux a été obtenue par les études des prix Nobel Goldstein et Brown qui ont montré que dans ces conditions prolifératives, la voie d’endocytose des LDL et la voie du mévalonate sont toutes deux suractivées (Brown and Goldstein, 1980). Des études menées par déprivation du mévalonate ont mis en évidence un blocage en phase S, qui n’est pas relancé par l’ajout de cholestérol, mais l’est par un ajout de mévalonate, montrant qu’un ou des produits de la voie du mévalonate autres que le cholestérol soit impliqués dans la prolifération cellulaire, au niveau de la réplication de l’ADN (Siperstein, 1984). La perte de rétrocontrôle du cholestérol sur la voie du mévalonate est attribuée à une mutation ponctuelle de SCAP qui
augmente son activité de stimulation du clivage de SREBP (Hua et al., 1996). Comme SREBP module l’expression de toutes les enzymes cholestérogéniques, c’est la voie du mévalonate dans sa globalité qui est suractivée dans les processus cancéreux (Brown and Goldstein, 1997).
La voie du mévalonate produit des dolichols et de l’ubiquinone en plus du cholestérol (fig. 12). Ces molécules ont des rôles physiologiques très importants mais n’ont pas de lien connu avec le cancer. Les ARNt sont eux impliqués dans la synthèse protéique, et leur isoprénylation sert à l’interaction avec l’ARN messager traduit. Par contre, le farnésyl-pyrophosphate (et le géranylgéranylfarnésyl-pyrophosphate) est lui impliqué dans la prénylation des protéines, modification post-traductionnelle de certaines protéines (petites protéines G, protéines G hétérotrimériques et certaines kinases) qui favorise leur ancrage à la membrane et leur activité. L’exemple le plus connu est du proto-oncogène Ras qui est impliqué dans des voies de signalisation cruciales pour le contrôle de la prolifération cellulaire, et dont les formes mutées oncogènes sont retrouvées dans 30% des cancers (Cox and Der, 1997; Goldstein and Brown, 1990). Dans les cellules cancéreuses, il a été montré qu’une plus grande partie du pool de farnésylpyrophophate est utilisé pour la formation de composés dits « non-stérols » (farnésylpyrophosphate, dolichols, ubiquinone) que dans les cellules saines (fig. 13). La formation de cholestérol est proportionnellement défavorisée mais reste plus importante dans les tissus cancéreux par rapport aux cellules.
La dérégulation de la voie du mévalonate due à la perte de rétrocontrôle du cholestérol sur la HMGCR est un phénomène précoce particulièrement important dans le processus de cancérogénèse (Siperstein, 1984), qui est retrouvé dans la quasi-totalité des cancers. Ainsi donc, il est probable que cette dérégulation affecte le métabolisme cellulaire à d’autres niveaux et par d’autres mécanismes que la prénylation de Ras.
Figure 12. Les différents produits de la voie du mévalonate. Le potentiel de transformation de certaines protéines par le farnésylpyrophosphate est également indiqué.
Figure 13. Activité différentielle de la voie du mévalonate entre état sain et cancéreux. L’épaisseur des flèches est proportionnelle à l’activité de synthèse. La flèche en pointillés représente le contrôle transcriptionnel du cholestérol sur la HMGCR. D’après (Mo ExpBiolMed2004)
4.3 Le cholestérol comme biomarqueur de l’état cancéreux
A partir des indices qui relient les dérégulations du métabolisme du cholestérol dans les processus cancéreux, quelques équipes de cliniciens ont cherché à déterminer des facteurs permettant le suivi de l’évolution de la maladie. Pour ce faire, différents facteurs
comme le taux global de cholestérol (TC), de triglycérides (TG), de cholestérol au sein des HDL (HDL-C), des LDL (LDL-C) ainsi que l’abondance des Apoprotéines Apo-AI et -B100 ont été suivis et comparés entre prélèvements de patients atteints de cancers du sang et de témoins sains. Les différents facteurs évoluent entre tissus sain et cancéreux. Cependant, le seul qui varie toujours dans le même sens est le taux de HDL-C, aussi bien dans les cas de leucémies (tableau 1) que de tumeurs solides (tableau 2). Le taux de HDL-C apparaît alors comme un bon marqueur de l’état cancéreux.
Evolution des facteurs étudiés entre état sain et cancéreux
Références
TC TG HDL-C LDL-C Apo-AI Apo-B100
=/- =/+ - = - = (Dessi et al., 1991; Dessi et
al., 1992; Dessi et al., 1995)
- + - =/- - = (Baroni et al., 1994; Baroni
et al., 1996)
- + - - - n.d. (Juliusson et al., 1995)
- n.d. - = - = (Aixala et al., 1997)
= + - = - = (Halton et al., 1998)
- n.d. - - n.d. n.d. (Allampallam et al., 2000)
Tableau 1. Comparatif du taux de triglycérides, de cholestérol et d’apoprotéines circulants entre des témoins sains et des patients atteints de leucémies. + : augmentation, - : diminution, = : pas de variation.
Le seul facteur qui évolue de la même façon dans chaque cas étudié est le taux de cholestérol dans les HDL, qui diminue chez les malades par rapport aux témoins sains. Les cercheurs ont alors postulé que le taux de HDL-C pourrait être un facteur de suivi de l’évolution du cancer. L’évolution des autres facteurs ne permet pas de suivi de ce type. Certaines de ces études ont ainsi pu montrer que suite à une chimiothérapie, le taux de HDL-C retourne à la normale. Il apparaît également que cette diminution ne soit pas une cause mais plus probablement une conséquence métabolique du cancer. Il est important également de noter que cette baisse de HDL-C a été diagnostiquée également chez des patients atteints de tumeurs bénignes, ce qui pourrait faire de cette baisse du taux de HDL-C un marqueur précoce de risque.
Cancer
Evolution des facteurs étudiés entre état sain et cancéreux
Références TC TG HDL-C LDL-C Apo-AI Apo-B100 Poumon = = - = n.d. n.d. (Dessi et al., 1992) - = - = n.d. n.d. (Umeki, 1993) n.d. n.d. - - -n.d. n.d. n.d.
(Siemianowicz et al., 2000a; Siemianowicz et al., 2000b; Siemianowicz et al., 2000c) Tractus gastro-intestinal - n.d. - n.d. n.d. n.d. (Dessi et al., 1984) = = - n.d. n.d. n.d. (Bayerdorffer et al., 1993) Adénome
colorectal n.d. + - + n.d. n.d. (Boyd and McGuire, 1990)
Sein
+ + - n.d. n.d. n.d. (Knapp et al., 1991)
- + - = n.d. n.d. (Kumar et al., 1991)
+ + - + n.d. n.d. (Araki and Yamamoto, 1992)
= n.d. - n.d. n.d. n.d. (Kokoglu et al., 1994)
- + - - n.d. n.d. (Schreier et al., 1999)
n.d. + - n.d. n.d. n.d. (Subbaiah et al., 1997)
Ovaires - n.d. - n.d. n.d. n.d. (Kanel et al., 1983)
Foie
n.d. n.d. - n.d. n.d. n.d. (Ahaneku et al., 1991)
n.d. n.d. - n.d. - =/+ (Ostroumova et al., 1986)
- n.d. - n.d. n.d. n.d. (Alexopoulos et al., 1987)
Tous cancers - + - - n.d. n.d. (Fiorenza et al., 2000)
Tableau 2. Comparatif du taux de triglycérides, de cholestérol et d’apoprotéines circulants entre des témoins sains et des patients ayant des tumeurs solides. + : augmentation, - : diminution, = : pas de variation.
4.4 Corrélation entre taux de HDL-C et métabolisme du cholestérol dans le tissu tumoral
Le métabolisme du cholestérol dans l’organisme est réglé via une série complexe de transporteurs et de mécanismes biosynthétiques qui dépendent des échanges continus entre le sang et les tissus, il convient de se demander dans quelle mesure une altération notable du métabolisme du cholestérol peut avoir des répercussions sur le taux plasmatique HDL-C.
Une corrélation négative a été établie entre le taux de cholestérol dans les HDL et la capacité de prolifération dans les cellules tumorales étudiées. Un faible taux de LCAT et de
HDLchol a été également trouvé chez le rat foetal et néonatal (Argiles and Herrera, 1981). Le taux de HDLchol est décrit pour varier en fonction de la pousse tumorale chez le rat atteint d’un hépatome, diminuant lors de la progression de la tumeur et retournant à la normale lors de la phase stationnaire de la tumeur (Dessi et al., 1992). L’action des HDL dans le maintien de l’homéostasie du cholestérol par stimulation du transport inverse, bien décrit dans le processus d’athérosclérose (Eisenberg, 1984) varie selon la capacité proliférative des cellules. En effet, il a été montré que la stimulation de l’efflux du cholestérol induit par un traitement avec des HDL naissants est plus faible pour des cellules à prolifération rapide ou des cellules transformées (Gebhard et al., 1987; Pittman et al., 1987). De plus, les récepteurs de surface permettant l’ancrage de l’ApoA1 des HDL à la membrane sont moins exprimés lorsque la cellule est soumise à un facteur de croissance et cet effet est estompé lorsqu’elle est soumise à un inhibiteur de prolifération cellulaire (Oppenheimer et al., 1988). Le traitement avec des inhibiteurs d’ACAT empêche également la diminution de l’efflux de cholestérol par les HDL observée dans les processus prolifératifs (Anchisi et al., 1990; Batetta, 1991) Les auteurs de ces études ont assimilé ces altérations du transport par les HDL dans le plasma à un reflet des altérations du métabolisme du cholestérol dans les cellules (Muntoni et al., 2009).
4.5 Rôle de l’estérification du cholestérol
Les résultats précédemment exposés montrent qu’une dérégulation globale de la cholestérogénèse apparaît dans les cellules pendant leur transformation en cellules tumorales, celle-ci est accompagnée d’une capacité accrue à capter le cholestérol circulant dans les LDL et d’un efflux très faible exprimé par une baisse notable du transport inverse vers le foie. Une accumulation importante de cholestérol a donc lieu dans les cellules.
Dans les tissus, l’ACAT-1 a pour role de maintenir un taux constant de cholesterol libre en accumulant l’excès de cholesterol sous forme d’ester (Lada et al., 2004). Ceci pour éviter une accumulation de cholestérol libre qui pourrait être délétère dans le cas où l’efflux ne permettrait plus de maintenir l’homéostasie (Willner et al., 2003). Dans une situation cancéreuse, l’import de cholestérol peut être augmenté, sa biosynthèse est accrue et son efflux fortement diminué. Le rôle de l’ACAT est alors crucial. D’un point de vue chimique, les
esters de cholestérol, issus du couplage du cholestérol avec un acide gras à longue chaîne, sont plus hydrophes que le cholestérol, ce qui rend impossible leur insertion dans les membranes des cellules, et ils s’accumulent sous forme de gouttelettes lipidiques dans le cytoplasme et sont retrouvés au coeur des lipoprotéines dans la circulation. Dans les cellules normales, on retrouve 65-90% de cholestérol libre dans la membrane et 5 à 10% d’esters de cholestérol dans les gouttelettes lipidiques. L’accumulation des esters de cholestérol dans les tissus produisant les lipoprotéines (foie, intestin) est normale ainsi que dans les organes sécrétant les hormones stéroïdes. Dans le reste de l’organisme, cette accumulation est associée à des pathologies et notamment l’athérosclérose.
Différentes études de la littérature cherchant à élucider les disfonctionnements du métabolisme du cholestérol dans les tumeurs montrent une accumulation d’esters de cholestérol et une activité ACAT accrue au sein des cellules cancéreuses (Boyd and McGuire, 1990; Umeki, 1993). Ces phénomènes ont été reliés à la croissance cellulaire.
Les premières études menées sur des tissus normaux placés dans des conditions de prolifération importante lors du développement, par chirurgie (hépatectomie partielle) ou par voie chimique (empoisonnement au plomb) on permis de mettre en évidence une forte augmentation du taux d’esters de cholestérol sans variation significative du taux de cholestérol (Dessi et al., 1984; Dessi et al., 1985). Dans ces deux situations, le retour à une prolifération normale est accompagné de manière synchrone d’un retour à la normale du métabolisme des esters de cholestérol (Dessi et al., 1986). Des résultats similaires ont été obtenus pour le rein chez l’homme (Ledda-Columbano et al., 1987), le pancréas, le cerveau et la moëlle épinière (après hémolyse) chez le rat ou encore au niveau de l’aorte chez le fœtus de lapin (Cayatte and Subbiah, 1989; Dessi et al., 1990; Rao et al., 1986). Ces résultats vont dans le sens d’un rôle fonctionnel de l’estérification du cholestérol pendant la prolifération cellulaire.
La prolifération continue des cellules est une des caractéristiques de l’état tumoral. Comme le métabolisme des esters de cholestérol paraissait plus important dans des conditions de prolifération rapide, quelques études ont évalué son évolution dans les processus cancéreux afin de déterminer si un lien pouvait appraître entre les deux phénomènes. Un comparatif entre deux lignées tumorales issues d’un même tissu ayant des vitesses de prolifération différentes ont permis de déterminer une corrélation positive entre
accumulation d’esters de cholestérol et vitesse de prolifération (Gebhard et al., 1987; Rao et al., 1986). Des résultats similaires ont été obtenus in vivo à partir de nombreuses autres tumeurs implantées chez le rat, la souris ou le hamster. Le taux d’estérification du cholestérol est ainsi jusqu’à 300 fois plus important dans une tumeur testiculaire de Leydig implantée chez le rat que dans un testicule sain (Konishi et al., 1991).
Finalement, des dérégulations du même ordre sont retrouvées chez l’homme dans certains cancers. Dans chaque cas, l’activité ACAT est accrue dans ces cellules et s’accompagne d’une accumulation des esters de cholestérol et d’une augmentation du ratio CE/TC (cholestéryl ester / cholestérol total), ce qui va dans le sens d’une estérification préférentielle du cholestérol dans les processus cancéreux (Batetta et al., 1999; Gebhard et al., 1987; Jeng and Klemm, 1984; Kokoglu et al., 1994; Nygren et al., 1997; Omsjo and Norum, 1985). Des liens ont été établi par ailleurs entre l’accumulation d’esters de cholestérol d’une part et le grade tumoral (Tosi and Tugnoli, 2005) et l’agressivité des tumeurs d’autre part. Cependant aucun lien de causalité n’a jamais été établi, ces études ont porté uniquement sur la recherche d’un outil prédictif et/ou descriptif de suivi de l’évolution des cancers.
Il apparaît au vu de ces résultats que le métabolisme du cholestérol est fortement modulé lors des processus cancéreux. Différents points sont particulièrement mis en avant :
- Le taux de HDL-C dans le sérum des patients cancéreux est plus bas que chez les témoins sains. Lors de la rémission de la tumeur, cette différence tend à disparaître.
- Les auteurs de ces études ont conclu que les variations du HDL-C étaient corrélées à une modification du métabolisme du cholestérol fortement modifié dans la tumeur, notamment au niveau de l’estérification du cholestérol
- La dérégulation du métabolisme du cholestérol est présente également dans les tumeurs bénignes ou dans les maladies prolifératives non tumorales, suggérant qu’il s’agit d’un marqueur précoce de transformation cellulaire
Globalement, les processus prolifératifs sont caractérisés par des changements dans le métabolisme du cholestérol, à la fois dans le plasma et dans les cellules en prolifération. Les changements plasmatiques semblent être une résultante des changements intracellulaires. Dans l’organisme, les stocks de cholestérol endogènes et exogènes sont finement régulés et les flux de cholestérol sont modulés en fonction de la demande des cellules. Comme les HDL transportent l’excès de cholestérol des cellules vers le foie pour sa dégradation, l’inhibition de ce transport inverse, couplée à l’augmentation de l’estérification au sein des cellules en prolifération apparaît comme un moyen de réduire l’efflux de cholestérol qui serait nécessaire au maintien de la croissance et de la division cellulaires.
Nous nous sommes posé la question du rôle effectivement joué par les ester de cholestérol dans les processus prolifératifs. L’étude du métabolisme du cholestérol dans les cellules tumorales et saines nous a permis de montrer une forte augmentation de l’estérification du cholestérol et de l’accumulation des esters dans les cellules cancéreuses par comparaison aux cellules saines issues au départ d’une même lignée.
Nous avons pu montrer qu’un ester de cholestérol, l’oléate est capable de promouvoir la prolifération et l’invasivité de cellules non tumorales et qu’ils sont impliqués dans la cascade de signalisation d’un récepteur de surface tumorigène activé de façon constitutive (Paillasse et al., 2009). C’est la première mise en évidence d’un rôle actif des esters de cholestérol dans les processus cancéreux.
II. Les oxystérols
Dans cette partie seront assimilés aux oxystérols les molécules comportant 27 ou 30 carbones issues de l’oxydation du cholestérol ou du lanostérol par addition d’un ou plusieurs atomes d’oxygène (fig. 14). Les oxystérols peuvent être fonctionnalisés par une fonction hydroxyle, hydroperoxyle, aldéhyde, cétone ou époxyde.
L’oxydation du cholestérol ou du lanostérol peut être causée par différentes voies, soit par auto-oxydation, soit par action d’une enzyme (Schroepfer, 2000). L’auto-oxydation pourra être causée directement par différentes espèces oxygénées réactives (tableau 3) ou par des espèces lipidiques peroxydées intermédiaires. Les espèces formées dépendent de l’espèce oxydante (Murphy and Johnson, 2008; Smith, 1987). Comme le cholestérol, les oxystérols peuvent s’agencer dans les membranes, mais en raison de leur hydrophilie considérablement accrue, ils se déplacent beaucoup plus rapidement entre les membranes des organelles intracellulaires et sont plus facilement accessibles aux récepteurs présents dans le compartiment cytosolique (Yan and Olkkonen, 2008). Cette hydrophylie leur permet également une plus grande solubilité dans le plasma, une capacité accrue au transport vésiculaire et un export facilité des tissus périphériques vers le foie (notamment au niveau cérébral pour le 24-hydroxycholestérol) (Vejux et al., 2008). Ces composés sont trouvés dans les tissus et la circulation chez les mammifères, en très faibles quantités par rapport au cholestérol. L'enrichissement en oxystérols est associé à certaines situations pathologiques, notamment l’athérosclérose et le cancer. Cependant de plus en plus d’études mettent en avant leurs rôles physiologiques qui apparaissent chaque jour plus importants (Gill et al., 2008; Schroepfer, 2000; Souidi et al., 2004).
1. origine
A l’instar du cholestérol, les oxystérols présents dans l’organisme peuvent être soit d’origine alimentaire, soit formés de manière endogène (Schroepfer, 2000). On distinguera alors les oxystérols formés par auto-oxydation et ceux formés de manière enzymatique.
Les oxystérols d’origine alimentaires sont retrouvés dans les aliments riches en cholestérol. La durée de stockage, la déshydratation, les hautes températures et la cuisson en présence d’oxygène favorisent la formation d’oxystérols (Leonarduzzi et al., 2002). Les oxystérols les plus communément détectés sont les 7α - et 7β-hydroxycholestérol, les 5,6α - et 5,6β-époxycholestanols (α- et β-EC) et le cholestane-3β,5α,6β-triol (CT). On retrouve également en faible quantité des oxystérols hydroxylés sur la chaîne latérale (19-, 20α- et 25-hydroxycholestérol). Des formes aldéhydes ou cétones sont également retrouvées comme par exemple les 3β-hydroxycholestan-6-one et 3β,5α-dihydroxycholestan-6-one et le 7-oxocholestérol (Leonarduzzi et al., 2002; Schroepfer, 2000).
Dans les cas étudiés dans la littérature, les oxystérols hydroxylés suivent la même voie d’absorption que le cholestérol au niveau de l’intestin et sont véhiculés dans les lipoprotéines, principalement sous forme estérifiée (Babiker and Diczfalusy, 1998). Cependant les oxystérols non estérifiés peuvent être véhiculés par l’albumine (Babiker and Diczfalusy, 1998). Ils sont assimilés par l’intestin, transportés et captés par les tissus périphérique plus rapidement que le cholestérol (Krut et al., 1997).
In vivo, les oxystérols peuvent être formés par auto-oxydation dans tous les tissus,
lorsqu’ils sont en présence d’espèces oxygénées réactives ou de radicaux libres. Leur formation enzymatique elle, diffère beaucoup selon les organes et le type d’oxystérol formé (Schroepfer, 2000).
HO HO HO HO 22(R)-hydroxycholestérol OH 25-hydroxycholestérol 24(S)-hydroxycholestérol OH OH HO OH HO OH HO O HO O HO O 7α-hydroxycholestérol 7β-hydroxycholestérol 5,6α-époxycholestanol 5,6β-époxycholestanol OH 20(S)-hydroxycholestérol HO OH 4β-hydroxycholestérol O cholest-4-èn-3,6-di-one 7-oxoocholestérol HO O 24(S)-24,25-époxycholestérol HO OH OH HO OH 27-hydroxycholestérol cholestane-3β,5α,6β-triol O
2. formation
2.1 Auto-oxydation
Le cholestérol est sujet à l’oxydation. Lorsque celle-ci est réalisée sans intervention d’une enzyme, on parle d’auto-oxydation. Elle se produit rapidement dès qu’il est en présence d’oxygène, pour donner naissance aux oxystérols (Smith, 1987).
La nature et les proportions des différents oxystérols dépend du type d’espèce réactive de l’oxygène à laquelle le cholestérol est exposé dans le tableau 3 (Smith and Jaworski, 1988). Il apparaît dans ce tableau que l’anion superoxyde n’est pas capable d’oxyder par lui-même le cholestérol. Cependant, au sein de membranes lipidiques, il est capable de former des radicaux peroxyde de lipides qui vont eux être capables d’oxyder le cholestérol (Murphy, JBC 2008).
Site de l’attaque Oxystérols produits Espèce oxydante
O3 O2+ 3O2 1O2 O2.- O22- HO. C3 Dérivés 3-oxo - - + - - - - Δ 5-hydroperoxyde 5,6 - - - + - - - EC β>α + β>α - - β>α α>β CT - - + - - + + 5,6-sécostérol + - - - - C7 7-hydroperoxydes - - β>α - - - - 7-hydroxydes - + β>α - - - 7-oxo - + + - - - +
Chaîne latérale Divers hydroxydes - - + - - - -
Tableau 3. Formation des oxystérols en fonction des espèces oxydantes impliquées. Certaines espèces oxydantes sont stéréosélectives. EC : 5,6-époxycholestanols, CT : cholestane-3β,5α,6β-triol. + : formation, - : pas de formation, β>α : rapport de formation des deux épimères.
Les principaux radicaux capables de se former à température ambiante sont ceux en C7 et en C25. Au contact de l’oxygène de l’air, ceux-ci donnent les radicaux peroxydes correspondants. Cette voie donne naissance aux oxystérols oxydés sur le cycle B, 7α - et 7β-hydroxycholestérol, 7-oxocholestérol, 5,6α- et 5,6β-époxycholestanol (α e t βEC
respectivement) et au cholestane-3β,5α,6β-triol (CT) (fig 15). Il existe deux autres voies minoritaires, d’une part la possibilité d’une déshydratation en position 3, donnant la cholest-5-èn-3-one, qui peut être hydroperoxydée en position 6 (6α ou 6β-hydroperoxycholest-4-èn-3-one). Ces hydroperoxydes vont ensuite se dégrader en hydroxyde puis s’oxyder en cétone pour donner la cholest-4-ène-3,6-dione (Smith, 1987) (Schroepfer, 2000).
D’autre part l’hydroperoxydation de la chaîne latérale peut avoir lieu. Les hydroperoxydes formés sont également sujets à dégradation en hydroxyde et donnent notamment les hydroxystérols 20(S)- ; 25- ; 25(S),26- et 27-hydroxycholestérol ainsi que la cholest-5-èn-3β-ol-24-one. Au total, ce sont 66 oxystérols qui ont été caractérisés comme produits de l’auto-oxydation du cholestérol à l’air libre (Smith, 1987) et le nombre de positions oxydables par les différents oxydants sur le cholestérol permet de donner d’innombrables autres structures possibles.
Au sein de la bicouche lipidique, l’oxydation du cholestérol peut également être réalisée par des radicaux peroxydes de lipides. Dans ce cas, quatre types de produits sont majoritairement formés : les 7-hydroperoxydes, les 7-hydroxydes, les 5,6-époxycholestanols (α et β dans chaque cas) et le 7-oxocholestérol. La génération de radicaux HO° par différents mécanismes (réaction de Fenton, action du péroxynitrite ou radiation ionisante) peut mener à la création d’un radical sur le carbone 7 par l’abstraction d’un proton allylique sur cette position. En effet cette liaison carbone – hydrogène est relativement faible, et est donc préférentiellement propice à la réaction avec des radicaux péroxyde lipidiques dérivés d’acides gras insaturés ou des radicaux alkoxyde. Le radical centré sur le carbone 7 est suffisamment stable pour rencontrer une molécule de dioxygène à l’état triplet (3O
2) pour former un radical hydroperoxyde. Ce mécanisme peut rendre compte de la peroxydation du cholestérol dans les membranes par propagation radicalaire classique. Le radical hydroperoxyde va propager la réaction en captant un hydrogène d’une molécule voisine et donner naissance aux 7α- et 7β-hydroperoxydes de cholestérol (fig 16A). Ces hydroperoxydes peuvent être réduits par du fer II en radicaux alkyloxyde qui vont soit donner les 7α- et 7β-hydroxycholestérol en arrachant un H° soit donner le 7-oxocholestérol en interagissant avec un radical hydroperoxyde lipidique pour terminer la cascade radicalaire (Murphy and Johnson, 2008).
Les oxystérols peuvent également être formés par des mécanismes non radicalaires impliquant l’oxygène singulet, l’ozone ou encore l’acide hypochloreux. L’oxygène singulet provoque la formation de 7-hydroperoxydes décrits précédemment et de 6-hydroperoxydes de choles-4-èn-3β-ol, précurseur de la cholest-4-ène-3,6-dione (Figure 16B). L’ozone peut donner entre autres du βEC après réarrangement et des produits issus de la coupure du cycle B entre les carbones 5 et 6 selon le mécanisme proposé à la figure 16C (Pulfer and Murphy, 2004). Le HOCl quand a lui est capable de donner une multitude de produits par action sur le cholestérol. Les plus abondants sont les α et βEC et les chlorhydrines obtenues par ouverture des époxydes pour donner des dérivés 5-chloro-6-hydroxylés ou 6-chloro-5-hydroxylés. D’autres dérivés du cholestérol du type hydroxyles ou cétones sont également décrits (van den Berg et al., 1993). Les α et βEC sont formés par action d’un hydroperoxyde lipidique sur le cholestérol, comme décrit dans la figure 16D (Watabe et al., 1984).
Figure 15. Formation des principaux oxystérols en contact avec l’oxygène de l’air. α -EC : 5,6α- et 5,6β- époxycholestanols. CT : cholestane-3β,5α,6β-triol
Fig 16. Mécanismes de formation des principaux oxystérols. A : formation des dérivés oxydés en position ; B : mécanisme de l’oxydation par l’oxygène singulet. LOO. : radical hydroperoxyde de lipide ; LOH : lipide hydroxylé ; LOOH : hydroperoxyde de lipide ; LO. : radical alkoxyde de lipide. Murphy and Johnson, 2008.
B
Fig 16. Mécanismes de formation des principaux oxystérols. C : mécanisme d’oxydation par l’ozone. D : formation des dérivés oxydés en postition 5 et 6. LOO. : radical hydroperoxyde de lipide ; LOH : lipide hydroxylé ; LOOH : hydroperoxyde de lipide ; LO. : radical alkoxyde de lipide. Murphy and Johnson, 2008.
Des études menées dans les années 70 et 80 ont mis en évidence le fait que toutes les formes d’oxygène actif ne sont pas capables de former tous les oxystérols. Au contraire, certains oxystérols ne sont formés que par une espèce oxygénée et peuvent donc être considérés comme une signature de la présence de cet oxydant in vivo (Smith and Jaworski, 1988). D’autre part, cette étude a permis de mettre en évidence une stéréosélectivité pour la formation des hydroperoxydes et des hydroxydes en position 7 et des 5,6-époxycholestanols (tableau 3). Ceci semble montrer que la dissymétrie entre les faces α et β du cholestérol pourrait avoir des répercussions biologiques.
2.2 Formation enzymatique
La synthèse enzymatique des oxystérols a été pour la première fois abordée en 1956 (Fredrickson and Ono, 1956) et concernait la formation de 25- et 27-hydroxycholestérol dans une fraction enrichies en mitochondries. Il est apparu depuis que de nombreux oxystérols sont formés de cette façon, principalement par des cytochromes P450. Des enzymes sont présents dans le foie ou les autres tissus pour former les oxystérols, et ceux impliqués dans leur dégradation via la voie des acides biliaires sont eux présents uniquement dans le foie (tableau 4).
A ces enzymes il faut ajouter CYP11A1, impliquée dans la formation de la prégnénolone dans les tissus stéroïdogènes (Pikuleva, 2006) et du 22(R)-hydroxycholestérol, et le cas particulier du 24,25-époxycholestérol qui est formé par une voie parallèle de la voie du cholestérol post-lanostérol (Nelson et al., 1981). Cet époxyde est issu de l’époxydation du 2,3(S)-monooxydo-squalène en 2,3(S)-22(S),23-dioxydo-squalène par l’OSC.
On obtient le schéma métabolique présenté à la figure 17. Elle met en évidence des formations localisées d’oxystérols dans les tissus dans lesquels ils vont avoir des fonctions particulières, et leur devenir commun après leur retour dans le foie, en tant que précurseurs d’acides biliaires. Il apparaît également des cas particuliers comme ceux du αEC qui ne suivent pas cette voie et sont métabolisés de manière très différente, qui sera exposée par ailleurs.
Enzyme Localisation substrats Fonction Principal oxystérol formé CYP7A1 Foie - cholestérol in vivo - 20(S)-, 24(S)-, 25, 27-hydroxycholestérol in vitro et COS7 7α-hydroxylation (Pikuleva, 2006; Schroepfer, 2000)
CYP27A1 Ubiquitaire - cholestérol 27-hydroxylation (Pikuleva, 2006; Schroepfer, 2000) CYP46A1 Cerveau - cholestérol in vivo - très large spectre in vitro (Mast N biochemistry 2003) 24(S)-hydroxylation (Pikuleva, 2006; Schroepfer, 2000)
25-hydro-xylase Ubiquitaire cholestérol 25-hydroxylation (Lund et al., 1998)
CYP3A4/5 Foie,
intestin cholestérol Oxydation en 3 (Bodin et al., 2001)
Mono-oxygénase
Glandes
surrénales cholestérol Epoxydation en α de la Δ
(Watabe and Sawahata, 1979) 5 CYP7B1 -Tissus stéroïdogéni ques - Foie, reins, intestin - 25- et 27-hydroxycholestérol - Hormones steroïdes : DHEA, E2, A2 7α-hydroxylation voie
« acide » des acides biliaires (Pikuleva, 2006)
CYP39A1 Foie - 24(S)-hydroxycholestérol >> 25- et 27-hydroxycholestérol 7α-hydroxylation d’oxystérols et d’hormones stéroïdes pour voie acides biliaires (Pikuleva, 2006) CYP8B1 Foie - 7α-hydroxycholestérol - dérivés oxydés en 27 du 7α-hydroxycholestérol 12α-hydroxylation (Pikuleva, 2006) 11β-HSD 1 7-HSD Foie 7α-hydroxycholestérol Oxydo-réduction en
position 7 (Song et al., 1998)
Tableau 4. Principales enzymes impliquées dans la formation des oxystérols. 11β-HSD-1 : 11β-hydroxystéroïde deshydrogénase de type 1 ; 7-HSD : 7- hydroxystéroïde deshydrogénase.