Délivré par l’université Toulouse III – Paul Sabatier Discipline ou spécialité : Sciences et Génie des Matériaux
Présentée par Laurent BAZIN
Anodes nanostructurées pour
microbatteries 3D Li-ion
JURY
F. FAVIER Chargé de recherche CNRS IGC, Université de Montpellier 2 Rapporteur A. WALCARIUS Directeur de recherche CNRS LCPME, Université de Nancy 2 Rapporteur R. DEDRYVERE Maitre de conférence IPREM, Université de Pau Examinateur M.J. MENU Professeur CIRIMAT, Université de Toulouse Directeur de thèse P. SIMON Professeur CIRIMAT, Université de Toulouse Directeur de thèse
Sommaire
Introduction 1
Chapitre 1- Les accumulateurs Li-ion, principes, amélioration de
l’anode et microbatterie 3D 5
I- LES ACCUMULATEURS LITHIUM-ION 6
I-1. Généralités sur les accumulateurs électrochimiques 6
I-1-1. Historique des générateurs électrochimiques 6
I-1-2. Principe des accumulateurs électrochimiques 8
I-2. L’accumulateur au lithium 13
I-2-1. Intérêt du lithium 13
I-2-2. L’anode de Li métal 14
I-2-3. La technologie lithium-ion : l’anode d’insertion 16
I-2-4.Anodes basées sur les alliages de lithium 18
II- L’INTERFACE : VERROU TECHNOLOGIQUE ET SCIENTIFIQUE 19
II-1. Les mécanismes aux interfaces 20
II-1-1. Contributions résistives 20
II-1-2. La couche passive (SEI) à l’interface MA/El 22
II-1-3. Expansion volumique aux interfaces 23
II-2.Utilisation des matériaux à l’échelle nanométrique 25
II-2-1. Les nanoparticules et les films minces (L< 1µm) 26
II-2-2. Le confinement (matrices, alliages intermétalliques, Li2O) 26
II-2-3. La nanostructuration 28
III- LES MICROBATTERIES 3D 30
III-1. Contexte 30
III-2. Géométries envisageables pour les microbatterie 3D - Performances 33
III-3. Le collecteur de courant nanostructuré de cuivre 37
III-3-1. Principe 38
III-3-2. Amélioration des performances électrochimiques par l’utilisation d’électrodes basées sur le collecteur de courant nanostructuré 39
IV- BIBLIOGRAPHIE 41
Chapitre 2- Partie expérimentale 45
I- THEORIE DU DEPOT ELECTROPHORETIQUE (EPD) 45
I-1. Principe 46
I-1-1. Histoire 46
I-2. La suspension colloïdale 46
I-2-1. Le choix du solvant 47
I-2-2. Charge des particules 47
I-2-3. Stabilité de la suspension 49
I-3. Le dépôt électrophorétique 51
I-3-1. Migration électrophorétique 52
I-3-2. Mécanismes de dépôt 52
I-3-3. Vitesse de dépôt 53
II- LE COLLECTEUR DE COURANT NANOSTRUCTURE DE CUIVRE 54
II-1. Préparation du collecteur de courant nanostructuré 54
II-1-1. Polissage du substrat de cuivre 54
II-1-2. Préparation de l’assemblage pour le dépôt 54
II-1-3. Le bain électrolytique et les conditions de dépôt 56
II-1-4. Retrait de la membrane 56
II-2. Géométrie 57
II-2-1. Images SEM 57
II-2-2. Gain de surface 58
III- SYNTHESES, GREFFAGES ET REVETEMENTS 59
III-1. Synthèse de nanoparticules d’étain 59
III-1-1. Synthèse hydrothermale 59
III-1-2. Synthèse par microémulsion inverse 59
III-2. Greffage d’organosilane sur les particules de SnO2 et SiO2 61
III-3. Dépôt électrophorétique EPD) 62
III-4. Dépôt électrolytique (ELD) 64
III-5. Fabrication des piles bouton 65
IV- TECHNIQUES UTILISEES 66
V BIBLIOGRAPHIE 69
Chapitre 3- Dépôt électrolytique d’étain sur le collecteur de
courant nanostructuré de cuivre 70
I- UTILISATION DE L’ETAIN COMME ANODE DE BATTERIE Li-ION 71
I-1. Intérêt de l’étain 71
I-2. Performances électrochimiques 72
I-3. Stratégie d’amélioration 74
I-3-1. Utilisation à l’échelle nanométrique 74
I-3-2. Confinement dans une matrice hôte : SnO2, SnS 75
I-3-3. Alliages à base d’étain. 78
I-3-4. Notre travail 79 II- DEPOT ELECTROLYTIQUE D’ETAIN SUR LE COLLECTEUR DE COURANT
II-3-2. Le type de contre électrode : étain, acier inox ou cuivre 84
II-3-3. La durée du dépôt 85
II-3-4. Observation en microscopie électronique en transmission 86
III- CARACTERISATIONS ELECTROCHIMIQUES DES ELECTRODES Cu/Sn 87
III-1. Courbes de cyclage galvanostatique 88
III-2. Tenue en cyclage, comparaison entre électrode nanostructurée et l’électrode plane épaisse 90
III-3. Tenue en cyclage, comparaison entre électrode nanostructurée et l’électrode plane mince 93
III-4. Comportement en puissance de l’électrode Cu/Sn 95
IV- CONCLUSIONS 96
V- BIBLIOGRAPHIE 97
Chapitre 4- Dépôt électrophorétique de SiO
2sur le collecteur de
courant nanostructuré 100
I- INTRODUCTION : INTERET DU DEPOT ELECTROPHORETIQUE 101
I-1. Les limitations du dépôt électrolytique 101
I-2. Les avantages du dépôt électrophorétique 101
I-3. Le défi technique 103
II- LA SILICE : COMPOSE MODELE POUR L’EPD 104
II-1. SiO2, considérations générales et synthèse 104
II-2. SiO2, stabilité des suspensions 106
II-2-1. Stabilisation électrostatique 106
II-2-2. Stabilisation stérique 107
II-2-3. Stabilisation électrostérique 107
II-3. SiO2, fonctionnalisation de surface, greffage 108
II-4. Stratégie 110
II-5. Paramètres d’EPD 111
III- SUSPENSION AQUEUSE DE NANOPARTICULES DE SILICE: GREFFAGE ET DEPOT ELECTROPHORETIQUE 112
III-1. Protocole 112
III-2. Fonctionnalisation des nanoparticules 113
III-3. Dépôt électrophorétique de SiO2-APTMS1 116
III-4. Bilan 120
IV- SUSPENSION ALCOOLIQUE DE NANOPARTICULES DE SILICE : GREFFAGE ET DEPOT ELECTROPHORETIQUE 121
IV-1. Protocole 121
IV-2. Fonctionnalisation des nanoparticules 121
IV-3. Dépôt électrophorétique 123
IV-3-. Dépôt électrophorétique de particules SiO2-APTMS2 123
IV-3-2. Dépôt électrophorétique de particules non greffées 127
IV-4. Bilan 128
V- CONCLUSIONS 129
Chapitre 5- Dépôt électrophorétique de SnO
2sur le collecteur de
courant nanostructuré 132
I – INTRODUCTION 133
I-1. SnO2, généralités, greffage 133
I-2. Objectifs 134
II– DEPOT ELECTROPHORETIQUE DE SnO2 A PARTIR DE NANOPARTICULES PREPAREES PAR SYNTHESE HYDROTHERMALE 135
II-1. Synthèse des nanoparticules 135
II-2. Dépôt électrophorétique des particules SnO2-SHT 137
III-2-1. Essais préliminaires 137
III-2-2. Greffage des nanoparticules SnO2-SHT avec les chlorure de N-triméthoxysilylpropyl-N,N,N-triméthylammoniun 140
III-2-3. Dépôt éléctrophorétique de SnO2-SHT 142
III- DEPOT ELECTROPHORETIQUE DE SnO2 A PARTIR DE NANOPARTICULES COMMERCIALES 143
III-1. Utilisation de nanoparticules commerciales de SO2 143
III-2. Caracterisation des poudres, greffage 144
III-2-1. Etude de la poudre SnO2-com 144
III-2-2. Fonctionnalisation des nanoparticules 145
I III-2-3. Etude des propriétés de dispersion en fonction du solvant utilisé 145
III-3. Dépôt électrophorétique des particules SnO2-com 147
III-3-1. Tests préliminaires 147
III-3-2. Influence de la tension appliquée 148
III-3-3. Influence de l’ajout d’additifs 151
III-3-4. Bilan 154
III-4. Caractérisations électrochimiques des électrodes obtenues 155
III-4-1. Courbe de cyclage de l’électrode Cu/SnO2 156
III-4-2. Tenue en cyclage de l’électrode Cu/SnO2 157
III-4-3. Tenue en cyclage : Comparaison avec l’électrode Cu/Sn obtenue au chapitre 3 158
IV- CONCLUSIONS 161
V- BIBLIOGRAPHIE 162
Conclusion générale 164
Au cours des derniers siècles, l’humanité a connu un développement technologique rapide, qui a conduit à une explosion des besoins en énergie et matières premières. Aujourd’hui, les conséquences de cette consommation sont clairement visibles : pollution, réchauffement de la planète, diminution des ressources fossiles. La solution à ces problèmes passe par une meilleure maîtrise de notre consommation d’énergie, de sa production et de son stockage. Ainsi, l’amélioration des systèmes d’accumulateurs électrochimiques, qui alimentent les équipements électroniques portables ou les véhicules électriques, est un enjeu particulièrement important. De plus, la production de batteries plus performantes va aussi permettre l’essor de nouvelles technologies. En particulier, la réalisation de batteries miniaturisées (microbatteries) est une condition nécessaire au développement des systèmes microélectroniques, ouvrant ainsi la voie à des applications innovantes dans les domaines de la santé, de l’informatique et la microélectronique. En termes d’énergie et de puissance, la technologie lithium semble la meilleure réponse à ces nouveaux besoins. Néanmoins, de nombreux progrès restent à accomplir aussi bien en terme de sécurité, de coût que de performance.
Dans cette perspective, les travaux présentés dans ce manuscrit concernent l’amélioration des performances de la batterie lithium-ion et plus particulièrement celles des électrodes négatives. Les recherches des dernières décennies ont montré que l’utilisation des matériaux actifs à l’échelle nanométrique (nanoparticules) pouvait donner lieu à de meilleures performances d’électrodes, via l’amélioration de la cyclabilité, de la puissance ou par l’apparition de nouvelles réactions. L’étape suivante a conduit à envisager la nanostructuration de l’électrode elle-même ; le passage à une géométrie tridimensionnelle de dimension nanométrique permet d’augmenter la surface de l’électrode, son accessibilité et donc ses performances électrochimiques.
Néanmoins, cette approche soulève de nombreux problèmes concernant la préparation et la mise en forme de l’électrode. En effet, le dépôt de couches minces de matériaux est un
L’objectif de ce travail est de réaliser de nouvelles électrodes négatives pour microbatteries 3D Li-ion, basées sur l’utilisation d’un collecteur de courant nano-architecturé, constituée de plots de cuivre nanométriques, alignés verticalement. Plus précisément, le challenge consiste à déposer une couche conforme de matériau actif, épaisse de quelques dizaines de nanomètres, sur toute la surface des plots par des techniques de dépôts électrolytique et électrophorétique. Des tests électrochimiques permettront ensuite de comparer les performances de ces électrodes par rapport à des électrodes planes et d’expliquer les effets bénéfiques de la nanostructuration.
Ce mémoire s’articule autour de cinq chapitres.
Dans le chapitre I, des considérations générales sur les accumulateurs électrochimiques seront rappelées. Les batteries Li-ion seront ensuite présentées ; on détaillera leur fonctionnement et les principales voies de recherche suivies dans l’optique de leur amélioration. Ensuite seront abordées les problématiques des interfaces, qui gouvernent le fonctionnement de ces batteries. La description de ces phénomènes sera suivie d’une revue des solutions proposées dans la littérature pour améliorer le fonctionnement des électrodes négatives. Enfin, nous nous intéresserons au cas particulier des microbatteries 3D. Ce concept sera décrit et un état de l’art des performances des matériaux actuels permettra de comparer les différentes approches envisagées pour la réalisation d’un tel système.
Le chapitre II proposera une rapide présentation de la théorie du dépôt électrophorétique. Ensuite, le procédé de préparation du collecteur de courant de cuivre sera décrit exhaustivement ainsi que les différents protocoles et méthodes de synthèse, de caractérisation et de dépôt utilisées.
Au cours du troisième chapitre, les résultats concernant le dépôt électrolytique d’étain sur la nanostructure de cuivre seront présentés. Une étude bibliographique de l’utilisation de l’étain en tant qu’anode de batterie Li-ion permettra de situer l’étude dans son contexte. Ensuite, l’élaboration de la couche d’étain et ses performances électrochimiques seront discutées.
L’objet du chapitre IV sera le dépôt électrophorétique de silice sur la nanostructure. Cette étude va permettre de prouver la faisabilité d’un revêtement conforme sur le collecteur nanostructuré par la technique de dépôt électrophorétique. Après une brève revue bibliographique concernant la suspension et la fonctionnalisation de nanoparticules de silice, l’influence des conditions expérimentales et d’une fonctionnalisation de surface des nanoparticules sur la morphologie sera discutée.
La compréhension du dépôt électrophorétique ainsi acquise nous permettra dans le chapitre V de réaliser le dépôt EPD de SnO2. Ici aussi, on s’intéressera à l’influence des
paramètres expérimentaux sur la morphologie et le comportement électrochimique des anodes préparées.
Chapitre 1
Les accumulateurs Li-ion,
principe, amélioration de
l’anode et microbatterie 3D
Dans cette partie, on s’attachera à replacer le sujet de cette thèse dans son contexte scientifique. Quelques points essentiels concernant les accumulateurs seront d’abord présentés, en insistant particulièrement sur l’accumulateur Li-ion et les raisons de son succès actuel. Ensuite, le rôle crucial des interfaces sera abordé avec les principaux problèmes qu’elles engendrent ainsi que les solutions adoptées pour les résoudre. Nous nous intéresserons enfin à la problématique de l’alimentation électrique des systèmes à l’échelle microscopique avec la présentation du concept de microbatteries 3D.
I- Les accumulateurs Lithium-Ion
I-1. Généralités sur les générateurs électrochimiques
I-1-1. Historique des générateurs électrochimiques
Dans un premier temps, il convient d’apporter une définition générale d’un générateur électrochimique. Cette notion désigne un système qui permet de fournir de l’énergie électrique à partir d’énergie chimique ; l’histoire des générateurs électrochimiques est donc à relier à celle de l’électricité. Les premiers progrès dans la compréhension des phénomènes électriques (du grec elektron : ambre jaune) datent du XVIIIeme siècle ; on peut par exemple citer la découverte des charges électriques par Du Fay en 1733 , les travaux de Franklin sur la nature de ces charges ainsi que l’énonciation des lois de l’électrostatique par Coulomb.
Dans ce contexte, le physicien italien Alessandro Volta propose en 1800 à la Royal Society le premier modèle de générateur électrochimique : la pile Volta. Celle-ci se compose d’un empilement de couples de disques de zinc et de cuivre, chaque couple étant séparé des autres par une épaisseur de tissu imbibé d’eau saumâtre. Lors de la décharge, les disques de zinc sont dissous et du dihydrogène se dégage sur les disques de cuivre.
En 1835, Daniell conçoit la pile Daniell, constituée d'une anode de zinc (électrolyte Zn SO4) et d'une cathode de cuivre (électrolytes CuSO4), reliées par un pont salin. Cette pile
présente deux avantages sur la pile Volta : une meilleure stabilité du courant délivré et l’absence de dégagement gazeux puisque les deux électrodes sont consommées (et non l’électrolyte).
Trente ans après Daniell, l’industriel français Leclanché met au point et brevette la pile Leclanché, à base de zinc et d’oxyde de manganèse, qui sera améliorée par ses héritiers jusqu’à devenir la pile saline cylindrique telle que nous la connaissons.
Le premier accumulateur (générateur électrochimique rechargeable) a été conçu par Planté en 1859. Sa batterie plomb/acide met en jeu deux couples redox du plomb PbSO4(s)/Pb
et PbO2(s)/PbSO4(s). Cent cinquante ans après son invention, cet accumulateur reste le plus
utilisé, notamment dans l’industrie et dans l’automobile, même si ses performances sont faibles en comparaison d’autres systèmes plus récents.
Les accumulateurs nickel-cadmium ont été découverts en Suède par Waldemar Jungner, qui déposa un premier brevet en 1899. L'invention mettait en œuvre de l'hydroxyde de nickel NiO(OH) à l'électrode positive, une électrode négative de cadmium et un électrolyte aqueux, KOH.
Aucun nouveau modèle de générateur électrochimique marquant n’apparaîtra durant environ cinquante ans, durant lesquels l’amélioration des générateurs existants est prépondérante, si l'on excepte la pile à combustible, qui n'est pas à proprement parler un générateur électrochimique puisqu'elle joue plus un rôle de système de conversion que de stockage de l'énergie.
Un autre type de batterie va prendre la suite des batteries Ni-Cd à partir des années 1970 : la batterie nickel hydrogène. D’abord, dans les années 1970, une batterie Ni-H2(g) va
équiper les satellites de la NASA. A partir de 1989, cette technologie évolue vers le Nickel Metal Hydrure (NiMH) dans laquelle l’électrode à hydrogène est remplacée par un matériau formant un alliage d’insertion avec l’hydrogène (oxyde). Des batteries NiMH équipent actuellement certains véhicules « hybrides » (Toyota Prius, Honda Civic).
Les derniers types d’accumulateurs, qui nous intéressent plus particulièrement, sont basés sur le lithium, utilisant des réactions d’insertion, de conversion ou d’alliage. Ces
systèmes, étudiés à partir des années 1970 et développés depuis 1992, seront décrits plus largement dans les paragraphes suivants.
Dans le tableau suivant (Tableau I-1), un aperçu des performances des accumulateurs mentionnés précédemment est présenté :
Electrode positive Electrode négative Tension de cellule (V)
Energie spécifique (Wh/g)
Plomb/acide PbO2 Pb 2 20-40
Ni/Cd NiO(OH) Cd 1,3 20-55
Ni/MH NiO(OH) H2 (adsorbé) 1,3 50-80
Li-ion (anode C) Li(1-x)MnO2 LixC 3,6 100-200
Tableau I-1 : Aperçu des performances de différentes technologies d’accumulateur1.
I-1-2. Principe des accumulateurs électrochimiques
Les générateurs électrochimiques se repartissent en deux catégories :
- les générateurs primaires, qui fournissent de l’énergie pendant une seule décharge et ne sont pas rechargeables ;
- les générateur secondaires, plus communément appelés accumulateurs ou batteries, qui fonctionnent grâce à des réactions réversibles et qui, après décharge, peuvent être rechargés en imposant un courant de sens opposé aux bornes du générateur.
Comme il a déjà été dit, un générateur électrochimique convertit l’énergie chimique en énergie électrique. Cette énergie chimique provient de la réaction d’oxydoréduction, c’est à dire du potentiel redox E des matériaux mis en jeu. En effet, ce potentiel est défini par l’équation :
Le potentiel redox se définit pour un couple redox, c’est à dire un couple de deux composés chimiques contenant le même élément chimique à un degré d’oxydation différent, appelés forme oxydée (plus haut degré d’oxydation) et forme réduite (plus bas degré d’oxydation). Ainsi, le potentiel redox ou potentiel redox standard E0 décrit la réactivité des composés redox entre eux. La valeur des potentiels redox est définie par rapport à un couple de référence (classiquement le couple H2O/H2 sur Pt), c’est à dire sa tendance à accepter ou à
céder des électrons. Lors d’une réaction redox spontanée classique, le transfert d’électrons se fait par contact direct entre les deux espèces concernées. Dans ce cas-là, l’énergie libérée par la réaction est difficilement utilisable, principalement dissipée sous forme de chaleur.
A l’opposé, la conception d’un générateur électrochimique se fait de telle sorte que les électrons produits par la réaction redox spontanée ne soient pas échangés directement entre les espèces réactives mais par l’intermédiaire d’un circuit électrique. Cela impose de placer les réactifs dans deux compartiments différents, séparés par un séparateur qui assure la conduction ionique entre les électrodes. Ce séparateur permet d’éviter le court-circuit ; les électrons mis en jeu par la réaction transitent alors par le circuit électrique, fournissant ainsi un courant.
Ces « contraintes », séparation des réactifs et conduction d’un courant électrique, permettent de définir plusieurs éléments principaux, entrant en compte dans la conception de tout générateur électrochimique :
Les électrodes : elles sont au minimum deux, de polarité différentes et seront le siège de chacune des deux demi-réactions de la réaction redox globale. Lorsque le générateur débite, l’oxydation se produit à l’anode (électrode positive) alors que, par opposition, la cathode, négative, sera le siège d’une réduction (dans le cas de réactions spontanées).
Ox1 + n1 e- → Red1
Red2 → Ox2 + n2 e-
Ces deux demi-équations mettent en jeu des électrons, qui doivent être transportés dans le circuit électrique. Ainsi il apparaît qu’une électrode se doit de contenir une partie solide, conductrice électrique, au contact de la solution contenant les réactifs. Il s’agit le plus souvent d’un métal, plongeant au contact de la solution. La zone de contact entre la partie solide et liquide d’une électrode, l’interface, gouverne son fonctionnement.
Le séparateur : celui-ci doit répondre à un double impératif, séparer les deux électrodes pour éviter les courts circuits électrique mais aussi assurer une conduction ionique
entre les deux électrodes. Le séparateur est imprégné d’électrolyte, solvant contenant un sel qui assure le transport des charges entre les électrodes. Le séparateur peut être un solide ou un polymère contenant une concentration suffisante d’espèces ioniques pour assurer la bonne conductivité de celui-ci. En pratique, le séparateur consiste souvent en une membrane (cellulose, polycarbonate) imbibée d’électrolyte support.
La figure suivante (Figure I-2) propose un schéma du fonctionnement d’un accumulateur électrochimique.
Figure I-2 : Représentation schématique d’un accumulateur électrochimique en fonctionnement (décharge).
Deux grandeurs fondamentales apparaissent sur la Figure I-2 :
- U : la tension de cellule du générateur
Cette grandeur est une tension électrique exprimée en Volt. Elle est reliée à un facteur thermodynamique : la différence de potentiel entre les couples redox cathodique et anodique à courant nul ou force électromotrice du générateur. A l’opposé, la tension de cellule est diminuée par deux facteurs, l’un cinétique et l’autre résistif. Le facteur cinétique provient de la surtension η, qui traduit la nécessité d’une activation de chaque électrode pour générer le courant consécutif à la réaction redox. Cette surtension est due à des phénomènes se déroulant
U = (E0c – E0a) – (ηc +ηa) – r.I
- I : le courant débité par le générateur (en Ampère)
Le courant est relié à la charge traversant le circuit, selon la loi de Faraday. Ce nombre dépend de la quantité de matière réagissant aux électrodes ainsi que de la quantité d’électrons n, libérée par mole de matière ayant réagi, selon l’équation :
Q = ∫ I(t)dt Q= nF nreagi
Q = quantité de charge ayant traversé le circuit F = constante de Faraday
nreagi = nombre de moles consommées pendant la décharge
Autour de ces paramètres, on peut ensuite définir des grandeurs dimensionnantes plus spécifiques aux batteries.
- Q : la capacité du générateur (en mAh)
Q représente la charge électrique totale délivrée par le générateur. Elle s’exprime relativement à la masse (capacité massique (mAh.g-1)) ou au volume du système (capacité volumique (mAh.L-1)). La valeur de la capacité dépend du courant de décharge. En effet, l’accessibilité du matériau actif n’est pas forcement homogène et la réaction d’électrode implique souvent une étape de diffusion dans certaines zones de l’électrode. A fort courant, la diffusion peut être trop lente et la zone concernée ne contribue plus à la capacité de la pile. La tension de coupure de la décharge va jouer, elle aussi, un rôle prépondérant ; plus la gamme de tension est large, plus nombreuses seront les réactions thermodynamiquement possibles au sein de l’électrode et plus nombreuses seront les charges extraite des électrodes.
- E : l’énergie électrique délivrée par le générateur (en Wh)
L’énergie est le produit de la capacité par le carré de la tension de cellule du générateur. Elle représente l’énergie totale qu’il peut fournir durant sa décharge. On
s’intéresse surtout à l’énergie relativement à la masse et au volume du générateur ; on parle alors d’énergie massique (en Wh.g-1) ou de d’énergie volumique (en Wh.cm-3), respectivement.
- P : la puissance électrique du générateur (en Watt)
La puissance est le produit de la tension de cellule du générateur par sa capacité. L’accumulateur doit être capable de fournir une quantité de puissance donnée sous un courant I et de maintenir cette valeur au cours de sa durée de vie. Elle s’exprime en regard de la masse et du volume du générateur : on parle alors respectivement de puissance spécifique (en W.g-1) et de densité de puissance (en W.cm-3).
Pour décrire l’évolution de ces deux paramètres, on peut tracer le diagramme de Ragone, qui donne l’énergie délivrée par une batterie en fonction de la puissance sous laquelle celle-ci travaille (Figure I-3).
Enfin, il convient de mentionner des grandeurs qui ne concernent pas directement les performances électriques du générateur mais qui décrivent ses limites de fonctionnement.
- L’autodécharge
L’autodécharge est la quantité de capacité (ou d’énergie ) perdue par unité de temps durant laquelle le générateur n’est pas en service (exprimée en %Q/an ou %Q/s). En effet, lorsqu’il ne débite pas, le générateur peut voir sa capacité diminuer à cause de réactions internes non désirées.
- La durée de vie en cyclage
Cette grandeur, caractéristique des accumulateurs, correspond au nombre de cycles de charge/décharge que peut effectuer la batterie sans que la valeur de la capacité ne diminue en déça d’un certain pourcentage de la capacité initiale (en general, 20%).
I-2. L’accumulateur au lithium
I-2-1. Intérêt du lithium
Pendant les deux dernières décennies du XXe siècle, la technologie Lithium a pris une place de plus en plus importante sur le marché du stockage de l’énergie. La principale raison de son succès commercial est l’augmentation rapide des besoins en sources d’alimentations pour les objets électroniques portables (téléphones, ordinateurs, etc.)
Ce succès prend ses origines avant tout dans les qualités du matériau qui est au cœur de cette technologie, le lithium. Ce métal, alcalin de numéro atomique 3, possède en effet des propriétés extrêmement intéressantes pour la conception d’accumulateurs.
Tout d’abord, le couple électrochimique Li+/Li possède un potentiel standard de -3,04 V par rapport à l’électrode standard à hydrogène (ESH), ce qui en fait une des anodes les plus négatives dans la nature. Ce potentiel permet d’atteindre, en association avec une cathode judicieusement choisie, des tensions de cellule supérieures à 3,5 V. Si on se souvient que l’énergie et la puissance d’une batterie croissent avec la tension de cellule, on comprend l’intérêt d’un tel matériau.
Ensuite, une autre caractéristique du métal lithium est sa faible masse molaire : 6,94 g.mol-1. En comparaison, la masse volumique d’autres matériaux de batteries est au moins dix
fois supérieure (Zn : 65, Pb : 207, Ni : 58, Cd : 112, …). Cette faible valeur permet un gain de masse et l’énergie, la puissance et la capacité massiques s’en trouvent sensiblement améliorées (la capacité théorique du lithium est de 3200 mAh.g-1)
Par ailleurs, si le lithium n’est pas thermodynamiquement stable dans la plupart des électrolytes, sa corrosion est néanmoins stoppée par la formation d’une couche de passivation à sa surface, la SEI (Solid Electrolyte Interphase) dans les solvants contenant un groupement carbonate. Ce phénomène sera décrit plus amplement dans la seconde partie de ce chapitre.
- Enfin, le lithium métal étant bon conducteur électrique (σ=107 S), il n’est pas nécessaire de lui associer un collecteur de courant.
I-2-2. L’anode de Li métal
Le premier générateur électrochimique primaire utilisant du lithium en tant que matériau actif est un système qui utilise une anode de lithium métallique. Bien que les propriétés électrochimiques du lithium soient connues depuis 1912, ce n’est que dans les années 1970 que les recherches pour l’utilisation de ce matériau ont pris leur essor.
Ces recherches aboutirent à la pile MnO2/Li. Un schéma de cette pile est donné à la
Figure I-4.
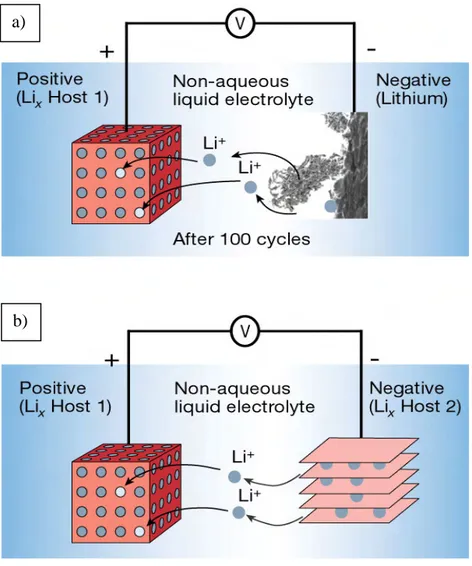
Cependant, lorsque l’on tente de réaliser un générateur secondaire utilisant une électrode de Li, un important problème apparaît au niveau de l’électrode de lithium. Lors de la phase de recharge, le lithium se redépose à l’anode non plus sous forme de couche uniforme et compacte mais sous forme dendritique3 (Figure I-5-a). Ce phénomène entraîne deux conséquences néfastes :
a) D’abord, le lithium dendritique a une plus forte tendance à réagir avec l’électrolyte ce qui diminue progressivement sa surface active et donc sa durée de vie en cyclage.
b) Ensuite, ces dendrites croissent au travers du séparateur et créent des micro courts-circuits en entrant en contact avec la cathode. Ceci a pour effet de provoquer un échauffement de la cellule pouvant parfois entraîner une explosion.
Pour pallier ces problèmes de croissance dendritique, Armand4 a proposé la batterie Li/polymère, qui utilise un électrolyte solide, qui empêche mécaniquement la création de courts-circuits. Cette technologie va conduire aux accumulateurs Li-polymères. Néanmoins, cette solution va entraîner d’autres problèmes, notamment concernant le transport de l’ion lithium entre les deux électrodes, ce qui implique un fonctionnement à 80°C.
Figure I-5 : a) Principe de la technologie Lithium , b) Principe de la technologie Lithium-ion ou technologie ‘’Rocking Chair’’.
I-2-3. La technologie lithium-ion : l’anode d’insertion
Pour pallier le problème de la croissance dendrite du lithium, la technologie lithium-ion a été développée. L’idée directrice est d’utiliser à la place de l’anode de lithium métal un composé « hôte », qui va subir une réduction électrochimique compensée par l’insertion d’ions lithium dans sa structure (Figure I-5-b), similairement à ce qui se produit à la cathode de MnO2. Ce type de composé est appelé ‘‘matériau d’intercalation’’. Dans ces matériaux,
a)
proposés, entre autres par Whittigham9, l’inventeur du concept de la batterie Li-ion et par Broussely10.
Le choix du matériau d’anode obéit donc à certains critères ; il doit pouvoir intercaler un grand nombre d’ions lithium et avoir une activité redox à des potentiels aussi bas que possible. Le carbone répondant à ces spécifications, de nombreux matériaux carbonés ont ainsi été étudiés11 (fluorures de carbone12, fibres de carbone13) mais c’est le carbone graphite14,15 qui a donné les meilleurs résultats : l’espace entre les feuillets de graphène permet au mieux l’intercalation d’un Li+ pour 6 atomes de carbone. De plus, son activité électrochimique se situe à des potentiels proches de celui de Li, entre 0,210 et 0,085 V vs Li+/Li16,17. La capacité théorique du graphite est donc de 372 mAh/g.
Basée sur la découverte de propriétés de la cathode LiCoO218 et l’anode de graphite19,
la première batterie Li-ion commerciale, en 199120, associe une anode de carbone avec une cathode d’oxyde de cobalt.
Les réactions d’électrodes sont :
C + x Li+ + e-↔ LixC (x= 0.133)
LiCoO2 ↔ Li(1-y) CoO2 + y Li+ + y e- (y= 0,5)
Cet accumulateur fournit un énergie de 100-150 Wh.kg-1 pour une tension de 3,6 V. Similairement à ce qui est observé sur l’anode de lithium, l’électrolyte se réduit sur l’électrode de carbone à son potentiel de fonctionnement (environ 0,2V vs Li+/Li), entraînant la formation d’une couche de passive (SEI). Cette couche, une fois formée, bloque la réaction de l’électrolyte mais donne lieu à une importante capacité irréversible. Autre inconvénient, l’intercalation d’ions lithium solvatés entre les feuillets de graphène exerce une contrainte mécanique (expansion volumique du graphite) qui provoque l’exfoliation des feuillets, faisant chuter la capacité de l’électrode.
Un autre problème associé aux anodes de graphite est leur faible capacité. En effet, les nouveaux besoins sociétaux en énergie transportable (véhicule hybrides, micro batteries, électronique) exigent une amélioration de la capacité des électrodes négatives au-delà des 372 mAh.g-1 actuels. Pour ces raisons, une nouvelle voie de recherche a émergé, afin de remplacer les anodes d’intercalation ; il s’agit des anodes en alliage de lithium.
I-2-4.Anodes basées sur les alliages de lithium
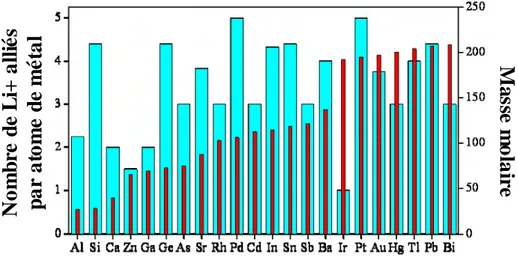
Dans cette approche, une nouvelle classe d’électrodes négatives est utilisée, basée sur les matériaux formant des alliages avec le lithium. En effet, certains matériaux peuvent accepter le lithium dans leur structure non plus par simple insertion mais par formation d’un alliage à bas potentiel, ce qui se traduit par un plus grand nombre de Li+ inséré par atome de matériau actif. Par exemple, parmi les composés les plus étudiés, l’étain et le silicium peuvent allier 4,4 Li+ par atome, l’antimoine 3, l’aluminium 2,2 (le graphite, pour mémoire, ne peut insérer que 0,133 Li+ par C). Une vue générale des possibilités des matériaux d’alliage est proposée à la Figure I-6.
Figure I-6 : Li+ inséré et masse molaire des métaux susceptibles de s’allier au lithium.
Les capacités spécifiques théoriques de ces alliages atteignent des valeurs très élevées, jusqu’à 4200 mAh.g-1 pour le silicium, largement satisfaisantes en terme d’énergie stockée.
Si ces alliages sont connus depuis les années 197021,22
,
leur utilisation pose encore des problèmes. Notamment, l’expansion volumique que subissent ces matériaux lors de la réaction d’alliage avec le lithium (400% pour Si23, 140% pour Sb24) va entraînerN
o
m
b
re
d
e
L
i+
a
ll
ié
s
p
a
r
a
to
m
e
d
e
m
é
ta
l
M
a
ss
e
m
o
la
ir
e
N
o
m
b
re
d
e
L
i+
a
ll
ié
s
p
a
r
a
to
m
e
d
e
m
é
ta
l
M
a
ss
e
m
o
la
ir
e
Figure I-7 : a) Schéma de l’expansion volumique du matériau actif lors de l’alliage de Sn avec Li ; b) Image MEB des craquelures formées à la surface d’une anode Si-Sn après
cyclage25 .
Beaucoup de travaux de recherches ont été consacrés à ces anodes avec pour objectif de leur conférer une énergie, une puissance ou une durée de vie en cyclage plus grande. Dans le paragraphe suivant, nous allons analyser l’origine des problèmes liés à l’utilisation de ces matériaux ainsi que les solutions qui ont été proposées pour y remédier.
II- L’interface : verrou technologique et scientifique
Un grand nombre de travaux, réalisés au cours des 20 dernières années mettent en lumière l’extrême importance des interfaces dans les électrodes d’accumulateurs, et plus particulièrement dans les anodes pour batteries Li-ion.
Une anode d’alliage classique comprend deux interfaces, l’interface matière active/collecteur de courant et l’interface matière active/électrolyte (notées respectivement MA/CC et MA/El). Ces deux interfaces vont être le siège de phénomènes physiques et chimiques très différents. La compréhension et le contrôle de ces interfaces va permettre d’améliorer le fonctionnement des anodes de batteries Li-ion26.
II-1. Les mécanismes aux interfaces
En premier lieu, il convient d’identifier et de comprendre les mécanismes d’interface qui provoquent la diminution des performances des anodes.
II-1-1. Contributions résistives
L’existence d’une interface implique toujours une résistance. Dans notre cas, trois résistances doivent être prises en considération : la résistance MA/CC, la résistance MA/El et la résistance de diffusion dans la matière active. Leur nature et les paramètres les influençant doivent être détaillés.
A la jonction MA/CC, la résistance est de nature ohmique, elle est donnée par la relation :
R = ρ . l . S-1
avec R= résistance électrique de l’interface (Ω), ρ = résistivité de l’interface (Ω.m), l = épaisseur de l’interface (m) et S, surface de l’interface (m²). L’épaisseur de l’interface étant faible et difficile à étudier, on utilise plus volontiers la formule:
R = ρ’. S-1
Avec ρ’= résistivité corrigée
D’après cette formule, plus la surface d’interface est faible, plus sa résistance est importante. Il convient donc de maximiser la valeur de S. Par ailleurs, la résistance augmente avec la résistivité de l’interface. Celle-ci va dépendre de plusieurs paramètres. La qualité du contact physique entre MA et CC va fortement influencer ρ’. Une soudure ou un électrodépôt de la matière active sur le collecteur de courant permettra un meilleur contact entre les deux
A la jonction MA/El, une résistance de nature électrochimique est à prendre en compte. Elle regroupe les impédances faradiques, dues à la réaction électrochimique et les impédances de transport, de double couche ou d’adsorption. Plus la surface de l’interface sera importante, plus sa résistance sera faible. A ces considérations s’ajoute aussi une notion de qualité du contact, décrite par la mouillabilité dans le cas d’une interface liquide/solide.
Il convient aussi de mentionner ici la résistance due à la matière active (ionique et électrique). Cette résistance n’est pas due aux interfaces elles-mêmes mais plutôt à la distance qui les sépare ; néanmoins, par souci de simplification, elle sera rangée dans cette catégorie. Dans une anode d’alliage de batterie lithium-ion, la matière active ne réagit pas uniquement en surface mais aussi au cœur de la couche de matériau : l’ion lithium doit donc diffuser au sein du matériau actif. De plus, les électrons fournis par la réaction électrochimique doivent eux aussi être conduits, jusqu’au collecteur de courant sur une certaine distance. Plus les interfaces MA/CC et MA/El sont rapprochées, plus les distances de diffusion sont raccourcies et plus la résistance de diffusion dans la matière active est faible.
Ces concepts sont résumés sur la Figure I-8.
La valeur de la résistance des interfaces va s’ajouter aux contributions de l’électrolyte et du séparateur dans le calcul de la chute ohmique. Celle-ci va gouverner la tension de la cellule, lui-même corrélé à la puissance de l’électrode par l’équation P= U.I.
Les effets de résistance dus aux interfaces ont donc une influence directe sur la puissance de l’électrode. Plus ces résistances seront minimisées, meilleures seront les performances en puissance de l’électrode.
II-1-2. La couche passive (SEI) à l’interface MA/El
Le potentiel standard d’une électrode de Li est de –3,04 V/ESH. Lorsque celle-ci est plongée dans un solvant organique de type carbonate, une réaction redox spontanée se produit, entraînant la dissolution du lithium et la réduction du solvant à sa surface. De même, la plupart des matériaux utilisés en anode, lorsque leur potentiel est proche de celui de Li+/Li (inférieur à –0,5 V/Li+/Li), catalysent eux-aussi des réactions de dégradation du solvant. Ces
réactions font que ces matériaux ne devraient pas être utilisables dans d’aussi larges domaines de tension (3,5 V pour les batteries Li-ion actuelles).
Mais ces considérations thermodynamiques ne tiennent pas compte d’un autre phénomène. En effet, les produits de ces réactions sont solides et vont s’accumuler à la surface du matériau actif pour former la « Solid Electrolyte Interphase » ou SEI. Cette couche possède deux propriétés qui permettent l’utilisation des anodes :
c) elle est un isolant électrique : la réaction de dégradation de l’électrolyte va être stoppée lorsque la SEI aura atteint une épaisseur suffisante.
d) elle conduit très bien les ions lithium, les réactions d’électrodes ne sont donc pas contrariées.
Les questions de SEI ont été particulièrement étudiées dans le cas de l’anode d’insertion de carbone27,28,29,3031 où la formation de la SEI est responsable de l’importante capacité irréversible observée lors du premier cycle. Une étude très complète de Aurbach et
Dans le cas des anodes d’alliages ou de conversion, on trouve aussi une importante littérature concernant la SEI. La composition de la SEI est identique à celle observée sur le carbone33, ce qui indique que le matériau actif joue un rôle de catalyseur et ne participe pas à la réaction. L’épaisseur de la SEI est de l’ordre de quelques nanomètres: Choi et al.34 observent une SEI de 2 à 7 nm d’épaisseur sur des particules de Sn, composée principalement de Li2CO3 et ROCO2Li ; Dolle et al.35 la mesurent à 1,7 nm sur le carbone. Une spécificité de
la SEI des anodes d’alliages est que celle-ci peut être détruite et reformée au cours du cyclage, à cause des changements de volume de l’électrode qui entraînent la fissuration de la couche. Inaba et al.36 reportent ce phénomène sur une électrode d’étain et le groupe de K.Edstrom37 le met en évidence dans le cas d’électrodes à base d’antimoine. Ce phénomène de destruction/reformation est responsable d’une importante capacité irréversible38.
La formation d’une SEI à l’interface matière active électrolyte est un phénomène complexe mais crucial pour le fonctionnement des batteries Li-ion. Il présente l’inconvénient d’engendrer des capacités irréversibles mais présente l’avantage de stopper la décomposition catalytique du solvant. Son importance est directement reliée à la surface de l’électrode : plus la surface de l’électrode est importante, plus la formation de SEI sera importante (en regard de l’activité électrochimique redox)Erreur ! Signet non défini..
II-1-3. Expansion volumique aux interfaces
Un défaut majeur des anodes d’alliages vient de l’expansion volumique du matériau actif. Lors de la phase d’alliage, l’ion lithium vient s’allier au métal actif et pénètre ainsi dans le réseau cristallin du matériau actif39. L’occupation de ces espaces entraîne une augmentation du paramètre de maille et donc du volume du matériau. Comme la quantité d’ion lithium alliées peut être très importante, cette expansion peut atteindre jusqu’à 400% ; elle crée un stress mécanique important au sein de l’électrode. Lors de la dé-lithiation, la maille retrouve progressivement son volume initial et cette contraction entraîne aussi des contraintes mécaniques. Le résultat de ces variations est une perte d’intégrité physique de l’électrode, qui s’effrite.
La variation des paramètres de mailles des composés Sn, Al et Si durant la lithiation est donnée sur le tableau I-2.
Nombre de Li+ alliés Volume par atome de
métal non allié (Å3)
Volume par atome de
métal allié (Å3) Expansion volumique (%) Sn 4,4 27,0 96,7 26040 Al 2,25 16,6 55,7 9641 Si 4,4 20,0 82,4 38023 Sb 3 - - 15042
Tableau I-2: Variation de volume durant la lithiation de Sn, Al et Si en Å3.
Le matériau subit donc une ‘‘respiration’’, se gonflant et se dégonflant au fil des cycles de charge décharge. Le résultat de ces changements de volume se fait sentir au niveau des deux interfaces MA/CC et MA/El.
A l’interface MA/El, c’est-à-dire à la surface de l’électrode apparaissent des craquelures. Ce phénomène a été extensivement étudié par Brousse et al.43 dans le cas de films d’oxyde d’étain et par Beaulieu et al.25 dans le cas d’un film SiSn. Beaulieu et al., en s’appuyant sur des observations d’AFM in situ et des images MEB décrivent les phénomènes de la manière suivante. Lors de la première lithiation, le film s’étend, perpendiculairement au substrat, sans expansion latérale. Lors de l’extraction du lithium, le film se contracte à la fois perpendiculairement et parallèlement au substrat et des fissures se créent à sa surface. De petits grains/particules sont formés, séparés les unes des autres par les fissures. La seconde lithiation semble refermer les fissures mais celles-ci sont encore visibles à fort grandissement. Les petites particules formées peuvent ainsi cycler réversiblement tant que la quantité de Li+ insérée ne les fait pas grossir suffisamment pour qu’elles se touchent. Si la quantité de Li+ alliée augmente, ces particules ne peuvent plus grossir sans entrer en contact entre elles. Ce contact crée de fortes contraintes mécaniques et provoque l’extension du film dans le sens latéral et la pulvérisation des particules, qui sont arrachées à l’électrode.
A l’interface MA/CC, le problème de l’expansion latérale de la couche va créer des contraintes mécaniques d’expansion. De plus, à l’échelle atomique, les modifications du
Pour résumer, l’expansion volumique va entraîner principalement une perte de matériau actif par pulvérisation de l’électrode. Cette perte va avoir un effet direct sur la capacité de l’électrode (moins de matière active implique moins de mAh) et donc sur l’énergie totale que peut stocker l’électrode. Ici, encore, la quantité de lithium alliée va avoir une forte influence sur la dégradation de l’électrode.
II-2. Utilisation des matériaux à l’échelle nanométrique
En réponse aux problèmes d’interface cités précédemment, des solutions ont été proposées pour améliorer les performances des anodes. Une de ces solutions implique l’utilisation des matériaux actifs à l’échelle nanométrique44. Nous donnerons ici des exemples concernant les matériaux d’anode les plus courants, le cas de l’étain faisant l’objet d’une étude bibliographie exhaustive dans le chapitre III.
Une taille nanométrique confère aux matériaux des propriétés bien spécifiques. Bruce et al.45 dressent une liste des avantages des nanomatériaux :
1) existence de réactions qui n’ont pas lieu à l’échelle micrométrique, y compris des réaction de décomposition de l’électrolyte,
2) distance de diffusion du lithium plus courte, ce qui augmente la vitesse d’insertion/dé-insertion du lithium,
3) transport électronique et ionique amélioré, 4) grande surface interfaciale,
5) meilleure accommodation des contraintes mécaniques.
Il existe plusieurs moyens d’obtenir cet ‘‘effet’’ nanométrique. L’utilisation de nanoparticules et nano-films est la plus évidente. Il est aussi possible d’obtenir des nanoparticules confinées au sein d’une matrice massive inactive ; ce type de composite peut être obtenu in ou ex-situ. Enfin, une troisième approche est celle de la nanostructuration des électrodes.
II-2-1. Les nanoparticules et les films minces (L< 1µm)
La caractéristique la plus intéressante des nanoparticules est qu’elles sont nanométriques dans les trois dimensions. Elles sont composées d’une grande fraction d’atomes de surface, à l’énergie et aux propriétés différentes. De plus, leur petite taille implique des contraintes mécaniques moins fortes durant la lithiation-délithiation, ce qui permet de limiter voire d’éviter la pulvérisation du matériau actif. Ce propos est illustré par Li et al.46 qui constatent une amélioration de la cyclabilité dans le cas d’une électrode nano-Si/KB (25 cycles au lieu de 5 cycles dans le cas de particules de quelques microns) et par Choi34 et al. qui obtiennent une capacité de 200 mAh.cm-1 pendant plus de 40 cycles avec des particules d’étain submicrométriques.
De la même manière, l’utilisation de couches minces permet aussi une amélioration de la cyclabilité si leur épaisseur est nanométrique. Les travaux de Bryngelsson et al.47 concernant les anodes à base d’antimoinemontrent le gain de cyclabilité (300 mAh.g-1 sur 60 cycles) qui accompagne l’utilisation de couches minces (300nm), composées de grains nanométriques. Kasavajjula et al. indiquent que la cyclabilité et la puissance sont améliorées lorsque l’épaisseur du film est suffisamment faible, notamment grâce au caractère amorphe qu’acquiert le silicium à l’échelle nanométrique23,48. Un résultat impressionnant est obtenu par Ohara et al.49 avec un film très mince (50 nm) de Si qui présente une capacité de 2,5 Ah.g-1 pendant 200 cycles, alors que l’anode de silicium massive perd toute sa capacité en quelques dix cycles.
L’utilisation de film minces est donc une solution probante aux problèmes de cyclabilité des anodes de batteries Li-ion.
II-2-2. Le confinement (matrices, alliages intermétalliques, Li2O)
Des particules seront dites confinées si elles sont incluses dans une matrice épaisse d’un autre matériau. Cette matrice va jouer un rôle de tampon mécanique et permettre le
La première consiste à choisir une méthode de préparation qui crée les particules au sein de la matrice. L’utilisation d’une matrice de carbone se prête particulièrement bien à ce type de composites. Par exemple, le groupe de Scrosati50 produit des particules de Sn dans une matrice de carbone et note l’effet de tampon de la matrice de carbone sur l’expansion volumique de l’étain (500 mAh.g-1 pendant 200 cycles). L’effet stabilisant de la matrice de graphite a aussi été exploité avec les anodes au silicium par Wu51et al. et Dimov et al.52. Dans ces deux cas, la matrice carbonée contribue à la capacité de l’électrode (800 mAh.g-1 durant 35 cycles) en plus de son effet de tampon mécanique. Mais des matrices inactives peuvent aussi être choisies. Ainsi, des composites Si/TiB, Si/SiC, Si/TiC53,54,55 ont permis l’amélioration de la stabilité des anodes à base de silicium. Notons que la matrice doit être conductrice électronique et ionique, pour pouvoir assurer le transport de l’ion Li+ et celui des électrons ; cette exigence limite l’utilisation de polymères. Par ailleurs, cette stratégie implique une absence de contact entre l’électrolyte et le matériau actif : la SEI est donc différente de celle des particules non confinées et dépendra de la nature de la matrice.
La seconde méthode concerne l’utilisation d’alliages ou de composés intermétalliques. Lorsque deux métaux sont alliés, l’un actif et l’autre inactif, le matériau actif subit une lithiation et son expansion volumique est accommodée par le réseau du métal inactif. Généralement, le composé inactif est extrudé autour du matériau actif et amortit les contraintes générées par la réaction d’alliage du lithium. Cette stratégie permet par exemple de réduire l’expansion de l’antimoine de 150 à 42%, ce qui donne lieu à une capacité stable sur plus de 60 cycles (300 mAh.g-1 sur 60 cycles)56. Les composés intermétalliques de silicium ont aussi été largement étudiés, mettant en évidence l’importance des qualités mécaniques du matériau inerte. Ainsi, un composé CaSi57 n’offre pas un réseau suffisamment résistant pour empêcher le délaminage alors que Si3N458 est au contraire trop dur, empêchant
l’intégration des ions Li+ dans sa structure Ce type de structure présente néanmoins un inconvénient : la capacité massique de l’électrode est notablement plus faible puisque l’électrode contient un matériau inactif, qui augmente sa masse sans modifier sa capacité. Ainsi, Sb a une capacité théorique de 660 mAh.g-1 alors que Cu2Sb ne peut délivrer que 323
mAh.g-1.
Enfin, le confinement peut être créé par une réaction in situ, durant le fonctionnement de l’électrode. Cette réaction dite de conversion permet de former à partir d’oxydes, des nanoparticules de métal dispersées dans une matrice d’oxyde de lithium. C’est le cas par
exemple pour Cu2O, CoO, Fe2O3 ou RuO245. Précisons que cette réaction est différente d’une
réaction d’alliage mais qu’un composé peut présenter à la fois une réaction d’alliage et une réaction de conversion. Par exemple, SnO2 subit une réaction de réduction irréversible qui
conduit à la formation de nano-particules d’étain dans Li2O. Sn forme ensuite des alliages
avec le lithium (ces alliages seront décrits dans le chapitre 3). Autre exemple, l’oxyde de ruthénium, étudié par le groupe de Maier59 présente un comportement similaire mais avec une réaction de conversion pleinement réversible (qui contribue à la capacité de l’électrode : 1100 mAh.g-1). Néanmoins, l’oxyde de lithium présente l’inconvénient d’être peu conducteur. De plus, les nanoparticules formées par la réaction ont tendance à coalescer et perdre leur caractère nanométrique. Ces particules plus grosses vont donner lieu à des contraintes mécaniques plus fortes, susceptibles de briser la matrice de Li2O, comme le suggèrent Sandu
et al.60. Ajoutons que d’autres types de composés (fluorures, sulfures, phosphures) donnent lieu à des réactions de conversion (formation de matrices de Li2S, LiF, Li3P), permettant de
diminuer l’hystérésis observée entre charge et décharge61,62,63.
II-2-3. La nanostructuration
Enfin, une dernière voie d’amélioration est la nanostructuration des électrodes. Traditionnellement, le matériau actif d’électrode se trouve sous forme de particules plus ou moins sphériques, plaquées en film sur un substrat plan. La nanostructuration consiste alors à modifier la forme du matériau afin qu’il adopte une géométrie qui améliorera ses performances. Ce type de structure va donner lieu à deux avantages principaux:
1) l’interface MA/El sera beaucoup plus grande : le transfert de l’ion Li+ sera facilité, ce qui améliorera la puissance de l’électrode,
2) une géométrie non plane va créer plus d’espace pour accommoder les variations de volume de la structure.
Une première possibilité consiste à nanostructurer le matériau actif. L’exemple le plus courant est l’utilisation de matériau actif sous forme de nanotubes ou de nanofibres. Ici, la
nanotubes, l’expansion peut aussi se faire vers l’intérieur du tube. De plus, les nanofibres permettent un transport de matière tout aussi rapide que dans les nanoparticules et améliorent la conductivité dans le matériau actif puisque celui-ci contient moins d’interconnexions entre particules. Si, dans le cas du carbone, l’utilisation de nanotubes donne lieu à des performances inférieures à celles du graphite64, d’autres matériaux tirent profit d’une structure en tube. TiO2, notamment, a fait l’objet de nombreuses études, qui montrent une amélioration de ses
performances65 (56 µAh.cm-2 après 50 cycles). Chan et al.66 ont étudié les nanofibres de silicium et montrent que leur nanostructuration est responsable de l’amélioration de la stabilité en cyclage (2750 mAh.g-1 pendant 30 cycles). La très grande surface développée des structures de types nanotubes ou nanofibres permet des gains importants en terme de puissance. Erjavec et al.67 et Qiao et al.68 le démontrent pour le cas de nanotubes de TiO2 avec
seulement 50% de perte de capacité entre 2C et 60C.
Une autre stratégie consiste à nanostructurer l’électrode en elle-même, en créant des structures auto-supportées ordonnées. Cette stratégie implique d’abandonner le mode de préparation traditionnel des électrodes (poudres mises en forme de films et enduites sur le collecteur de courant). Ici, le matériau actif est directement déposé sur le collecteur de courant. La géométrie du matériau est conçue à l’échelle nanométrique. Les deux architectures d’électrodes nanostructurées les plus répandues sont les mousses et les piliers.
Une électrode de type mousse est construite autour d’un réseau de pores et de canaux désordonnés. Elle présente une très grande surface spécifique (428 cm2/cm3 pour les mousses de nickel de Yang et al.69) et des chemins de diffusion plus courts. Huang et al.70 proposent une anode de NiO de type mousse qui délivre 600 mAh.cm-2 pendant 50 cycles.
La nanostructure de type pilier est constituée d’une forêt de plots de matériau actif, orientés perpendiculairement au substrat. En fonction de la méthode de préparation, ces piliers sont plus ou moins bien alignés, de tailles et de diamètres choisis. Une description détaillée de ce type d’électrode sera proposée dans le paragraphe suivant, puisque cette géométrie est largement utilisée pour les applications en microbatteries 3D. L’utilisation d’anodes nanostructurées apporte des avantages certains par rapport aux stratégies précédentes, comme une grande surface de contact entre le collecteur de courant et la matière active (résistances d’interfaces plus faibles), une excellente conductivité (puisque le matériau est monobloc) et une organisation spatiale de l’électrode (modélisation plus facile, transferts mieux compris).
Néanmoins, la très grande surface spécifique de ces structures ou des nano-matériaux va engendrer un inconvénient : le phénomène de SEI va prendre une grande importance (relativement aux réactions redox de pile) et donner lieu à d’importantes capacités irréversibles qu’il faudra contrôler.
III- Les microbatteries 3D
III-1. Contexte et principe
Le concept de microbatterie désigne un accumulateur dont l’épaisseur est compris entre 0,1 et quelques millimètres pour une surface projetée de l’ordre du centimètre carré. C’est donc un générateur de très petite taille destiné à fournir de l’énergie aux équipements miniaturisés. En effet, le besoin en microbatteries est alimenté par plusieurs applications qui entrent dans l’ère du ‘‘nano’’. D’abord, dans le domaine médical, l’alimentation énergétique des pacemakers ou des appareils auditifs requièrent des batteries de faible masse et peu encombrantes. De plus, les technologies de délivrance de médicaments in-vivo seront bientôt matures et peuvent exiger des sources d’énergie adaptées à leur taille et à leur milieu. Ensuite, les systèmes micromécaniques (regroupés sous l’acronyme MEMS : Micro ElectroMechanical Systems) sont de plus en plus développés et le problème de leur alimentation reste peu étudié71.
Dans les systèmes de type microbatterie, une grandeur prend une importance toute particulière : la surface projetée (« footprint area » en anglais). En effet, l’intégration de la microbatterie aux micro-systèmes électroniques demande une certaine compatibilité de forme avec ceux-ci, c’est-à-dire une surface la plus proche possible de celle du composant alimenté. Cette surface projetée devient une grandeur critique alors que le volume du système (en particulier son épaisseur) a souvent une importance moindre. En conséquence, on s’intéressera à la capacité surfacique (mAh.cm-²), à l’énergie surfacique (Wh.cm-²) et à la
l’augmentation de la puissance) et l’augmentation de la quantité de matériau actif dans la batterie (améliorant ainsi la capacité et donc l’énergie).
Pour diminuer les distances de diffusion de l’ion Li+, les microbatteries 2D ont été développées. Celles-ci s’appuient sur les techniques de couches minces. Sur la surface plane des collecteurs de courant sont déposées de fines couches de matériau actif et la distance inter-électrode est minimisée. Obtenues par pulvérisation magnétron72, photolithographie73, PVD74 ou électrodépôt, ces couches ont des épaisseurs comprises entre 0,1 et quelques microns. L’avantage de cette minceur est que le transport de matière au sein des électrodes et de l’électrolyte est largement facilité. Les accumulateurs ainsi conçus utilisent pour la plupart des technologies Li-ion (réaction d’insertion et d’alliage), même si d’autre systèmes sont étudiés comme Zn-Ag2O et Zn/air. Le design en couche mince apporte alors un autre
avantage : elle permet de diminuer les contraintes mécaniques dues à la formation de l’alliage entre lithium et matériau actif. Le Tableau I-3) donne un aperçu de ce qui a été réalisé dans le domaine des microbatteries dans les 15 dernières années.
1995 1999-2000 1999-2000 1996-1999 19996-1999 2004 2007 2001 2007
Process Sputtering Th. Evap. Pulsed laser deposition LPCVD Electrodépot PVD Cathode V2O5 LiCoO2 Mn2O4 LiCoO2 LiMn2O4 nc Pt NiOOH AgO
Anode Li Li Li Li Li Cu2Sb Al Zn Zn
Electrolyte LIPON LIPON Liglass LiClO4 LiClO4 EC/DEC KOH/KCl KOH/ZnO KOH
Tension (V) 3,75 4 4-4,5 3,2-3,5 3,2-3,6 2,4-4 0,8-1,3 1,7-1,8 1,55 Epaisseur (µm) 7-15 2,2-15 0,3 0,2-1,5 0,2-1,5 0,25 6 100 25 Surface (cm²) 1,21 1-3,22 1 3 3 1 1 0,02 1 Capacité (µAh) 13,5-18,5 160 15 216 252 300 64 1,9-6,8 100 Courant décharge (µA) 242-484 100 10 30 30 40 240 250 Capacité surfacique (µAh/cm²) 20-120 60-150 20-60 72 84 300 400 277-970 100
Tableau I-3 : Revue des performances des quelques microbatteries (d’après F. Albano et al.74).
Le tableau I-3 montre que l’énergie surfacique des microbatteries 2D est inférieure au mWh.cm-2 et leur capacité spécifique proche de la centaine de µAh.cm-2. Néanmoins, ces résultats ont été obtenus pour des régimes de charge peu élevés, entre C et C/10. Ces valeurs ne sont pas encore suffisantes pour satisfaire les besoins en énergie des MEMS par exemple.
En fait, lors de la conception d’une microbatterie 2D, il existe toujours un compromis entre puissance et capacité d’une électrode. Une couche très mince présente de bonnes performances en terme de puissance et régime de décharge mais sa capacité est limitée par la faible quantité de matériau actif. A l’inverse, si on augmente l’épaisseur de la couche, la capacité augmente mais les performances en puissance s’effondrent. Pour améliorer cette technologie, il est nécessaire de développer des électrodes à la fois puissantes et énergétiques.
A partir de ce constat le concept de microbatterie 3D a été proposé. Cette approche consiste à élaborer des électrodes tridimensionnelles, de très grande surface développée. Pour cela, on va tirer profit de l’importance moindre de l’épaisseur du système et développer des nano-architectures qui créeront de la surface dans la troisième dimension, perpendiculairement au substrat. On passe ainsi d’une électrode plane à une électrode nano-architecturée, souvent basée sur des nano-piliers ou des nano-tubes, conçue de manière à augmenter la surface d’électrode réelle (surface développée), tout en maintenant une faible surface projetée. L’intérêt de cette nanostructuration est que le matériau actif se trouve toujours en couche mince, mais en quantité plus importante puisque la surface de l’électrode est augmentée. L’importance de l’épaisseur L de la batterie par rapport au rayon de sa surface projetée d est donnée par le facteur de forme (aspect ratio) qui vaut L/d. Plus celui-ci est grand, plus l’espace est judicieusement utilisé et plus l’énergie surfacique de la batterie sera grande. Cette approche propose d’importants challenges scientifiques et techniques comme la réalisation de collecteurs de courant ou d’électrodes nanostructurés, le dépôt conforme de matériaux actifs sur ces nanostructures, l’ajout d’électrolyte, les contacts électriques et parfois la préparation simultanée de l’anode et de la cathode.
III-2. Géométries envisageables pour les microbatteries 3D et leurs
performances
Long et al.75 ont rassemblé, dans un récent article de revue, les différentes stratégies que l’on peut envisager pour concevoir la géométrie d’une microbatterie 3D :
a) réseau alterné d’anodes et de cathodes cylindriques interconnectées b) réseau de feuillets (anodes et cathodes) interdigités
c) réseau de cathodes cylindriques recouvertes d’un dépôt conforme de polymère conducteur d’ions, incorporé dans une matrice anode
d) architecture apériodique (éponge) : réseau poreux de particules cathode, recouvertes de polymère conducteur d’ions dans une matrice anode
La Figure I-9 présente un schéma descriptif de ces quatre principes :
Figure I-9 : Quatre géométries pour les microbatteries 3D (d’après Long et al.75).
Les géométries a) et b) sont interdigitées, c’est à dire qu’elles sont constituées de deux réseaux distincts d’anodes et de cathodes, imbriqués l’un dans l’autre. Les deux types d’électrodes possèdent la même géométrie (piliers ou feuillets).
Pour obtenir la géométrie a), on alterne sur un même substrat une ligne de cathodes et une ligne d’anodes cylindriques. Typiquement, le diamètre des piliers varie entre 5 et 100 µm
a)
d)
c) b)