DOCTORAT DE L'UNIVERSITÉ DE TOULOUSE
Délivré par :
Institut National Polytechnique de Toulouse (INP Toulouse) Discipline ou spécialité :
Chimie Organométallique et de Coordination
Présentée et soutenue par :
M. JAMAL EL KARROUMIle samedi 10 mai 2014
Titre :
Unité de recherche : Ecole doctorale :
REACTIONS DE CARBONYLATION DE SUBSTRATS NATURELS DE
PLANTES
Sciences de la Matière (SM)
Laboratoire de Chimie de Coordination (L.C.C.) Directeur(s) de Thèse :
MME MARTINE URRUTIGOITY M. AHMED BENHARREF
Rapporteurs :
M. MATHIEU SAUTHIER, ECOLE NATIONALE SUP DE CHIMIE DE LILLE M. MOHAMED AKSSIRA, UNIVERSITE HASSAN II CASABLANCA MAROC
Membre(s) du jury :
1 M. HASSAN BIHI LAZREK, UNIV. DE CADI AYYAD MARRAKECH MAROC, Président
2 M. AHMED BENHARREF, UNIV. DE CADI AYYAD MARRAKECH MAROC, Membre
2 Mme MARTINE URRUTIGOITY, INP TOULOUSE, Membre
2
Remerciements
Ce travail de thèse a été réalisé au Laboratoire de Chimie de Coordination de Toulouse dans l’équipe «Catalyse et Chimie Fine », et au Laboratoire de Chimie Biomoléculaire, Substances Naturelles et Réactivité de Marrakech.
Je tiens à remercier en premier lieu, Monsieur le Professeur Philippe Serp pour m’avoir accueilli au sein de l'équipe.
J’adresse mes remerciements à Madame le Professeur Martine Urrutigoïty pour son encadrement et son soutien durant ces trois années de thèse. Vos explications, vos précieux conseils et vos connaissances bibliographiques m'ont beaucoup apporté. Je vous remercie énormément pour votre aide, votre grande patience, votre disponibilité et aussi pour votre gentillesse.
Je tiens à remercier Monsieur le Professeur Ahmed Benharref. J’ai beaucoup appris en vous côtoyant. Je vous remercie pour la confiance que vous m'avez accordée pour mener à bien ce projet de thèse. J’espère que nos souhaits en ce qui concerne la recherche au Maroc se réaliseront.
Je souhaite ensuite remercier le Docteur Madame Maryse Gouygou qui m’a aussi aidé à réaliser ce travail. Je vous adresse toute ma gratitude et je vous remercie pour votre disponibilité, vos conseils, votre sympathie et pour avoir été présente chaque fois que j’en avais besoin.
Je remercie Monsieur le Professeur Hassan Bihi Lazrek de l’Université Cadi Ayyad de Marrakech d’avoir accepté d’être président de mon jury de thèse. Ainsi que mes rapporteurs Messieurs les Professeur Mathieu Sauthier et Mohamed Akssira pour le temps qu’ils ont accordé à la lecture de cette thèse et à l' élaboration de leur rapport. L’intérêt qu’ils ont porté à mes travaux ainsi que leurs critiques ont permis d’améliorer ce mémoire.
Je souhaite remercier aussi tous les services analytiques de l’ENSIACET, du LCC, et de l’UPS qui m’ont aidé à avancer ce travail.
3
Un grand merci à tous les membres de l'équipe pour leur soutien quotidien: Philippe K., Odile, Rosa, Carole, Jérôme D., Jérôme V., Marc, Christelle, Lucie, Idaline, Nora, Laurent, Jasmine, Sakhayna, Sylvie, Liping, Trang, Mustapha, Bruno, Revathi, Anas, Xiaojiang, Delphine, Jenny, Hanh, Romain, Pierre, Abedrrahim, Florence, Sandrine. Enfin une pensée spéciale pour Nabila, Abdelouahd, Hassan, Kevin, Ganna, Stéphane, Mohamed et Valentin.
Je souhaite aussi remercier Monsieur le Professeur Eduardo Dos Santos de m’avoir accueilli dans son laboratoire pendant mon séjour au Brésil. Merci aussi à tous les autres membres du laboratoire notamment Kelly, Matheus, Evelisy, Roberta, Rochel.
Je n'oublie pas de remercier ma famille et tous mes amis qui m’ont accompagné et soutenu pendant tout le long de mon parcours d’études.
4 Abréviations dppm dppe dppp dppb dcpb dppf TPP DBP TMP DMP PPP PCy3 PMes3 t Bu nBu p-iBu Me Et Ar COD APTS T.C Rdt S/C P/Pd P/Rh t mL GC HPLC IR RMN J SM HRMS PCO DCM DCE DMF THF e.d Isom. TOF TON PM 1,2-bis(diphénylphosphino)méthane 1,3-bis(diphénylphosphino)éthane 1,4-bis(diphénylphosphino)propane 1,4-bis(diphénylphosphino)butane 1,4-bis(dicyclohexylphosphino)butane 1,1'-(diphénylphosphino)ferrocène triphénylphosphole dibenzphosphole tétraméthylphosphole diméthylphosphole pentaphénylphosphole tricyclohexylphosphine trimésithylphosphine ter-butyl n-butyl para-isobutyl méthyl éthyl aryl cycloctadiène
acide para-Toluène sulfonique taux de convérsion
rendement
rapport substrat sur catalyseur rapport phosphore sur palladium
rapport phosphore sur rhodium temps de travail
millitre
chromatographie en phase gazeuse chromatographie liquide à haute performance
infra rouge
resonance magnetique nucléaire constante de couplage spectrometrie de masse
spectrométrie de masse à haute résolution pression de CO dichlorométhane dichloroétghane diméthylformamide tétrahydrofurane excès diastéréoimérique isomérisation turn over frequency
turn over number poids moléculaire
5
Résumé:
La fonctionnalisation par voie catalytique de substrats naturels a été menée dans le but d’accroître l’activité biologique reconnue de la molécule naturelle de départ ou de découvrir de nouvelles activités. Des réactions catalytiques telles que l'hydroformylation et l'alcoxycarbonylation mettant en jeu le monoxyde de carbone et catalysées par des complexes du rhodium et du palladium ou encore la réaction de cycloisomérisation catalysée par des complexes d'or ou du platine ont déjà permis d'accéder sélectivement à de nouvelles molécules intéressantes.
Dans une première partie nous nous sommes intéressés à l'huile essentielle du cèdre de l'atlas (cedrus atlantica) qui est constituée d’une partie hydrocarbure et d’une partie oxygénée. Notre étude s'est focalisée sur la partie oxygénée de l'huile essentielle du cèdre de l'atlas qui est composée de deux cétones isomères, les Z- et E-α- atlantones. La réaction de cyclocarbonylation des alcools allyliques dérivés des α-atlantones a été étudiée. Cette réaction catalysée par des complexes du palladium permet d'obtenir des lactones à 5 et à 6 chainons. Plusieurs systèmes catalytiques de type [PdCl2L2]/SnCl2.2H2O ont été testés, lors de cette
étude et nous avons montré que la regiosélectivité de la réaction peut être contrôlée par la nature du ligand utilisé. Ainsi, les ligands monophosphines favorisent la formation des lactones à 6 chainons obtenues sous forme de deux diastéréoisomères et les ligands diphosphines favorisent la formation de celles à 5 chainons obtenues sous forme de quatre diastéréoisomères. Ces lactones sont complètement caractérisées par RMN 1D et 2D et la spectrométrie de masse. Des monocristaux ont été obtenus et analysés par diffraction des rayons X.
Dans une deuxième partie, la synthèse d'aldéhydes par réaction d'hydroformylation à partir de l'estragol, un allylbenzène extrait de l'huile essentielle de l'estragon, a été étudiée en présence du système catalytique [Rh(cod)(OMe)]2/ligand phosphole. Ainsi, nous avons évalué l'activité
de plusieurs ligands phospholes dans cette réaction d'hydroformylation. Tous les ligands phospholes testés se sont révélés actifs et chimiosélectifs dans la réaction d'hydroformylation d'estragol pour donner majoritairement l'aldéhyde linéaire correspondant.
Dans une étude préliminaire, nous avons étudié la réaction de cycloisomérisation d'énynes oxygénés dérivés d'α-atlantones catalysée par des complexes d'or ou du platine.
6
Summary
The catalytic functionnalisation of the natural substrates have been developped to increase their own biological activity or to give them new biological properties. The reactions such as hydroformylation, alkoxycarbonylation and cyclocarbonylation in presence of carbon monoxide catalyzed by rhodium or palladium complexes or cycloisomerisation catalyzed by gold or platinium complexes give an access to new interesting molecules with high selectivity.
In first part we have been interested in study of the essential oil of the Atlas Cedar (Cedrus
Atlantica). We focused in this study on the oxygenated fraction, which contains the two
sesquiterpenic ketone,isomers Z- and E-α-atlantone. Starting from allylic alcohols derived from α-atlantone, the cyclocarbonylation reaction catalyzed by palladium complexes have been investigated. This reaction provide a mixture of five and six membered ring lactones with excellent conversion and excellent chemioselectivity. Different catalytic systems [PdCl2L2]/SnCl2.2H2O or [Pd(OAc)2]/L have been studied. The regiochemical control
depends on the nature of the ligand L. The monophosphine ligands favor the formation of the six-membered ring lactones obtained as two diastereomers, while the diphosphine ligands allow the formation of the five- membered ring lactone obtained as four diastereomers. These new lactones were fully characterized by 1D and 2D NMR and mass spectrometry. Monocrystals of the six- and five-membered ring lactones suitable for X-ray diffraction analysis have been obtained.
In a second part the hydroformylation reaction of estragol, a natural allylbenzene extracted from the essential oil of estragon, have been studied with the catalytic system [Rh(cod)(OMe)]2/phospholes. All the phosphole ligands show good activities and
chemoselectivities in the hydroformylation of estragol and affords the linear aldehyde corresponding as a major product.
In a preliminary study, we have investigated the cycloisomerisation reaction of o-tethered enynes derived from α-atlantones catalyzed by gold or platinum complexes.
7
Sommaire
Introduction générale: ... 10
Chapitre I: Rappels bibliographiques sur la fonctionnalisation des monoterpènes et des allylbenzènes. I-Introduction ... 15
II-Réaction d'alcoxycarbonylation et de cyclocarbonylation... 15
II-1-Réaction d'alcoxycarbonylation des monoterpènes et des dérivés d'allylbenzènes…….. 16
II-2-Réaction de cyclocarbonylation... 23
II-3-Mécanisme de la réaction... 29
III-Réaction d'hydroformylation de terpènes et de dérivés naturels d’allybenzène... 31
III-1-Hydroformylation des monoterpènes avec une double liaison exocyclique... 32
III-2-Hydroformylation des monoterpènes avec une double liaison endocyclique... 36
III-3-Hydroformylation des monoterpènes avec un groupement hydroxy... 40
III-4-Hydroformylation des dérivés naturels de type allybenzène... 42
III-5-Mécanisme de la réaction d'hydroformylation... 46
IV-Réaction d'hydroaminométhylation... 48
IV-1-Hydroaminométhylation des monoterpènes et des allylbenzènes...49
IV-2-Mécanisme de la réaction d'hydroaminométhylation... 53
V-Réaction de cycloisomérisation d'énynes dérivés de monoterpènes……….. 54
V-1- Cycloisomérisation d'énynes issus de monoterpènes... 55
IV-2-Mécanisme de la réaction de cycloisomérisation...59
VI-Conclusion………...………... 59
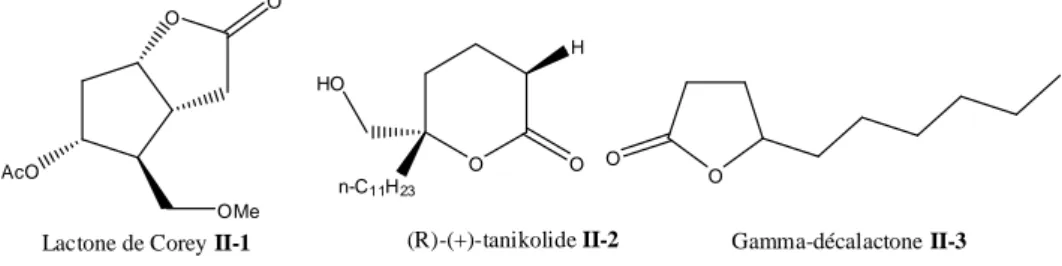
Chapitre II: Cyclocarbonylation d'alcools homoallyliques dérivés d'α-atlantones catalysée par le système catalytique [PdCl2L2/SnCl2].2H2O. I-Introduction... 63
II-Transformation chimique des α-atlantones (Z et E)... 67
II-1-Synthèse du 6-méthyl-2-(4 méthylphényl)heptan-4-one, II-15... 67
II-2-Synthèse du 4-isobutyl-6-para-tolylhept-1-ène-4-ol II-17... 68
II-3- Synthèse du 4-isobutyl-2-méthyl-6-para-tolylhept-1-ène-4-ol II-18... 68
III-Réaction de cyclocarbonylation des alcools homoallyliques II-17 et II-18... 69
III-1-Cyclocarbonylation de l’alcool II-17 ……….………..….. 69
III-1-1-Etude du système [PdCl2(PPh3)2]/SnCl2.2H2O... 72
III-1-2-Synthèse des complexes de palladium (II)... 75
III-1-3-Etude du système [PdCl2(L)2]/SnCl2.2H2O, L= monophosphine... 77
III-1-4-Etude du système [PdCl2L2] /SnCl2.2H2O, L= diphosphine... 80
III-1-5-Caractérisation des lactones II-19 et II-20... 83
III-1-6-Mécanisme de la réaction... 85
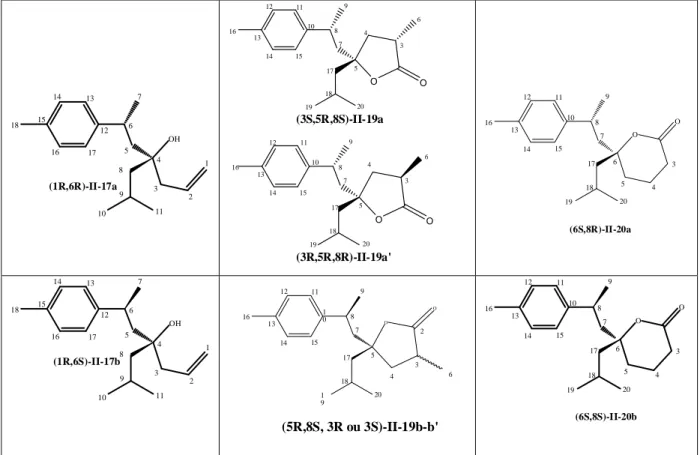
III-2-Cyclocarbonylation de l'alcool II-18... 86
III-2-1-Etude du système [PdCl2(L)2]/SnCl2.2H2O, L= monophosphine... 86
III-2-2-Etude du système [PdCl2L2] /SnCl2.2H2O, L= diphosphine ... 87
IV-Conclusion……….. 88
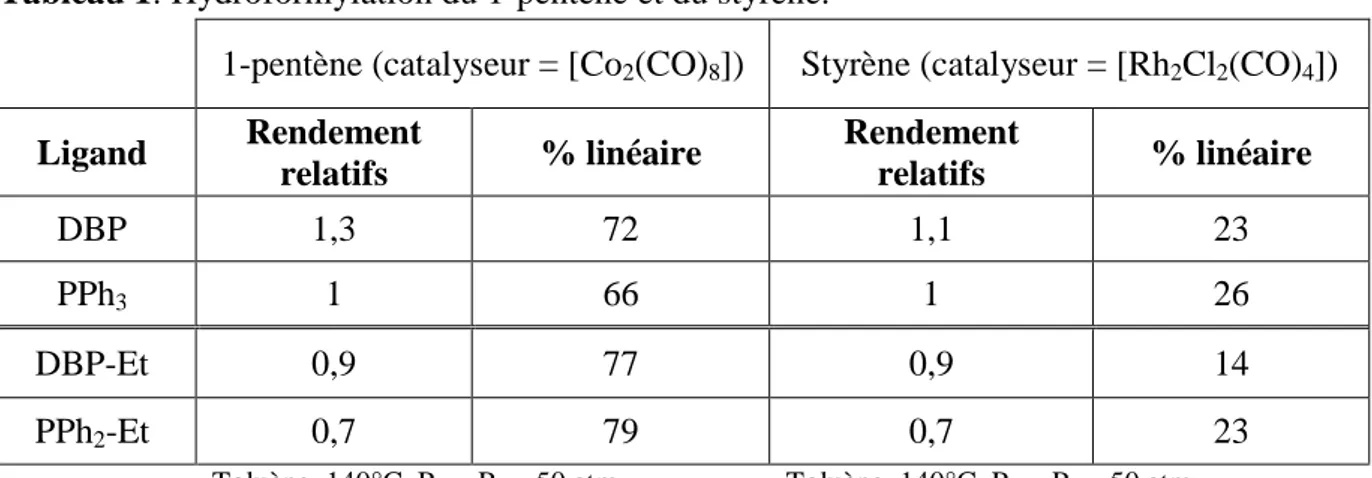
Chapitre III: Hydroformylation du styrène et de l'estragol catalysée par des complexes du rhodium comportant des ligands phospholes. I-Introduction... .91
8
III-Synthèse des ligands phospholes et phospholyl(phosphino)méthane……….... 98
III-1-Stratégie de synthèse... 98
III-2-Synthèse des 1-phénylphospholes... 99
III-3-Synthèse des ligands phospholyl(phosphino)méthane... 101
IV. Hydroformylation du styrène... 102
IV.1-La réaction... 102
IV.2-Précurseurs du rhodium utilisés...103
IV.3-Evaluation des ligands phospholyl(phosphino)méthane III-10... 103
IV.4-Evaluation des ligands phospholyl(phosphino-borane)méthane III-11... 107
IV.4-Evaluation des ligands phospholes……… 110
IV-5-Conclusion...111
V-Hydroformylation de l'estragol... 111
V.1. La réaction……….. 112
V-1-Etude de l'effet du ligand... 112
V-2-Effet du rapport P/Rh... 113
V-3-Effet de la température... 114
V-4-Influence de l'utilisation d'un mélange de deux ligands sur l'hydroformylation de l'estragol... 115
VI-5-Conclusion………..……….. 117
Chapitre IV: Projets en cours et perspectives I-Cyclocarbonylation d'alcools homoallyliques dérivés d'α-atlantones catalysée par le système catalytique [Pd(OAc)2]/L/APTS... 121
I-1-Cyclocarbonylation de l'alcool II-18 avec le système catalytique [Pd(OAc)2]/L2/ATPS (L2=diphosphine)... 122
II-2-Cyclocarbonylation de l’alcool II-17 avec le système catalytique [Pd(OAc)2]/L/ATPS (L= monophosphine, diphosphine)... 124
II-Cycloisomérisation d' 1,6-énynes oxygénés dérivés d'α-atlantones catalysée par des complexes d'or et de platine... 126
II-1-Synthèse des 1,6-énynes oxygénés... 126
II-1-1-Synthèse du 5-(para-tolyl)-3-allyloxy-3-isobutyl-1-phénylhexyne IV-1... 126
II-1-2-Synthèse de 6-paratolyl-4-(propyl-3-oxy)-2-méthyl-hept-2ène IV-2... 127
II-2-Réaction de cycloisomérisation de l’ényne oxygéné IV-1...128
II-2-1-Etude de complexes d'or(I)... 129
II-2-2-Etude de complexes complexes de platine(II)... 130
III-Conclusion et perspectives……… 131
Conclusion générale...133
Chapitre V: Partie Expérimentale... 137
I-Considérations générales...139
II-Synthèses des dérivés des Z- et E-α-atlantones...142
III-Synthèses des ligands phospholes...153
IV-Synthèses des complexes...161
V-Réactions catalytiques...166
9
Introduction générale:
Depuis plusieurs années l'utilisation des matières premières renouvelables, notamment celles issues de l'agriculture, a pris une place prépondérante dans l'industrie chimique1 du fait de la nécessité d'évoluer vers un développement durable associé à l'émergence des principes de la chimie verte2 pour une production plus respectueuse de l'environnement. Plusieurs recherches sont consacrées à l'utilisation des ressources renouvelables pour la production de biocarburants3 et de produits de base pour l'industrie chimique.4 Parmi les ressources renouvelables qui sont largement étudiées nous trouvons les acides gras d'huiles végétales, et leurs esters méthyliques, ainsi que les terpènes.
La carbonylation des oléfines est une transformation attractive et efficace qui permet d'introduire une molécule de CO pour accéder à des esters, aldéhydes, cétones ou encore des lactones en une seule étape économe en atomes. En effet, la carbonylation des acides et esters gras insaturés 5,6,7donne accès à des composés fonctionnalisés d'intérêt en chimie fine. Les monoterpènes sont aussi largement étudiés en carbonylation. Certains terpènes sont extraits par rectification des essences végétales dont ils sont les constituants odorants et d'autres sont obtenus par hémisynthèse à partir des précédents. En effet, la fonctionnalisation des terpènes par voie catalytique en présence des métaux de transitions a attiré l’atention dans ces dernières années8,9,10 car elles permettent à partir de produits moins couteux de produire des synthons à haute valeur ajoutée.
Nous nous sommes intéressés tout au long de ce travail à la fonctionnalisation d’un sesquiterpène extrait de l’huile essentielle du cèdre de l’Atlas, l’α-atlantone et d'un dérivé naturel de l'allylbenzène, l’estragole extrait de l'estragon, afin d'accéder à de nouvelles molécules qui peuvent présenter une activité biologique potentielle.
1 U. Biermann, U. Bornscheur, M.A.R. Meir, J. O.Metzger, H. J. Schäfer, Angew. Chem.Int. Ed. 2012,50, 3854-3871.
2
P. T. Anastas, M. M. Kirchhof, Acc. Chem. Res. 2002,35, 686-694. 3
a) D. Casanave, J-L. Duplan, E. Freund, Pur Appl. Chem. 2007, 79, 2071-2081. b)D. Martin Alonso, J. Q. Bond, J. A. Dumesic, Green Chem., 2010, 12,1493-1513.
4 a) A. Corna, S. Iborra, A. Velty, Chem. Rev. 2007, 107, 2411-2502; b) R.Diercks, J.-D. Arndt, S. Freyer, R. Geier, O. Machhammer, J. Schawartze, M. Volland, Chem. Eng. Technol. 2008, 31, 631-637.
5 EnFrankel, S. Meltin S, W.K. Rohwedder, I. Wender, J. Am. oil Chem. Soc. 1969, 46, 133-138. 6
Kandanarachchi P, Guo A, Petrovic Z J. Mol. Catal A. Chem. 2002, 184, 65-71. 7
C. Jimenez-Rodriguez , Eastham GR, D.J. Cole-Hamilton, Inorg. Chem. Commun. 2005, 8, 878-881. 8
P. J. Teisseire "Chimie des substances odorantes", 1991, Lavoisier (Paris).
9
K. A. D. Swift Topics in Catalysis, 2004, 27, 143-155.
10
10
Dans le premier chapitre, nous décrivons les principaux résultats de la littérature sur les réactions de carbonylation de monoterpènes et d'allybenzènes naturels telles que l’alcoxycarbonylation, la cyclocarbonylation et l’hydroformylation qui donnent accès à des esters, des lactones ou des aldéhydes. La réaction d’hydroaminomethylation qui est une réaction tandem de carbonylation-hydrogénation, y est également présentée. La réaction de cycloisomérisation a été également étudiée sur les monoterpènes transformés en énynes et montre qu’elle permet d’accéder à des molécules cycliques complexes en une seule étape répondant ainsi au souci d'économie d'atomes.
Les travaux de thèse sont présentés ensuite dans les chapitres suivants. Dans un deuxième chapitre nous exposons les résultats obtenus dans la réaction de cyclocarbonylation des alcools homoallyliques dérivés d'α-atlantones et catalysée par le système catalytique de palladium(II) [PdCl2L2/SnCl2.2H2O] comportant des ligands phosphine.
Le troisième chapitre est consacré à la réaction d'hydroformylation catalysée par des complexes de rhodium coordonné à des ligands phospholes. Dans un premier temps, nous présentons la synthèse des ligands phospholes y compris les ligands phosphole-phosphine utilisés pour cette étude. Ensuite, nous discutons les résultats catalytiques obtenus dans la réaction d'hydroformylation tout d'abord sur le styrène, choisi comme substrat modèle et par la suite sur l'estragol.
Enfin, le quatrième chapitre traite des projets en cours de réalisation. La première partie est consacrée à la réaction de cyclocarbonylation des alcools homoallyliques catalysée par un autre système catalytique de palladium(II) de type [Pd(OAc)2]/L/APTS. La deuxième partie
de ce chapitre concerne une étude préliminaire de la réaction de cycloisomérisation d' 1,6-énynes dérivés de l'α-atlantones catalysée par des complexes d'or(I) et de platine(II). Nous abordons quelques perspectives dans la suite de ce travail.
12
Chapitre I:
Rappels bibliographiques sur la fonctionnalisation
des monoterpènes et des allylbenzènes.
14
I-Introduction :
Les monoterpènes sont une famille de produits naturels qui présentent un intérêt chimique considérable. Connus depuis un peu plus d’un siècle, les monoterpènes constituent les composants d’un très grand nombre d’huiles essentielles tirées de fleurs, de feuilles ou de fruits. Ils comportent dix atomes de carbone et correspondent à deux unités isopréniques et sont classés en trois groupes, acycliques, monocycliques et bicycliques. Ces substrats ont fait l’objet d’études variées sur leur transformation chimique dû à la présence de groupements fonctionnels dans leur squelette qui peuvent être impliqués dans des réactions chimiques.
Dans cette partie bibliographique, nous nous sommes attachés à présenter les résultats les plus pertinents obtenus dans les réactions de carbonylation telles que l’alcoxycarbonylation, l’hydroformylation ainsi que l’hydroaminométhylation qui est une réaction tandem de carbonylation et d’hydrogénation.
Nous présentons aussi les résultats de la littérature concernant les dérivés naturels d’allylbenzène qui sont des substrats intéressants à fonctionnaliser dans les réactions de carbonylation.
Enfin une dernière partie est consacrée à la réaction de cycloisomérisation d’énynes dérivées de monoterpènes. Cette réaction est une réaction intéressante au point de vue économie d’atomes et qui permet d’accéder à des molécules complexes bicycliques et tricycliques contenant des motifs diéniques ou des cyclopropanes.
II-Réaction d'alcoxycarbonylation et de cyclocarbonylation:
L'alcoxycarbonylation est une réaction entre une oléfine et un alcool en présence de monoxyde de carbone (schéma 1). Elle permet de préparer des esters linéaires ou ramifiés qui présentent de nombreuses applications industrielles dans la synthèse de plastifiants et de détergents1. Les esters ramifiés ont une large application en industrie pharmaceutique2,
1
Ph. Kalck, M. Urrutigoïty, O. Dechy-Cabaret In: “Catalytic Carbonylation Reactions” Top. Organomet. Chem. Matthias Beller Ed., Springer-Verlag, 2006, 18, 97-123.
15
puisqu'ils peuvent représenter des précurseurs importants pour la synthèse de différents agents anti-inflammatoires non stéroïdiens, comme le naproxène, et l’ibuprofène.3,4
R CO R'OH + + R OR' O + R OR' O [Pd] *
Schéma 1: Réaction d'alcoxycarbonylation.
Cette réaction est catalysée par des complexes du palladium dans des conditions douces. De nombreux catalyseurs tels que le cobalt, le nickel ont aussi été utilisés pour cette réaction5,6mais dans des conditions de pressions et températures très sévères. L'enjeu était de contrôler la chimiosélectivité de la réaction afin de favoriser la regioséléctivité. Plusieurs études ont montré que les complexes du palladium sont très actifs dans ce type de réaction et permettent de travailler en conditions plus accessibles.7,8,9
II-1-Réaction d'alcoxycarbonylation des monoterpènes et des dérivés d'allylbenzènes:
Cette réaction d’alcoxycarbonylation a été largement étudiée au sein de notre laboratoire sur différents monoterpènes et allylbenzènes naturels en utilisant le complexe de palladium (II) de type [PdCl2(PPh3)2] en présence de 2.5 équivalent de SnCl2, dans des conditions
relativement douces de température, 97°C, et sous 40 bars de pression de CO pendant 16h (schéma 2).10,11 Les différents résultats obtenus ont montré que ce système catalytique est très actif et donne de bonnes conversions (84 à 95%) avec une chimio- et une regioséléctivité importantes avec certains monoterpènes. En effet dans le cas du limonène I-1, la réaction permet d'accéder à l'ester correspondant I-4 avec un rendement de 74%. Lors de cette réaction, la formation du produit d'isomérisation I-2 et du produit I-3 issu de l'addition du méthanol sur l'oléfine, 3% et 8% respectivement, a été observée. Dans les conditions utilisées l'ester linéaire est sélectivement formé. Pour la (-)- carvone I-5, la sélectivité en ester I-6
3 a) H. Sonawane, N. S. Bellur, J. R. Ahuja, D. G. Kulkarni Tetrahedron: asymmetry 1992, 3, 163-192; b) J. P. Rieu, A. Bouchrele, H. Cousse, G. Mouzin Tetrahedron 1986, 42, 4095-4131.
4 W. Gerhazt (Ed) Ullman's Encyclopedia Of Industrial Chemistry, Vol. 3, VCH Weinhein, 1985, p. 41. 5 R. F. Heck, J. Am. Chem. Soc., 1969, 91, 6707-6714.
6
H.M. Colquoun, D.J. Thompson, M.V. Twigg, Carbonylation, 1991, Plenum Press, New York 7
G.P. Chiusoli, C. Venturello, S. Merzoni, Chem.Ind. 1968, 29, 977. 8 C. Botteghi, G. Consiglio, P. Pino, Chimia 1973, 27, 477-478. 9 J. F. Knifton, J. Org. Chem, 1976, 41, 2885-2890.
10
a) R. Naigre, T. Chenal, T. Ciprès, Ph. Kalck, J.C. Daran, J. Vaisserman, J. Organomet. Chem. 1994, 480, 91-102. b) T. Chenal, T. Ciprès, I. Jenck, Ph. Kalck, Y. Perez, J. Mol. Catal. 1993, 78, 351-366
11
a) T. Chenal, thèse de l’ INP de Toulouse, 1992. b) G. Lenoble, R. Naigre, T. Chenal, M. Urrutigoïty, J.C. Daran, Ph. Kalck, Tetrahedron : Asymmetry 1999, 10, 929-936.
16
augmente à 90% et le sous produit I-7 est obtenu avec 5% de rendement. Par contre, dans le cas de la dihydrocarvone I-8 seulement 60% de sélectivité en ester est obtenue, les produits secondaires I-10 et I-11 sont formés avec des rendements de 14% et 10% respectivement. Il est dommage que les auteurs n'expliquent pas les résultats différents obtenus entre la carvone et la dihydrocarvone qui ne diffèrent que par la présence d'une double liaison endocyclique pour I-5.
17 O O COOMe O OMe + PdCl2(PPh3)2, MeOH PPh3, SnCl2, Toluène (-)-carvone I-5 90% O O COOMe O OMe + PdCl2(PPh3)2, MeOH PPh3, SnCl2, Toluène dihydrocarvone I-8 O + I-6 (90%) I-7 (5%)
I-9 (60%) I-10 (14%) I-11 (10%) (+)-limonène I-1
OMe
COOMe
+ +
PdCl2(PPh3)2, MeOH
I-2 (3%) I-3 (8%) I-4 (74%)
PPh3, SnCl2,Toluène O COOMe PdCl2(PPh3)2, MeOH PPh3,SnCl2, Toluène OAc PdCl2(PPh3)2, MeOH PPh3,SnCl2, Toluène OAc OMe OAc + COOMe (+)-Pulégone I-15 acétate d'isopulégyle I-12
I-16 (30%)
I-13 (7%) I-14 45%)
Schéma 2: Alcoxycarbonylation de quelques monoterpènes.
Avec le même système catalytique, les auteurs ont élargi cette réaction à des oléfines plus complexes, telles que la (+)-pulégone I-15 et l'acétate d'isopulégyle I-12 (schema 2). Dans le cas de la (+)-pulégone, le rendement en esters n'est que de 30% du à l'encombrement stérique de la double liaison tétrasubstituée. Dans le cas de l'acétate d'isopulégyle le rendement en
18
ester est meilleur, 45%, mais reste encore faible. Les auteurs expliquent ce résultat par l'encombrement stérique du groupement acétate qui gêne la coordination du substrat au palladium.
L'alcoxycarbonylation de monoterpènes bicycliques comme le β-pinène et le (-)-camphène12 a été reportée dans la littérature avec le même système catalytique et dans des conditions opératoires similaires à celles décrites précédemment. Les conversions obtenues pour le β-pinène sont faibles, de l’ordre de 8 à 16%. Le β-pinène I-20 n'est pas transformé en ester mais la réaction produit un mélange de produits issus de réactions de réarrangement donnant d'autres terpènes comme le limonène I-1, l'α-terpinolène I-2, l'α-terpinène I-21 et réactions d’additions nucléophiles avec le chlore I-22 et un groupement méthoxy I-3 (Schéma 3). Les auteurs ont remarqué qu’en absence de complexe du palladium, la présence de SnCl2
comme acide de Lewis suffisait pour obtenir les sous-produits de transformation du β-pinène. Par contre, le camphène I-17 est transformé à 61% de conversion avec une chimiosélectivité de 90% en faveur de l'ester avec une régiosélectivité en linéaire (I-18) de 100% (Schéma 3). Ce dernier est obtenu sous forme de deux isomères avec un rapport 55/45 en faveur de l'isomère endo. L'utilisation des diphosphines comme la dppe (1,2-bis-diphényl-phosphinoéthane), la dppp (1,3-bis-diphénylphosphinopropane), ne donne aucune activité catalytique. Seule la dppb (1,4-bis-diphénylphosphinobutane) donne une conversion de 14% avec une sélectivité de 45% en faveur de l'ester I-18.
COOMe +
MeO
(-)-camphène I-17 I-18 I-19
PdCl2(PPh3)2/SnCl2 CO/MeOH PdCl2(PPh3)2/SnCl2 CO/MeOH Cl + + (-)-β-pinène I-20
I-2 I-21 I-22
Schéma 3: Alcoxycarbonylation du camphène et du pinène.
12
L. L. da Rocha, A. O. Dias, E. N. dos Santos, R.i Augusti, E. Gusevskaya J. Mol. Catal A: Chemical 1998, 132, 213–221.
19
Plus récemment, Kalck et al.13 ont décrit l'alcoxycarbonytion du dihydromyrcénol I-23 (schéma 4), un monoterpène contenant à la fois une double liaison et une fonction alcool, avec le même système catalytique [PdCl2(PPh3)2/SnCl2]. Lors de cette réaction, trois esters
linéaires sont formés. En effet, la déshydratation du dihydromyrcénol conduit à la formation du dihydromyrcène I-25, ce dernier subit à la fois une réaction d'alcoxycarbontlation suivi de la réaction d’addition du méthanol donnant lieu à la formation des deux esters I-26 et I-28. L'ester I-27 est obtenu par alcoxycarbonylaltion du dihydromyrcénol. La sélectivité en esters peut atteindre 75% selon les conditions opératoires utilisées. La présence de traces d'eau dans le milieu réactionnel entraine la réaction d'hydroxycarbonylation du dihydromycène I-25 pour donner l'acide 29. L'isomérisation du dihydromyrcènol ne dépasse pas les 20% (composé
I-24) mais la déshydratation du substrat en dihydromyrcène, produit I-25, est par contre très
présente et peut atteindre les 40%. Enfin, il est intéressant de noter que la réaction donne la lactone à neuf chainons I-30 avec un rendement qui peut atteindre 14%. Afin d’obtenir cette lactone avec un meilleur rendement, les auteurs ont joué sur plusieurs paramètres expérimentaux que nous développerons dans la partie cyclocarbonylation.
Lorsque la réaction est effectuée en absence de méthanol, la formation d'ester ou d'éther n’est pas observée, la déshydratation reste toujours importante. L'eau formée dans le milieu réactionnel réagit rapidement pour conduire à l'acide avec de bons rendements variant entre 60% et 90%. La formation rapide de l'acide qui suppose la formation initiale d'une espèce palladium-alkyle explique le faible pourcentage observé d'isomérisation qui résulte d'une β-élimination de cette espèce intermédiaire.
20 OH OH COOMe COOH O O OMe COOMe PdCl2(PPh3)2,CO MeOH dihydromercénol I-23
I-24 I-25 I-26
I-28 I-29 I-30
OH COOMe
I-27
+ + +
+ +
Schéma 4: Alcoxycarbonylation du dihydromyrcénol
Les mêmes auteurs ont étudié l'alcoxycarbonylation sur deux terpènes bicycliques présentant une double liaison interne, le (+)-∆-3-carène I-31 et le (-)-nopol I-37 avec ce système catalytique PdCl2(PPh3)2/SnCl2 (schéma 5)14. Pour le (+)-∆-3-carène, les taux de
conversion obtenus se situent entre 30 et 44%. L’isomérisation du substrat en (+)-∆-2-carène
I-32 est importante et ne facilite pas la sélectivité en esters I-34, I-35 et I-36 qui n’est que de
16%. L’augmentation de la pression de CO à 100 bar ou de la température à 100°C n’améliore pas les résultats. L’utilisation du mélange de gaz CO/H2 (79/21) permet
d’augmenter légèrement la conversion à 50% mais l’isomérisation reste importante. La présence de H2 dans le mélange de gaz devait favoriser la formation de l’espèce hydrure de
palladium qui semble être l’espèce active dans cette réaction15.
Pour le (-)-nopol 37, les résultats montrent surtout la formation de plusieurs isomères
I-39, I-40 dû à la migration de la double liaison et d’un éther I-41 par addition nucléophile de
l’alcool. Aucun ester n’est formé lors de cette réaction malgré les modifications des paramètres de la réaction qui ne permettent que d’améliorer les taux de conversion et d’isomérisation.
14 G. Lenoble Thèse de l’Université Paul Sabatier, Toulouse, 2000 15 H. Brunner, H. Alper, J. Org. Chem. 1997, 62, 7565-7568.
21
MeOOC COOMe
[PdCl2(PPh3)2/SnCl2]
CO/MeOH
(+)-D-3-carène I-31 I-32 I-33
COOMe
I-34 I-35 I-36
+ + + + OH [PdCl2(PPh3)2/SnCl2] CO/MeOH OH OH CHO OH + MeO (-)-nopol I-37
I-38 I-39 I-40 I-41
+
Schéma 5: alcoxycarbonylation du (+)-∆-3-carène et du (-)-nopol.
D’autres alcènes d’origine naturelle tels que les allylbenzènes ont été impliqués dans l’étude de la réaction d’alcoxycarbonylation mais nous trouvons peu de références bibliographiques. Les principaux travaux sur l’estragol 42a, l’eugénol 42b ou le safrole
I-42c sont décrits par l’équipe de Kalck et al16. En utilisant le même système catalytique [PdCl2(PPh3)2]/SnCl2 sous 40 bar de CO à une température de 97°C, les allybenzènes sont
complètement transformés au bout de 6h et les esters I-43a-c, I-44a-c et I-45a-c sont obtenus avec des rendements de 75-82% avec une sélectivité en ester linéaire d’environ 70-75% (schéma 6). Une réaction d’isomérisation de l’alcène terminal en alcène interne qui varie de 15 à 23% diminue la chimiosélectivité de la réaction. L’étude n’a été focalisée que sur la régiosélectivité de la réaction et non sur la stéréosélectivité de l’ester en β.
16 a) I. Ciprès, J. Jenck, Ph; Kalck J. Mol. Catal 1990, 58, 387-392; b) T. Chenal, I. Ciprès, J. Jenck, Ph; Kalck J. Mol. Catal 1993, 78, 351-366.
22 R1 R2 R1 R2 R1 R2 O OMe MeO O R1 R2 R1 R2 OMe O PdCl2(PPh3)2, MeOH PPh3, SnCl2,Toluène PdCl2(PPh3)2, MeOH PPh3, SnCl2,Toluène isomérisation +
estragol I-42a: R1=MeO, R2=H
eugénol I-42b: R1=OH, R2=MeO
saf role I-42c: R1=R2= OCH2O
(γester) I-43a-c
(αester) I-45a-c
(β-ester) I-44a-c
Schéma 6: alcoxycarbonyaltion des allylbenzènes.
Dans cette partie, nous avons montré que la réaction d'alcoxycarbonylation de monoterpènes et d’allylbenzènes est intéressante pour obtenir directement de nouveaux esters mais elle demeure difficile à réaliser avec certains monoterpènes encombrés et la réaction d’isomérisation qui intervient au niveau des allybenzènes empêche une transformation totale en esters. Néanmoins le système catalytique [PdCl2L2]/SnCl2 se montre efficace tant en
termes de chimiosélectivité qu’en regiosélectivité et permet de travailler dans des conditions relativement douces de pression et de température.
II-2-Réaction de cyclocarbonylation:
La réaction de cyclocarbonylation se déroule en deux étapes, la carbonylation puis la cyclisation qui en fait une réaction tandem. Elle peut être considérée comme une réaction d'alcoxycarbonylation d'un substrat contenant à la fois une oléfine et une fonction -ZH (Z= O, N) qui réagira avec l’intermédiaire acyle formé pendant le cycle catalytique. (schéma 7)
O
ZH Z
CO, [M]
Z= O, N
Schéma 7: Réaction de cyclocarbonylation
Cette réaction représente une stratégie intéressante pour produire des molécules relativement sophistiquées qui peuvent présenter des activités biologiques. Certaines de ces
23
molécules ont trouvé une large application dans les industries chimique, pharmacologique, et agrochimique comme, par exemple, les lactones qui sont des composés très utilisés dans les aliments et les boissons.17 Ainsi, il est possible de combiner ces deux étapes successives pour transformer des monoterpènes relativement complexes en produits fonctionnalisés intégrant un motif lactone ou un motif cyclopentanone.
L'utilisation du système catalytique [PdCl2(L)2]/SnCl2.H2O (L monophosphine ou
diphosphine) décrit précédemment s'est avérée très utile pour la réaction de cyclocarbonylation de différents monoterpènes comme le (+)-isolimonène, la (+)-isopulégol, le dihydromyrcénol, le géraniol, et l'alcool périllyque. La cyclocarbonylation du (+)-isolimonène I-1 et du (+)-isopulégol I-49 effectuée à 80°C sous 40 bar de CO dans le toluène donne respectivement une cyclopentanone et une lactone à six chainons avec des taux de conversion proche de 100% après 16h de réaction18,19 (schéma 8).
PdCl2/L2/SnCl2.2H2O CO H H O + + (+)-isolimonéne I-1 9% OH PdCl2/L2/SnCl2.2H2O CO O O (+)-isopulégol I-49
I-46 I-47 I-48 (86%)
I-50 (100%)
Schéma 8: cyclocarbonylation de l'isolimonéne et de l'isopulégol.
Dans le cas du (+)-isolimonène I-1, le rendement en cyclopentanone I-48 est de 86% et une faible isomérisation qui ne dépasse pas 9% est observée. Pour la (+)-isopulégol I-49, la lactone I-50 a été obtenue avec un rendement de 100%. Tous les produits ont été caractérisés par les méthodes spectroscopiques usuelles. Les auteurs ont confirmé que les mêmes résultats sont obtenus que ce soit en présence ou en absence du méthanol.
17 L. Dufosse, A. Latrasse, H. E. Spinnler, Sci. Aliments 1994, 14, 17-50. 18
G. Lenoble, R. Naigre, T. Chenal, J.C. Daran, M. Urrutigoïty, Ph. Kalck, Tetrahedron Asymmetry 1999, 10,
929-936.
24
La formation de la cyclopentanone à partir du (+)-isolimonène entraine la création de deux carbones asymétriques avec la perte de chiralité de l’un des carbones de départ (C4). Seuls deux diastéréoisomères ont été obtenus (schéma 8 ). Au niveau de la formation de la lactone
I-50, un carbone asymétrique supplémentaire est créé donnant deux diastéréoisomères
(schéma 8 ). La présence de carbones asymétriques sur les substrats de départ permet en absence de ligand chiral d'obtenir des excès diastéréoisomériques de l'ordre de 12 à 24% avec PPh3 comme ligand. Cet excès peut être augmenté jusqu’à 64-69% pour la cyclopentanone et
60% pour la lactone en utilisant les ligands diphosphines comme dppb et dppf (1,1’bis-diphénylphosphinoferrocène). Les auteurs proposent que le contrôle de cette diastéréoséléctivité ne peut provenir que du substrat lui même. Afin de valider cette hypothèse ils ont étudié l'influence d'une diphosphine chirale, la DIOP.
Le tableau 1 reporte les principaux résultats obtenus dans cette étude.
Tableau 1: cyclocarbonylation du (1R,4R)-(+)-isolimonène et 1R,2S,5R)-(-)-isopulégol.
substrat catalyseur T.C (%) Sélectivité (%) Isom. (%) e.d (%)
(1R,4R)-(+)-isolimonène [PdCl2PPh3] 95 86 9 12 [PdCl2(dppb)] 75 49 26 64 [PdCl2(dppf)] 71 48 23 69 [PdCl2(-)-DIOP] 77 43 34 64 [PdCl2(+)-DIOP] 77 43 34 64 1R,2S,5R)-(-)-isopulégol [PdCl2PPh3] 100 100 - 24 [PdCl2(dppb)] 83 99 - 60 [PdCl2(dppf)] 95 90 - 44 [PdCl2(-)-DIOP] 97 96 - 62 [PdCl2(+)-DIOP] 97 96 62
e.d: excès diastéréoisomérique
D'après le tableau, les conversions baissent légèrement dans le cas de l'isopulégol et fortement dans le cas de l'isolimonène en présence des diphosphines (de 95 à 77%). Comme pour les diphosphines achirales utilisées précédemment, l’utilisation des deux stéréoisomères du ligand DIOP permet une augmentation de l’excès diastéréoisomérique avec un résultat
25
identique. Les auteurs en concluent que le caractère chiral de la DIOP ne rentre pas en jeu lors de la réaction de cyclocarbonylation et l’excès diastéréoisomérique est amené par l'effet stérique de la diphosphine qui favorise l'un ou l'autre isomère lors de l'attaque du substrat sur le complexe du palladium.
Récemment, les travaux de thèse de Lisa Diab20,21 sur la cyclocarbonylation de l'isolimonéne et l'isopulégol ont montré que cette réaction de cyclocarbonylation dépend de plusieurs paramètres comme l'effet de SnCl2 et du solvant. SnCl2 joue un rôle important dans
cette réaction car il rentre dans la sphère de coordination du catalyseur pour obtenir une bonne activité. L’utilisation de solvants peu coordonnants comme le CH2Cl2 et du
1,2-dichloroéthane améliore les performances du catalyseur en termes d’activité et de diastéréosélectivité car l’excès diastéréoisomérique est augmenté jusqu’à 70% pour l’isopulégol.
Une étude approfondie de la réaction de cyclocarbonylation du dihydromyrcénol, du géraniol, et de l'alcool périllyque a également été menée22,23. Nous avions vu dans la partie I1 que la réaction d'alcoxycarbonylation du dihydromyrcénol produit majoritairement l’ester
I-26 et l'acide I-29 (Cf Schéma 4). Dans le but d'améliorer la sélectivité en lactone I-30 tout en
conservant le même système catalytique PdCl2(PPh3)2/SnCl2.H2O, les auteurs ont utilisé du
tamis moléculaire pour piéger l'eau qui se forme dans le milieu réactionnel et éviter ainsi la réaction parasite d'hydroxycarbonylation (Schéma 9).
OH OH
COOH
O O PdCl2(PPh3)2,CO
dihydromercénol I-23 I-24 I-25 I-29 I-30
+ + +
Schéma 9: Cyclocarbonylation du dihydromyrcénol.
La présence de 3g de tamis moléculaire dans le milieu réactionnel améliore considérablement le rendement et la sélectivité en lactone I-30 de l’ordre de 79% et diminue
20 L. Diab, Thèse de l’Université Paul Pabatier, 2007.
21 L. Diab, M. Gouygou, E. Manoury, Ph. Kalck, M.Urrutigoïty, J. Mol. Catal. A: Chemical, 2007, 278, 102–106 22
G. Lenoble, M. Urrutigoïty, P. Kalck, Tetrahedron. Lett., 2001, 42, 3697-3700.
23 D. H. Nguyen, F. Hebrard , J. Duran, A. Polo, M. Urrutigoïty, P. Kalck, Appl. Organometal. Chem. 2005 ,19, 30–34.
26
les produits de déshydratation. En absence de tamis moléculaire, 80% d’acide sont obtenus comme produit majoritaire. L’augmentation de la quantité de tamis moléculaire permet d’augmenter encore la sélectivité en lactone (92%) mais au détriment de la conversion qui baisse (60%). Les auteurs ont aussi réalisé cette réaction en présence de formiate de méthyle qui devait donner un résultat comparable à celui obtenu avec le tamis moléculaire. Mais, expérimentalement la réaction ne donne que le produit d'isomérisation I-24 avec un taux de conversion faible de 21%.24 Les auteurs supposent que le tamis moléculaire joue non seulement le rôle d'agent absorbant, mais il est capable aussi de piéger l'acide chlorhydrique libéré lors de la génération de l'espèce catalytique active.
La cyclocarbonylation du géraniol I-51, qui est un alcool allylique disubstitué en position
γ, est réalisée avec le même système catalytique. La lactone à six chainons I-53, l'ester I-54,
et l'acide I-52 constituent les principaux produits obtenus lors de cette transformation (Schéma 10). OH OH O O O O O PdCl2(L)2/SnCl2 + +
géraniol I-51 I-52 I-53 I-54
CO
Schéma 10: cyclocarbonylation du géraniol.
Dans le cas du géraniol, une pression de CO de l'ordre de 80 à 100 bar est nécessaire pour obtenir les trois produits carbonylés. En effet, à 80°C, sous 100 bar pression de CO et pendant 16h un taux de conversion de 72% est observé avec des sélectivités en ester I-54 de 65%, en acide I-52 de 13% et en lactone I-53 de 2%. La formation de la lactone implique une isomérisation de la double liaison dans cette réaction tandem de cyclocarbonylation23. D’autres sous-produits non carbonylés issus de réactions parasites comme l’isomérisation en position interne de la double liaison C=C, la déshydratation de la fonction allylique ou l’étherification du substrat ont également été identifiés avec une contribution de 20%. L'augmentation du temps de la réaction de 16h à 40h ne permet pas d'améliorer le taux de conversion (74,2 %), seule la sélectivité en ester I-54 est améliorée (75,5%). Ce système
24
27
catalytique s’avère néanmoins décevant puisqu’il ne favorise pas la formation de la lactone mais plutôt l’ester I-54. L'utilisation de ligands diphosphines tels que le dppf et le dppb n'améliore pas les taux de conversion mais réoriente la sélectivité de la réaction. Ainsi, avec dppf à 100°C et sous 80 bar de pression de CO seule la formation de l’acide I-52 est favorisée avec une sélectivité de 20%. Dans les mêmes conditions mais avec un temps de réaction plus long (24h), le ligand dppb permet la formation de la lactone I-53 avec une sélectivité de 25%. L’utilisation du système catalytique [Pd(OAc)2/dppb] décrit par Alper15 pour la carbonylation
des alcools allyliques à 100°C et sous 80 bar de pression d'un mélange CO/H2 (1/1) ne
provoque que la formation de l'acide I-52 avec un rendement de 90%.
L’alcool périllyque I-55 est un alcool allylique cyclique di-substitué en position β et γ- possédant une double liaison endocyclique. La réaction de cyclocarbonylation est représentée dans le schéma 11. OH O O PdCl2(PhCN)2/dppb/SnCl2 CO/H2
Alcool périllyque I-55 I-56
Schéma 11: cyclocarbonylation de l'alcool périllyque.
Ce substrat s’est avéré plus difficile à carbonyler dans les conditions de catalyse décrites précédemment puisque l'utilisation du système [PdCl2(PPh3)2]/SnCl2.2H2O ou
[PdCl2(dppb)]/SnCl2.2H2O ne donne aucune conversion. Les auteurs ont utilisé un autre
système catalytique [PdCl2(PhCN)2]/dppb/SnCl2.2H2O sous un mélange de CO et H2, dans le
dichlorométhane, et d'une façon surprenante, 80% de conversion sont obtenus après 20h avec une sélectivité de 40% en faveur de la lactone I-5625. L'utilisation du ligand dppf au lieu de dppb entraine une diminution de la conversion ainsi que la sélectivité (50% et 20% respectivement).
L'alcool périllyque a été étudié en cyclocarbonylation par Alper et son groupe15 en utilisant le système catalytique de type [Pd(OAc)2]/dppb sous 55 bar d'un mélange de pression
25
28
CO/H2 (1/1) à 110°C dans le dichlorométhane. Ils obtiennent la lactone I-56 avec un
rendement de 56%.
Nous retiendrons de cette partie bibliographique que cette réaction de cyclocarbonylation catalysée par des complexes du palladium est une voie intéressante pour la synthèse directe de lactones à partir de monoterpènes porteurs d’une fonction alcène et hydroxyle.
Ainsi, l'alcoxycarbonylation et la cyclocarbonylation des terpènes ouvrent des perspectives séduisantes pour la synthèse de nouvelles molécules à partir de produits naturels. Il est intéressant de regarder le mécanisme de formation de ces produits et les espèces impliquées dans le cycle catalytique.
II-3-Mécanisme de la réaction:
Le mécanisme de la réaction d'alcoxycarbonylation a été largement étudié et les premières études sur le mécanisme au palladium remontent au début des années 70.6,26,27Deux mécanismes sont généralement proposés.
Le premier met en jeu l'hydrure de palladium B1 comme espèce active de départ dans la première étape du cycle catalytique puis l'insertion de l'oléfine dans la liaison Pd-H pour former l'espèce palladium alkyl B2 suivie de la coordination et l'insertion de CO pour donner l'acylpalladium B3. L'alcoolyse de ce dernier permet de régénérer l'hydrure de palladium et engendrer l'ester.
Le deuxième mécanisme fait intervenir l'espèce alcoxycarbonylpalladium A1 puis l'insertion de l'oléfine dans cette espèce suivie de la protonolyse de A2 pour former le produit. L'étape finale permet de régénérer l'espèce active A1 par coordination de CO sur l'espèce alcoxypalladium [(Pd(OR')(X)(Ln)] A3 suivi d'une insertion dans la liaison Pd-OR'. (Schéma
12).
26 J. Tsuji, Palladium Reagent and catalyst, 1999, John Willey& Sons.
29 [Pd(COOR')(X)(Ln)] [Pd(H)(X)(Ln)] R Ln(X)Pd OR' O O Ln(X)Pd R Ln(X)Pd R R [Pd(OR')(X)(Ln)] R R'OH R OR' O CO CO A3 A1 A2 B1 B2 B3
Schéma 12: Les deux cycles proposés pour la réaction d’alcoxycarbonylation
Des études approfondies menées par Toniolo et ses collaborateurs ont proposé la coexistence des deux mécanismes sans vraiment privilégier l'un par rapport à l'autre.28,29 Il a fallu attendre le début des années 1990 pour que plusieurs auteurs tranchent finalement dans le sens d'un mécanisme impliquant un complexe hydruropalladium.30 En effet, cette proposition repose sur l'observation expérimentale en infra rouge de deux bandes caractéristiques ν(Pd-H) à 2040 cm-1 et ν(Sn-Cl) à 315 cm-1 du complexe [Pd(H)(SnCl3)(PPh3)2] synthétisé à partir de l’addition de SnCl2 à [PdCl2PPh3] sous 100 bar
de CO.31
Récemment, au sein du laboratoire32, des travaux menés sur la synthèse de complexes hydrures du palladium du type [Pd(H)(X)L2] ont permis la caractérisation pour la première
fois par RMN multinoyaux à basse température du complexe pentacoordoné [Pd(H)(SnCl3)(PCy3)2]. Les auteurs ont réussi à effectuer la synthèse de complexes hydrures
du palladium [Pd(H)(X)L2] ou [Pd(H)(X)L2(L')]+. Le rôle du co-catalyseur SnCl2 a été
précisé : il forme l'espèce [Pd(H)(SnCl3)(L)2] capable de coordonner l'alcène et d'enclencher
le cycle catalytique. La mise en œuvre directe de ce complexe dans l'autoclave de
28 G. Cavinato, L. Toniolo, J. Organomet. Chem. 1993, 444, C65-C66. 29 G. Cavinato, L. Toniolo, J. Organomet. Chem. 1990, 398, 187-195. 30
A. Seayad, A. A. Kelkar, L. Toniolo, J. Mol. Catal. 2000, 151, 47-59. 31
J.F. Knifton, J. Org. Chem. 1976, 41, 2885-2890
32 a) D. H.Nguyen, G. Laurenczy, M. Urrutigoïty, Ph. Kalck, Eur. J. Inorg. Chem. 2005, 4215–4225; b) D. H.Nguyen, Y. Coppel, M. Urrutigoïty, Ph. Kalck, Journal of Organomet Chem 2005, 690, 2947–2951.
30
carbonylation, à la place des précurseurs qui le forme in situ, permet d’effectuer une catalyse sous conditions beaucoup plus douces (60°C, 10 bar de CO).32b
III-Réaction d'hydroformylation de terpènes et de dérivés naturels d’allybenzène:
La réaction d’hydroformylation, également appelée synthèse oxo, consiste en l’addition d’hydrogène et de monoxyde de carbone (CO) sur une double liaison C=C pour créer une fonction aldéhyde CHO (schéma 12). Les aldéhydes représentent des synthons particulièrement intéressants du fait de leurs réactivités ou de leurs activités biologiques et olfactives intéressantes. Une application majeure des aldéhydes obtenus industriellement par hydroformylation reste la préparation d’alcools qui trouvent de multiples débouchés selon la longueur de la chaine33. De même, cette réaction revêt aussi une importance dans la production d’arômes34. En effet de nombreuses substances naturelles contiennent un groupe aldéhyde dont la présence est importante pour la perception olfactive de la molécule. Les monoterpènes qui possèdent une double liaison dans leur squelette constituent un réservoir de produits importants pour l’industrie des parfums et des arômes. Les aldéhydes issus de l’hydroformylation des monoterpènes peuvent être utilisés directement ou après transformation pour leurs propriétés odoriférantes comme composants de parfums.34
R [M] R CHO CO/H2 + R CHO
Schéma 12: Réaction d'hydroformylation
Cette réaction peut être catalysée par plusieurs métaux de transition tels que le cobalt, le platine ou le rhodium.35 Le cobalt et le platine sont peu utilisés dans cette transformation à
33 a) Rhodium catalyzed Hydroformylation, (C. Claver, P.W.N.M. van Leeuwen Eds), Kluwer Academic Publishers, Dordrecht , 2000, b) C.D. Frohning, C. W. Kolpaintner in Applied Homogeneous Catalysis with Organometallic compounds, 2nd Edition, ( B. Cornils, W.A. Herrman, Eds), Wiley-VCH,Weiheim, 2002, pp 31-83.
34
E. Gusevskaya, J. Jimenèz-Pinto, A. Borner, ChemCatChem 2013, 5, 1-31
35
a) K.D Wiese, D. Obst In: “Catalytic Carbonylation Reactions” Top. Organomet. Chem. Matthias Beller Ed., Springer-Verlag, 2006, 18, 1-35.; b) R. Franke, D. Selent, A. Börner, Chem. Rev. 2012, 112, 5675−5732.
31
cause d'une réaction d'hydrogénation compétitive qui fait baisser la chimiosélectivité de la réaction. Le rhodium est le métal qui permet le meilleur compromis activité/sélectivité dans des conditions de réactions douces.
L’hydroformylation de terpènes et de dérivés d’allylbenzène a été très étudiée ces dernières années et nous allons présenter dans la partie ci-dessous les principaux résultats obtenus dans cette réaction catalysée essentiellement par des complexes du rhodium et du platine.
III-1-Hydroformylation des monoterpènes avec une double liaison exocyclique:
Les premiers résultats d'hydroformylation des monoterpènes remontent aux années 80 et concerne principalement le limonène I-1 et le β-pinène I-20. Les brevets publiés36 montrent que la double liaison exocyclique est préférentiellement carbonylée dans des conditions de catalyse variant entre 100 et 400 bar de pression à 70-180°C. Les systèmes catalytiques utilisés sont constitués de complexes de rhodium(I) comme [Rh(µ-Cl)(cod)]2,
[Rh(acac)(CO)2]2 ou [RhCl(CO)(PPh3)2]. L’isomérisation de la double liaison du limonène
(I-1) donne l’α-terpinolene (I-2) et le γ−terpinolène (I-47) considérés comme sous-produits, comme l’α-pinène (I-60) pour le β-pinène (I-20) (Schéma 13 et 14). Les travaux de van Leeuwen et son équipe37 relatent qu’il est possible de réaliser l’hydroformylation du limonène dans des conditions beaucoup plus douces avec de bonnes vitesses de réaction et une sélectivité totale en aldéhyde. La réaction se déroule entre 3 et 14 bars à 80°C en présence du complexe [Rh(OAc)(cod)]2. L'aldéhyde linéaire est obtenu sélectivement du fait que
l'insertion du CO se fait exclusivement sur le carbone non substitué de la double liaison du substrat. La nature du ligand phosphoré ajouté modifie la vitesse de réaction : elle est beaucoup plus grande avec le ligand phosphite P(O-otBuPh)3
(tris(o-terbutylphényl)phosphite) qu’avec PPh3. Une autre équipe reporte que l’utilisation du
complexe [RhCl(CO)(PPh3)2] en présence de dix équivalents de P(O-otButPh)3 entre 70 et
110°C sous 20 à 90 bar d'un mélange CO/H2 (1.25/1) donne des rendements 10 fois plus
élevés que lorsque le complexe est utilisé avec un excès de triphénylphosphine (PPh3/Rh=12)
et l'isomérisation de la double liaison reste faible. Les auteurs proposent une action concertée
36
a) J. Hagen, K. Bruns (Henkel KGaA), DE 2849742, 1980.; b) J. Hagen, R. Lehmann, K. Bansemir (Henkel
KGaA), DE 2914090, 1980, EP 0018504, 1980. 37
32
de la phosphine et du phosphite sur le rhodium pour expliquer l'activité et la sélectivité particulière du système catalytique38.
[Rh] CO/H2
CHO
(-)-limonène I-1 I-57
+ +
α-terpinolène I-2 γ-terpinolène I-47 Schéma 13: hydroformylation du limonène
L'hydroformylation du (-)-limonène a été réalisée avec le complexe [HRh(CO)(PPh3)3]39, et
une conversion totale est obtenue en 24h à 65°C sous 40 bar de CO/H2, avec un rendement de
64% en aldéhyde linéaire. De même l'hydroformylation du β-pinène I-20 en 10-formylpinane
I-59a-b a été menée avec le même complexe à 110°C sous 34 bar de CO/H2. 78% de
conversion sont obtenus avec un rendement de 75% en aldéhyde linéaire et seulement 3% de produit secondaire en α-pinène ont été observés.40 (schéma 14).
OHC
[Rh] H2/CO
(-)-β-pinène I-20 10-formylpinane I-59a-bcis et trans I-60 +
Schéma 14: hydroformylation du β-pinène.
38 V. M. Tormyshev, G. A. Skripko, V.D. Shteingarts, VI Russian Conference on Organometallic Chemistry,
1995, Rhodium Express, p26
39
J. C. Chalchat, R. Ph. Garry, E. Lecomte, A. Michet, Flavour and Fragrance J. 1991, 6, 179-182.
40 a) A. J. Chalk, in "Flavors and Fragrances : A world perspective" , Ed. B. M. Lawrence, B. D. Mookherjee, B. J. Willis Elsevier, Amsterdam, 1988, ou Dev. Food Sci., 18 (Flavors Fragrances), 1988,867. b)A. J. Chalk, in P. N. Rylander, H. Greenfield, R. L. Augustine (eds), Calatlysis of Organic reaction, M. Dekker, New York, 1988, P. 43
33
D'autre études ont montré que cette transformation peut se faire d'une manière
diastéréoséléctivité.41 En effet, en partant du (1S,5S)-(-)-β-pinène ou du (1R,5R)-(+)-β-pinène, le trans-(1S,2R,5S)-(-)- ou le trans-(1R,2S,5R)-(-)-10-formylpinane est obtenu avec une diastéréosélectivité de 93% en impliquant le système catalytique [Rh4(CO)12]/dppe mais les
conversions sont faibles, de l’ordre de 30%, suite à la forte réaction d’isomérisation du β- en
α-pinène. Les auteurs expliquent cette diastéréosélectivité par l’approche du centre métallique par la face énantiotopique la plus encombrée du-(-)-β-pinène. L'isomère cis du 10-formylpinane I-59 est généralement majoritaire. Sa formation est favorisée par des températures plus basses et des pressions plus élevées42. Cette diastéréoséléctivité s'explique par les contraintes stérique lors de la coordination du catalyseur à la double liaison du substrat.
De la même façon, l'hydroformylation du (-)-β-pinène a été effectuée en présence de système [Rh(cod)(OAc)]2/L avec L= PPh3 ou L= P(Bn)3.43 Les meilleurs résultats en terme de
selectivité et diastéréosélectivité, 87% et rapport cis/trans de 6,3/1 respectivement, sont obtenus en présence de la tribenzylphosphine (P(Bn)3. La nature du ligand joue un rôle
important dans ces résultats puisque les auteurs montrent que l'utilisation du ligand tripentafluorophénylphosphine (P(C6F5)3) entraine une sélectivité de 46% et une
diastérosélectivité fortement en faveur de l'isomère trans (cis/trans=1/10,5)
Des travaux antérieurs44 au sein du laboratoire ont permis de carbonyler la double liaison exocyclique d'autres monoterpènes, dans des conditions plus douces de pression (5 à 13 bar) et de température (84°C) en utilisant le catalyseur de type [Rh(µ-StBu)(CO)(P(OPh)3)]2 en
présence d’un excès de PPh3. Les complexes dinucléaires du rhodium(I) à ponts thiolato
[Rh(µ-StBu)(CO)PR3)]2, (PR3 étant une phosphine ou un phosphite) sont très actifs pour
l’hydroformylation d’alcènes terminaux45.
Les différents substrats étudiés et les résultats obtenus sont regroupés dans le tableau 2.
41 F. Azzaroni, P. Biscarini, S. Bordoni, G.Longoni , E. Venturini, J. Organomet. Chem. ,1996, 508, 59-67. 42
a) W. Himmele, H. Siegel, Tetrahedron Lett., 1976, 12, 907-910; b) W. Himmele, H. Siegel, Tetrahedron Lett., 1976, 12, 911-911.
43
H.J.V. Barros, M. L. Ospina, E. Arguello, W. R. Rocha, E. Gusevskaya, E. N. dos Santos,J. Organometal. Chem. 2003, 671, 150-157
44 S. Sirol,Ph. Kalck, New J. Chem. 1997, 21, 1129-1137.
45 a) J.M. Frances, A. Thorez, Ph. Kalck, New J. Chem. 1984, 8, 213-216. b) Ph. Kalck, D.C. Park, F. Serein, A. Thorez, J. Mol. Cat. 1986, 36, 349-357.
34
Tableau 2: Hydroformylation de quelques monoterpènes naturels.
Substrat S/C P/Rh P CO/H2 T.C (%) Sélectivité en aldéhyde % (+)-limonènea 600 6 5 88 98 (-)-alcool périlliqueb 300 2,5 12 100 100 (+)-carvoneb 300 2 13 89 98 acétate d'isopulégolb 600 2 13 70 100 a
Substrat: 48 mmol, bSubstrat: 60 mmol, CO/H2 (1/1), T(bain)= 84°C, Toluène 40mL, T.C=
taux de conversion, S/C= Substrat/Catalyseur, P/Rh= Phosphore/Rhodium, t(h)=18.
Comme il est décrit dans le tableau, les conversions obtenues varient entre 70 et 100%, avec des sélectivités quasi totales en aldéhydes. La regioséléctivité en faveur de l'aldéhyde linéaire est totale. Les réactions d’isomérisation ou d’hydrogénation n’ont presque pas été observées.
Des essais d’hydroformylation asymétrique du (+)-limonène ont été menés en présence de complexes dinucléaires [Rh2(µ-StBu)2(CO)2(diphos)] et de diphosphines achirales et chirales
mais les excès diastéréoisomériques obtenus sont du même ordre voire plus faible que celui obtenu avec le complexe [Rh(µ-StBu)(CO)(P(OPh)3)]2.
L’utilisation de systèmes bimétalliques à base de Pt/Sn dans la réaction d’hydroformylation du limonène et du β-pinène a été également étudiée46. Les systèmes catalytiques [Pt(Cl)2(PPh3)2/SnCl2/PPh3] et [Pt(Cl)2(diphosphine)/SnCl2/PPh3] avec les
diphosphines dppe, dppp, dppb transforment à 100°C sous 90 bar de CO/H2(1/1)
chimioselectivement le -(+)-limonène en aldéhyde linéaire dans un mélange des deux diastéréoisomères (90%) mais les conversions sont faibles, de l’ordre de 10-20%. Des résultats similaires avaient été déjà reportés dans la littérature.47 Si la réaction est faite à température plus élevée (130°C) et un temps de réaction plus long (>4h) les conversions augmentent fortement (85-95%) mais l’adéhyde se transforme en un dérivé bicyclique, le 4,8-diméthyl-bicyclo{3.3.1]non-7-en-2-ol I-61 suite à une cyclisation intramoléculaire
46 E.V. Gusevskaya, E.N. dos Santos, R. Augusti, A. de O. dias, C.M. Foca, J. Mol. Catal.A. 2000, 152, 15-24. 47
35
diastéréosélective catalysée par l’espèce Pt/SnCl2 (Schéma 15). La formation des produits
d’hydroformylation et de cyclisation dépend fortement du rapport Sn/Pt. De plus un rapport supérieur à 1 favorise l’isomérisation de la double liaison qui devient non réactive en hydroformylation.
CO/H2
CHO
(-)-limonène I-1 I-58
+ PtCl2L2/SnCl2
OH
I-61
Schéma 15: hydroformylation du limonène en présence du [PtCl2L2]/SnCl2.
Le (-)-β-pinène , quant a lui, est transformé majoritairement en trans-10-formylpinane avec un excès diastéréoisomérique de 94%. Le meilleur résultat en terme de conversion (70%) et de sélectivité (84%), est obtenu en présence du ligand diphosphine dppb.
III-2-Hydroformylation des monoterpènes avec une double liaison endocyclique.
D'autre études décrivent l'hydroformylation de quelques monoterpènes cycliques avec une double liaison interne tels que l'α-pinène, le ∆-2-et ∆-3-carène et le camphene.48,49,50,51
Le (+)-α-pinène est transformé majoritairement en (+)-2-formyl-bornane I-63, suite à un réarrangement de Wagner-Meerwein, en présence de [Co2(CO)8] comme catalyseur48 et en
(+)-3-formyl-pinane I-64 et 10-formyl-pinane I-59a, I-59b dans un rapport cis/trans de 8/1 en présence de [Rh(cod)(Cl)]249,50,51 (schéma 16). Le 10-formyl-pinane est obtenu sous sa forme
de deux diastéréoisomères résultant de l’isomérisation de l’α-pinène en β-pinène. L'utilisation des catalyseurs [Co2(CO)8]et [Rh(cod)Cl]2 nécessite une température de 110°C et une haute
pression de CO/H2 (1/1) entre 200-650 bar. Par contre, l'utilisation du précurseur
[Rh(cod)(OMe)]2 en présence du ligand tris(o-terbutyl)phosphite permet de travailler dans des
conditions de température et de pression plus basses (100°C et 80 bar) ; le trans
48 W. Himmele, H. Siegel, Tetrahedron Lett 1976, 17, 907–910. 49
M. C. de Freitas, C. G. Vieira, E. N. dos Santos, E. V. Gusevskaya, ChemCatChem 2013, 5, 1884 –1890.
50
a) S. Pfohl, J. Paust, H. Siegel, W. Himmele, W. Hoffmann, K. v. Fraunberg(BASF AG), DE 2404305, 1975. b) W. Hoffmann, W. Himmele, J.Paust, K. v. Fraunberg, H. Siegel, S. Pfohl (BASF AG), DE 2453283, 1976.
51
36
pinane I-63 est obtenu avec un rendement de 60% et les deux diastéréoisomères du 10-formyl-pinane avec un rendement de 30%
CHO CHO CHO + + (+)-α-Pinène I-62 CO[Rh] /H 2 CHO Co2(C O8) CO/H 2 I-63
I-64 I-59a I-59b
Schéma 16: Hydroformylation du (+)-α-Pinène.
L'hydroformylation du camphène I-17 a été reportée en présence de différents catalyseurs métalliques. Les premières études reportent que le catalyseur [Co2(CO)8]sous 207 barà
120-150°C transforme le camphène en aldéhyde linéaire avec un rendement de 65% (schéma 18).52 Les systèmes catalytiques PtCl2(L)/PPh3/SnCl2 avec L=PPh3, dppe, dppp, dppb se
montrent, dans les mêmes conditions utilisées précédemment pour le limonène et le β-pinène, efficaces et produisent exclusivement l'aldéhyde linéaire exo I-65 et endo I-66 dans les mêmes proportions (70% de conversion, 96% de sélectivité, exo/endo= 42/58) (schéma 18). Les auteurs précisent que la régiosélectivité est due à l'encombrement stérique du produit de départ et à celui du complexe du platine mais cela ne joue pas sur la diastéréosélectivité.46 L'absence de SnCl2 entraine une inactivité du système catalytique mais un excès de SnCl2
provoque la formation de sous produits d’isomérisation et d’hydrogénation à hauteur de 50%.
52
![Tableau 1: Cyclocarbonylation de l'alcool II-17 catalysé par le complexe [PdCl 2 (PPh 3 ) 2 ] /SnCl 2 .2H 2 O](https://thumb-eu.123doks.com/thumbv2/123doknet/3287009.94298/69.892.125.758.289.445/tableau-cyclocarbonylation-alcool-catalysé-complexe-pdcl-pph-sncl.webp)
![Tableau 2: Réaction de cyclocarbonylation de l'alcool II-17 catalysée par [PdCl 2 (L) 2 ]/ SnCl 2 ,2H 2 O](https://thumb-eu.123doks.com/thumbv2/123doknet/3287009.94298/74.892.100.798.266.657/tableau-réaction-cyclocarbonylation-alcool-ii-catalysée-pdcl-sncl.webp)

![Tableau 5 : Cyclocarbonylation du II-17a et II-17b catalysé par le système catalytique [PdCl 2 L 2 / SnCl 2 ,2H 2 O]](https://thumb-eu.123doks.com/thumbv2/123doknet/3287009.94298/79.892.118.755.159.440/tableau-cyclocarbonylation-ii-catalysé-système-catalytique-pdcl-sncl.webp)
![Tableau 10: Réaction de Cyclocarbonylation de l'alcool II-18 catalysé par [PdCl 2 (L) 2 / SnCl 2 ,2H 2 O], L =diphosphine](https://thumb-eu.123doks.com/thumbv2/123doknet/3287009.94298/86.892.142.744.522.713/tableau-réaction-cyclocarbonylation-alcool-catalysé-pdcl-sncl-diphosphine.webp)

![Tableau 7 : Hydroformylation du styrène en présence du système catalytique [Rh(CO) 2 (acac)]/phospholyl(phosphino)méthane a](https://thumb-eu.123doks.com/thumbv2/123doknet/3287009.94298/104.892.183.714.408.581/tableau-hydroformylation-styrène-présence-système-catalytique-phospholyl-phosphino.webp)
