VARIATION GÉNÉTIQUE CHEZ L’ÉPINETTE
NOIRE (PICEA MARIANA) : RELIER L’EXPRESSION
GÉNIQUE À LA DIVERSITÉ GÉNÉTIQUE
Mémoire
GUILLAUME TESSIER
Maîtrise en Sciences forestières Maître ès sciences (M.Sc.)
Québec, Canada
iii
Résumé
Les arbres doivent s’adapter aux variations climatiques afin de survivre aux changements saisonniers, et ce, de façon multi-annuelle. Plusieurs études ont montré qu’il y a de la variabilité génétique à l’intérieur des espèces et que cette variabilité aide les espèces à survivre et à s’acclimater à différents changements environnementaux. Vingt-quatre gènes potentiellement reliés à l’adaptation ont préalablement été identifiés chez l’épinette noire (Picea mariana [Mill.] B.S.P.). L’objectif de cette étude était d’acquérir des évidences du rôle potentiel de ces gènes dans l’acclimatation et l’adaptation en étudiant leur expression. Pour ce faire, l’expression des gènes sera étudiée au niveau spatio-temporelle (la variation de l’expression selon le stade physiologique la variation de l’expression selon le tissus) et aussi selon les différents génotypes.
En général, il a été montré dans cette étude qu’il y avait beaucoup de variabilité dans l’expression des gènes candidats selon les tissus ou l’organe étudié, mais aussi selon le stade physiologique de l’arbre. Les gènes étaient exprimés majoritairement dans des tissus pouvant être reliés plus directement dans l’acclimatation et l’adaptation, par exemple la tige primaire. De plus, pour certains gènes, une interaction entre le génotype et le stade physiologique de l’arbre a été observée, c’est-à-dire que certains génotypes pourraient réagir dans le temps alors que d’autres le feraient moins.
v
Table des matières
Résumé ... iii
Table des matières ... v
Liste des tableaux ... vii
Liste des figures ... ix
Remerciements ... xiii
Avant-propos ... xv
Chapitre 1 - Introduction ... 1
1.1 La physiologie et l’acclimatation des arbres en réponse aux facteurs climatiques ... 1
1.2 Adaptation climatique et diversité génétique ... 5
1.3 Génétique et mécanismes moléculaires reliés à l’adaptation ... 8
1.4 Mise en contexte du projet ... 12
1.5 Hypothèses et objectifs de recherche ... 14
Chapitre 2 ... 17
2.1 Résumé ... 17
2.2 Abstract ... 17
2.3 Introduction... 18
2.4 Materials and methods ... 20
Plant material ... 21
Experiment 1 ... 21
Experimental design ... 21
RNA extractions and quality control ... 22
cDNA preparation and quantitative PCR ... 22
Experiment 2 ... 23
DNA extractions, genotyping and gene expression ... 23
Statistical analysis ... 24
2.5 Results... 25
Accumulation profiles of candidate gene RNA transcripts in vegetative tissues ... 25
Development of a genetically diverse study population ... 26
Comparison of expression levels between genotypes at different sampling dates ... 27
Relationship between candidate genes and growth and phenology ... 28
2.6 Discussion ... 28
Tissue expression profiles of candidate genes ... 28
Temporal variation ... 30
Genotypic variation of expression, height growth, timing of budset ... 31
2.7 Acknowledgments ... 33
Chapitre 3 - Conclusion générale ... 43
3.1 Profils d’expression des gènes candidats ... 43
3.2 Variation temporelle ... 45
3.3 Variation génotypique, croissance en hauteur et aoûtement ... 47
3.4 Critique des résultats ... 50
3.5 Retombées de présent travail et applications potentielles des résultats ... 51
Bibliographie ... 53
vii
Liste des tableaux
Chapitre 1
Tableau 1.1 : Nombre de plants livrés annuellement au Québec pour la période 2001-2007 pour les forêts publiques (Fortier 2008)………23
Chapitre 2 :
Tableau 2.1 : Description and accession numbers of the candidate genes potentially involved in acclimation and genotyping results and study population genotypic frequencies ………...53 Tableau 2.2 : Statistical analyses of transcript abundance comparing genotypes and sampling dates………...54
Annexes :
Tableau S1 : Expression level of candidate genes for each genotype………...58 Tableau S2 : Correlation between the genotype and the phenotype……….59
ix
Liste des figures
Chapitre 1
Figure 1.1 : Phases, processus et facteurs déterminants du cycle annuel de croissance et de dormance chez les arbres et les plantes pérennes………..11 Figure 1.2 : Représentation des différentes composantes d’un gène………...20 Figure 1.3 : Transcription d’une molécule d’ADN en molécule d’ARN...21
Chapitre 2 Figure 2.1 :
Hierarchal clustering of genes based on transcript abundance in different vegetative tissues……….………...53 Figure 2.2 :
Difference in the number of RNA transcripts between the two sampling dates for the 24 candidates genes…………..……….……….54 Figure 2.3 :
Expression levels of GRAS family transcription factor differentially expressed between genotypes………...55 Figure 2.4 :
Expression levels for the two sampling dates for each genotype………56 Figure 2.5 :
Phenotypes progression over time in 2011………..………...57
Annexes
xi À tous ceux qui m’ont aidé
xiii
Remerciements
Je tiens d’abord à remercier mon directeur de recherche, John Mackay, pour m’avoir donné la chance et l’opportunité d’entreprendre un projet de maîtrise dans son laboratoire. Ce projet m’as permis de développer mes connaissances et mon expertise scientifique et de m’épanouir dans un environnement de travail agréable et enrichissant.
Je remercie également les Fonds de recherche du Québec sur la nature et les technologies (FRQ-NT) pour une subvention à Jean Bousquet et John Mackay ayant permis la concrétisation de ce projet et une bourse d’étude pendant deux ans.
Je veux aussi remercier les professionnels de recherche du laboratoire Isabelle Giguère, Brian Boyle, Sébastien Caron et Claude Bomal qui ont grandement contribué à mon apprentissage en répondant patiemment à mes questions et en me transmettant avec plaisir leur bagage de connaissances ainsi que leur grande expérience de travail. J’exprime ma reconnaissance envers Jean Bousquet pour avoir accepté de me co-diriger, et Julien Prunier pour avoir été un mentor pour moi pendant toute la durée de ce projet de recherche.
Je ne veux surtout pas oublier ma compagne de vie, Krystina, qui dans les dernier moments m’a aidé à garder la motivation nécessaire.
Finalement, il ne faut surtout pas oublier mes parents pour leur soutien et leur amour.
xv
Avant-propos
Le projet de recherche faisant l’objet de ce mémoire a été financé par le CRSNG, Génome Québec et Génome Canada dans le cadre du projet ARBOREA II sous la direction du Dr John Mackay et en co-direction avec le Dr Jean Bousquet.
Ce mémoire est présenté sous forme de trois chapitres. Le premier chapitre présente une revue bibliographique décrivant l’acclimatation des espèces forestières au cours des changements climatiques. Il relate l’implication de certains gènes dans l’acclimatation et l’adaptation climatique. Ce chapitre fait aussi part des hypothèses et objectifs de recherche discutés au cours de ce mémoire.
Le deuxième chapitre a été rédigé en anglais et sous forme de manuscrit puisqu’il sera soumis dans un avenir proche à une revue scientifique. Dans ce chapitre, le choix des gènes candidats a été effectué en collaboration avec Dr Julien Prunier de l’équipe du Dr Jean Bousquet. L’extraction des ARNs et les RT-qPCR ainsi que leur analyse ont quant à elles été réalisées par l’auteur (Guillaume Tessier).
Enfin, le troisième chapitre contient un bref résumé des conclusions de ce mémoire et propose des perspectives de recherche possibles suite à ce projet de maîtrise.
1
Chapitre 1 - Introduction
De nombreuses études ont montré qu’il existe de la variabilité génétique à l’intérieur des espèces d’arbre. Cette variation génétique peut occasionner des variations phénotypiques et être reliée aux phénomènes d’acclimatation des arbres chez ces derniers. Cette variabilité peut causer un changement de la protéine issue d'un gène ou modifier le niveau d’expression de ce même gène. Dans cette étude, on s’attardera à analyser la modification du niveau d’expression de gènes potentiellement reliés à l’adaptation chez l’épinette noire (Picea mariana). Dans un premier temps, les impacts de la physiologie et l’acclimatation des arbres en réponse aux facteurs climatiques seront abordés. Ensuite, l’adaptation climatique et la diversité génétique seront traitées et enfin, la génétique et les mécanismes moléculaires reliés à l’adaptation seront abordés.
1.1 La physiologie et l’acclimatation des arbres en réponse aux
facteurs climatiques
Les facteurs environnementaux ont une grande influence sur la croissance et le développement des arbres (au niveau hormonal et génétique) et leur phase cyclique. Certains exemples de processus physiologiques sont la germination des graines, l’architecture de la cime vivante, la floraison, le débourrement ou l’aoûtement. Les arbres doivent s’acclimater aux variations des facteurs climatiques au fil des saisons. Pour ce faire, des changements rapides et réversibles du métabolisme ou de la morphologie ont lieu (El Kayal. 2011). L’acclimatation a comme conséquence une modification du phénotype sans changer le génotype.
Dans les régions aux climats tempérés et nordiques, les plantes et les arbres alternent entre les phases de croissance active et de dormance. Ce processus est illustré à la figure 1. La température joue un rôle important dans le passage d’une phase du cycle à l’autre, et ce, pour toutes les transitions. La photopériode joue chez les arbres un rôle tout aussi important mais plus circonscrit dans le temps (figure 1).
2
Figure 1.1. Phases, processus et facteurs déterminants du cycle annuel de croissance et de dormance chez les arbres et les plantes pérennes.
L’écodormance se produit environ de la mi-juillet jusqu’en septembre et implique un arrêt de la croissance et la formation du bourgeon terminal en réponse à un raccourcissement de la photopériode et/ou une diminution de la disponibilité en eau (stress hydrique) et d’azote. L’écodormance automnale est réversible ; une augmentation de la température vers la fin de l’été peut faire retourner les arbres en croissance active. L’endodormance (approfondissement de la dormance) se produit de septembre à novembre et permet d’acquérir la résistance au gel afin de survivre à l’hiver. Ce processus est induit par un raccourcissement de la photopériode et une baisse de la température. Ensuite, l’accumulation de degrés-jours lors de l’augmentation de température au printemps fait sortir les arbres de leur écodormance en leur faisant reprendre le cycle saisonnier de croissance active (Raven 1999).
La synchronisation de l’entrée en dormance d'un arbre avec la fin de la période propice à la croissance est reliée à la date d’apparition du bourgeon terminal lors de l’aoûtement (Bigras et Colombo 2001). Cette partie de l’arbre contient une des structures les plus vulnérables au froid, c'est-à-dire le méristème apical. Le développement et la protection du méristème apical est un phénomène physiologique et développemental complexe qui comprend la formation du bourgeon, l’acquisition de la tolérance au froid et à la dessiccation et l’acquisition de la dormance (Rohde et Bhalerao 2007; Ruttink et al. 2007). Les méristèmes apicaux sont aussi responsables de la croissance en hauteur et jouent un rôle important dans la coordination du développement normal des arbres. Plus tôt le
3 bourgeon terminal apparaît, plus tôt l’arbre acquiert la résistance au froid nécessaire pour hiverner sans dommages et plus tôt la croissance s’arrête (Bigras et Colombo 2001; Howe et al. 2003). La formation du bourgeon terminal et l’arrêt de la croissance sont induits par des changements de l’environnement liés à la fin de l’été et à l’automne, tels qu’une baisse de la photopériode et de la température. Les jours plus courts et les basses températures semblent être nécessaires pour compléter le développement normal des bourgeons, incluant le passage en dormance (Heide et Prestrud 2005; Lagercrantz 2009; Owens et Molder 1977). Comme l'aoûtement, le niveau de tolérance au gel des végétaux n’est pas le même tout au long de l’année. Suivant l'aoûtement, il augmente en effet progressivement en automne, via un processus actif (Weiser 1970) appelé l’endurcissement (Levitt 1980a; Levitt 1980b; Sakai et Larcher 1987; Sakai 1987). L'aoûtement est donc une caractéristique phénologique importante de l'acclimatation au froid chez les arbres. On connaît déjà plusieurs gènes reliés à l’aoûtement chez d’autres espèces végétales comme le peuplier et Arabidopsis. Chez le peuplier, le phytochrome-B2 (PHYB2) et Abelson interactor 1-B (ABI1B) sont reliés à l’aoûtement par exemple. PHYB2 est un gène impliqué dans la perception de la photopériode alors que ABI1B est impliqué dans la transduction du signal de réponse à l’acide abscissique (Frewen et al. 2000). De plus, le gène Vernalization 2 (TaVRN2) peut réguler le temps de floraison et l’induction de la tolérance au froid chez Arabidopsis, mais peut aussi avoir les mêmes effets chez d’autres espèces (Diallo et al. 2010). De plus, la première étude de « microarray » comparant la croissance et la dormance sur le méristème chez Populus tremula a révélé une grande variation de l’expression des gènes entre ces deux stades développementaux et a permis d'identifier certains facteurs de transcription, notamment CBF (CRT binding factor), LEC-1 (Leafy cotyledon 1) et FIE (Fertilization-independant endosperm), apparemment reliés à l’acclimatation au froid (Schrader et al. 2004). Des profils d’expression de gènes ont été obtenus sur des pousses et des racines chez le pin sylvestre (Pinus sylvestris) (Joosen et al. 2006) et sur des feuilles chez l’épinette de Sitka (Picea sitchensis) (Holliday et al. 2008). Chez le pin sylvestre, des activateurs de transcription de la famille d’AP2-domain nommés CBF (Cold binding factor) et DREB1 (Drought responsive element) se lient à l’élément CRT/DRE et activent la transcription à la suite d’un stress dû au froid ou à la déshydratation (Joosen et al. 2006).
4
Chez l’épinette de Sitka, plusieurs gènes ont aussi été reliés avec l’aoûtement et la résistance au froid, notamment EARLI1, GIGANTEA (GI), CAX1 (cation exchanger), PHYA (phytochrome 1), CBL2 (Casitas B-cell lymphoma 2), LEA (Late embryogenesis abundant) et MPK6 (Mitogen activated protein kinase 6) (Holliday et al. 2008). EARLI1 est un gène impliqué dans le métabolisme lipidique et la paroi cellulaire et a été identifié comme ayant un rôle important dans les transferts de lipides chez Medicago truncatula. GIGANTAE est un gène connu pour être impliqué dans le signal des phytochromes sous certains stimuli environnementaux (ex. floraison) et dans la régulation du cycle circadien chez Arabidopsis thaliana (Huq et al. 2000; Mizoguchi et al. 2005). Le calcium joue un rôle primordial dans plusieurs cascades métaboliques. Plusieurs transporteurs de calcium sont nécessaires pour la transmission des signaux suite au froid. CAX1, un antiporteur Ca2+/H+ vacuolaire est reconnu chez Arabidopsis pour avoir cette fonction (Català et al. 2003; Xiong et al. 2002). Le rôle de PHYA est démontré chez des peupliers hybrides (Populus tremula x tremuloides) où la surexpression de PHYA bloquait la cessation de croissance et l’acclimatation au froid sous jours courts (Olsen et al. 1997). Les protéines CBL (calcineurin B-like) interagissent spécifiquement avec CIPK (CBL-interacting protein kinase) lors de l’influx de calcium (Shi et al. 1999) et chez Arabidopsis, le complexe CBL/CIPK est une composante cruciale du signal de stress face au froid (Albrecht et al. 2003; Cheong et al. 2003). De plus, le gène LEA est impliqué dans la réponse au stress osmotique chez Pseudotsuga menziesii (Jarvis et al. 1997). FLOWERING LOCUS T (FT) a été identifié comme ayant un rôle potentiel dans la cessation de la croissance reliée à la baisse de la photopériode (Holliday et al. 2008) et dans le cycle de la croissance chez l’épinette de Norvège (Gyllenstrand et al. 2007).
En bref, le nombre et la nature des gènes identifiés par ces études suggèrent l'existence de différents mécanismes moléculaires sous-jacents à l’acclimatation. Ces mécanismes agissent en modifiant le métabolisme et la paroi cellulaire et permettent la perception des signaux environnementaux (photopériode) ainsi que la transduction des signaux. Ces études montrent que les variations de l’expression des gènes jouent un rôle dans l’acclimatation au froid chez les plantes.
5
1.2 Adaptation climatique et diversité génétique
Plusieurs études ont montré l’existence de variations génétiques reliées à la date d’aoûtement, qui dans certains cas, sont corrélées avec des gradients latitudinaux (Beaulieu et al. 2004; Li et al. 1997; Frewen et al. 2000; Hall et al. 2007; Ingvarsson et al. 2008; Jaramillo-Correa et al. 2001). La date d’aoûtement reflète donc bien l’adaptation des espèces arborées. Toutefois, un aoûtement trop précoce peut limiter la croissance à une courte période et donc nuire au développement de l’espèce (Li et al. 1993; Bigras and Colombo 2001; Howe et al. 2003). Il a été montré que la date d’aoûtement est en partie déterminée génétiquement chez l’épinette noire (Morgenstern 1969a). Ces études ont montré que la date d’aoûtement est liée à la latitude, avec une date d’aoûtement précoce reliée à une meilleure résistance au froid. Ce phénomène est connu pour être induit par plusieurs changements physiologiques et phénologiques chez les plantes et les arbres (Bigras et Colombo 2001; Clapham et al. 1998; Clapham et al. 2001; Guy 1990; Thomashow 1999).
L’adaptation climatique est un caractère quantitatif, c’est-à-dire que sa variation résulte de l'action de plusieurs gènes (polygénique) et elle est caractérisée par une distribution continue. L'action de ces nombreux gènes est plus ou moins fortement modulée par l’environnement (Li et al. 1993; Scheiner 1993). La plupart des caractères d’intérêt qui sont ciblés par l’amélioration génétique sont des caractères polygéniques, i.e. la croissance, la rusticité, la densité du bois, etc.
La variation du phénotype, tel qu’un caractère quantitatif comme l’aoûtement, peut être modélisée en fonction de la variation génétique et l'environnement.
Phénotype (P) Génotype (G) + Environnement (E) + Interaction génotype x environnement (GxE)
Cette équation montre que le phénotype est expliqué en partie par le génotype, par l’environnement et par l'interaction de ces deux facteurs. Parmi les facteurs environnementaux, on peut noter le climat et la photopériode (Namkoong 1979).
6
Une démarche qui permet de connaître la part de l'effet de la variabilité génétique et de celle de la variabilité environnementale dans la variabilité totale d'un caractère quantitatif est l’estimation de la variance totale (σ2T) observée du caractère, et d’estimer les
composantes génétique (σ2G) et environnementale de cette variance. Ceci se fait en
contrôlant les sources de variations dues aux effets génétiques et celles dues aux effets environnementaux. Connaissant VG par rapport à VT, une proportion qui sera variable d'un
caractère à un autre, on pourra évaluer la part de contrôle génétique du caractère (White et al. 2007).
Par exemple, la plante Hieracium umbellatum est présente en Suisse dans deux habitats différents. Un de ses habitats est rocheux en bordure de falaise et l’autre habitat est sablonneux. L’habitat de Hieracium umbellatum détermine le phénotype. La survie et la fitness d’une population ou d’une espèce dépendent 1) de la plasticité phénotypique et 2) de la diversité génétique de celle-ci, qui permet l’adaptation de l’espèce au fil des générations ("Botany online: Evolution: The Modern Synthesis - Phenotypic and Genetic Variation; Ecotypes", consulté le 2012-04-04).
La plasticité phénotypique est la capacité d’un organisme à exprimer des phénotypes différents pour un génotype donné. Longtemps considérée comme indépendante du génotype, la plasticité phénotypique possède une base génétique (Agrawal 2001). Les réponses plastiques contribuent à une occupation plus complète des divers habitats dans la nature (Sultan 2000) et l’étude de l’évolution de la plasticité pourrait permettre de prédire la réponse des populations à des changements écologiques. On peut donc penser que la sélection naturelle affecterait l’évolution de la plasticité phénotypique en sélectionnant des individus à haute plasticité (Nussey et al. 2007; Sarkar et Fuller 2002).
La diversité génétique peut être très importante pour qu’une espèce puisse s’adapter à un changement dans son environnement. En fait, plus la diversité génétique est grande au sein d'une population, plus il y aura de variations d’allèles pouvant potentiellement intervenir dans l'adaptation de l'espèce à un changement environnemental. (National Biological Information Infrastructure, consulté le 14 octobre 2011). Ce phénomène d’adaptation au niveau génétique se fait via la sélection naturelle qui, au fil des générations, retient les individus étant le mieux adaptés à l’environnement local. Un phénomène qui
7 peut diminuer cette diversité génétique est la dérive génétique. La dérive génétique est la modification de la fréquence d’un allèle ou d’un génotype par le fait du hasard. Plus la population est petite, plus les effets de la dérive génétique sont importants.
Chez les conifères, plusieurs études ont montré qu’il existe une importante variation génétique à l’intérieur des espèces. Par exemple, chez l’épinette blanche (Picea glauca). on a montré qu’il y a de la variation génétique à l’intérieur et entre les différentes provenances (Li et al. 1993). Cette étude a montré que la variation génétique est corrélée positivement avec le nombre de branche chez des arbres d’un an, avec le débourrement et l’aoûtement chez des arbres de trois ans et avec la croissance en hauteur chez des arbres de huit ans. De plus, une importante variation génétique reliée à l’adaptation a aussi été déjà observée chez l’épinette noire (Beaulieu et al. 2004).
Un autre moyen utilisé par les espèces arborées pour survivre à un changement dans leur environnement local est la migration vers un habitat adéquat. Cependant, tandis que les analyses initiales ont montré que la vitesse de migration des arbres à l’ère post-glaciaire pouvait atteindre près de 200 m par année, des études plus récentes proposent que cet estimé doit être révisé à une vitesse de migration actuelle de moins de 100 m par année (McLachlan et Clark 2004; McLachlan et al. 2007). De plus, le taux de migration nécessaire pour suivre le déplacement de leur habitat relié à l'augmentation prévue de la concentration en CO2 et de la température qui devrait doubler au cours du prochain siècle,
serait de 1000 m par année (Malcolm et al. 2002). Donc la vitesse de migration prédite est grandement inférieure à la vitesse nécessaire pour que les espèces forestières migrent pour suivre leur optimum climatique. Par exemple, les biomes situés en haute latitude nécessiteront une vitesse de migration sans précédent à cause d’un réchauffement climatique plus extrême vers les pôles (IPCC 2014). De plus, une analyse de la végétation au nord du Canada utilisant l’imagerie satellitaire infrarouge montre une très faible expansion des forêts depuis les 25 dernières années malgré un réchauffement régional de 0.6°C (Masek 2001). Les hypothèses avancées pour expliquer ce lent déplacement sont le manque de sources de graines, les barrières physiques qui empêchent la dispersion de ces dernières et les mauvaises conditions des sols (Aitken et al. 2008).
8
Les grandes adaptations des écotypes des essences forestières peuvent se résumer par l’adaptation à quelques facteurs essentiels dont la température et la longueur de la saison de croissance d’une part, et l’aridité d’autre part. Ces deux facteurs sont ceux qui repartitionnent les essences végétales qui recouvrent la terre (Sakai et Larcher 1987). Le facteur température est majoritairement déterminé par la latitude et l’altitude (Morgenstern 1992) et il est souvent limitant dans la zone boréale et tempérée. Plus on monte en latitude, plus la saison de croissance est courte et la température est basse, non seulement pendant l’hiver, mais aussi pendant la saison de croissance. Cette relation est très similaire à celle relative à l’altitude : plus on monte en altitude, plus la saison de croissance et la température diminuent (Nanson 2004).
1.3 Génétique et mécanismes moléculaires reliés à l’adaptation
La régulation de l’expression des gènes est un mécanisme essentiel au bon fonctionnement d’un organisme vivant. Elle sert à exprimer les gènes au bon moment, permettant le développement, la différenciation et la croissance des cellules. Cette régulation permet aussi aux organismes d’ajuster leur machinerie enzymatique pour réagir face aux changements de l’environnement (Heintzman et Ren 2007). L’expression d’un gène renvoie à la fonction et à l’activité biologique du produit qui découle de ce gène. La régulation de l’expression peut impliquer divers mécanismes et étapes, parmi lesquels la transcription et la traduction sont les plus importantes. Les paragraphes qui suivent résument les structures et les mécanismes majeurs en cause.Un gène est une partie d’ADN qui peut être transcrit en ARN (figure 2). Si le gène est traduit en protéine, la séquence est dite codante. Cette partie du gène est délimitée par la présence d’un premier codon traduit, dit « start » en 5’ et d’un codon de terminaison ou « stop » en 3’ de ce gène. La région codante dans les ARNs messagers est délimitée par une région 5’ UTR (région non traduite) et une région 3’ UTR qui font aussi partie de la région codante. Les régions 5’ et 3’ UTR jouent des rôles importants dans la traduction, la régulation, la stabilité et/ou la localisation des ARNs (Gerstein et al. 2007). Dans la majorité des cas, le gène commence par une séquence de nucléotides constituant le promoteur. Le rôle du promoteur est de permettre l’initiation et la régulation de la
9 transcription de l’ADN en ARN. Le gène se termine par une séquence terminatrice appelée terminateur qui marque la fin de la transcription. La molécule d’ARN produite peut être soit traduite en protéine ou soit être immédiatement fonctionnelle (ARN ribosomiques et ARN de transfert).
Figure 1.2. Représentation des différentes composantes d’un gène.
Le promoteur permet l’initiation et la régulation de la transcription de l’ADN en ARN. Le premier codon traduit est dit « start » et le dernier codon traduit est le codon « stop ». La région codante est délimitée par une région 5’UTR et une région 3’UTR. Enfin, le RBS (Ribosome binding site) est le site de fixation du ribosome pour initier la transcription (http://nitro.biosci.arizona.edu, consulté 14 juillet 2014).
La synthèse de protéines se déroule en deux étapes qui sont la transcription de l’ADN en ARNm (ARN messager) qui a lieu dans le noyau, et la traduction de l’ARN messager en une protéine dans le cytoplasme de la cellule.
À l'étape de la transcription, il est possible de multiplier les copies d'ARNm pour la phase de traduction et donc de synthétiser la protéine en quantité plus importante. Pour synthétiser l’ARNm, un complexe protéique appelé l'ARN polymérase est nécessaire. Pour débuter la transcription, une protéine spécifique au gène à transcrire se fixe au promoteur, permettant à l’ARN polymérase de se lier à la molécule d’ADN et de la parcourir afin de synthétiser la molécule d'ARN complémentaire. L’ARN polymérase déroule d’abord la double hélice d’ADN, sépare les deux brins et assemble les nucléotides en se servant du brin complémentaire. Derrière l’ARN polymérase, les deux brins se rapparient et l’ADN s’enroule de nouveau. Enfin, lorsque l’ARN polymérase rencontre le site de terminaison, elle se sépare de l’ADN et l’ARN est libéré (Figure 3).
10
Figure 1.3. Transcription d’une molécule d’ADN en molécule d’ARN.
Une protéine se fixe au promoteur et permet à l’ARN polymérase de se lier à la molécule d’ADN et de la parcourir pour la lire. L’ARN polymérase déroule la molécule d’ADN, sépare les deux brins et assemble les nucléotides en se servant du brin complémentaire. Derrière l’ARN polymérase, les deux brins se rapparient et l’ADN s’enroule de nouveau. Enfin, l’ARN polymérase se sépare de l’ADN et l’ARN est libéré (Beljanski 2013).
Ensuite, la traduction permet de passer de la molécule d’ARN à une protéine grâce à un complexe protéine/ARN qu'est le ribosome et la présence d'ARNt liés à des acides aminés. La première étape de la traduction de l’ARNm est l’initiation. Lors de cette étape, l’ARNm se fixe à un ribosome qui assemble une séquence d’acides aminés selon les codons lus. Ensuite, il y a l’étape d’élongation dans laquelle le ribosome se déplace codon par codon et ajoute un acide aminé à la protéine en synthèse par l’intermédiaire d’un ARN de transfert (ARNt). Enfin, lorsque l’on atteint le codon-stop (UAA, UGA, UAG), la protéine est complète et le ribosome se détache de la protéine et de l’ARNm (Campbell et Reece 2004).
Un point de contrôle majeur de la régulation de l’expression des gènes se situe au niveau de la transcription. Les régions régulatrices des gènes se situent en amont du site d’initiation de la transcription et sont appelées séquences promotrices. La régulation se fait
11 par l’intermédiaire de protéines se liant à des séquences spécifiques de l’ADN du promoteur. Cette liaison permet d’activer ou de réprimer la transcription d’un gène. L’accumulation des transcrits d’ARNm fourni une indication indirecte de la régulation de la transcription. Elle est aussi influencée par la stabilité et la dégradation active des ARNm qui conditionnent leur durée de vide dans le cytoplasme. Ce paramètre fréquemment utilisé comme indicateur de l’expression.
Parmi les protéines régulatrices de la transcription, il y a les facteurs de transcription (Heintzman et Ren 2007; Pabo et Sauer 1992). Les facteurs de transcription sont des protéines régulatrices de la transcription et jouent un rôle important dans la biologie d’une cellule. Chez les plantes, la régulation transcriptionnelle est réalisée par l’intermédiaire de plus de 1500 facteurs de transcription, chacun contrôlant l’expression d’environ 10-1000 gènes cibles (Guo et al. 2008; Riechmann et al. 2000).
Parmi les facteurs de transcription, les gènes KNOX ont été identifiés comme ayant un rôle important dans certains processus physiologiques. Une étude de « microarray » et de qRT-PCR a démontré que les gènes KNOX (KNOX1, KNOX3 et KNOX4) étaient surexprimés pendant le milieu et la fin de la formation du bourgeon chez l’épinette blanche (Picea glauca), coïncidant avec l’initiation du primordia (Kayal et al. 2011). Un autre exemple de facteur de transcription impliqué dans des processus physiologiques importants reliés à l’acclimatation chez Arabidopsis est le gène FT1 (FLOWERING TIME 1) (Greenup et al. 2009). FT1 est impliqué dans la régulation de la floraison. De plus, le gène VERNALIZATION INDEPENDANCE 4 a été identifié comme ayant un rôle dans la vernalisation (Helliwell 2011). La vernalisation est le processus par lequel la plante passe du stade végétatif au stade reproductif (i.e. la floraison). Ces deux exemples montrent l'importance des facteurs de transcription dans la physiologie des plantes, notamment dans l'acclimatation à son environnement.
12
1.4 Mise en contexte du projet
Les conifères sont de très grande importance dans la forêt boréale couvrant plus des deux tiers (67%) des forêts canadiennes, la plupart étant dominées par les épinettes ou ayant les épinettes comme composantes majeures. Au Canada, de 2000 à 2007, 650 000 000 semences ont été plantées en moyenne par année, couvrant une surface totale de 3715 km2 annuellement. Les épinettes comptent pour 58 % du nombre de semences plantées (380 000 000) (National Forestry Database). Dans cette étude, l’épinette noire (P. mariana) sera l’espèce d’intérêt. C'est une essence majeure sur le plan économique (Fortier 2008; Springer 1991) (Tableau 1). En effet, l’épinette noire (EPN) est une essence de très grande importance pour le reboisement au Québec. Dans le but de potentiellement assister cette espèce dans son acclimatation aux changements climatiques à venir, il serait intéressant d’identifier les polymorphismes génétiques qui sont reliés à son adaptation (Prunier et al. 2011; Prunier et al. 2012).
Tableau 1.1 : Nombre de plants livrés annuellement au Québec pour la période 2001-2007 pour les forêts publiques (Fortier 2008)
Essences 2001 2002 2003 2004 2005 2006 2007 Nobles 105 733 271 326 364 171 427 614 164 350 134 000 95 415 EPB 11 502 744 13 052 770 27 550 449 12 033 560 12 923 269 14 133 058 13 929 019 EPN 70 474 886 72 997 910 71 804 280 58 017 022 54 064 892 73 257 824 65 906 580 EPO 727 112 866 336 1 766 797 1 582 374 1 562 068 2 386 156 2 772 316 PIB 581 648 588 299 1 639 103 974 259 562 022 506 819 357 432 PIG 30 191 637 26 662 160 23 847 727 26 180 692 25 302 908 32 424 023 33 664 018 MEL 219 414 142 146 567 715 1 008 130 158 24 938 1 185 MEX 673 281 678 517 805 056 572 185 612 422 331 968 536 389 PEH 249 058 425 871 1 432 162 627 783 619 048 636 169 594 594
De nos jours, l’adaptation peut être étudiée directement au niveau génomique (Storz 2005). Par exemple, la méthode de détection d’« outliers » permet d’identifier des polymorphismes géniques représentant des variations dans la séquences d’ADN potentiellement responsables d’adaptation dans un ensemble de polymorphismes neutres
13 (Namroud et al. 2008). En statistiques, un « outlier » est une observation qui est significativement distante du reste des données. À l’aide de ces méthodes et de diverses méthodes de biologie moléculaire parfois couplées à des mesures quantitatives telles que la date d’aoûtement et la croissance en hauteur, 25 SNPs (Single Nucleotide Polymorphims) ont été identifiés comme étant potentiellement reliés à l’adaptation climatique chez l’épinette noire (Prunier et al. 2011; Prunier et al. 2012; Prunier et al. 2013). En effet, plusieurs méthodes ont été utilisées pour valider ces 25 SNPs, incluant l’identification de QTLs et des tests d’associations génétiques (Prunier et al. 2012; Prunier et al. 2013).
Certains des SNPs qui ont été reliés à des adaptations climatiques présentent des substitutions non synonymes pouvant entraîner des variations fonctionnelles des protéines. Les variations alléliques représentées par ces SNPs peuvent pourraient aussi être associées à des variations du niveau de l’expression génique (causés ou pas par les SNPs eux-mêmes). Par exemple, l’accumulation des transcrits pourrait varier entre les différents génotypes présents pour chacun de ces SNPs (Prunier et al. 2011).
14
1.5 Hypothèses et objectifs de recherche
Premièrement, cette étude devait avant toute chose évaluer les sources autres que génétiques de variation des transcrits chez l’épinette noire. Ainsi, on devait tenir compte de la variation spatio-temporelle (tissu spécifique à un temps donné. par exemple pendant la saison de croissance) des transcrits à l’intérieur des arbres afin d’évaluer le plus précisément possible le lien potentiel entre la séquence d’ADN et l’expression. Il est aussi bien connu que l’accumulation des transcrits d’ARN peut varier entre les organes, les tissus et les cellules, ainsi qu'en fonction de conditions environnementales, incluant la photopériode. Il était donc important de cerner où et quand sont exprimés les ARN messagers pour chacun des gènes visés. Cette information aura permis d’élaborer un plan d’échantillonnage visant à comparer les différents génotypes rencontrés pour chacun des SNPs des gènes candidats examinés, et ainsi à mieux isoler toute source de variation génétique qui serait causée par des variations d’un génotype à l’autre pour un même tissu et à un même temps donné par exemple.
Par conséquent, le premier objectif de ce mémoire était de dresser le profil d’accumulation des transcrits au travers des différents tissus. L’hypothèse était que l’expression des gènes est préférentielle dans les tissus les plus affectés par les changements associés à l’acclimatation au froid (apex et bourgeons). Pour vérifier cette hypothèse, les profils d’accumulation des transcrits dans neuf tissus différents sont ont été établis à certaines dates spécifiques de la saison de croissance.
Le second objectif était de déterminer l’accumulation des transcrits des différents gènes dans le temps. Deux expériences ont été menées : 1) les profils d’accumulation des transcrits pour les différents gènes ont été établis à quatre dates différentes au cours de l’aoûtement et 2) les profils d’accumulation des transcrits en fonction des génotypes ont été établis à deux dates différentes pendant la période proximale à l’aoûtement. Une des hypothèses était qu’il y avait de la variation dans l’expression des gènes à différentes dates de la période de cessation de la croissance. L’autre hypothèse était que pour certains gènes reliés à l’acclimatation, il y avait une interaction entre le génotype et le stade
15 physiologique, c’est-à-dire que certains génotypes pourraient réagir dans le temps alors que d’autres le feraient moins.
Le troisième objectif de ce projet était de déterminer si le génotype pouvait être relié à des variations de l’expression des gènes associés à l’acclimatation au froid. L’hypothèse testée était que le niveau d’expression des transcrits de certains gènes potentiellement reliés à l’acclimatation variait selon le génotype observé. Pour vérifier cette hypothèse, le profil d’accumulation des transcrits a été établi pour chacun des génotypes de chaque gène. Ces facteurs ont donc été tenus en compte lors de l’évaluation des différences d’expression entre génotypes.
Bref, la première expérience vise à étudier la variation spatio-temporelle des profils d’accumulation des transcrits chez des jeunes épinettes noires. La deuxième expérience, quant à elle, vise à établir une relation entre le génotype et l’expression génique chez des gènes candidats pour la croissance et l’acclimatation.
17
Chapitre 2
Genetic variation in black spruce (Picea mariana): linking genetic expression to genetic variation
Guillaume Tessier, Julien Prunier, Jean Bousquet and John Mackay
2.1 Résumé
Les arbres doivent s’acclimater aux variations climatiques afin de survivre aux changements saisonniers, et ce, de façon multi-annuelle. Plusieurs études ont montré qu’il y a de la variabilité génétique à l’intérieur des espèces et que cette variabilité aide les espèces à survivre et à s’acclimater à différents changements environnementaux. Vingt-quatre gènes potentiellement reliés à l’adaptation ont préalablement été identifiés chez l’épinette noire (Picea mariana [Mill.] B.S.P.). L’objectif de cette étude était d’acquérir des évidences du rôle potentiel de ces gènes dans l’acclimatation et l’adaptation en étudiant leur expression. Pour ce faire, l’expression des gènes a été étudiée au niveau spatio-temporelle, soit la variation de l’expression selon le stade physiologique et selon le tissus végétatif à l’intérieur de l’arbre, et aussi selon les différents génotypes.
Une grande variabilité dans l’expression des gènes candidats a été observée entre les tissus ou l’organe étudié, et aussi entre les stades physiologiques de l’arbre. Les gènes étaient exprimés majoritairement dans des tissus pouvant être reliés plus directement à l’acclimatation et l’adaptation, par exemple la tige primaire. Pour certains gènes, une variation significative dans l’expression des transcrits d’ARN a été observée entre les différents génotypes. De plus, pour l’expression de certains gènes, une interaction entre le génotype et le stade physiologique de l’arbre a été observée, c’est-à-dire que certains génotypes pourraient réagir dans le temps alors que d’autres le feraient moins.
2.2 Abstract
Trees must acclimate to change in climatic conditions in order to survive to seasonal variations, and this process must occur of the course of several years. Several studies have shown that genetic variability within species helps species to survive and to acclimate to
18
different environmental changes. Twenty-four genes potentially related to adaptation have previously been identified in black spruce (Picea mariana [Mill.] BSP). The objective of this study was to acquire evidence supporting their potential role in acclimation and adaptation by studying their expression. To do this, the variation of gene expression was studied as a function of the physiological stage during the annual growth cycle and tissue type, and among several different genotypes.
Considerable variability in the expression of candidate genes was observed between different vegetative tissues or organs, and between different physiological stages of the trees. The genes were expressed predominantly in tissues that can be linked more directly in acclimation and adaptation, for example the primary stem. For some genes, a significant variation of transcript accumulation has been showed between genotype. Furthermore, for some genes, an interaction between the genotype and physiological state of the tree was observed, that is to say that some genotypes might react in time while others would less.
2.3 Introduction
In order to survive, perennial plant species from the temperate and boreal areas have to acclimate to winter conditions each year. Trees from these regions are able to withstand winter conditions including deep and prolonged frosts by acquiring season cold and dehydration tolerance (Bigras and Colombo 2001). These physiological changes are reversible and may be monitored by following the development of vegetative buds. The timing of this phenomenon – called budset - is directly related to the adaptation to local temperature conditions. As such, the earlier the vegetative buds develop, the earlier trees cease to growth and acquire cold tolerance required for winter acclimation (Bigras and Colombo 2001; Howe et al. 2003).
Several changes in metabolism or morphology occur during acclimation. Protection of the apical meristem is a key change. It is a complex physiological and developmental phenomenon including formation a vegetative terminal bud, cold tolerance acquisition of and desiccation, and the acquisition of dormancy (Rohde and Bhalerao 2007; Ruttink al. 2007). The cessation of growth and the formation of a terminal bud are induced
19 by changes in the environment during summer or early fall. A decrease in the photoperiod and in temperature are key factors for initiation and completion of bud formation, respectively (Rohde and Bhalerao 2007 and El Kayal 2011).
Several genes related to budset have been identified in trees and plants such as poplar and Arabidopsis thaliana. In poplar, the phytochrome-B2 (PHYB2) gene is involved in the photoperiod perception and an Abelson interactor 1-B (ABI1B) gene is involved in signal transduction response to abscisic acid (Frewen et al. 2000). In Arabidopsis, the Vernalization 2 (TaVRN2) gene can regulate flowering time and the induction of cold tolerance; it was shown to have the same effect in other species like wheat (Diallo et al. 2010). Variation in the accumulation levels of a large number of transcripts has been shown in white spruce in relation to abrupt or gradually shortening of the photoperiod (El Kayal et al. 2011).
Genetic variation within trees species has been reported in the timing of bud set for a number of forest tree species including conifers and broadleaf trees (e.g. Li et al. 1993; Frewen et al. 2000; Jaramillo-Correa, Beaulieu and Bousquet 2001; Beaulieu et al. 2004; Hall et al. 2007; Ingvarsson et al. 2008), which is correlated with latitudinal gradients (Li et al. 1997; Beaulieu et al. 2004; Hall et al. 2007). Several polymorphisms in DNA sequences have been associated with climatic variations by comparing populations from different sites in various tree species, and several of the polymorphisms that have been identified were located in genes encoding transcription factors (e.g. Jump et al. 2006; Namroud et al. 2008; Eveno et al. 2008; Eckert et al. 2009; Prunier et al. 2011).
Genetic variation also enabled the identification of genes related to the timing of winter acclimation in trees by comparing gene expression and cold hardiness among different populations at different times of the year. For instance, in Sitka spruce (Picea sitchensis), genes that were thus linked with acclimation included EARLI1, GIGANTEA (GI), CAX1 (cation exchanger), PHYA (phytochrome 1), CBL2 (Casitas B-cell lymphoma 2), LEA (Late embryogenesis abundant) and MPK6 (map kinase 6) (Holliday et al. 2008).
20
While the above reports have linked acclimation and bud formation with sequence variations in genes including transcription factors or with variation in transcript levels in trees, the relationship between DNA sequence polymorphisms and gene expression remained to be tested. Thus, the main objective of this study was to explore the link between gene expression and the genetic diversity impacting upon acclimation. Recent studies in the boreal conifer black spruce (Picea mariana [Mill.] B.S.P.) identified 24 genes related to climate adaption by using outlier detection (Prunier et al. 2011. 2012) and bulk segregant analyses followed by genetics association tests (Prunier et al. 2013). One or more SNPs within each of the genes were significantly associated with temperature, precipitation, budset or juvenile growth. In this report, gene expression for these 24 candidate genes was investigated by assessing variation in transcript level accumulation as a function of tissue type within trees, date during the growth season and tree genotype. Allelic variations represented by these SNPs may be directly or indirectly linked to cis- or trans- effects and thus to be related to genetic variation in expression.
2.4 Materials and methods
Identification of candidate genes for climatic adaptation in black spruce
Twenty-four candidate genes were identified based on previous reports on the genetic diversity in natural populations of the boreal conifer black spruce (P. mariana [Mill.] B.S.P.) (Table 1). One of the studies investigated the distribution of SNPs among natural populations and identified a set of SNPs significantly related to precipitation and/or temperature variation, representing 26 genes potentially linked to climate adaptation (Prunier et al. 2011). The second report described two experiments: 1) a bulk segregant analysis comparing individuals representing extreme phenotypes from 26 populations that identified 37 SNPs potentially related to the timing of budset and juvenile growth and, 2) an association study which tested all of the above SNPs identified 24 genes significantly associated with these same traits (Prunier et al. 2013), with some of these SNPs in overlap with the first study (Table 2.1).
21 Plant material
Two major experiments were carried out to investigate the accumulation of RNA transcripts in young black spruce tissues under controlled conditions for these 24 candidate genes. For the first experiment, two years old plants were obtained from a local nursery and were transferred to three-liter pots in May 2010. The trees were obtained from open-pollinated seed lots from one geographic region in Québec (See supplemental materials Figure S1). The trees were grown in a plastic greenhouse at Université Laval in 2010, under natural photoperiod and temperature regimes of Quebec City. They were watered daily and fertilization was applied to all the trees.
For the second experiment, a population of 1800 two year-old black spruce trees was grown for one full year at the Université Laval nursery under natural photoperiod and temperature before the experiment was started. The objective was to gather a wide variety of genotypes representing a large portion of Quebec territory while avoiding areas of potential introgression areas with red spruce (P. rubens) (Perron and Bousquet 1997).This was achieved by assembling a population of 300 open-pollinated plants from six different seed orchards in Quebec representing distinct geographic regions away from the zone of contact with red spruce (Supplemental Material Figure S1). The plants were grown in a plastic greenhouse from June 2010 to September 2011 and were overwintered outside. Fertilization regime was used for optimal nutrition and growth.
Experiment 1
The first experiment aimed at assessing the temporal and spatial variation of transcript accumulation profiles in young black spruce trees.
Experimental design
Two different samplings and analyses were conducted in this experiment to assess the variation of transcripts accumulation in different organs and tissues, at different dates and hours of sampling. In a first analysis, nine different tissues were sampled on August 4th. 2010 for 10 randomly selected plants for the purpose of comparing different vegetative tissue types. The tissue samples were: apex (AX), primary elongating stem (ES), root periderm (including secondary phloem) (PR), stem phelloderm (including secondary
22
phloem) (PS), root tips (RT), root secondary xylem (XR), stem secondary xylem (XS), current year branch needles (YNB), current year needles on primary stem (YNS). For a second analysis, four of the same tissues (AX, YNS, ES and YNB) were sampled in addition to branch apex (AXB) and branch stem (ESB) on ten other young black spruces on August 11th, 2010 at 9 AM and 3 PM, in order to compare gene expression at different times during the day, and between branches and the main stem. All samples were then stored at -80 °C to preserve RNA integrity.
RNA extractions and quality control
Samples were immersed in liquid nitrogen and ground to a fine powder in jars of 50 ml with stainless steel balls (25 mm) using the “Mixer Mill” (Mixer Mill MM300 Retsh GmbH, Haan, Germany). RNA was extracted following Chang et al. (1993) adapted by Pavy et al. (2008) and concentration was measured using a spectrophotometer (Nanodrop ND-1000, Thermo Scientific, Wilmington. USA). RNA quality was determined with the “Bioanalyzer 2100” (Agilent Technologies, Santa Clara, CA, USA) based on the RIN quality index (RNA Integrity Number) as above 1.7 and the 28S/18S ratio above 1.8 for use in RT-qPCR .
cDNA preparation and quantitative PCR
Transcript levels were determined by RT-qPCR (reverse transcription-quantitative polymerase chain reaction). The reverse transcription of total RNA (500 ng per sample) was carried out with the "First strand cDNA synthesis system" according to the manufacturer's instructions (Invitrogen. Carlsbad. CA. USA). The amplification primers specific to each of the candidate genes were designed based on the genomic DNA sequences reported by Prunier et al. (2013) using the Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and primers properties (self-annealing or potential hairpin formation) were ensured using OligoCalc (http://www.basic.northwestern.edu/biotools/oligocalc.html). The reaction mixture for qPCR was composed of QuantiFast SYBR Green 1X (Qiagen, Toronto, ON, Canada), amplification primers at a concentration of 300 nM, and 5 ng of cDNA. The qPCR reaction mixtures were assembled using an epMotion 5075 pipetting robot (Eppendorf, Hamburg,
23 Germany) and amplifications were performed using a LightCycler 480 (Roche, Toronto, ON, Canada) with the following program: an activation step of five minutes at 95°C, followed by 50 amplification cycles of 94°C for ten seconds and 62°C for one minute. A melting curve analysis at the end of amplification was used to verify that a single product was amplified (Boyle et al. 2009).
The fluorescence readings of the qPCR thermal cycler were converted to number of transcript molecules by using the LRE method (Rutledge and Stewart 2008) with minor modifications adapted for Excel as described by Boyle et al. (2009). Excel formulas, macros and tutorial were available in the publication section of Arborea project website (http://www.arborea.ulaval.ca/publications/index.html). The number of molecules was converted to log2 scale for statistical analyses. The geNorm software was then used to normalize the number of transcript molecules (log2 scale) (Vandesompele et al. 2002) and allowed the identification of the following reference genes for optimal normalization: CDC2 (cyclin dependent protein kinase, BT106071). EF1a (elongation factor 1, BT102965), GQ0071_B06 (ribosomal protein, BT115036) and GFP (green fluorescent protein).
Experiment 2
The second experiment aimed at assessing the relationship between genotype and gene expression at candidate gene loci for growth and acclimation.
DNA extractions, genotyping and gene expression
In order to represent all possible genotypes for the 24 candidate SNPs studied, a genetically diverse study population was developed. DNA was extracted from a few needles (20-100 mg of fresh tissue) using a NucleoSpin 96 Plant II kit (Macherey-Nagel, Düren, Germany) from 1152 trees randomly selected with equal representation among the plants from each of the six seed orchards. Extraction efficiency was assessed based on DNA concentrations evaluated on a subset of 10 samples randomly selected in each plate with a spectrophotometer (Nanodrop ND-1000, Thermo Scientific, Wilmington, USA). Genotyping was carried out by Sequenom technology (McGillUniversity- Genome Québec Innovation Center) as described by Prunier et al. (2013). Among the 24 SNPs tested, 23
24
SNPs yielded adequate results with Sequenom technology. The remaining SNP was genotyped with the "TaqMan ® SNP Genotyping Assays” (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Once all the genotyping results were obtained, the population structure was tested with the STRUCTURE software (Pritchard et al. 2000). Using a burn-in period of 30000 and a running length period of 100 000 in a model with correlated frequencies among populations, population structure was tested 10 times for 1 to 10 putative groups (K).
Based on the frequency of genotypes in each genotypic class (three classes, one heterozygous and two homozygous) for each of the 24 SNPs among the 1152 trees above, a subset of 142 trees was assembled and retained to further study gene expression within each genotypic class for each gene, with each class represented by a minimum of 10 trees. During the summer of 2011, height growth and terminal budset timing were assessed following the methods of El Kayal et al. (2011) on each of the 142 trees two times per week from the beginning of June 2011 to the end of August 2011 (Figure 5). A preliminary analysis (see results from the third analysis of the first experiment) showed that transcript levels in a tissue were very similar among the lateral branches and the main stem (there was no significant difference between the transcript levels in the branch and in the main stem). Hence, it was possible to perform non-destructive sampling of shoot tip tissues by removing the tip of one lateral branch (1 cm) comprising the meristem, current-year buds, needles and stem for each of the 142 trees. This procedure offered the opportunity to resample the same individuals on June 22nd and July 27th for gene expression analyses, while avoiding biases related to genetic background variations among individuals and potential epistatic consequences. RNA extractions, quality control, cDNA preparation and quantitative PCR were performed as described for experiment 1.
Statistical analysis
Data analysis was carried out using the R statistical environment (release 2.13.0). Differences between the numbers of RNA transcripts for each of the experiments were tested using ANOVAs followed by Waller-Duncan’s multiple range tests to assign differential expression between different dates and tissues. The associated p-values were also adjusted for multiple testing using Bonferroni correction for multiple comparisons
25 unless specified. The significance of the differential expression between 9 AM and 3 PM samples was tested with a student t-test. P-values were corrected using the Bonferroni Step-down (Holm) procedure for multiple comparisons.
2.5 Results
Accumulation profiles of candidate gene RNA transcripts in vegetative tissues
We hypothesized that adaptive genes significantly related to climate or growth and phenology may be preferentially expressed in tissues that are most affected by photoperiod and climatic variations during the growth season. For example, the apex of newly formed stems responds to changes in photoperiod and temperature by altering their metabolism and forming dormant buds in which the shoot meristem may overwinter. Similarly, the cambial zone undergoes significant cellular changes but the regulation of this process has been less studied. We investigated this hypothesis by monitoring RNA transcript accumulation in nine different vegetative tissues for each of the 24 candidate genes. Data were obtained from ten young greenhouse trees that already formed a terminal bud (sampled August 4th, 2010) by using RT-qPCR determinations with gene specific probes (for details see methods).
RNA transcripts for each of the 24 candidate genes were detected in all of the nine vegetative tissues and displayed diverse expression profiles. Statistically significant differences between tissues were observed for most of the genes (21/24) (Fig. 2.1). Only four genes showed no difference between tissues (AP2 domain [Genbank accession #BT100365], WRKY DNA binding domain [BT103112], Myb domain protein 36 [BT100400] and B-box zinc finger family [BT105756]). The fold difference between the lowest and the highest transcript level ranged from 1.82 for a tubby like gene to 41.36 for a LEA protein (BT102742) (log2 fold difference of 0.86 and 5.37 respectively). The within tissue transcript averages were grouped into four different classes (according to Waller Duncan’s multiple range test). A majority of the genes showed preferential transcript accumulation in primary stems and many of the same genes also accumulated strongly in the stem secondary phelloderm and in the root periderm (Fig. 2.1). Two genes had
26
preferential transcript accumulation in the root tips or in the needles, and none had a transcript preference for secondary xylem of the main stem or the roots.
Additionally, RNA transcripts for each of the 24 candidate genes were monitored at different dates during the growth season and a few of the genes varied significantly between the four dates tested. Six genes were differentially expressed between dates in the elongating stem tissue, two genes in the apex and one gene in current-year needles of the stem. The fold difference between the lowest and the highest transcript level ranged from 2.31 for GRAS family transcription factor (BT114502) in primary stem to 18.77 for bZIP transcription factor (PgBZIP-8, BT118997). According to Waller Duncan’s multiple range test, these transcript levels grouped into two different classes. The comparison of RNA transcripts levels for the 24 candidate genes between morning and afternoon produced no significant differences.
Development of a genetically diverse study population
A black spruce population was developed to enable gene expression studies that aimed at comparing the different genotypes of 24 candidate SNPs for climatic adaptation. Young trees were obtained from open-pollinated seed collected in populations spanning the geographic area previously studied by Prunier et al. (2011). In total, 1152 black spruces from six different regions of Quebec (Supplemental materials, Figure S2) were genotyped for each of the 24 SNPs by using Sequenom technology and Taqman genotyping (for details see methods).
The number of trees representing each of three potential diploid genotypes (two homozygotes and a heterozygote) at each locus varied widely between the SNPs, as reflected by the minor allele frequency ranging from 0.01 to 0.49 in the sample of 1152 black spruces (Table 2.1). Based on the genotypic distributions, a study population of 142 trees was assembled so as to obtain a minimum of 10 trees for each genotypic class for use in the gene expression monitoring and analysis.
27 As genetic covariance related to hierarchical population structure among the individuals of the study population could bias comparisons among genotypic classes, the STRUCTURE software (Pritchard et al. 2000) was applied to verify that there was no population clusters in our data set. As expected from a previous study in the same area (e.g. Prunier et al. 2011), the partition yielding the highest posterior probability was for K=1, indicating no significant hierarchical population structure (results not shown).
Comparison of expression levels between genotypes at different sampling dates
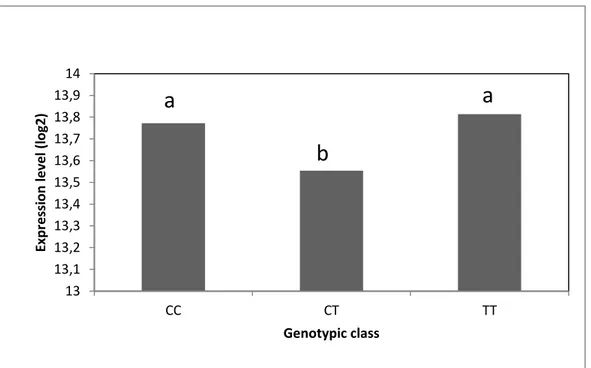
Transcript levels of the 24 candidate genes were assessed in shoot tips of each of the 142 trees at two distinct growth stages. i.e. during active height growth (date 1: June 22nd, 2011) and after the completion of the height growth phase when plants had well-formed buds (date 2: July 27th, 2011). An analysis considering all of the data (two-factor ANOVA) indicated that the transcript levels varied significantly (P < 0.05) between sampling dates for a majority of genes (21), and between genotypes for two genes (Table 2.2). In addition, a significant interaction among dates and genotypes was observed for two genes (Table 2.2). However, transcript levels for one gene (GRAS family transcription factor. BT114502) did not vary significantly between dates but between genotypes (Figure 2.2). Based on these observations, genotypic effects were also tested for each date separately using one-factor ANOVA. Three genes, a leucine-rich receptor-like protein kinase, a zinc finger C2H2 (DV997502) and a zinc finger C3HC4 (BT106728), presented transcript levels varying significantly between the genotypic classes (Table 2, P < 0.05). These three genes with significant transcripts variations between genotypic classes were observed at the first date and two of them were nearly significant at the second date (Figure 2.3).
Of the 21 genes that varied between the two sampling dates, all except two showed higher transcript levels at the second sampling date (Figure 2.4). Transcript levels increased by two-fold or more for nine of the genes and increased by as much as 64-fold for two of them.
Interactions between the sampling date and the genotype were observed for two of the genes that also presented significant genotype effects (Zinc finger C2H2 type and zinc
28
finger C3HC4 domain) (Table 2.2). The strongest interactions were observed for the zinc finger C2H2 gene where the homozygous genotype AA had lower transcript levels than the heterozygous and the homozygous genotypes (AG and GG) at date 1 but appeared only slightly higher at date 2 (Figure 2.3).
Relationship between candidate genes and growth and phenology
The relationship between candidate SNPs and growth and phenology phenotypes and was assessed at two time points during the growth season, which were the sampling dates for RNA isolations (June 22th,2011 and July 27th, 2011). We calculated a cumulative score for budset and growth from the beginning of the phenotypic measurements until the date of the second sampling, and a cumulative score for budset and growth from the first sampling date to the second sampling date. A significant effect of the candidate SNP genotype on budset was observed for two genes (Zinc finger C2H2 type, 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily [BT102917]) at date 1 (Figure 2.5) (ANOVA, P < 0.05). At date 2, no significant effect of a candidate SNP genotype on budset was observed.
2.6 Discussion
The aim of this study was to gain evidence of the potential role of a set of genes in acclimation and adaptation by investigating their expression profiles. These were obtained by comparing different vegetative tissues and organs, and different phases of growth. These analyses served to delineate spatio-temporal variations in regard to the acclimation process involving budset. Secondly, the relationship between genotypic variations on gene expression was examined in regard to adaptation. Together, these observations may help to link genetic variation and mechanisms of climate adaptation.
Tissue expression profiles of candidate genes
The RNA transcript profiling showed that all but four of the 24 candidate genes were differentially expressed among the nine different vegetative tissues. Genes were generally more highly expressed in the primary stem (ES), the main stem phelloderm (PS)