UNIVERSITE DE GENEVE FACULTE DE MEDECINE Section Médecine clinique Département de Médecine Interne
Division d’Endocrinologie
Thèse préparée sous la direction de la Dresse Ursula Lang P.D.
MODULATION PAR L’OESTRADIOL ET L’ALDOSTERONE DU
CONTRÔLE HORMONAL DE L’EXPRESSION DE LA COX-2 DANS
LES CARDIOMYOCYTES
Thèse
Présentée à la Faculté de Médecine de l’Université de Genève
pour obtenir le grade de Docteur en médecine
par
Romina Claudia CAPOCCIA
de
Neuchâtel
Thèse n° 10294
Remerciements
Un grand Merci tout d'abord à la Dresse Ursula Lang, pour m'avoir accueillie dans son équipe, ainsi que pour ses encouragements, sa disponibilité et son enthousiasme.
Je remercie également sincèrement Christine Gerber-Wicht ainsi que Manuella Rey pour leur précieuse aide technique, leur soutien et le temps qu'elles m'ont accordé toujours avec le sourire.
Je tiens à remercier Le Professeur Jacques Philippe pour m'avoir permis de réaliser ce travail au sein de la Division d'Endocrinologie et de Diabétologie. Enfin, merci à tout le laboratoire pour son accueil et son aide dans ce monde nouveau de recherche fondamentale
RESUME
MODULATION PAR L’OESTRADIOL ET L’ALDOSTERONE DU CONTRÔLE HORMONAL DE L’EXPRESSION DE LA COX-2 DANS LES CARDIOMYOCYTES
Selon un nombre croissant d’études, la COX-2, isoenzyme inductible, exprimée de manière transitoire en réponse à des facteurs de croissance, des cytokines, ou des promoteurs de tumeurs et prédominant dans des conditions inflammatoires, aurait un effet protecteur sur les cardiomyocytes.
Plusieurs études rapportent une expression augmentée de COX-2 dans différents tissus (le muscle lisse, les cellules mésangiales, les cellules endothéliales), en présence de certaines hormones, tel que l’endothéline-1 et l’angiotensine II. En revanche, il existe très peu d'informations au niveau du cœur dans ce domaine.
Les objectifs de travail étaient les suivants:
1) Déterminer la cinétique de l'effet de l'AngII et de l'ET-1 sur l'expression de la COX-2 au niveau des cardiomyocytes ventriculaires du rat nouveau-né.
2) Etudier les effets de l'aldostérone et de l'oestradiol sur l'expression de la COX-2 dans des cardiomyocytes stimulés avec de l'Ang-II et de l'ET-1. Les résultats obtenus montrent que l'Ang-II et l'ET-1 induisent la COX-2 en fonction du temps. Ces deux hormones induisent une augmentation rapide de l'expression de la COX-2 qui atteint des valeurs maximales après 1 h de stimulation. Quant à l'aldostérone et l'oestradiol, nos observations montrent que ces deux stéroïdes exercent un effet modulateur négatif sur l'expression de la COX-2 dans les cardiomyocytes ventriculaires.
MODULATION PAR L’OESTRADIOL ET L’ALDOSTERONE DU CONTRÔLE HORMONAL DE L’EXPRESSION DE LA COX-2 DANS LES
CARDIOMYOCYTES
TABLE DES MATIERES
I. INTRODUCTION
p.11) CARDIOMYOCYTES p.1
2) PROSTAGLANDINES p.3
2.1) Sécrétion de PGI2 dans des cultures de cardiomyocytes p.3
2.2) Phospholipase A2 p.3 2.3) Cyclooxygénase p.4 3) ANGIOTENSINE II (AngII) p.9
4)
ENDOTHELINE-1 (ET-1) p.11 5) ALDOSTERONE (aldo) p.13 6) OESTRADIOL (E2) p.157) Régulation du système cardio-vasculaire par l'AngII p.16bis et l'ET-1 et influence de l'ostradiol et de l'aldostérone
II. MATERIEL ET METHODES
p.18 1) COMPOSITION DES MILIEUX ET DES TAMPONS p.182) METHODES p.19
2.1) Mise en culture des cardiomyocytes ventriculaires de
rats nouveau-nés p.19
2.2) Stimulation des cardiomyocytes ventricualires p.21 2.3) Analyse des protéines par électrophorèse et
imunodétection (western blot) p.21
III. RESULTATS
p.231) EFFETS DE L’ANGIOTENSINE II ET DE L’ENDOTHELINE-1
SUR L’EXPRESSION DE LA COX-2 p.23
2) EFFETS DE L’ALDOSTERONE SUR L’EXPRESSION
DE LA COX-2 DANS LES CARDIOMYOCYTES VENTRICULAIRES p.26
2.1) stimulation avec l’ET-1 p.26
2.2) stimulation avec l’AngII p.28
2.3) stimulation des cellules avec l’AngII et expression de la
COX-2 en fonction de la variation de concentration d’aldostérone p.30 3) EFFETS DE L’OESTRADIOL SUR L’EXPRESSION DE LA
COX-2 DANS LES CARDIOMYOCYTES VENTRICULAIRES p.33
3.1) stimulation avec de l’ET-1 p.33
3.2) stimulation avec de l’AngII p.34
3.3) stimulation des cellules avec l’AngII et expression de la
COX-2 en fonction de la variation de concentration de p.36 l’oestradiol
IV. DISCUSSION
p.381) INDUCTION DE LA COX-2 PAR L’ANGIOTENSINE
II ET L’ENDOTHELINE-1 p.38
2)MODULATION DE L’EXPRESSION DE LA COX-2 PAR DES HORMONES STEROÏDIENNES : ALDOSTERONE
I. INTRODUCTION
1) CARDIOMYOCYTES
Le muscle cardiaque est constitué de cellules, les cardiomyocytes, liés les uns aux autres par des disques intercalaires. Les embranchements des cardiomyocytes forment un réseau complexe tridimentionnel. Tous les cardiomyocytes sont capables de dépolarisations rythmiques spontanées. Mais, c’est un groupe de myocytes de l’atrium qui constitue le pacemaker et propage les stimulations électriques à travers le myocarde par des gap junctions situées entre les myocytes. C’est pourquoi, malgré le fait qu’il soit constitué par des cellules séparées, le myocarde se comporte comme un syncytium [1]. Le cardiomyocyte, en plus de sa fonction contractile propre, possède des propriétés endocrines en ce sens qu’il est capable de synthétiser et libérer dans son environnement immédiat et dans la circulation générale des facteurs autacoïdes tels que certaines prostaglandines, comme la prostaglandine E2 et la prostacycline, des dérivés de phospholipides membranaires et des hormones peptidiques, les peptide natriurétique auriculaire ou ANP et le BNP (Brain natriuretic peptide).
Le cardiomyocyte possède dans sa membrane cellulaire des récepteurs à plusieures hormones : les catécholamines, l’endothéline, l’angiotensine ll et la vasopressine, l’aldostérone, des hormones thyroïdiennes, l’insuline entre autres. L’activation des récepteurs à l’angiotensine ll (Ang II) et à la vasopressine aboutit à des effets variés, comprenant des effets inotropes et chronotropes, des effets de croissance par l’intermédiaire de la stimulation d’oncogènes ainsi qu’à la stimulation de la fonction endocrine des cardiomyocytes. L’angiotensine ll et l’endothéline-1 peuvent provoquer une réponse hypertrophique chez le rat [2],
une réponse qu’on observe également in vivo durant le développement du coeur. En effet, dans la période post-natale, les cellules musculaires cardiaques perdent leur capacité à se diviser et dès ce moment, la croissance du coeur se fait par hypertrophie : phénomène caractérisé par une augmentation de la quantité de protéine par cellule [3].
2) PROSTAGLANDINES :
2.1) Sécrétion de prostacyclines dans des cultures de cardiomyocytes
L’augmentation de la sécrétion de prostacycline (PGI2) dans le cœur après des lésions ischémiques peut résulter de la production de PGI2 par différents types de cellules, notamment les cellules endothéliales. Des expériences effectuées dans des cellules soumises d’abord à l’ischémie puis à une réoxygénation ont montré une libération de prostacycline plus grande lors de la réoxygénation [4], [5]. Des travaux effectués par notre laboratoire ont montré une augmentation de la sécrétion de prostacycline par l’Ang ll et la vasopressine (AVP) dans les cardiomyocytes ventriculaires, par la voie de l’activation de la PKC et la phospholipase A2 [6, 7]
La synthèse de prostacycline est régulée par deux étapes limitantes : la libération d’acide arachidonique des phospholipides membranaires par la phospholipase A2 (PLA2) et la conversion de cet acide arachidonique en précurseurs de prostanoïdes (PGH2) par la cyclooxygénase.
2.2) Phospholipase A2 :
La phospholipase A2 est impliquée dans différentes réponses cellulaires, dont la défense de l’hôte, la coagulation, la digestion. De nombreuses études effectuées dans différents types cellulaires ont montré que la PLA2 joue un rôle clé dans la régulation des voies de transduction des signaux médié par les lipides.
1. La PLA2 sécrétoire qui demande du Ca++ en concentration millimolaire pour accomplir sa fonction enzymatique. Elle est elle-même subdivisée en deux sous-groupes basés sur sa sélectivité tissulaire. On trouve le groupe 1 principalement dans le pancréas. Et le groupe dans les tissus non pancréatiques [8].
2. La PLA2 cytosolique qui requiert du Ca++ en concentration submicromolaire pour une activité cytosolique optimale. On la trouve dans le compartiment cytosolique de la plupart des cellules [9].
3. La PLA2 qui est indépendante du Ca++.
2.3) Cyclooxygénase :
Le Prostaglandine endoperoxyde H synthase ou cyclooxygénase catalyse une étape de la formation des prostaglandines et du thromboxane. Elle possède deux fonctions enzymatiques : une fonction dite cyclooxygénase, reponsable de l’oxydation de l’acide arachidonique en PGG2 suite à l’insertion de deux molécules d’O2 et une fonction peroxydase qui réduit les PGG2 en PGH2 [10]. Il existe 2 types de PGHS : PGHS-1 et PGHS-2 ou COX-1 et COX-2 qui catalysent toutes les deux la formation des PGH2 à partir de l’acide arachidonique.
PLA2 : phospholipase A2 AA : acide arachidonique COX : cyclooxygénase PG : prostaglandine TX : Thromboxane
Schéma de la synthèse d’éicosanoïdes
La PLA2 et la COX transforment les phospholipides membranaires en PGH2, qui est ensuite
converti par des enzymes spécifiques des tissus en prostacycline, prostaglandines et thromboxane. La PGH2, après avoir été synthétisé par les COX, est transformé par des
synthases différentes en PGD2, PGE2, PGF2a ou Txa2
Les deux isoenzymes de la cyclooxygénase possèdent essentiellement la même structure primaire avec 60% d’identité ainsi qu’une structure cristallographique semblable [11, 12] et elles possèdent des propriétés cinétiques très proches. Les deux sont inhibées par des antiinflammatoires non stéroïdiens (AINS) bien que des inhibiteurs spécifiques de chacune aient été développés. COX-1 et –2 se trouvent dans les cellules sous forme d’homodimères attachés par leur domaines hydrophobes aux membranes du réticulum endoplasmique et du noyau [13, 14].
Phospholipides membranaires AA PGG2 PGH2 PGD2 PGE2 PGF2 PG2 TXA2 TXB2 PLA2 COX-1 COX-2 DIFFERENTES SYNTHASES
Malgré leurs points communs structuraux et fonctionnels, les deux isoformes possèdent des fonctions biologiques différentes.
COX-1 et COX-2 sont codées par des gènes distincts [15].
La COX-1 est considérée comme l’enzyme constitutive ; et son expression est régulée tout au long de son développement ; elle est responsable de la production physiologique, constitutive de prostaglandines. Une fois que l’enzyme a formé les prostanoïdes dans le réticulum endoplasmique, ces derniers agissent par des récepteurs de surface, pour effectuer des tâches d’entretien de l’organisme ; par exemple, l’équilibre hydrosodé par le rein, ou la protection gastrique contre l’acidité, ou encore l’hémostase.
La COX-2 au contraire est l’enzyme dite inductible, qui dans des condition standards est habituellement absente des cellules et qui est exprimée de manière transitoire en réponse à des facteurs de croissance, des cytokines (Il-1, IL-6, PDGF, TNF-α) ou des promoteurs de tumeur ; elle prédomine dans les conditions inflammatoires. La COX-2 synthétise les prostanoïdes sur l’enveloppe nucléaire et ces derniers semblent agir au niveau nucléaire durant la réplication ou la différenciation nucléaire [16].
Les éicosanoïdes ont des effets biologiques variés malgré leur structures semblables. Ils sont impliqués dans les fonctions respiratoires, puisqu’ils provoquent une bronchoconstriction. Les PGE2 stimulent la production de mucus gastrique qui avec les bicarbonates inhibent la rétrodiffusion des H+. En outre ils tempèrent la sécrétion acide de l’estomac par une action directe sur les cellules pariétales. Les PGE2 et les PGI2 permettent une vasodilatation rénale. Les PGI2 permettent une relaxation des cellules musculaires lisses vasculaires (VSMC), mais provoquent une contraction des cellules musculaires lisses du tractus digestif. Ils sont en outre connus pour leur effet proinflammatoire. On utilise
depuis le début du siècle les antiinflammatoires non stéroïdiens, qui inhibent les deux isoenzymes et empêchent ainsi la production de prostaglandines et de thromboxanes proinflammatoires [16]. Mais cette supression complète d’eicosanoïdes est à l’origine de nombreux effets indésirables de ces médicaments, notamment l’insuffisance rénale et l’ulcère gastrique. C’est pourquoi on développe de plus en plus des inhibiteurs spécifiques de la COX-2. Ainsi, l’inflammation est diminuée, mais la production basale d’eicosanoïdes nécessaire au maintien de l’homéostasie gastrique et rénale est conservée [17, 18].
Les COX ont en outre un intérêt thérapeuthique certain au vu de leur implication dans les thromboses coronaires , dans l’inflammation, dans le cancer du côlon et dans la maladie d’Alzheimer [19].
Les premières études considéraient la COX-2 comme délétère tandis que la COX-1 était vue comme protectrice, mais actuellement, la distinction n’est plus aussi claire.
Plusieurs études récentes ont montré des effets variés de la COX-2 au niveau du cœur.
Adderley et al. (1999) [20] ont montré que les lésions des cardiomyocytes dues au H2O2 et à la doxorubicine sont limitées par la formation de prostacycline, qui reflète l’induction de COX-2.
De même, Noreen P. Dowd ont montré que le degré de lésion cardiaque chez un animal traité avec de la doxorubicine et du SC236 était diminué par l’administration préalable d’iloprost, un analogue de la PGI2 [21].
Une étude canadienne [22] a démontré l’induction de la COX -2, en association au processus inflammatoire et à la cicatrisation, dans le myocarde lors de l’infarctus du myocarde . En revanche, le rôle exact de cet enzyme dans cette maladie fatale n’a pas été déterminé [22].
Une étude menée chez le lapin a montré que l’augmentation de la COX-2 joue un rôle essentiel dans la protection du coeur lors de la phase tardive du préconditionnement ischémique (cycle d’occlusion/reperfusion coronaire de 4 minutes). En effet, dans cette expérience, le préconditionnement ischémique montre une augmentation rapide de l’ARMm de la COX-2, une induction importante de la COX -2 dans les 24 heures et une augmentation du contenu cardiaque en prostaglandines, produits enzymatiques de la COX-2. Dans cette expérience, l’utilisation d’inhibiteurs sélectifs de la COX-2 (NS -398 et celecoxib) bloquent totalement l’effet cardioprotecteur, tant pour les lésions ischémiques réversibles (ischémie), que pour les lésions irréversibles (infarctus) Cette étude semble donc définir la COX-2 comme un enzyme cardioprotecteur [23].
Une augmentation de l’enzyme COX-2 ainsi que de son mRNA a été démontré lors d’un rejet d’allogreffe cardiaque. Néanmoins, l’inflammation et l’apoptose des cardiomyocytes n’ont pas été réduits de manière significative par un traitement d’inhibiteurs de la COX-2 [24].
Une fibrose cardiaque a été rapporté par Wong et al (1988) [25] chez la souris dont le gène de la COX-2 a été inactivé, suggérant que la COX-2 aurait des propriétés cardioprotectrices.
3) ANGIOTENSINE II (Ang ll)
L’Ang ll est un octapeptide et le composant actif principal du système rénine-angiotensine. Ce système est important pour le contrôle de la pression artérielle et l’équilibre ionique, par l’effet direct de l’Ang ll et par l’intermédiaire de la sécrétion d’aldostérone. La diminution de la pression artérielle moyenne de même qu’une augmentation du K+ plasmatique ou une restriction sodée stimule respectivement les barorécepteurs ou les chémorécepteurs de l’appareil juxtaglomérulaire du rein, ce qui provoque la sécrétion de la rénine, un enzyme de la zone de la Macula Densa, qui agit pendant 30 minutes à une heure en transformant l’angiotensinogène en angiotensine l au niveau du foie. Ensuite, l’enzyme de conversion de l’angiotensine convertit l’angiotensine l en angiotensine ll au niveau des poumons ; cet enzyme est détruit en quelques minutes dans le sang par l’action d’angiotensinogénases. L’Ang ll permet une constriction artérielle directe, la réabsorption rénale de Na+, une augmentation de la précharge cardiaque, de l’inotropisme et du chronotropisme ainsi qu’une stimulation de la soif. L’effet final est une augmentation du volume plasmatique et de la résistance vasculaire périphérique, donc une augmentation de la pression artérielle. L’Ang ll permet également une augmentation de la sécrétion d’aldostérone par la zone glomérulaire du cortex surrénalien. L’aldostérone, un minéralocorticoïde, qui au niveau rénal augmente la réabsorption du sodium, entraîne donc une rétention d’eau, mécanisme qui permet également une augmentation de la pression artérielle par augmentation de la précharge cardiaque, mais de manière moins importante que l’angiotensine elle-même. En outre, l’aldostérone provoque une augmentation de l’excrétion du K+. Ensuite, un rétrocontrôle négatif permet un arrêt de la sécrétion de rénine, ce qui permet une stabilisation de la pression artérielle et de l’équilibre ionique [26].
Le rôle de l’Ang ll a été étudié dans plusieurs pathologies cardiaques en utilisant des inhibiteurs du système rénine-angiotensine (RA). Dans nombre de ces études cliniques, l’efficacité des inhibiteurs de l’enzyme de conversion de l’angiotensine dans l’hypertension artérielle et l’insuffisance cardiaque a été démontrée [27, 28]. Habituellement, ce système rénine-angiotensine est décrit comme un système général avec une Ang ll distribuée dans tout le corps. Pourtant, il existerait d’après plusieurs auteurs [29]., des systèmes RA localisés dans des organes spécifiques comme par exemple le cœur qui fonctionne par voie autocrine ou paracrine.
L’Ang ll se lie à deux types de récepteurs AT1 et AT2 qui sont exprimés dans de nombreux tissus dont le cœur [30]. Ces récepteurs appartiennent à une famille de récepteurs à 7 domaines transmemembranaires et se distinguent les uns des autres par leur affinité pour les antagonistes non peptidiques [31]. Ces récepteurs sont augmentés après un infarctus du myocarde [32] et une hypertrophie [33]. Dans le cœur, ce sont les récepteurs AT1 qui sont responsables de la plupart des effets physiologiques de l’hormone [34]. D’après une étude menée aux Pays-bas [35]., il semble que lors d’hypertrophie cardiaque due à des stress physiques, l’Ang ll, ainsi que l’endothéline-1 et le TGF-β libérés par les cellules cardiaques et vasculaires participeraient à ce phénomène par une action autocrine et paracrine.
4) ENDOTHELINE-1 (ET-1)
La famille des endothélines comporte trois membres : ET-1, ET-2, ET-3, qui ont été identifiés par clonage [36]. Une préproendothéline est clivée par des endopeptidases spécifiques en grande endothéline, puis l’enzyme de conversion de l’endothéline la convertit en endothéline
L’ET-1 est un grand peptide qui compte 21 acides aminés. La transcription de son gène peut être régulée dans la cellule endothéliale au niveau de l’ARNm par la thrombine, l’adrénaline, l’Ang ll, l’ADH, le TGF-β, l’ester de phorbol ; sa libération peut être inhibée par le NO et l’ANP [37]. On la trouve à la surface des cellules endothéliales de la plupart des vaisseaux. Le stimulus nécessaire à sa sécrétion est la lésion de l’endothélium vasculaire. Le rôle physiologique de cette hormone n’est pas encore élucidé, mais on la trouve augmentée dans certaines pathologies, comme la toxémie gravidique, l’insuffisance rénale aiguë ou chronique [38]. L’endothéline administrée de manière exogène a montré une augmentation de la résistance vasculaire périphérique. Mais durant les premières minutes d’administration intraveineuse, l’endothéline diminue la résistance vasculaire périphérique ; probablement dû à une libération de composés vasodilatateurs comme le NO, le PGH2 ou l’ANP [37].
On a découvert que l’ET-1 possédait un rôle de facteur de croissance dans certaines cellules de mammifères, comme les VSMC et les fibroblastes [39, 40]. En ce qui concerne le cœur, Kojima et al., 1995 [41] ont démontré qu’un antagoniste non spécifique du récepteur à l’endothéline diminuait la taille de l’infarctus chez le rat, le lapin, le chien, quand on l’administre avant ou après un infarctus aigu du myocarde. L’ET-1 a également un effet inotrope positif dans des cardiomyocytes ventriculaires en culture [41], en outre, l’ET-1 induit une hypertrophie des cardiomyocytes [42, 43, 44]. L’ET-1 est également produite par
des cellules non endothéliales comme les VSMC, les cellules épithéliales rénales, les cellules mésangiales glomérulaires et certaines lignées de cellules cancéreuses [45, 46, 47, 48]. Ces observations ont mené à l’hypothèse que l’endothéline endogène produite par les cardiomyocytes pourrait être impliquée dans la pathogenèse de l’hypertrophie cardiaque par un mécanisme autocrine et paracrine. Dans ce contexte, Ito et al. (1996) [49] ont rapporté une augmentation de l’ARNm de la préproendothéline-1 et de la libération de l’ET-1 et ils ont suggéré que cette activation endogène peut, au moins en partie, médier la réponse hypertrophique induite par l’hypoxie. La synthèse myocardique accélérée d’ET-1 contribue également à la dysfonction du ventricule gauche durant la transition de la phase d’hypertrophie du ventricule gauche à celle d’insuffisance cardiaque congestive chez le rat hypertendu [50]. En outre, Kobayashi et al., (1999) [51] ont rapporté que le niveau de l’ARNm de la préproendothéline, celui de l’ET-1 ainsi que celui du récepteur étaient significativement plus hauts chez les rats en insuffisance cardiaque chronique que chez les rats contrôles.
5) ALDOSTERONE :
L’aldostérone est un minéralocorticoïde très puissant qui est responsable du le 90% de l’activité de tous les minéralocorticoïdes. L’absence de sa sécrétion par le cortex surrénalien entraînerait la mort en jours à deux semaines. Sans minéralocorticoïdes, la concentration plasmatique de K+ augmenterait à l’inverse du Na+ et du volume liquidien extracellulaire. En effet, l’aldostérone agit sur l’échangeur Na+/K+ des cellules épithéliales tubulaires rénales.
La sécrétion d’aldostérone est stimulée par : 1) L’augmentation du K+ extracellulaire ; 2) L’activation du système RA ; 3) la sécrétion basale d’ACTH, indispensable, mais qui ne provoque que peu de variation dans le taux sécrété d’aldostérone. La sécrétion d’aldostérone est diminuée en présence d’une hypernatrémie.
L’aldostérone permet par son action d’augmenter la tension artérielle et de diminuer le K+, d’effectuer un rétrocontrôle négatif pour atteindre un équilibre tensionnel et ionique [52].
Comme d’autres hormones stéroïdes, l’aldostérone commence son action en se liant à ses récepteurs intracellulaires qui ensuite sont capables de contrôler la transcription de plusieurs gènes. Dans les cardiomyocytes de rats nouveau-nés et adultes, des concentrations physiologiques d’aldostérone ont montré une augmentation de trois fois des isoformes principales des pompes Na+/K+- ATPase [52b]. En outre, dans les cellules cardiaques de rats nouveau-nés, un traitement de 24 heures d’aldostérone stimule l’activité de l’antiport Na+/H+ et de l’échangeur Cl-/HCO3- [53]. Récemment, il a été rapporté dans les cardiomyocytes de rats adultes, que l’aldostérone induisait une expression augmentée du courant de Ca2+ [54]. L’événement initial de l’action de l’aldostérone est sa liaison à son récepteur. Ce récepteur a la même affinité pour l’aldostérone et pour les glucocorticoïdes. C’est pourquoi la sélectivité in vivo de
ce récepteur requiert la présence d’un enzyme, le 11β-hydrostéroïde déshydrogénase, qui métabolise les glucocorticoïdes en dérivés inactifs. Lombès et al (1995) [55] ont montré une coexpression de cet enzyme et du récepteur à l’aldostérone dans le cœur, qui contient donc les outils cellulaires nécessaires à l’action de l’aldostérone.
Un excès chronique d’aldostérone est associé à un dépôt de collagène interstitiel cardiaque et périvasculaire marqués [56, 57, 58]. La spironolactone, antagoniste de l’aldostérone, prévient cette fibrose cardiaque, ce qui indique qu’un récepteur aux minéralocorticoïdes est impliqué dans cette réponse [56]. Une étude récente nous montre l’implication de l’angiotensine-2 et de son récepteur dans le mécanisme de la fibrose cardiaque, suggérant que le récepteur cardiaque AT1 serait une cible pour l’aldostérone [59].
6) OESTRADIOL :
Le 17β-oestradiol est le plus puissant et le plus important des oestrogènes. Sa source principale est la conversion extraglandulaire de l’androstènedione, au niveau des tissus périphériques.
Les oestrogènes permettent le développement des caractères sexuels secondaires chez la femme, la croissance utérine, l’épaississement de la muqueuse vaginale, la fluidification du mucus cervical et le développement du système canalaire des seins.
Toutes les hormones stéroïdiennes sont dérivées du cholestérol provenant de trois sources : les lipoprotéines circulantes (surtout les LDL), le cholestérol synthétisé de novo dans l’ovaire et le cholestérol libéré des esters stockés dans les gouttelettes lipidiques.
La conversion du cholestérol en prégnénolone est l’étape limitante de la stéroïdogenèse ovarienne, qui à lieu au niveau des cellules de la granulosa, des cellules thécales et surtout au niveau du corps jaune.
Pour être transporté dans le sang, l’oestradiol est lié à l’albumine à 60%, à la globuline liant la testostérone à 38% et 2-3% est libre.
Les hormones stéroïdiennes ont un faible poids moléculaire et pénètrent facilement dans la cellule en diffusant à travers la membrane, bien qu’il puisse y avoir un transporteur qui participe à ce transport ; elles entrent dans le noyau et se lient à un récepteur nucléaire. L’affinité, la spécificité et la grande concentration des récepteurs aux stéroïdes permettent à une petite quantité d’hormone de produire un effet biologique. La spécificité des récepteurs aux oestrogènes est partiellement déterminée par le nombre de récepteurs dans la cellule. Par exemple, dans l’utérus, l’action biologique des oestrogènes est grande grâce à la concentration élevée des récepteurs dans une cellule. Il se produit une transformation active du complexe de l’hormone-récepteur après la liaison de
l’hormone au noyau. Cette liaison implique un changement de la conformation qui, soit rend accessible des sites DNA, qu’on appelle steroid response elements (SREs), soit altère des complexes de la régulation de la transcription, pour leur permettre d’interagir avec des SREs pour activer ou réprimer des gènes [60]. Aucune étude n’a été publiée sur l’effet de l’oestradiol sur l’expression de la COX-2 dans les cardiomyocytes. Mais nous savons qu’il existe sur les cardiomyocytes des récepteurs aux oestrogènes.
En effet, la différence qu’il existe dans les maladies cardiovasculaires entre les deux sexes a suggéré que les oestrogènes agissaient directement sur le tissu cardiaque. C’est pourquoi Grohe et al. (1997) [61] ont cherché à savoir si les cardiomyocytes et les fibroblastes possédaient des récepteurs aux oestrogènes. Ils ont découvert par une méthodes d’immunofluorescence que le récepteur protéique aux oestrogènes était exprimé dans les cardiomyocytes et les fibroblastes cardiaques. La translocation nucléaire du récepteur protéique aux oestrogènes a été démontrée après stimulation des cardiomyocytes avec du 17-β-oestradiol (E2) [61].
Dans d’autres cellules que les cellules cardiaques, par exemple, les cellules musculaires lisses vasculaires, l’E2 stimule l’activité de la COX-2 et de la PGI2 synthase [62].
Ghanam K. et al. (2000) [63] ont vu avec l’aorte du lapin hypercholestérolémique que l’E2 restore l’effet relaxant de l’Acétylcholine (Ach) qui était inhibé par l’hypercholestérolémie.
Dans l’endomètre également, on a constaté que le pic de l’expression de la COX-2, exprimé durant les trois phases du cycle menstruel, correspond à la phase diestrus, qui correspond au niveau maximal d’E2, ce qui suggère une induction de la COX-2 dépendant de l’E2 [64].
7) REGULATION DU SYSTEME CARDIO-VASCULAIRE PAR L’ANGIOTENSINE-II ET
L’ENDOTHELINE-I ET INFLUENCE DE L’OESTRADIOL ET DE L’ALDOSTERONE
-Infarctus
-hypertrophie
ALDO
ALDO
Augmentation:Stress oxydatif: -Doxorubicine lésions -Infarctus -inflammation -lésions ischémniques -AngII -Et-1 Augmentation: -COX-2 -PGI2 (réduit également les extrasystoles ventriculaires lors
ET
ET
-
-
1
1
-Inotrope positif -Hypertrophie >> insuffisance cardiaque -augmentation taille infarctus Croissance des cellules musculaires lisses vasculaires et des fibroblastesVAISSEAUX
VAISSEAUX
Baisse de la résistance vasculaire dans les premières minutesANGII
ANGII
-réabsorption Na+ -excrétion K+ Augmentation : -Fluide extracellulaire -Volume sanguin -Précharge cardiaque -Baisse Pression Artérielle -Restriction Na+ RAA RAA vasoconstriction Lésion vasculaireE2
E2
COX-2 / PGE2,PGI2COEUR
COEUR
Inotrope + Augmentation: Augmentation du débit -Résistance vasculaire périphérique -Pression artérielleII) MATERIEL ET METHODES:
1) COMPOSITION DES MILIEUX ET DES TAMPONS :
ENZYMES:
Dnase Type IV ou I: pointe de spatule (Sigma D 4138) (dessicateur -20°C) Trypsine 1:250: 125mg/50ml (invitrogen 27250-042)
filtration de la solution avec filtre type milipore 0.22 µ ANTICORPS : COX-2 murine Polyclonal Antiserum, Cayman Anticorps anti-rabbit , Covalab
MILIEU DE CULTURE:
FCS: Fœtal calf serum (seromed N°SO115, chez Fakola) Hanks, sans Mg++, sans Ca++:( invitrogen 14175-073)
Antibiotiques: Pénicilline/streptomycine (invitrogen 15140-122)
Antimycotique: Fungizone conc. finale 250µg/500 ml(Pharmacie HCUG)
Facteur de croissance ITS (Insulin-Transferin-Sodium Selenite) Sigma I 1884, conc. finale: 1%
Milieu de culture: Milieu Mc Coys 5A (modifié) (Gibco 21500-046= poudre pour 5l de milieu)
Mc Coys complet= 500 ml de milieu + 1ml de solution de Fungizone + 5ml de Pénicilline/Streptomycine + 5 mlITS + 50 ml de FCS
Mc Coy’s sans rouge de phénol : Cell culture technologies GmbH TAMPON DE PHOSPHATE (PBS) :
TAMPON DE LYSE:
50 mM de Tris-base, 150mM de NaCl, 10% de glycérol, 1% de Triton X-100, 2 mM d'EDTA Triple, 2 mM d'EGTA, 40 mM de β-glycérophosphate, 50 mM de fluorure de sodium, 10 mM de pyrophosphate de sodium, 200 µM d'orthovanadate de sodium (ajouté au dernier moment), 10µg/ml de leupeptine, 0,3 TIU/ml de trasylol, 1µM de pepstatine A, 1mM de PMSF (ajouté au dernier moment), 100 nM de calyculine.
TAMPON DE TRANSFERT:
25 mM de Tris-base, 190 mM de glycine, méthanol 20% TAMPON DE LAVAGE:
50 mM detris-base, 200 mM de NaCl, Tween 20 (0,2%), ajuster à pH 7.5 TAMPON DE BLOCAGE:
Tampon de lavage et lait maigre en poudre 5% TAMPON D'INCUBATION:
Tampon de lavage et lait maigre en poudre 1%
2) METHODES :
Chez des rats Winstar de 24-48h, prélévement du ventricule in situ, placé dans du tampon Hanks, sans Mg++ et sans Ca++, à température ambiante. Contrôle sous binoculaire du prélévement des ventricules pour déceler la présence d'oreillettes. Le prélévement est réalisé rapidement et la dispersion enzymatique est réalisée le plus précocément possible. Les ventricules sont rincés pour éliminer les globules rouges dans du Hanks sans Mg++ et sans Ca++. Aspiration de la solution saline. Emincer aux ciseaux les ventricules de la manière la plus fine, pendant environ 5 minutes et commencer la dispersion enzymatique. 1ère dispersion: mettre les morceaux de tissu dans 10 ml de solution d'enzymes, dans un tube conique, dans le bain-marie, sur l'agitateur. Le barreau doit tourner réguliérement, pour éviter d'abîmer les cellules. Durée: 8 minutes
Retirer du bain-marie, laisser décanter quelques secondes, prélever le surnageant avec une pipette en veillant à ne pas aspirer de gros morceaux et jeter cette première dispersion (globules rouges, déchets cellulaires).
Ajouter 8 à 13 ml de solution d'enzyme dans le tube contenant les ventricules et remettre dans le bain-marie sur l'agitateur. A la fin des 8 minutes, laisser décanter quelques secondes, aspirer le surnageant au moyen d'une pipette, mettre dans un tube conique de 15 ml avant de centrifuger 4 minutes à 1200 rpm (la dispersion se répète 10 à 12 fois jusqu'à ce que tous les morceaux de tissus soient dispersés.)
Pendant ce temps, préparer un tube de 50 ml avec environ 20 ml de Mc Coy's complet dans lequel on gardera les cellules centrifugées après chaque étape de la dispersion.
Lorsque la centrifugation est terminée, aspirer le surnageant, récolter le pellet dans le milieu Mc Coy's complet du tube préparé à cet effet et garder au bain marie à 37°C.
Lorsque la totalité des cellules a été récoltée, on centrifuge le tout 5 minutes à 1200 rpm. On aspire le surnageant et on resuspend le pellet dans le milieu de
culture Mc Coy's complet + FCS 10%, puis on répartit les cellules dans les pétris. Les cardiomyocytes commencent à se contracter spontanément 24 à 48h après la mise en culture. Pour effectuer nos expériences, nous avons utilisé les cellules 3 jours après la mise en culture.
2.2.) Stimulation des cardiomyocytes ventriculaires:
24 heures avant la stimulation par des hormones, les cardiomyocytes sont déprivés en étant mis dans du milieu Mc Coy's dépourvu de FCS. Dans le même temps, les stéroïdes sont ajoutés dans les pétris déstinés aux longues incubations, c'est-à-dire 20-24 heures. 20 heures après, le milieu est changé et les hormones sont ajoutées pour une durée de trois heures.
2.3.) Analyse des protéines par électrophorèse et immunodétection (Western blot) :
Les cardiomyocytes, cultivés dans des boîtes de Pétris de 6 cm de diamètre contenant 2,5 ml de Mc Coy's sans FCS, sont lavées avec 5 ml de tampon phosphate froid (PBS) et lysées avec 100µl de tampon de lyse conservé à 4°C. Après avoir gratté les boîtes de Pétris, le lysat est récolté dans des tubes Eppendorfs, vortexé et déposé 10 minutes sur le la glace (4°C). Ces deux dernières étapes sont répétées une seconde fois. Les tubes sont ensuite centrifugés 10 minutes à 4°C à 10'000 rpm et le surnageant, contenant le lysat total, est récupéré. La quantité de protéines présente dans chaque tube est déterminée par spectrophotométrie à 595 nm grâce à la méthode de Bradford (Kit BIORAD Protein Assay). Après avoir ajouté la solution stop dénaturante de
Laemmli (Tris 60mM pH 6.8; SDS 2% ; β-mercapto 5% ; glycérol 10% ; bromophénol blue 0.01%) et après avoir porté les échantillons 5 minutes à ébulition, les protéines, chargées à quantité égale (25µg/puit), sont séparées selon leur poids moléculaire par électrophorèse sur un gel de sodium de dodecylsulfate polyacrylamide de 8%. Après une heure de migration à 150 mV les protéines du gel sont transférées sur une membrane de nitrocellulose pendant 2 heures à 100 mV, à 4°C. Pour éviter des liaisons non spécifiques, ces membranes sont incubées 1 heure à température ambiante dans du tampon de blocage. Elles sont ensuite lavées 3x10 minutes dans du tampon de lavage, puis incubées avec un anticorps polyclonal anti-COX-2 spécifique (stock:0.5 mg/ml) dilué (1:1000) dans du tampon d'incubation pendant 2 heures. Les membranes sont à nouveau lavées et incubées 1 heure avec un anticorps secondaire anti-lapin couplé à une perroxydase (HRPeroxydase) et dilué (1:2000). Avant de révéler les bandes immunoréactives par chemioluminescence (ECL), une étape de 6 fois 10 minutes de lavages est nécessaire.
2.4.) Statistiques :
Les tests statistiques suivants ont été utilisés : test de T de Student-Fisher, pour paires non-appariées, le “one-way analysis of variance” (ANOVA). Une probabilité p<0.05 est acceptée comme statistiquement significative.
III. RESULTATS
1) EFFET DE L’ANGIOTENSINE II ET DE L’ENDOTHELINE SUR L’EXPRESSION DE LA COX-2
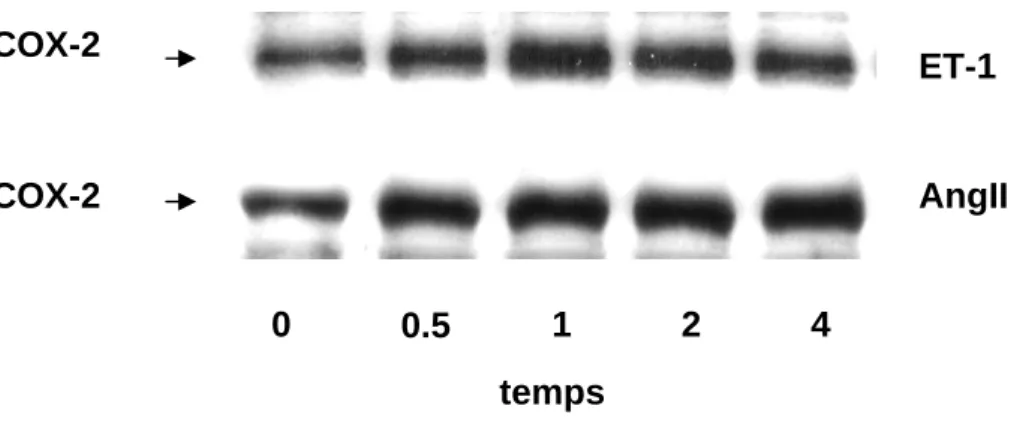
L’angiotensine et l’endothéline sont deux hormones qui stimulent la production de prostacyclines dans les cardiomyocytes ventriculaires [65, 66]. Les analyses de notre laboratoire par électrophorèse et immunodétection (Western blot) de l’expression de la COX-2 montrent que ces deux hormones augmentent l’expression de la COX-2 dans ces cellules cardiaques. Ces résultats sont illustrés dans les figures 1a et 1b qui présentent la cinétique de l’induction de la COX-2 par l’AngII (2x10-7 M) et par l’ET-1 (10-7 M).
Cette induction est dépendante du temps. Après une heure d’incubation avec l’ET-1 et l’AngII, on observe une augmentation de l’expression de la COX-2 de 208 ± 21% du contrôle et de 171 ± 12% du contrôle respectivement, par rapport aux cellules non stimulées. Dans le cas de la stimulation par l’ET-1, la stimulation descend légérement dès deux heures. En ce qui concerne l’AngII, l’effet stimulant qu’elle exerce sur l’expression de la COX-2 atteint un plateau dès une heure de stimulation.
100 150 200 CO X-2 (% d u c o n tr ô le ) temps (h)
*
*
*
*
*
*
*
0 1 2 3 4 AngII Et-1 ContrôleFig. 1a et 1b : Cinétiques de l’expression de la COX-2 induite par l’AngII et l’ET-1 dans les cardiomyocytes ventriculaires. Analyse des Western blots par densitométrie
Les cellules cardiaques ont été incubées pendant 30 min, 1h, 2h et 4h avec de l’AngII (2x10-7M) ou avec de l’ET-1 (10-7M). La quantité de COX-2 présente dans les cellules a été déterminée par l’analyse densitométrique du western blot. Les résultats sont exprimés en % du contrôle et représentent les moyennes + sem de 4 expériences indépendantes. *=p<0.05 par rapport au contrôle.
COX-2
ET-1
COX-2 AngII
0 0.5 1 2 4
temps
Fig. 1c : Western blot illustrant la cinétique de l’expression de la COX-2 induite par l’AngII (2x10
-7 M) et l’ET-1 (10-7 M) dans les cardiomyocytes ventriculaires. Cette figure est
2) EFFETS DE L’ALDOSTERONE SUR L’EXPRESSION DE LA COX-2:
2.1) Stimulation des cellules avec de l’ET-1 (Fig.2)
Dans les cardiomyocytes ventriculaires, nous avons observé une expression basale de la COX-2 (Fig 1b et 2b). L’analyse densitométrique des western blots démontre que cette expression est légèrement, mais pas significativement réduite après 3h d’incubation avec de l’aldostérone (10-6M). En revanche, après 20h d’exposition à l’aldostérone, l’expression de la COX-2 est réduite à 35 ± 5% des valeurs contrôles. Lorsque les cellules sont stimulées avec de l’ET-1, la préincubation avec de l’aldostérone pendant 3h abolit l’expression de la COX-2 induite par l’ET-1. Un prétraitement de 20h avec l’aldostérone n’abolit pas seulement l’effet de l’ET-1, mais réduit l’expression de la COX-2 à 36 ± 13% des valeurs contrôles, un résultat très semblable à celui obtenu avec des cellules non stimulées.
ctrl Et-1 0 100 200 basal aldo 3h aldo 20h C O X -2 (% d u co n tr ô le )
*
*
+*
+Fig.2a : Effet de l’aldostérone sur l’expression de la COX-2 dans des cardiomyocytes ventriculaires stimulées avec de l’ET- (101 -7 M). Analyse des Western blot par densitomé rie t Les cellules ont été incubées, soit avec, soit sans ET-1 (10-7M) pour une durée de 3h, en présence ou en absence d’aldostérone (10-6 M). Les cardiomyocytes ont été incubés pendant 3h ou 20-24h avec de l’aldostérone (10-6 M). Les colonnes représentent les valeurs moyennes ± SEM de 4-5 expériences indépendantes. *=p<0.05 par rapport aux valeurs basales, dans des cellules non stimulées. t=p<0.05 par rapport à la COX-2, dans des cellules stimulées par l’ET-1
COX-2
Ctrl ET-1 aldo 3h aldo 20h aldo 3h aldo 20h + ET-1 + ET-1
Fig 2b : Western blot représentant l’effet de l’aldostérone sur l’expression de COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’ET-1 10-7 M
2.2) Stimulation des cellules avec de l’angiotensine II (Fig.3)
L’analyse densitométrique des Western blots démontre que l’expression de la COX-2 est légèrement, mais pas significativement réduite après 3h d’incubation avec l’aldostérone (10-6M). Par contre, après 20h d’exposition à l’aldostérone, l’expression de la COX-2 est réduite à 35 ± 5 % des valeurs contrôles. Lorsque les cellules sont stimulées avec de l’AngII, la préincubation avec l’aldostérone pendant 3h abolit l’expression de la COX-2 induite par l’AngII. Un prétraitement de 20h avec l’aldostérone n’abolit pas seulement l’effet de l’AngII, mais réduit l’expression de la COX-2 à 38 ± 8% des valeurs contrôles, un résultat très semblable à celui obtenu avec des cellules non stimulées.
ctrl Ang II 0 100 200 basal aldo 3h aldo 20h C O X -2 ( % du c ont rô le ) + +
*
*
*
Fig.3a : Effet de l’aldostérone sur l’expression de la COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’AngII (2x10-7 M).
Les cellules ont été incubées, soit avec, soit sans AngII (2x10-7M) pour une durée de 3h, en présence ou en absence d’aldostérone (10-6 M). Les cardiomyocytes ont été incubés pendant 3h ou 20-24h avec de l’aldostérone (10-6 M). Les colonnes représentent les valeurs moyennes ± SEM de 4-5 expériences indépendantes. Analyse des Western blots par densitométrie. *=p<0.05 par rapport aux valeurs basales dans des cellules non stimulées. +=p<0.05 par rapport à la COX-2 dans des cellules stimulées par l‘AngII.
ctrl AngII aldo 3h aldo 20 aldo 3h aldo 20h +AngII +AngII
COX-2
Fig 3b: Western blot représentant l’effet de l’aldostérone (10-6 M sur l’expression de la COX
2 dans les cardiomyocytes ventriculaires stimulés avec de l’Angiotensine II (2x10
)
--7 M).
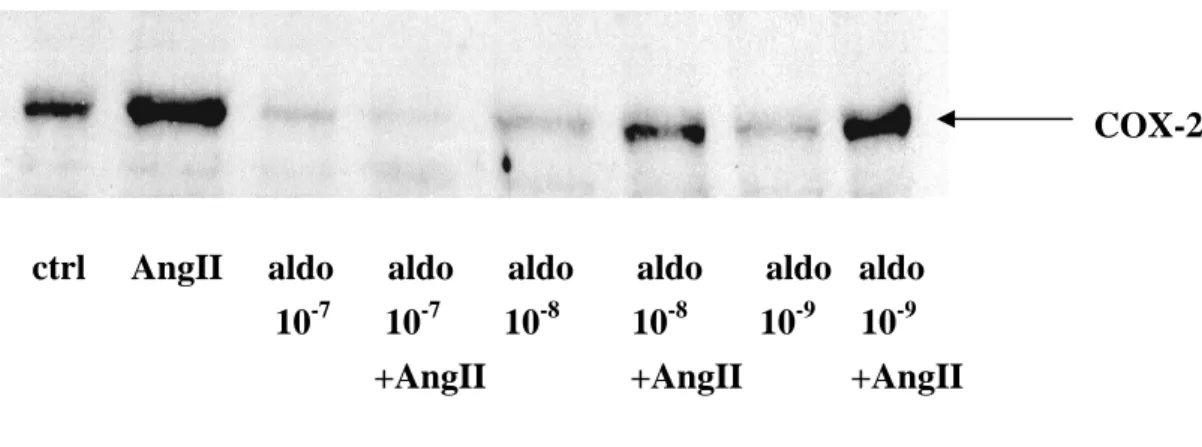
2.3) Stimulation des cellules avec de l’angiotensine II et expression de la COX-2 en fonction de la variation de la concentration d’aldostérone (Fig.4)
Les résultats exprimés dans la Fig. 4 montrent que l’expression de la COX-2 sous l’effet de l’aldostérone dépend de la concentration d’aldostérone utilisée. L’effet inhibiteur que l’aldostérone exerce sur l’expression de cet enzyme induit par l’AngII augmente de manière linéaire entre des concentration qui passent de 1 à 100 nM. L’aldostérone inhibe également l’expression de la COX-2 basale, mais de manière non linéaire.
0 1 10 100 0 100 200 aldo aldo + Ang II aldostérone (nM) CO X-2 (% d u c o n tr ô le )
Fig.4a : Variation de la concentration d’aldostérone et son effet sur l’expression de la COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’AngII (2x10-7 M). Les cellules ont été
incubées soit avec, soit sans AngII (2x10-7 M) pour une durée de 3h, en présence ou en absence de différentes concentrations d’aldostérone. Les cardiomyocytes ont été incubés pour une durée de 20-24h avec l’aldostérone, à des concentrations variant de 1 à 100 nM. La COX-2 a été révélée par électrophorèse et immunodétection (western blot).
COX-2
ctrl AngII aldo aldo aldo aldo aldo aldo
10-7 10-7 10-8 10-8 10-9 10-9 +AngII +AngII +AngII
Fig. 4b : Western blot représentant l’effet de l’aldostérone sur l’expression de la COX-2 en fonction de la variation de concentration d’aldostérone.
3) EFFETS DE L’OESTRADIOL SUR L’EXPRESSION DE LA COX-2 DANS LES CARDIOMYOCYTES VENTRICULAIRES:
3.1)Stimulation des cellules avec de l'ET-1 (Fig.5)
Contrairement à l'aldostérone, l'oestradiol (E2) ne produit aucun effet sur
l'expression basale de la COX-2. Toutefois, l’induction de la COX-2 par l’ET-1 est légèrement, mais pas significativement inhibée par l’oestradiol. Cet effet inhibiteur est le même pour 20h et 3h de traitement à l’oestradiol.
Fig. 5a : Effets de l'oestradiol sur l'expression de la COX-2 dans des cardiomyocytes ventriculaires stimulés avec de l'ET-1 (10-7 M).
Les cellules ont été incubées, soit avec, soit sans ET-1 (10-7M) pour une durée de 3h en présence ou en absence d’oestradiol (10-6M). Les cardiomyocytes ont été incubés pendant 3h ou 20-24h avec l’oestradiol (10-6M). Ces colonnes représentent les valeurs moyennes ± SEM de 5 expériences indépendantes. *=p<0.05 par rapport aux valeurs basales dans des cellules non stimulées.
Fig 5b : Western blot représentant l’effet de l’oestradiol sur l’expression de la COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’ET-1.
3.2) Stimulation des cellules avec de l'Angiotensine II (Fig.6)
Les résultats illustrés dans cette figure sont très semblables à ceux illustrés dans la figure 5: en absence de stimulation avec l'hormone, une incubation de 3h ou de 20h avec de l’E2 10-6M ne provoque pas de changement statistiquement significatif dans l'expression de la COX-2. En revanche, dans des cellules stimulées avec de l'AngII (2x10-7M), un prétraitement de 3h ou de 20h avec de l’E2 (10-6M) abolit l’expression de la COX-2 induite par l’AngII.
Fig.6a : Effets de l'oestradiol sur l'expression de la COX-2 dans des cardiomyocytes ventriculaires stimulés avec de l'AngII (2x10-7 M).
Les cellules ont été incubées, soit avec, soit sans AngII (2x10-7M) pour une durée de 3h en présence ou en absence d’oestradiol (10-6M). Les cardiomyocytes ont été incubés pendant 3h ou 20-24h avec l’oestradiol (10-6M). Les colonnes représentent les valeurs moyennes ± SEM de 6 expériences indépendantes. *=p<0.05 par rapport aux valeurs basales dans des cellules non stimulées. +=p<0.05 par rapport à la COX-2 dans des cellules stimulées avec de l’AngII.
Fig 6b : Western blot représentant l’effet de l’oestradiol sur l’expression de la COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’Ang II.
3.3) Stimulation des cellules avec de l'AngII et expression de la COX-2
en fonction de la variation de la concentration de l'oestradiol
Une expérience dans laquelle nous avons utilisé des concentrations de 1 à 1000nM indique que des concentrations de 10 à 100nM ont des effets similaires à la
concentration utilisée dans les expériences décrites dans ce travail (10-6 M); cette concentration n'est donc pas toxique pour les cellules musculaires cardiaques
ventriculaires. En revanche, une concentration de 1 nM ne semble provoquer aucun effet; elle est donc insuffisante.
Fig.7 : Variation de la concentration d’oestradiol et son effet sur l’expression de la COX-2 dans les cardiomyocytes ventriculaires stimulés avec de l’AngII (2x10-7M).
Les cellules ont été incubées soit avec, soit sans AngII (2x10-7M) pour une durée de 3h, en présence ou en absence de différentes concentrations d’oestradiol (1-1000 nM). Les cardiomyocytes ont été incubés pendant 20-24h avec les différentes concentrations d’oestradiol.
IV. DISCUSSION :
1) INDUCTION DE LA COX-2 PAR L’ANGIOTENSINE ET L’ENDOTHELINE
Pour la première fois, nous avons démontré dans les cardiomyocytes ventriculaires que l’AngII et l’ET-1 induisaient la COX-2 de manière dépendante du temps. Les premières expériences concernant l’induction de la COX-2 étaient réalisées avant tout avec des cytokines. Récemment, il a été découvert que des hormones telles que l’AngII et l’ET-1 pouvaient induire l’expression de la COX-2 dans certaines cellules. En effet, il a été démontré dans le muscle lisse vasculaire que l’AngII induisait la COX-2 [67].
Young et al (2000) ont montré dans ces mêmes cellules une cinétique de l’induction de la COX-2 semblable à celle vue dans des cellules stimulées avec une cytokine, comme le TNF-α, ou avec de l’AngII. [68].
L’AngII était déjà connue pour augmenter la production de PG dans les cellules mésangiales et les cellules endothéliales [69, 70].
Il a été démontré récemment par une équipe allemande qu’après un infarctus du myocarde, une activation des fibroblastes cardiaques par l’AngII entraînait une forte expression de la protéine COX-2, dépendante du temps [71].
En revanche, chez le rat, Cheng et al (1999) [72] ont observé qu’au niveau des cellules de la macula densa du rein, l’AngII inhibe l’expression de la COX-2 stimulée par l’ester de phorbol ; le mécanisme d’action n’a pas encore été élucidé En ce qui concerne l’ET-1, plusieurs groupes ont étudié l’effet de l’ET-1 sur l’expression de la COX-2 dans les cellules mésangiales [73, 74, 75]. Hughes et al (1995) [73] ont notamment démontré par une étude récente que l’ET-1 induisait de manière prolongée la production de PGE2 dans les cellules mésangiales et augmentait l’action de la COX-2 avec un net effet à une heure et un plateau d’une
durée d’environ 6 heures. Une étude japonaise [76] rapporte que l'endothéline active l'expression de la COX-2 dans les astrocytes en culture, ce qui suggère que l'ET est impliquée dans la régulation des fonctions des astrocytes.
2) LA MODULATION DE L'’EXPRESSION DE LA COX-2 PAR DES HORMONES STEROÏDIENNES : ALDOSTERONE ET OESTRADIOL :
Notre étude montre que dans les cardiomyocytes, la COX-2 n’est pas seulement induite, mais est également exprimée à l’état basal, dans des cellules qui durant 24 heures ont été déprivées de sérum. Cette observation est en accord avec deux autres études. Sorli et al (1998) [77] ont démontré que dans les îlots pancréatiques, la COX-2 est exprimée à l’état basal et après stimulation avec l’interleukine-1. De même, Cheng et al (1999) [72] ont observé que dans les cellules cTALH dans la région de la macula densa, la COX-2 est exprimée à l’état basal en absence de stimuli externes.
Dans cette étude, nous avons également démontré que des hormones stéroïdiennes telles que l’aldostérone et l’oestradiol entraînaient une modulation de l’expression de la COX-2.
Nous avons constaté que l’aldostérone exerçait un effet négatif sur l’expression de la COX-2 dans des cardiomyocytes ventriculaires. Un traitement de 3h avec de l’aldostérone n’a pas d’effet sur la COX-2 basale, tandis qu’il abolit la COX-2 induite par l’ET-1 et l’AngII. Des incubations à long terme (20-24h) avec de l’aldostérone réduisent à 35% l’expression de la COX-2 dans des cellules contrôles et stimulées.
A notre connaissance, aucune étude n’a été publiée sur l’effet de l’aldostérone sur la COX-2 dans le cœur, ni dans d’autres cellules.
Dans ce travail, nous avons également constaté que l’effet inhibiteur de l’aldostérone dépendait le la concentration de l’hormone. Une concentration de 10-8 M provoque un effet inhibiteur significatif, ce qui indique que cet effet est spécifique du récepteur aux minéralocorticoïdes. En effet, l’aldostérone active le récepteur aux glucocorticoïdes uniquement à des concentrations > 10-7 M [78]. Néanmoins, d’autres expériences utilisant des inhibiteurs spécifiques aux récepteurs corticoïdes seront nécessaires pour confirmer cette voie de signalisation.
Quant à l’oestradiol, il abolit uniquement la COX-2 induite par l’AngII, mais n’a pas d’effet significatif sur la COX-2 induite par l’ET-1. De même, l’oestradiol n’a pas d’effet sur l’expression basale de la COX-2.
L’effet de l’oestradiol sur l’expression de la COX-2 a déjà été étudié dans différents types cellulaires. Engstrom et al (2001) [79] ont démontré dans le myomètre du rat non gravide une augmentation de la production d’ARNm de la COX-2 sous l’influence de l’oestradiol. Par contre, dans l’utérus gravide de lapin, l’oestradiol semble inhiber la production de prostaglandines E2 après suppression de l’expression de la COX-2 stimulée par l’IL-1, ce phénomène permettrait donc d’inhiber le déclenchement du travail, par inhibition de la production de PGE2 [80]. L’effet de l’oestradiol semble donc être dépendant des conditions physiologiques d’un tissu spécifique.
Des études plus approfondies sont nécessaires pour déterminer à quel niveau l’aldostérone et l’oestradiol interviennent. Il s’agirait de voir si elles inhibent l’activité du promoteur de la COX-2 et/ou si elles accélèrent la dégradation de l’ARNm de la COX-2.
En conclusion, notre étude montre que dans le cardiomyocyte ventriculaire, la COX-2 est exprimée à l’état basal. L’AngII et l’ET-1 augmentent cette expression de manière dépendante du temps. Quant à l’aldostérone et à l’oestradiol, ils ont un effet modulateur négatif sur l’expression de la COX-2.
REFERENCES
1) Bloom and Fawcett: a textbook of histology. 12ème édition, (1994); chap 10, p 294-308
2) Thorburn J., McMahon M., Thorburn A. (1994) Raf-1 kinase activity is necessary and sufficient for gene expression changes but not sufficient for cellular morphology changes associated with cardiac myocytes hypertrophy. J. Biol. Chem., 269, 30580-30586.
3) Chien K.R., Knowlton K.U., Zhu H., Chien S. (1991) Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptative physiologic response. The FASEB Journal, 5, 3037-3046.
4) Pinson A., Tirosh R., Tremblover V., and Shoshami E., (1996) Oxygen deprivation and reoxygenation augment prostacyclin synthesis in cultured ventricular myocytes. Prostaglandins Leukot. Essent. Fatty Acids 54: 211-215. 5) Oudot F., Grynberg A., and Sergiel J.P. (1995) Eicosanoid synthesis in cardiomyocytes: influence of hypoxia, reoxygenation. And polyunsaturated fatty acids. Am J. Physiol 268: H308-315
6) Church D.J., Braconi S., Van der Bent V., Vallotton M. B., and U. Lang. (1994) Protein kinase C-dependent prostaglandin production mediates angiotensin II-induced atrial-natriuretic peptide release. Biochem. J. 298: 451-456
7) Van der Bent V., Church D.J., Vallotton M.B., Meda P., Kem D.C., Capponi A.M., and U. Lang (1994) [Ca++]I and protein kinas C in vasopressin-induced prostacyclin and ANP release in rat cardiomyocytes. Am. J. Physiol. 266: H57-H605
8) Dennis E.a. (1994) Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 269: 13057-13060.
9) Kramer R.M. and Sharp J.D. (1997) Structure, function and regulation ok Ca++ -sensitive cytosolic phosphlipase A2 (cPLA2 ). FEBS Lett. 410: 49-53.
10) Smith W.L. (1991) The aspirin and heme binding sites of PGG/H synthase. Adv. Prostaglandin. Thromboxane. Leukot. Res. 21A: 77-80
11) Picot D. (1994) Prostaglandin h synthase: implications for membrane structure: FEBS Lett., 346: 21-25
12) Luong C. (1996) Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2: Nat. Struct. Biol., 3: 927-933
13) Morita I. (1995) Different and intracellular locations for prostaglandinsendoperoxyde H synthase-1 and –2. J. Biol. Chem. 270: 10902-10908
14) Spencer A.G. (1998) Subcellular localisation of prostaglandin endoperoxyde H synthase-1 and –2 by immunoelectron microscopy: J. Biol. Chem 273: 9886-9893
15) Hia T., (1992), Human cyclooxygenase-2 cDNA: Proc Natl Acad Sci. U.S.A. 89: 7384-7388
16) Mitchell J.A. (1995) cycloxygenase-2: regulation and relevance in inflammation: biochem Pharmacol. 50: 1535-1542
17) Masferrer JL, (1994) Selective inhibition of inducible cyclooxygenase-2 in vivo is anti-inflammatory and nonulcerogenic. Proc. Natl. Acad Sci U.S.A. 91: 3228-3232
18) Seibert K. (1994) Role of inducible cyclooxygenase (COX-2) in inflammation 4: 17-23
19) Smith W.L., Dewitt, D.L.(1996) Prostaglandin Endoperoxide H Synthase-1 and –2. Advances in immunology, vol 62: 167-215. Review
20) Adderley SR, Fitzgerald DJ., (1999) Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases ½-mediated induction of cyclooxygenase-2. J.Biol. Chem 274(8):5038-46
21) Dowd NP, Scully M, Adderley SR, Cunningham AJ, Fitzgerald DJ. (2001) Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J. Clin. Invest. 108(4): 585-590
22) Wong SCY, Fukuchi M., Melnyk P, Rodger I. (1998) Induction of cyclooxygenase-2 and activation of nuclear factor-κB in myocardium of patients with congestive heart failure. Circulation 98: 100-103
23) Shinmura K., Tang XL., Wang Y., Xuan YT., Liu SQ., Takano H., Bhatnagar A., Bolli R. (2000) Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of preconditioning in conscious rabbits. Proc. Natl. Acad. SCI., USA, 97(18): 10197-10202
24) Yang X. Ma N. Szaboles MJ, Zhong J, Athan E Sciacca RR Michler R Anderson GD Wiese JF, Leahy K, Gregory S, Cannon PJ. (2000) Upregulation of COX-2 during allograft rejection. Circulation 1001(4): 430
25) Wong SCY. Fukuchi M. Melnik P. Rodger I. Giaid A. (1998). Induction of cyclooxygenase-2 and activation of nuclear factor κB in myocardium of patients with congestive heart failure. Circulation 98: 100-103
26) Guyton and Hall: Textbook of medical physiology, 9ème edition, (1996), unites IV. V, XIV.
27) Teerlink J.R. (1994) The evolving role of angiotensin-converting enzyme inhibition in heart failure: expanding the protective envelope. J. Cardiovasc. Pharmacol. 24:S32-7
28) Fouad-Tarazi F.M. (1994) Hemodynamic effects of inhibitors of the rennin-angiotensin system. J. Hypertens.12: S25-9
29) Baker K.M. , Booz G. W., and Dostal D.E. (1992) Cardiac actions of angiotesin II: role of an intracardiac renin-angiotensin system. Annu. Rev. Physiol. 54 : 227-241
30) Sechi L.A., Griffin C.A., Grady E.F., Kalinyak J.E., and Schambelan M., (1992) Characterization of angiotensin II receptor subtypes in rat heart. Circ. Res. 71: 1482-1489.
31) Timmerans P.B. Wong P.C., Chiu A.T. and Heberlin W.F. (1991) Nonpeptide angiotensin II receptors antagonists. Trends Pharmacol. Sci. 12: 55-62
32) Meggs L.G., Coupet J., Huang H., Li W.C.P., Capasso J.M., Homcy C.J., and Anversa P., (1993) Regulation of angiotensin II receptors on ventricular myocytes after myocardial infarction in rats. Circ. Res. 72: 1149-1162
33) Suzuki J., Matsubara H., Urakami M., and Inada M., (1993) Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circ. Res. 73: 439-447
34) Timmerans P.B., Wong P., Chiu A.T., Herblin W.F., Benfield P., Carini D.J., Lee R.J., Wexler R.R., Saye J.M., and Smith RD. (1993) Angiotensin II receptors and angiotensin II receptors antagonists. Pharmacol. Rev. 45: 205-251.
35) Pönicke K., Heinroth-Hoffmann I., Brodde OE. Becker K. (1997) Trophic effect of angiotensin II in neonatal rat cardiomyocytes: role of endothelin-1 and non-myocyte cells.
British journal of pharmacology 121: 118-124
36) Inoue A., Yanagisawa M., Kimura S., Kasuya Y., and Myauchi T. (1989) The human endothelin family – 3 structurally and pharmaologycally distinct isopeptides predicted by 3 separate genes. Proc. Natl acad. Sci. USA 86: 2863-2867
37) Krämer B.K. Ackermann M., Kohler S.M., and Riegger G.A.J. (1994) Role of endothelin in hypertension. Clin. Investig. 72: 88-93
38) Guyton and Hall, textbook of medical physiology, 9ème édition, 1996, p.
39) Hirata Y., Takagi Y., Fukuda Y., and Marumo F. (1989) endothelin is a potent mitogen for rat vascular smooth muscle cells. Atherosclerosis 78: 225-228
40) Takuwa N., Takuwa Y., Yanagisawa K., Yamashita K., and Masaki T. (1989 A novel vasoactive peptide endothelin stimulates mitogenesis through inositol lipid turnover in Swiss 3T3 fibroblasts. J. Biol. Chem. 264: 7856-7861
41) Krämer B.K., Nishida M., Kelly R.A., and Smith T.W. (1992) Endothelins. Myocardial action of a new class of cytokines. Circulation 85: 350-356.
42) Ito H., Hirata Y., Hiroe M., Tsujino M., Adachi S., Takamoto T., Nitta M., Taniguchi K., and Marumo F., (1991) Endothelin-1 induces hypertrophy with enhanced expression of muscle-specific genes in cultured neonatal rat cardiomyocytes. Circ. Res. 69: 209-215
43) Shubeita H.E., McDonough P.M., Harris A.N., Knowlton K.U., Glembotski C.C., Brown J.H., and Chien K.R. (1990) Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular
myocytes. A paracrine mechanism for myocardial cell hypertrophy. J. Biol. Chem. 265: 20555-20562
44) Suzuki T., Hoshi H., and Mitsui Y. (1990) Endothelin stimulates hypertrophy and contractility of neonatal rat cardiac myocytes in a serum-free medium. FEBS Lett. 268: 149-151
45) Hahn A.W., Resink T.J., Scott B.T., Powell J., Dohi Y., and Buhler F.R. (1990) Stimulation of endothelin mRNA and secretion in rat vascular smooth muscle cells: a novel autocrine function. Cell Regul. 1: 649-659
46)Ohta K., Hirata Y., Imai T., Kanno K., Emori T., Shichiri M., and Marumo F. (1990) Cytokine-induced release of endothelin-1 from porcine renal epithelial cell line. Biochem. Biophys. Res. Commun. 169: 578-584
47) Sakamoto H., sasaki S., Hirata Y., Imai T., Ando K., Ida T., Sakurai M., Yanagisawa T., Masaki T., and Marumo F., (1990) Production of endothelin-1 by rat cultured mesangial cells. Biochem Biophys. Res., Commun. 169: 462-468
48) Shichiri M., Hirata Y., Nakajima T., Ando K., Imai., Yanagisawa M., Masaki T., and Marumo F. (1991) endothelin-1 is an autocrine-paracrine growth factor for human cancer cell lines. J. Clin. Invest. 87: 1867-1871
49) Ito h., Adachi S., Tamamori M., Fujisaki H., Tanaka M., Lin M., Akimoto H., Marumo F., and Hiroe M. (1996) Mild hypoxia induces hypertrophy of cultured neonatal rat cardiomyocytes: a possible endogenous endothelin-1-mediated mechanism. J. Mol. Cell. Cardiol.28: 1271-1277
50) Iwanaga Y., Kihara Y., Hasegawa K., Inagaki K., Yoneda T., Kaburagi S., Araki M., and Sasayama S., (1998) Cardiac endothelin-1 plays a critical role in the functional deterioration of left ventricles during the transition from the compensatory hypertrophy to congestive heart failure in salt-sensitive hypertensive rats. Circulation 98: 2065-2073.
51) Kobayashi T., Miyauchi T., Sakai S., Kobayashi M., Yamaguchi I., Goto K., and Sugishita Y., (1999) Expression of endothelin-1, ETA and ETB receptors, and ECE and distribution of endothelin-1 in failling rat heart. Am. J. Physiol. 276: H1197-11206
52) Guyton and Hall: Textbook of medical physiology, 9ème edition, (1996), unité XIV.
52b) Ikeda U, Hyman R, Smith TW, Medford RM, Aldosterone-mediated regulation of Na+, K+-ATPase gene expression in adult and neonatal rat cardiomyocytes. J Biol Chem. 1991, 266: 12058-12066
53) Korichneva I., Puceat M., Millanvoye-Van Brussel E., Geraud G., Vassort G.(1995) Aldosterone modulates both the Na+/H+ antiport and Cl-/HCO3- exchanger in cultured neonatal rat cardiac cells. J. Mol. Cell. Cardiol. 27: 2521-2528
54) Benitah J.P. Vassort G. (1999) Aldosterone upregulates Ca++ current in adult rat cardiomyocytes. Circ. Res. 1999; 85: 1139-1145
55) Lombes M., Alfaidy N., Eugene E., Lessana A., Farman N., Bonvalet J.P. (1995). Prerequisite for cardiac aldosterone action. Mineralocorticoid receptor and 11 β-hydroxysteroid dehydrogenase in the human heart. Circulation. 92: 175-182
56) Brilla C.G., Matsubara L.S., Weber K.T (1993) Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary an secondary hyperaldosteronism. J. Mol. Cell Cardiol. 25: 563-574
57) Robert V., Thiem N.V., Cheav S.L., Mouas C., Swynghdauw B., Decayre C., (1994) Increased cardiac types I and II collagen mRNAs in aldosterone-salt hypertension. Hypertension 24: 30-36
58) Young M., Fullerton M., Dilley R., Funder J.W., (1994) Mineralocorticoids, hypertension and cardiac fibrosis. J. Clin. Invest. 93: 2578-2583
59) Robert V., Heymes C., Silvestre J.S., Sabri A., Swynghdauw B., Decayre C. (1999) Angiotensin AT1 receptor subtype as a cardiac target of aldosterone. Hypertension 33:981-986
60) Williams Textbook of endocrinology, 9ème edition, chapitre 15, p. 761-768 61) Grohe C. Kahlert S. Lobbert K. Stimpel M. Karas RH. Vetter H. Neyses L. (1997) Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett 416(1): 107-112
62) Chang WC, Nakao J, Orimo H, Murota SI. (1980) Stimulation of prostaglandin cyclooxygenase and prostacyclin synthase activities by estradiol in rat aortic smooth muscle cells. Biochim Biophys Acta 620(3): 472-482
63) Ghanam K, Lavagna C, Burgaud JL, Javellaud J, Ea-Kim L, Oudart N. (2000) Involvement of cyclooxygenase-2 in the protective effect of 17β-estradiol on hypercholesterolemic rabbit aorta. Biochem. Biophys. Res. Commun. 275(2): 696-703
64) Shoda T. Hatanaka K. Saito M. Majima M. Ogino M. Harada Y. Nishijima M. Katori M. Yamamoto S. (1995). Induction of cyclooxygenase type 2 (COX-2) in rat endometrium at the peak of serum estradiol during the estrus cycle. Jpn. J. Pharmacol. 69(3) : 289-291
65) Rebsamen MC, Church DJ, Morabito D, Vallotton MB, Lang U. (1997), Role of cAMP and calcium influx in endothelin-1-induced ANP release in rat cardiomyocytes. Am j Physiol. 273 : E922-931
66) Church DJ, Braconi-Quintaje S, van der Bent V, Vallotton MB, Lang U. (1994) Protein kinase c-dependent prostaglandin production mediates angiotensin II-induced atrial natriuretic peptide release. Biochem J. 298 :451-456
67) Ohnaka K, Numaguchi K, Yamakawa T, Inagami T. (2000) Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension 35: 68-75
68) Young W. Mahboubi K. Haider A. Li I Ferreri NR. (2000) Cyclooxygenase-2 is required for tumor necrosis factor-alpha- and angiotensin II-mediated proliferation of vascular smooth muscle cells. Circ. Res. 86(8): 906-914
69) Schlondorff D, DeCandido S. Satriano JA. (1987) Angiotensin II stimulates phospholipases C and A2 in cultured rat mesangial cells. Am. J. Physiol. 253:C113-C120
70) Gimbrone MAJr. Alexander RW. (1975) Angiotensin II stimulation of prostaglandin production in cultured human vascular endothelium. Science 189: 219-220
71) Scheuren N. Jacobs N. Ertl G. Schorb W. (2002) Cyclooxygenase-2 in myocardium stimulation by angiotensin-II in cultured cardiac fibroblasts and role at acute myocardial infarction. Journal of molecular and cellular cardiology. 34:29-37