Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation.
8
0
0
Texte intégral
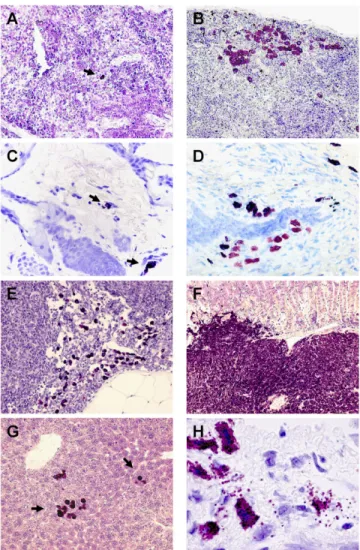
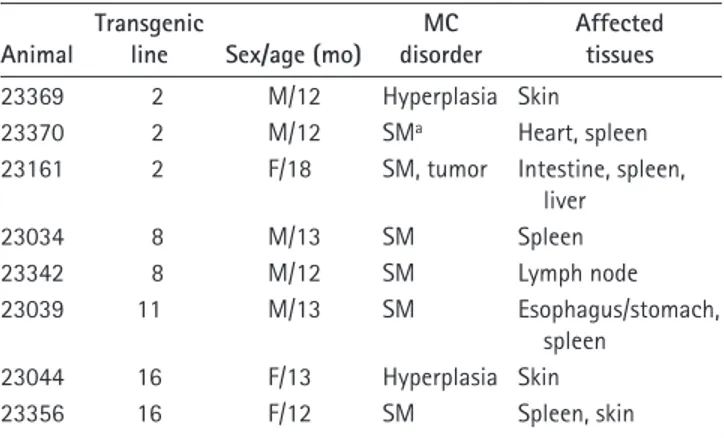
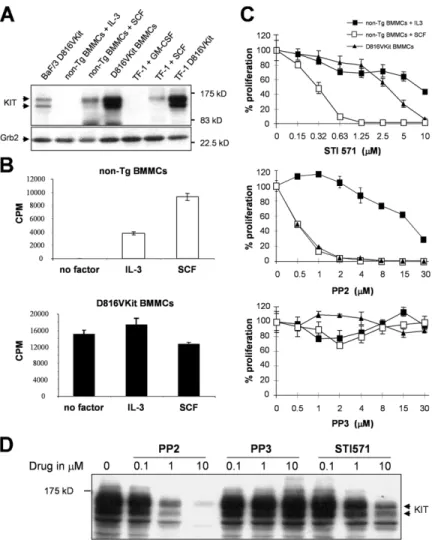
Figure
Documents relatifs