Sustainable hydrogen production via glycerol steam
reforming with and without in-situ CO2 removal:
materials development and application
Thèse
Marziehossadat Shokrollahi Yancheshmeh
Doctorat en génie chimique
Philosophiæ doctor (Ph. D.)
Production durable d'hydrogène par reformage à la
vapeur du glycérol couplé ou pas à la sorption in-situ du
CO
2: développement des matériaux et application
Thèse
Marziehossadat Shokrollahi Yancheshmeh
Sous la direction de :
Résumé
Au cours des dernières décennies, l'hydrogène a beaucoup attiré l'attention en tant que vecteur d'énergie verte. Actuellement, plus de 95% d'hydrogène est produit à partir de combustibles fossiles, ce qui a été remis en question par l'épuisement des ressources et l'augmentation des émissions de gaz à effet de serre. Par conséquent, les ressources renouvelables neutres en carbone telles que la biomasse et les produits chimiques dérivés de la biomasse suscitent un intérêt croissant comme alternative pour la production d'hydrogène. En tant que sous-produit principal du processus de fabrication du biodiesel, le glycérol est devenu une source prometteuse de production d’hydrogène. Bien que le reformage à la vapeur (« steam reforming », SR) soit reconnu comme une approche prometteuse pour convertir le glycérol en hydrogène, le procédé est confronté à un certain nombre de défis, notamment la présence de réactions limitées par l’équilibre chimique et la nécessité d'un système couteux de purification en aval. Pour remédier ces problèmes, une solution prometteuse est l’application du procédé de reformage à la vapeur couplé à la sorption spécifique in-situ (« sorption enhanced steam reforming », SESR), dans lequel les réactions de reformage, la réaction du gaz à l’eau (« water gas shift », WGS) et la capture du CO2 se
produisent simultanément en utilisant un catalyseur de reformage et un sorbant solide pour le CO2. Dans ce procédé, l'élimination du CO2 se produit simultanément à la réaction de
reformage, décalant la réaction du WGS vers la production d'hydrogène et produisant un flux de gaz enrichi en hydrogène en une seule étape. Les facteurs clés du succès de cette technologie sont principalement (i) les catalyseurs de reformage et les sorbants de CO2
pouvant fonctionner efficacement dans les conditions difficiles du procédé SESR et (ii) le moyen d’associer le catalyseur au matériau sorbant.
Cette thèse porte sur le développement de catalyseurs et de matériaux bifonctionnels catalyseur-sorbant efficaces pour la production durable d'hydrogène par le SR et le SESR du glycérol (SRG et SESRG). Plus spécifiquement, ce travail fait l’objet de quatre directions principales: (i) l’étude de l’effet de l’addition de vapeur pendant la carbonatation ou la calcination sur les performances du sorbant Ca9Al6O18-CaO lors de la capture du CO2, (ii) le
développement des matériaux bifonctionnels Ca9Al6O18−CaO/xNiO (x = 15, 20 et 25% en
iv
CeO2 sur la stabilité des matériaux en fonctionnement cyclique SESRG/régénération, (iii) le
développement d’une nouvelle méthode de synthèse du spinelle NiAl2O4 plus facilement
réductible et l’étude de l'effet de l'addition de CeO2 sur ses performances catalytiques, et (iv)
le développement d’une nouvelle méthode de synthèse de deux matériaux bifonctionnels catalyseur-sorbant à base de Ni-CaO pour obtenir une distribution très uniforme des sites actifs catalytiques.
(i) Les performances du sorbant Ca9Al6O18-CaO pour la capture du CO2 ont été étudiées
en présence de 2.3 et 9.5% en volume de vapeur. Les résultats obtenus ont révélé que la réactivité du sorbant était remarquablement améliorée pour les deux concentrations de vapeur injectée lors de l'étape de carbonatation. Dans le cas de l'addition de vapeur pendant la calcination, la performance de la capture a été influencée négativement ou positivement en fonction de la concentration de vapeur : pour 2.3%, la réactivité du sorbant a été diminuée, tandis que la présence de 9.5% a entraîné une augmentation de la capacité de capture pendant les 9 premiers cycles.
(ii) Deux séries de matériaux bifonctionnels catalyseur-sorbant ont été développées pour la production d’hydrogène de haute pureté par SESRG. L'utilisation des matériaux Ca9Al6O18-CaO/xNiO (x = 15, 20 et 25% en poids) pendant cinq cycles SESRG/régénération
a révélé que leur réactivité diminuait rapidement, principalement à cause du frittage du CaO et du dépôt de coke. De ce fait, la période de pre-breakthrough et le rendement en hydrogène ont diminué de façon notable pendant l’opération cyclique. Il est intéressant de noter que l’ajout de CeO2 au matériau le plus efficace (Ca9Al6O18−CaO/20NiO) a permis d’améliorer
considérablement sa stabilité. Le matériau bifonctionnel activé avec 10% (en poids) de CeO2
a démontré les meilleures performances: pureté et rendement en H2 de 98% et 91%,
respectivement, pendant 20 cycles SESRG/régénération.
(iii) Une nouvelle méthode impliquant la calcination en une ou deux étapes d'un alcoolate de métal mixte Ni-Al (« Ni-Al mixed-metal alkoxide », (Ni-Al)-Glycerate) a été développée pour la synthèse de spinelle de NiAl2O4. À des fins de comparaison, le spinelle de NiAl2O4
a également été synthétisé par la méthode classique de co-précipitation suivie de la technique de calcination en deux étapes. Les résultats de la caractérisation des matériaux ont révélé que la synthèse de spinelle de NiAl2O4 par la calcination de (Ni-Al)-Glycérate en deux étapes a
conduit à la formation d'un catalyseur plus facilement réductible et d'une structure poreuse plus développée. Cet échantillon représentait le rendement en H2 le plus élevé (76.38%) et la
conversion du glycérol en produits gazeux (95.42%) par rapport aux autres échantillons. Afin de réduire ou éviter la formation de coke, CeO2 (10% en poids) a été incorporé dans
l’échantillon préparé par la calcination de (Ni-Al)-Glycérate en deux étapes. L'analyse thermogravimétrique du catalyseur promu par CeO2 après la réaction de reformage a révélé
que la formation de coke était presque complètement supprimée.
(iv) La méthode développée pour la synthèse de spinelle de NiAl2O4 dans les travaux
précédents a été combinée au traitement du sorbant à base de CaO avec une solution d’éthanol/eau afin de synthétiser deux nouveaux matériaux bifonctionnels catalyseur-sorbant à base de Ni-CaO pour la production d'hydrogène via SESRG. Les expériences effectuées en opération cycliques SESRG/régénération ont montré une activité et une stabilité supérieures pour le matériau bifonctionnel Ca3Al2O6-CaO/NiO-CeO2 (pureté de l’H2 d’environ 96%
pendant 10 cycles), par rapport à NiAl2O4-CaO/NiAl2O4-CeO2 (pureté de l’H2 d’environ 90%
pendant les 6 premiers cycles, diminuant à 86% au cours des 4 derniers cycles).
En conclusion, les résultats présentés dans cette thèse montrent que le SESRG peut être une approche très prometteuse pour la production d’hydrogène de haute pureté en une seule étape, à condition que les matériaux bifonctionnels catalyseur-sorbant utilisés possèdent une distribution uniforme des sites actifs catalytiques et à sorption à l’échelle nanométrique et une résistance élevée au frittage de CaO et formation de coke. Pour préparer des matériaux bifonctionnels catalyseur-sorbant présentant ces caractéristiques, deux approches principales ont été utilisées dans ce travail: (i) le développement de nouvelles méthodes de synthèse permettant une distribution homogène des éléments ciblés (Ca, Ni, Al et Ce dans cette étude) et (ii) l'utilisation de CeO2 comme promoteur prometteur pour réduire ou supprimer la
vi
Abstract
Over the past few decades, hydrogen has attracted a great deal of attention as a green energy carrier. Currently, more than 95 % of hydrogen is produced from fossil fuels, which has been questioned by the depletion of resources and increase of greenhouse gas emissions. Therefore, renewable, carbon-neutral resources such as biomass and biomass-derived chemicals has been receiving a growing interest as an option to produce hydrogen. As a main byproduct in the biodiesel manufacturing process, glycerol has emerged as a promising source for hydrogen production. Although steam reforming (SR) is being recognized as a promising approach for converting glycerol to hydrogen, this process faces a number of challenges including the presence of equilibrium-limited reactions and the need of an expensive downstream purification system. To alleviate these problems, a promising alternative is sorption enhanced steam reforming (SESR) process, in which steam reforming, water gas shift (WGS), and CO2 capture reactions occur simultaneously using a reforming
catalyst and a CO2 solid sorbent. In this process, CO2 removal occurs simultaneously with
the reforming reaction, shifting the WGS reaction towards hydrogen production and producing a hydrogen-enriched gas stream in a single step. The key factors in the successful application of this technology are mainly: (i) reforming catalysts and CO2 sorbents that can
work efficiently under the harsh conditions of SESR process and (ii) mixing pattern of catalyst and sorbent.
This thesis focuses on the development of efficient catalyst and catalyst-sorbent bifunctional materials for sustainable hydrogen production by SR and SESR of glycerol (SRG and SESRG). More specifically, four main objectives of our work are: (i) investigating the influence of steam addition during either carbonation or calcination on the CO2 capture
performance of Ca9Al6O18-CaO sorbent, (ii) developing Ca9Al6O18−CaO/xNiO (x = 15, 20,
and 25 wt.%) and Ca9Al6O18−CaO/20NiO−yCeO2 (y = 5, 10, and 15 wt %) catalyst-sorbent
bifunctional materials and studying the influence of CeO2 on the material stability in cyclic
SESRG/regeneration operation, (iii) proposing a new method for the synthesis of a more readily reducible NiAl2O4 spinel and studying the influence of CeO2 addition on its catalytic
performance, and (iv) novel synthesis of two Ni-CaO-based catalyst-sorbent bifunctional materials with highlyuniform distribution of catalytic active sites.
(i) CO2 capture performance of Ca9Al6O18-CaO sorbent was investigated in the presence
of two concentrations of steam, 2.3 and 9.5 vol.%. The obtained results revealed that the sorbent reactivity was remarkably enhanced for both concentrations of steam injected during carbonation step. In the case of steam addition during calcination, the CO2 capture
performance was influenced negatively or positively depending on the concentration of steam. For 2.3 vol.% steam, the sorbent reactivity was worsened, while the presence of 9.5 vol.% steam led to an increase in the CO2 capture capacity during 9 initial cycles.
(ii) Two series of catalyst-sorbent bifunctional materials were developed for the sustainable production of high-purity hydrogen by SESRG. Using Ca9Al6O18−CaO/xNiO (x
= 15, 20, and 25 wt.%) materials during five SESRG/regeneration cycles revealed that their reactivity was rapidly deteriorated mainly due to CaO sintering and coke deposition. As a result, the pre-breakthrough time and hydrogen yield decreased notably over five cycles. Interestingly, the addition of CeO2 to the most efficient catalyst (Ca9Al6O18−CaO/20NiO) led
to a significant enhancement in material stability during cyclic operation. The bifunctional material promoted with 10 wt.% of CeO2 demonstrated the best performance, with a stable
H2 purity of ∼98% and H2 yield of ∼91% over 20 SESRG/regeneration cycles.
(iii) A novel method, involving one- or two-step calcination of Ni-Al mixed-metal alkoxide ((Ni-Al)-Glycerate), was developed for the synthesis of NiAl2O4 spinel. For
comparison purposes, the NiAl2O4 spinel was also synthesized through the conventional
co-precipitation method followed by two-step calcination technique. The characterization results revealed that the synthesis of NiAl2O4 spinel through two-step calcination of
(Ni-Al)-Glycerate resulted in the formation of a more easily reducible catalyst and a more developed porous structure. This sample showed the highest H2 yield (76.38 %) and glycerol conversion
into gaseous products (95.42 %) when compared to other two samples. In order to avoid or reduce coke formation, 10 wt.% of CeO2 was incorporated into the sample prepared by
two-step calcination of (Ni-Al)-Glycerate. The thermogravimetric analysis of the CeO2-promoted
catalyst after SRG reaction revealed that the coke formation was almost completely suppressed.
(iv) The method developed for the synthesis of NiAl2O4 spinel in the previous work was
Ni-viii
CaO-based catalyst-sorbent bifunctional materials for hydrogen production via SESRG. Cyclic SESRG/regeneration experiments showed that the Ca3Al2O6-CaO/NiO-CeO2
bifunctional material possessed higher activity and stability when compared to NiAl2O4
-CaO/ NiAl2O4-CeO2. The former one exhibited a high constant H2 purity of around 96 %
over 10 cycles, while the latter showed a H2 purity of approximately 90 % over the first 6
cycles, followed by the further decrease to 86 % over the last 4 cycles.
In conclusion, the results presented in this thesis show that SESRG can be a very promising approach for high-purity hydrogen production in a single step, providing that the employed catalyst-sorbent bifunctional materials possess uniform distribution of catalytic and sorption active sites on nanoscale and high resistance against CaO sintering and coke formation. To prepare catalyst-sorbent bifunctional materials with these characteristics, two main approaches were employed in this work: (i) developing new synthesis methods that provide a homogeneous distribution of targeted elements (Ca, Ni, Al, and Ce in this study) and (ii) using CeO2 as a promising promoter to reduce or suppress coke formation and
Table of Contents
Résumé ... iii
Abstract ... vi
Table of Contents ... ix
List of figures ... xiii
List of Tables ... xviii
Nomenclature... xx
Acknowledgement ... xxiii
Preface ... xxiv
Introduction ... 1
Chapter 1. Literature review... 9
1.1. High temperature CO2 sorbents ... 9
1.1.1. CaO-based sorbents ... 9
1.1.1.1. Application of various sources of calcium to produce CaO-based sorbents ... 12
1.1.1.2. Incorporation of inert materials into CaO structure ... 16
1.1.1.3. Reactivation and treatment of CaO-based sorbents... 28
1.1.2. Ceramic CO2 sorbents ... 40
1.1.2.1. Lithium zirconate (Li2ZrO3) ... 43
1.1.2.2. Lithium orthosilicate (Li4SiO4) ... 46
1.1.2.3. Sodium zirconate (Na2ZrO3) ... 49
1.1.2.4. Other ceramic materials ... 51
1.1.2.5. Kinetic models ... 53
1.2. Ni-based catalysts for steam reforming of glycerol ... 54
1.2.1. The nature of support ... 54
1.2.2. Metal-support interaction ... 59
1.2.3. Addition of different promoters ... 60
1.3. Sorption enhanced steam reforming of glycerol ... 65
1.4. Literature review conclusion ... 68
1.5. Objective of the work ... 70
1.5.1. General objective ... 71
x
Chapter 2. Methodology ... 73
2.1. Materials ... 73
2.2. Material preparation ... 74
2.2.1. CaO-based sorbents ... 74
2.2.1.1. Synthesis of pure CaO sorbent by thermal decomposition ... 74
2.2.1.2. Synthesis of pure CaO and Al-stabilized CaO-based sorbents by acid treatment ... 74
2.2.1.3. Synthesis of pure CaO sorbent by ethanol/water treatment ... 75
2.2.2. Ni-based catalysts ... 75
2.2.2.1. Synthesis of NiAl2O4 spinel by one- or two-step calcination of Ni-Al mixed-metal alkoxide ... 76
2.2.2.2. Synthesis of NiAl2O4 spinel by two-step calciantion of Ni-Al mixed-metal hydroxide ... 76
2.2.2.3. Synthesis of CeO2-promoted NiAl_G2 catalyst by the conventional impregnation method ... 77
2.2.3. Ni-CaO-based catalyst-sorbent bifunctional materials ... 77
2.2.3.1. Synthesis of Ca9Al6O18-CaO/xNiO (x = 15, 20, and 25 wt.%) and Ca9Al6O18-CaO/20NiO-yCeO2 (y = 5, 10, and 15 wt.%) bifunctional materials by Acidification method ... 77
2.2.3.2. Synthesis of Ca3Al2O6-CaO/NiO-CeO2 and NiAl2O4-CaO/NiAl2O4 -CeO2 bifunctional materials by a novel method ... 78
2.3. Materials characterization ... 78
2.3.1. X-ray diffractometry ... 78
2.3.2. X-ray photoelectron spectroscopy ... 79
2.3.3. Scanning electron microscopy ... 80
2.3.4. Transmission Electron Microscopy ... 80
2.3.5. Nitrogen physisorption ... 81
2.3.6. Thermogravimetric analysis ... 81
2.4. CO2 capture test ... 82
2.5. SRG and SESRG tests ... 83
2.6. Calculations ... 84
Chapter 3. Influence of steam addition during carbonation or calcination on the CO2 capture performance of Ca9Al6O18-CaO sorbent ... 87
Résumé ... 87
3.1. Introduction ... 89
3.2. Experimental section ... 92
3.2.1. Material preparation ... 92
3.2.2. Cyclic CO2 capture ... 93
3.2.3. Material characterization ... 94
3.3. Results and discussion ... 94
3.3.1. The effect of steam addition during carbonation ... 94
3.3.2. The effect of steam addition during calcination ... 101
3.4. Conclusion ... 105
Chapter 4. Sustainable production of high-purity hydrogen by sorption enhanced steam reforming of glycerol over CeO2-promoted Ca9Al6O18-CaO/NiO bifunctional material... 107
Résumé ... 107
Abstract ... 109
4.1. Introduction ... 110
4.2. Experimental Section ... 113
4.2.1. Materials ... 113
4.2.2. Preparation of Bifunctional Catalyst-Sorbent Materials ... 114
4.2.3. Material Characterization ... 114
4.2.4. CO2 Capture Test ... 115
4.2.5. SESRG Test ... 116
4.3. Results and Discussion ... 117
4.3.1. Characterization of Fresh Samples ... 117
4.3.2. CO2 Capture Characteristics ... 122
4.3.3. Sorption Enhanced Steam Reforming of Glycerol ... 127
4.3.4. Characterization of Spent Materials ... 134
4.4. Conclusion ... 139
Chapter 5. A novel synthesis of NiAl2O4 spinel from a Ni-Al mixed-metal alkoxide as a highly efficient catalyst for hydrogen production by glycerol steam reforming ... 142
Résumé ... 142
Abstract ... 143
5.1. Introduction ... 144
5.2. Experimental procedure ... 146
xii
5.2.2. Catalyst characterization... 147
5.2.3. Catalytic performance evaluation ... 149
5.3. Results and discussion ... 151
5.3.1. Characterization of fresh catalysts ... 151
Figure 5.2. Pore size distribution of the calcined catalysts obtained from N2 physisorption measurements. ... 154
5.3.2. Catalytic activity in steam reforming of glycerol ... 161
5.3.2.1. Influence of synthesis method on the catalytic performance of NiAl2O4 spinel in SRG ... 162
5.3.2.2. Influence of CeO2 addition on the catalytic performance of NiAl_G2 catalyst in SRG ... 170
5.3.2.3. Catalytic stability of Ce/NiAl_G2 ... 174
5.4. Conclusion ... 177
Chapter 6. A novel synthesis of Ni-CaO-based catalyst-sorbent bifunctional materials for high-purity hydrogen production via sorption enhanced steam reforming of glycerol ... 180
Résumé ... 180 Abstract ... 181 6.1. Introduction ... 182 6.2. Experimental procedure ... 185 6.2.1. Material preparation ... 185 6.2.2. Material characterization ... 186 6.2.3. CO2 capture test ... 187 6.2.4. SESRG test ... 188
6.3. Results and Discussion ... 189
6.3.1. Characterization of Fresh Materials ... 189
6.3.2. CO2 capture performance of developed materials ... 193
6.3.3. Sorption enhanced steam reforming of glycerol... 198
6.3.4. Characterization of used materials ... 208
6.4. Conclusion ... 210
General conclusions and future outlook ... 212
1. General conclusions ... 212
2. Future outlooks ... 214
List of figures
Figure I.1. Global energy-related CO2 emissions [1]. ... 1
Figure I.2. Average annual growth in energy demand by fuel [1]. ... 2 Figure I.3. Transestrification of triglyceride with methanol for biodiesel production [15]. .. 3 Figure I.4. European Union biodiesel production for the period 2004 to 2016 [8]. ... 3 Figure I.5. United States biodiesel growth pattern for the period 2006 to 2016 [9]... 4 Figure I.6. Biodiesel global production of top 16 countries in 2015 [10]. ... 4 Figure I.7. Schematic comparison of CO2 diffusion in physical mixing and catalyst-sorbent
bifunctional patterns [63]... 7 Figure 1.1. Sintering phenomenon of CaO; light grey: CaO phase and dark grey: CaCO3
phase [79]. ... 10 Figure 1.2. Typical weight changes vs. time for a repeated number of calcination/carbonation cycles of Piaseck limestone [76]. ... 11 Figure 1.3. Possible mechanism for the formation of different Al-based stabilizers (adapted from [127]). ... 21 Figure 1.4. Schematic representation of pore−skeleton model [162]. ... 35 Figure 1.5. Adsorption mechanism on Li2ZrO3 solid sorbent [194]. ... 44
Figure 1.6. CO2 sorption on Na2ZrO3 at different temperatures. (A) T ≤ 550 °C; the Na2CO3–
ZrO2 external shell is mesoporous, and the CO2 diffusion occurs through the mesoporous
structure. (B) T > 550 °C; the Na2CO3–ZrO2 external shell is not porous [227]. ... 50
Figure 1.7. Deactivation test of Ni-based catalysts supported on Al2O3 (dash), CeO2 (dash–
dot), and SiC (solid line) [47]. ... 57 Figure 1.8. Stability tests of nickel-based catalysts supported on Al2O3 (▪), CeO2 (▴), and
SiC (•) [47]. ... 57 Figure 1.9. H2 yield and selectivity of C-containing gas products of Ni/La2O3 and LCx
catalysts in SRG [53]. ... 60 Figure 1.10. Product distribution for steam reforming of glycerol over Ni catalysts (T = 873 K, S/C=33, P = 1bar) [36]. ... 61 Figure 1.11. Proposed redox mechanism of WGS reaction over ceria [12]. ... 62 Figure 1.12. Gas products molar ratio ( H2, CO2, CO, CH4), H2/Ni ratio (min−1) ( ),
H2 yield (mol%) ( ) and C (wt.%) in dry basis ( ) for the NiAl5Ce, NiAl10Ce and NiAl20Ce
catalysts and for thermodynamic equilibrium at steady state [39]. ... 62 Figure 1.13. Schematic representation of the reaction mechanism over NiSn/MgO-Al2O3
catalysts [50]. ... 65 Figure 2.1. Bragg diffraction by crystal planes ([300]). ... 79 Figure 2.2. A schematic diagram of the IGA microbalance [302]. ... 83 Figure 3.1. CO2 sorption activity of developed sorbent through 15 carbonation/calcination
cycles with steam present at different concentrations for carbonation: adsorption at 650 C, 15 vol.% CO2 and desorption at 800 C, 100 vol.% N2. ... 95
xiv
Figure 3.2. Adsorption data of sorbent during the 8th cycle at different concentrations of steam for carbonation: adsorption at 650 C, 15 vol.% CO2 and desorption at 800 C, 100 vol.%
N2. ... 97
Figure 3.3. SEM images of (a) fresh sorbent and spent sorbents after 15 carbonation/calcination cycles (b) without steam and with (c) 9.5 vol.% steam present in carbonation. ... 98 Figure 3.4. Pore size distribution of fresh sorbent and spent sorbents after 15 carbonation/calcination cycles without steam and with 9.5 vol.% steam present in carbonation. ... 98 Figure 3.5. XRD pattern of fresh sorbent. ... 99 Figure 3.6. CO2 sorption behavior of sorbent through 15 carbonation/calcination cycles
without and with 9.5% steam present for carbonation: adsorption at 550 C, 15 vol.% CO2
and desorption at 800 C, 100 vol.% N2. ... 100
Figure 3.7. Adsorption profile of sorbent during the 8th cycle without and with 9.5 vol.% steam present in carbonation: adsorption at 550 C, 15 vol.% CO2 and desorption at 800 C,
100 vol.% N2. ... 101
Figure 3.8. CO2 sorption activity of sorbent through 15 carbonation/calcination cycles with
steam present at two levels during calcination: adsorption at 650 C, 15 vol.% CO2 and
desorption at 800 C, steam balanced with N2. ... 102
Figure 3.9. Adsorption and regeneration data of sorbent at two levels of steam injected with calcination for the (a) 2nd and (b) 13th cycles: adsorption at 650 C, 15 vol.% CO2 and
desorption at 800 C, steam balanced with N2. ... 103
Figure 3.10. SEM images of sorbent after 15 carbonation/calcination cycles (a) without steam and with (b) 2.3 vol.% steam and (c) 9.5 vol.% steam present in calcination. ... 105 Figure 4.1. XRD patterns of calcined bifunctional materials. The following compounds were detected: ( ) CaO; ( ) CeO2; ( ) NiO; ( )Ca9Al6O18. (b) A magnified view of the shift of
diffraction peaks at 2 of 37.35 and 43.25. ... 118 Figure 4.2. SEM images of freshly calcined bifunctional materials. ... 120 Figure 4.3. Representative TEM images and Ni particle size distributions of reduced Ni20 and Ni20Ce10 bifunctional materials. ... 121 Figure 4.4. CO2 capture capacity (a) and molar conversion of CaO (b) for Nix and Ni20Cey.
Carbonation conditions: T = 550 C, flowrate = 150 mL/min containing 15 vol% CO2, 9.5
vol% H2O, and 75.5 vol% Ar; Calcination conditions: T = 800 C, 100 vol% Ar. ... 123
Figure 4.5. Conversion profiles of Nix and Ni20Cey as a function of time at the 1st cycle.
Carbonation conditions: T = 550 C, flowrate = 150 mL/min containing 15 vol% CO2, 9.5
vol% H2O, and 75.5 vol% Ar; Calcination conditions: T = 800 C, 100 vol% Ar. ... 124
Figure 4.6. CO2 capture capacity (a) and molar conversion of CaO (b) in the absence and
presence of water. Carbonation conditions: T = 550 C, flowrate = 150 mL/min containing 15 vol% CO2, 9.5 vol% H2O, and 75.5 vol% Ar under wet condition and containing 15 vol%
CO2 and 85 vol% Ar under dry condition; Calcination conditions: T = 800 C, 100 vol% Ar.
Figure 4.7. Typical gaseous product distribution during the first cycle of SESRG over Ni20 bifunctional material. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h
-1. ... 128
Figure 4.8. Breakthrough curves of (a) Ni15, (b) Ni20, and (c) Ni25 over 5 cycles. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h-1. ... 129 Figure 4.9. Average hydrogen yield as a function of cycle number for Nix bifunctional materials. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h-1. ... 130 Figure 4.10. Breakthrough curves of (a) Ni20Ce5, (b) Ni20Ce10, and (c) Ni20Ce15 over 5 cycles. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h-1. ... 132
Figure 4.11. Average hydrogen yield as a function of cycle number for Ni20Cey bifunctional materials. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h-1. ... 132
Figure 4.12. Average hydrogen purity and yield as a function of cycle number for Ni20Ce10 bifunctional materials. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.55 h
-1. ... 134
Figure 4.13. XRD patterns of spent Ni20 and Ni20Ce10 bifunctional materials upon 5 SESRG/regeneration cycles. The following compounds were detected: ( ) CaO; ( )CeO2;
( )Ni; ( ) Ca9Al6O18; ( )Graphite. ... 135
Figure 4.14. SEM images of spent Ni20 and Ni20Ce10 after 5 SESRG/regeneration cycles. ... 136 Figure 4.15. Representative TEM images and Ni particle size distributions of spent Ni20 and Ni20Ce10 after 5 SESRG/regeneration cycles. ... 137 Figure 5.1. XRD patterns of (a) freshly calcined and (b) reduced catalysts... 152 Figure 5.2. Pore size distribution of the calcined catalysts obtained from N2 physisorption
measurements. ... 154 Figure 5.3. H2-TPR profiles of freshly calcined catalysts. ... 156
Figure 5.4. XPS spectra of Ni 2p3/2 core level for calcined (a) NiAl_G1 and (b) NiAl_G2
catalysts. ... 157 Figure 5.5. XPS spectra of Ni 2p3/2 core level for reduced (a) NiAl_G1 and (b) NiAl_G2
catalysts. ... 158 Figure 5.6. SEM images of (Ni,Al)-Glycerate precipitate before and after two-step calcination. ... 159 Figure 5.7. TEM images and Ni0 particle size distributions of reduced (a) NiAl_G1, (b)
NiAl_G2, (c) NiAl_C, (d) and Ce/NiAl_G2 catalysts. ... 160 Figure 5.8. (a) Total glycerol conversion, glycerol conversion into gaseous products, and H2
yield, (b) gas products selectivity, and (c) H2/CO and CO2/CO molar ratios for the NiAl_G1,
NiAl_G2, and NiAl_C catalysts. Reaction conditions: T = 630 °C, P = 1 atm, W/G = 9, GHSV = 19600 cm3 gcat-1 h-1, glycerol solution to argon molar ratio = 1, and time on stream
= 4 h. ... 164 Figure 5.9. Possible reaction pathways occurring in SRG. ... 167 Figure 5.10. (a) TGA and derivative weight profiles, (b) rate of carbon deposition, and (c) TEM images and Ni0 particle size distributions for the used NiAl_G1, NiAl_G2, and NiAl_C
catalysts after 4 h of SRG reaction. ... 169 Figure 5.11. Raman spectra of the used catalysts after 4 h of SRG reaction. ... 170
xvi
Figure 5.12. (a) Total glycerol conversion, glycerol conversion into gaseous products, and H2 yield, (b) gas products selectivity, and (c) H2/CO and CO2/CO molar ratios for the
NiAl_G2 and Ce/NiAl_G2 catalysts. Reaction conditions: T = 630 °C, P = 1 atm, W/G = 9, GHSV = 19600 cm3 gcat-1 h-1, glycerol solution to argon molar ratio = 1, and time on stream
= 4 h. ... 171 Figure 5.13. (a) TGA and derivative weight profiles, (b) rate of carbon deposition, and (c) TEM images and Ni0 particle size distributions for the used NiAl_G2 and Ce/NiAl_G2 catalysts after 4 h of SRG reaction. ... 174 Figure 5.14. (a) Glycerol conversion into gaseous products and H2 yield, and (b) gas products
selectivity for the Ce/NiAl_G2 catalyst. Reaction conditions: T = 630 °C, P = 1 atm, W/G = 9, GHSV = 19600 cm3 gcat-1 h-1, glycerol solution to argon molar ratio = 1, and time on stream = 16 h. ... 175 Figure 5.15. (a) TGA and derivative weight profiles, (b) rate of carbon deposition, and (c) a representative TEM image for the used Ce/NiAl_G2 catalyst after 16 h of SRG reaction. ... 176 Figure 5.16. Raman spectra of the used Ce/NiAl_G2 catalyst after 16 h of SRG reaction. ... 177 Figure 6.1. XRD patterns of freshly (a) calcined and (b) reduced bifunctional materials. 191 Figure 6.2. Representative TEM images and Ni0 particle size distributions of reduced (a) CSBM_1 and (b) CSBM_2 bifunctional materials. ... 192 Figure 6.3. (a) Molar conversion of CaO and (b) CO2 capture capacity for S_1, S_2, and S_3
sorbents. Carbonation conditions: T = 550 °C, flow rate = 150 mL min-1 containing 15 vol.% CO2, 9.5 vol.% H2O, and 75.5 vol.% Ar. Calcination conditions: T = 800 °C, 100 vol.% Ar.
... 195 Figure 6.4. Conversion profiles of S_1, S_2, and S_3 sorbents as a function of time at the 1st cycle. Carbonation conditions: T = 550 °C, flow rate = 150 mL min-1 containing 15 vol.% CO2, 9.5 vol.% H2O, and 75.5 vol.% Ar. Calcination conditions: T = 800 °C, 100 vol.% Ar.
... 195 Figure 6.5. (a) Molar conversion of CaO and (b) CO2 capture capacity for S_3, CSBM_1,
and CSBM_2. Carbonation conditions: T = 550 °C, flow rate = 150 mL min-1 containing 15 vol.% CO2, 9.5 vol.% H2O, and 75.5 vol.% Ar. Calcination conditions: T = 800 °C, 100
vol.% Ar. ... 197 Figure 6.6. Conversion profiles of S_3, CSBM_1, and CSBM_2 as a function of time at the 1st cycle. Carbonation conditions: T = 550 °C, flow rate = 150 mL min-1 containing 15 vol.% CO2, 9.5 vol.% H2O, and 75.5 vol.% Ar. Calcination conditions: T = 800 °C, 100 vol.% Ar.
... 198 Figure 6.7. Molar composition of gaseous products for the first cycle of SESRG over (a) CSBM_1 and (b) CSBM_2. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.72 h-1. ... 200
Figure 6.8. H2 yield for the first cycle of SESRG over (a) CSBM_1 and (b) CSBM_2. SESRG
Figure 6.9. Evolution of (a-e) molar composition of gaseous products and (f) H2 yield over
10 cycles of SESRG/regeneration for CSBM_1 and CSBM_2. SESRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.72 h-1. ... 206 Figure 6.10. Evolution of (a) Molar composition of gaseous products and (b) glycerol conversion into gas phase and H2 yield over 3.3 h of SRG reaction for Ce/NiAl_G2 catalyst.
SRG conditions: P = 1 atm, T = 550 C, S/C = 3, WHSV = 1.72 h-1. ... 207
Figure 6.11. XRD patterns of used bifunctional materials after 10 SESRG/regeneration cycles. ... 209 Figure 6.12. Representative TEM images and Ni0 particle size distributions of used (a)
xviii
List of Tables
Table I.1. The most common side reactions in SRG. ... 5 Table 1.1. Summary of investigations on CaO-based sorbents synthesized from different precursors. ... 12 Table 1.2. Summary of investigations on metal oxide-stabilized CaO-based sorbents. ... 17 Table 1.3. Summary of investigations on the treatment of CaO-based sorbent via different methods. ... 29 Table 1.4. Summary of investigations on alkaline ceramic sorbents. ... 41 Table 1.5. Summary of investigations on employing Ni-based catalysts in SRG. ... 55 Table 1.6. Amounts of carbonaceous species deposited on NiCe, NiAl5Ce, NiAl10Ce and NiAl20Ce after SRG [27]. ... 63 Table 2.1. Chemical composition of Newfoundland limestone determined by XRF analysis. ... 74 Table 3.1. BET surface area and total pore volume of fresh and spent sorbents. ... 98 Table 4.1. Summary of literature results for hydrogen production from pure glycerol through SESR in fixed-bed reactors... 112 Table 4.2. Calculated values of d-spacing (nm) for three main reflections of CaO and NiO. ... 118 Table 4.3. Chemical composition and physical properties of the calcined Nix and Ni20Cey samples. ... 119 Table 4.4. Summary of the TGA results for the spent Ni20 and Ni20Ce10 bifunctional materials after 5 SESRG/regeneration cycles. ... 138 Table 5.1. Crystallite sizes of NiAl2O4, Ni, and CeO2 for the synthesized catalysts. ... 153
Table 5.2. BET surface area, total pore volume, and average pore diameter of the calcined catalysts obtained from N2 physisorption measurements. ... 154
Table 5.3. Reduction temperature and H2 consumption for the freshly calcined catalysts
obtained from H2-TPR analysis. ... 156
Table 5.4. Binding energies (eV) of core levels of the NiAl_G1 and NiAl_G2 catalysts. 158 Table 5.5. Binding energies of Ni 2p3/2 core level and surface reduction degree for the reduced
NiAl_G1 and NiAl_G2 catalysts. ... 158 Table 5.6. Average Ni0 particle size of the fresh catalysts reduced in 20 vol%H2/Ar at 800
°C. ... 161 Table 5.7. Detailed information on the blank test (without catalyst). ... 162 Table 5.8. Glycerol conversion into liquid products and liquid products selectivity for the NiAl_G1, NiAl_G2, and NiAl_C catalysts after 4 h of SRG reaction. ... 166 Table 5.9. Glycerol conversion into liquid products and liquid products selectivity for Ce/NiAl_G2 after 4 and 16 h of SRG reaction. ... 172 Table 6.1. BET surface areas of freshly calcined materials. ... 191 Table 6.2. Average Ni0 particle size of freshly reduced and used CSBM_1 and CSBM_2 determined from TEM images. ... 193 Table 6.3. Molar composition of gaseous products in the pre-breakthrough and post-breakthrough stages of SESRG over CSBM_1 and CSBM_2. ... 201
Table 6.4. Glycerol conversion into liquid and solid phases, as well as liquid products selectivity for the Ce/NiAl_G2 catalyst. ... 208
xx
Nomenclature
BET Brunauer-Emmett-Teller theory
CAMO Ca-Al mixed-metal oxides
CCS CO2 capture and sequestration
CLP calcium looping process
CSBM catalyst-sorbent bifunctional material
CTAB hexadecyl trimethyl ammonium bromide
DFR dual fluid reactor
FH2 the molar flow of produced H2
FGlyin the glycerol molar flow at the inlet
FID flame ionization detector
FSP flame spray pyrolysis
GC gas chromatography
GHSV gas hourly space velocity
HPLC high performance liquid chromatography
HTl hydrotalcite-like
HTLc hydrotalcite
∆Hr298 K heat of formation reaction at 298 k
IGA intelligent gravimetric analyzer
LDHs layered double hydroxides
MMO mesoporous metal oxides
MMT montmorillonite substance
OMPs organometallic precursors
PA pyroligneous acid
SEM scanning electron microscopy
SESR sorption enhanced steam reforming
Si gaseous products selectivity
Si' liquid products selectivity
SR steam reforming
t time
TEM transmission electron microscopy
TGA thermogravimetric analysis
TPR temperature-programmed reduction
wi initial weight of sample
W/G water to glycerol molar ratio
WGS water gas shift
wt weight of sample at time t
wt.% weight percent
𝑋𝐺𝑙𝑦𝑐𝑒𝑟𝑜𝑙,𝑔𝑎𝑠 glycerol conversion into gaseous products
𝑋𝐺𝑙𝑦𝑐𝑒𝑟𝑜𝑙,𝑙𝑖𝑞𝑢𝑖𝑑 glycerol conversion into liquid products 𝑋𝐺𝑙𝑦𝑐𝑒𝑟𝑜𝑙,𝑡𝑜𝑡𝑎𝑙 total glycerol conversion
XPS X-ray photoelectron spectroscopy
XRD X-ray powder diffraction
XRF X-Ray fluorescence spectrometry
xxii
To my husband, Mohammad Reza,
for his true love and friendship.
Acknowledgement
I would like to express my eternal gratitude to Prof. Maria Cornelia Iliuta, my Ph.D. supervisor, for her full support of my Ph.D. study and providing an opportunity for me to study at Université Laval in Canada. Her scientific knowledge and enthusiasm guided me throughout every step of my research project.
This thesis could not be accomplished without financial supports of The Natural Sciences and Engineering Research Council of Canada (NSERC), FQRNT Centre in Green Chemistry and Catalysis (CGCC), and The Fonds de recherche du Québec - Nature et technologies (FRQNT).
I would like to acknowledge all Chemical Engineering Department staffs, who considerably helped me through my study at Université Laval. I really appreciate all helps of technicians, especially Jean-Nicolas Ouellet and Jérôme Noël.
I am very grateful to all my friends and colleagues in Prof. Iliuta’s research group, whose motivations and supports assisted me through tough times during this period. I particularly thank Mahmoodreza Karimiestahbanati, Ommolbanin Ali Zadeh Sahraei, Mustapha Aissaoui, and Kang Gao. I wish them the best of luck in their future works. I would also like to thank Dr. Hamid Reza Radfarnia for his collaboration with us.
Last, but by far the most, I would like to thank my family for their unconditional love and supports during this period of my life, without whom I undoubtedly could not accomplish this doctorate. I cannot express in words my deep sense of gratitude to my beloved husband and best friend, Mohammad Reza, with his undying love, tremendous understandings and supports.
xxiv
Preface
This dissertation comprises 6 chapters, which come after an introduction regarding sustainable production of hydrogen from steam reforming and sorption enhanced steam reforming of glycerol. Chapter 1 provides a comprehensive literature review on Ni-based reforming catalysts, high temperature CO2 sorbents, and Ni-CaO-based catalyst-sorbent
bifunctional materials for sorption enhanced steam reforming of glycerol. Chapter 2 describes the materials used in this thesis, the methodology of material preparation and characterization, CO2 capture experiments, steam reforming and sorption enhanced steam
reforming experiments, and product analyses. Chapters 3-6 represent the main part of this thesis (results and discussions), which consists of the research findings and their analysis. Finally, the general conclusions and suggestions for future work are given in chapter 7.
This thesis was prepared based on the following published and submitted papers in/to scientific journals:
1. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, and M.C. Iliuta, High temperature CO2 sorbents and their application for hydrogen production by sorption enhanced
steam reforming process, Chemical Engineering Journal, 283 (2016) 420-444 (As a part of Chapter 1).
2. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, and M.C. Iliuta, Influence of steam addition during carbonation or calcination on the CO2 capture performance of
Ca9Al6O18-CaO sorbent, Journal of Natural Gas Science and Engineering, 36 (2016)
1062-1069 (Chapter 3).
3. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, and M.C. Iliuta, Sustainable production of high-purity hydrogen by sorption enhanced steam reforming of glycerol over CeO2-promoted Ca9Al6O18-CaO/NiO bifunctional material, ACS
Sustainable Chemistry and Engineering, 5 (2017) 9774−9786 (Chapter 4).
4. M. Shokrollahi Yancheshmeh, O. A. Sahraei, M. Aissaoui, and M.C. Iliuta, A novel synthesis of NiAl2O4 spinel from a Ni-Al mixed-metal alkoxide as a highly
efficient catalyst for hydrogen production by glycerol steam reforming, submitted (Chapter 5).
5. M. Shokrollahi Yancheshmeh and M.C. Iliuta, A novel synthesis of Ni-CaO-based catalyst-sorbent bifunctional materials for high-purity hydrogen production via sorption enhanced steam reforming of glycerol, submitted (Chapter 6).
The candidate has main contribution in all stages of the work presented in these papers, including design of the experimental set-up, design of experiment, synthesis and characterization of materials, planning and performing experiments, as well as writing the papers by considering the supervisor’s comments.
The results of the present thesis have also been presented in the following academic national and international conferences:
1. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. Catalyst development for glycerol steam reforming process. Chemical Engineering research day, Montreal, Canada, March 2019.
2. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Development of bifunctional catalyst-sorbent material for high-purity hydrogen production via intensified sorption-enhanced steam reforming of glycerol. CCVC Annual Conference, Quebec, Canada, May 2018.
3. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. High-purity hydrogen production by intensified sorption-enhanced steam reforming of glycerol over CeO2
Promoted Ca9Al6O18-CaO/NiO Hybrid Materials. 66th Canadian Chemical
Engineering Conference, Quebec, Canada, October 2016.
4. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. Metal-stabilized CaO-Nickel hybrid sorbent-catalysts for high purity hydrogen production by intensified sorption enhanced steam glycerol reforming. 252nd ACS National Meeting, Philadelphia, PA, USA, August 2016.
5. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. High-purity hydrogen production by sorption-enhanced steam glycerol reforming (SESGR) over a metal-stabilized CaO-Nickel hybrid sorbent-catalyst. Designing New Heterogeneous Catalysts: Faraday Discussion (Royal Society of Chemistry), London, United Kingdom, April 2016.
xxvi
6. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. Influence of steam addition in carbonator/calciner on the CO2 capture performance of Ca9Al6O18-CaO
sorbent. 65th Canadian Chemical Engineering Conference, Calgary, Canada, October
2015.
7. M. Shokrollahi Yancheshmeh, H.R. Radfarnia, M.C. Iliuta. Influence of steam addition in carbonator/calciner on the CO2 capture performance of Ca9Al6O18-CaO
Introduction
Despite recent progress in climate change mitigation, further efforts are still needed to reduce anthropogenic greenhouse gas emissions, of which CO2 emissions are considered to
be one of the most significant. Global energy-related CO2 emissions increased by 1.4 % in
2017 and reached a historic high of 32.5 Gt (Figure I.1.1). This growth, which came after three years of flat emissions and was faster than the 10-year average of 1.3 %, was issued from 2.1 % growth in global energy demand (Figure I.1.2). Global energy demand in 2017 reached around 14050 Mtoe, of which 81 % was supplied from fossil fuels. The increase in energy-related CO2 emissions in 2017 is a strong warning for global efforts to combat climate
change. According to the IEA’s Sustainable Development Scenario, increasing the share of low-carbon (e.g., natural gas) and/or free-carbon energy sources (e.g., biomass) for world energy supply and developing more efficient and cost-competitive technologies to capture and storage the CO2 (CCS) are the most effective and economic methods to meet the
long-term climate goals [1].
2
Figure I.1.2. Average annual growth in energy demand by fuel [1].
In this context, “hydrogen economy” represents a promising route towards a carbon-free energy system. The term “hydrogen economy” refers to the use of hydrogen as a fuel for energy storage and transfer. However, hydrogen does not occur naturally and it must be produced from other energy sources. Nowadays, nearly 48% of the worlds hydrogen is produced through the steam reforming of methane, while the reforming of naphtha/oil contributes with 30%, the coal gasification with 18%, and electrolysis with only 3.9%. Despite the large contribution of fossil fuel-based processes to the hydrogen production, these kinds of technologies face substantial challenges such as the increasing global warming effect and the drastic reduction of fossil fuel sources. Therefore, chemical and energy-related industries are studying alternative renewable sources such as biomass or biomass-derived oxygenates for hydrogen production. In this regard, hydrogen production from glycerol has been extensively investigated over the last two decades [2-7].
Glycerol is produced as a byproduct, with a mass yield of 10 %, in the transesterification process to produce biodiesel (Figure I.1.3). In recent years, production of biodiesel has become a controversial topic mainly due to its direct and indirect competition with food industries and its cost effectiveness. However, it’s benefits such as reduction of CO2
emissions and enhancement of energy security cannot be ignored. In fact, it can be said that the potential impact of biodiesel industry can differ widely according to national and local conditions and to the choice of specific technologies and feedstocks. Data on production clearly reveals that despite all the controversial issues related to this industry, biodiesel production has grown 31 % between 2011 and 2016. Biodiesel production in European Union
member countries was increased from 1.93 million tons in 2004 to 11.5 million tons in 2016, with Germany and France taking the lead (Figure I.1.4) [8]. Similarly, the United States biodiesel production grew from 250 million gallons in 2006 to 2.89 billion gallons in 2016 (Figure I.1.5) [9]. The world biodiesel production was 30.1 billion liters in 2015, with the United States, Brazil, Germany, France and Argentina being the top five producers (Figure I.1.6) [10]. The growing biodiesel production has resulted in an excess supply of glycerol. It is anticipated that the annual production of glycerol will reach to about 3 megatons in 2020, whereas the industries consume only 500 kilotons glycerol each year [11, 12]. The increasing availability of glycerol has led to the current in-depth research into converting glycerol to higher-value products, including hydrogen. Moreover, the conversion of glycerol to high value and useful products will enhance the economics of biodiesel production [13, 14].
Figure I.1.3. Transestrification of triglyceride with methanol for biodiesel production [15].
4
Figure I.1.5. United States biodiesel growth pattern for the period 2006 to 2016 [9].
Figure I.1.6. Biodiesel global production of top 16 countries in 2015 [10].
To date, various methods such as pyrolysis, partial oxidation, steam reforming, autothermal reforming, and aqueous-phase reforming have been proposed for converting glycerol to hydrogen. As mentioned earlier, steam reforming is the most commonly used method for hydrogen production from natural gas. Steam reforming of glycerol (SRG) provides the possibility of using current steam reforming plants, with little modification, for converting glycerol to hydrogen. This is the very reason why SRG is regarded as one of the most promising methods for converting glycerol to hydrogen [16, 17].
Steam reforming of glycerol can be represented by Eq. (I.1), which combines the pyrolysis (Eq. (I.2)) and water gas shift (WGS) reactions (Eq. (I.3)) in one reactor to produce hydrogen. As can be seen, hydrogen production from glycerol is potentially attractive considering that one mole of glycerol can theoretically produce 7 mole of hydrogen [17]. However, SRG involves complex reactions that result in the formation of many possible byproducts, including carbon monoxide, carbon dioxide, methane, ethane, ethylene, ethanol, acetaldehyde, acetic acid, acetone, methanol, acrolein, allyl alcohol, formaldehyde, and coke.
Table I.1.1 lists the most common side reactions that can occur during SRG. The complexity of SRG negatively affect the final purity of hydrogen in the produced gas stream. To achieve high hydrogen purity, it is therefore necessary to perform purification and separation steps in the downstream process, which increases the total capital cost and the complexity, and decreases the overall efficiency of the entire process [18, 19]. To overcome this obstacle, sorption enhanced steam reforming of glycerol (SESRG) represents a promising alternative to conventional SRG [11, 13, 20]. 298 1 3 8 3 3 2 3 2 7 2 r 128 . C H O + H O CO + H = kJ mol− (I.1) 298 1 3 8 3 4 2 3 r 251 . C H O H + CO = kJ mol− (I.2) 298 1 2 2 2 r 41 . CO+H OH +CO = − kJ mol− (I.3)
Table I.1.1. The most common side reactions in SRG.
Name of reaction Reaction ∆𝐇𝐫𝟐𝟗𝟖 𝐊(kJ.mol-1)
Methanation 2 4 2 3 CO+ H CH +H O -206 Methanation 2 4 2 4 2 2 CO + H CH + H O -165 CH4 steam reforming 4 2 3 2 CH +H OCO+ H +207 CH4 dry reforming 4 2 2 2 2 CH +CO CO+ H +247 CO disproportionation 2 ( ) 2COCO +Cs -172 CH4 decomposition 4 2 2 ( )s CH H +C +75 CO hydrogenation 2 2 ( )s CO+H H O C+ -131 CO2 hydrogenation 2 2 2 2 ( )s CO + H H O C+ +306
Sorption enhanced steam reforming (SESR) process offers the possibility to achieve a high purity of hydrogen from a single-rector process. The concept of SESR process was first
6
proposed by Carvill et al. from Air Products and Chemicals Inc. to achieve a high conversion of reactants to products for an equilibrium-controlled, endothermic reaction at a much lower temperature than would be necessary by a conventional catalytic reactor [21]. The fundamental of this process is to remove CO2 simultaneously with the reforming reaction by
adding a high-temperature solid CO2 sorbent such as CaO and therefore, shift the WGS
reaction equilibrium towards hydrogen production. In SESRG process, the combination of SRG and CO2 capture reactions, represented by Eq. (I.1) and (I.4), respectively, leads to a
new reaction for converting glycerol into highly pure hydrogen given in Eq. (I.5). As can be seen, the new reaction is exothermic and the heat released can be recovered by heat integration. SESRG process also provides the opportunity to increase the conversion of glycerol to gaseous products, suppress the side reactions such as methanation and coke formation, work at relatively low W/G ratios, and enhance the yield of highly pure hydrogen [11, 12, 22]. The efficiency of this process is strongly influenced by both the CO2 sorbent
and the reforming catalyst, as well as their mixing pattern.
298 1
( )s 2 ( )g 3 ( )s r 178 .
Cao +CO CaCO = − kJ mol− (I.4)
298 1
3 8 3 3 2 3 ( )s 7 2 3 3 ( )s r 407 .
C H O + H O+ CaO H + CaCO = − kJ mol− (I.5) A highly efficient sorbent for CO2 capture at high temperature should possess specific
properties such as: thermal stability at high operating temperatures (450-700 °C), adequate CO2 sorption capacity and kinetics, easiness of sorbent regeneration, long-term cyclic
stability, and reasonable production cost. The most promising high-temperature solid sorbents available in the literature mainly include ceramic alkaline-based and CaO-based sorbents. Hydrotalcite (HTLc) is another type of sorbent, but its CO2 uptake capacity is very
low in comparison to the other materials [23, 24]. The literature review related to the
present thesis will therefore focus on the properties and preparation methods of
CaO-based and alkaline-CaO-based sorbents as two promising solid sorbents for CO2 capture at
high temperatures.
The product distribution in SRG is strongly affected by catalyst formulation. For the selective hydrogen production from glycerol, the catalyst employed in steam reforming process would be expected to promote not only the rupture of C-C, O-H, and C-H bonds but
also the elimination of metal-passivating CO by the WGS reaction. Moreover, either C-O cleavage or CO/CO2 hydrogenation should not be promoted by such a catalyst [25-27]. To
date, almost all studies have been devoted to the noble metal-based catalysts, such as Pt-, Pd-, Ir-Pd-, Rh-Pd-, and Ru-based ones [25Pd-, 26Pd-, 28-35]Pd-, and non-noble metal-based catalystsPd-, such as Ni-based ones [33, 36-62]. Among them, Ni-based catalysts are the most investigated ones due to their high ability to break C-C bonds, high availability, and low cost. Therefore,
although many different catalysts can be used for SRG, the present thesis will focus only on the Ni-based catalysts. The literature review will also be limited to this subject.
Figure I.1.7. Schematic comparison of CO2 diffusion in physical mixing and catalyst-sorbent bifunctional patterns [63].
The mixing pattern of catalyst and sorbent is one of the key factors that strongly influences the successful application of SESR concept for large-scale hydrogen production. To date, two different mixing patterns have been proposed in the literature: (i) physical mixing and (ii) integrating catalyst and sorbent into a single particle (Figure I.1.7) [64]. In the former case, the CO2 generated during the reforming reaction must diffuse from the
interior surface of the catalyst to the neighboring sorbent where it can be adsorbed. In the latter case, however, the direct on-site sorption of CO2 can be realized by integrating the
catalyst and sorbent into a single particle, i.e., catalyst-sorbent bifunctional material. In this case, a higher sorption rate and in turn, a higher reaction rate can be achieved due to the higher concentration of CO2 over the sorbent active sites. Integrating the catalyst and sorbent
into a single particle can not only eliminate mass transfer limitations but also reduce the process complexity [65-68]. The present thesis will therefore focus on the synthesis of
8
catalyst-sorbent bifunctional materials for SESRG. The literature review related to the
Chapter 1. Literature review
1.1. High temperature CO
2sorbents
The following section will provide an overview of the most commonly used high-temperature CO2 sorbents, CaO-based and alkaline-based materials. The main objectives of
this section are threefold: (i) to provide an overview of the major characteristics of these sorbents; (ii) to describe the different types of synthesis methods that can be used for their preparation; and (iii) to provide an overview of the available techniques for improving their performance in severe operating conditions.
1.1.1. CaO-based sorbents
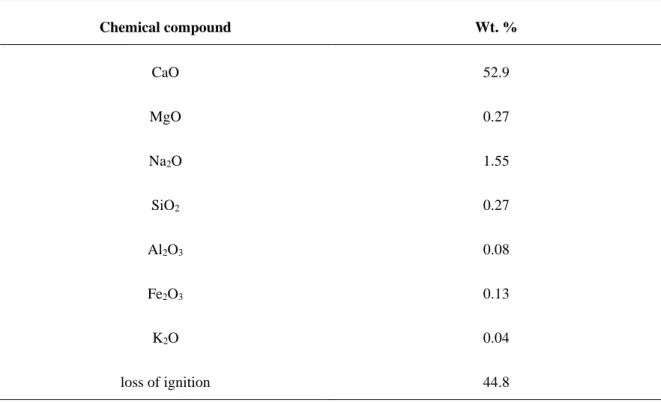
CaO is the most common naturally occurring CO2 sorbent that exists in nature in the
forms of limestone (CaCO3), dolomite (CaMg(CO3)2), and huntite (CaMg3(CO3)4). This
sorbent has attracted a lot of attention because of its low raw material cost, high CO2 sorption
capacity, and adequate kinetics of reactions. The cyclic carbonation/calcination of CaO occurs via the reversible reaction given in Eq. (1.1).
298 1
( )s 2 ( )g 3 ( )s r 178 .
CaO +CO CaCO = − kJ mol− (1.1) Theoretical CO2 capture capacity of limestone is as high as 0.786 gCO2 gsorbent-1.
Nevertheless, dolomite and huntite have lower CO2 sorption capacity (dolomite: 0.46 gCO2
gsorbent-1 and huntite: 0.25 gCO2 gsorbent-1) because MgO does not participate in CO2 adsorption.
The carbonation reaction of CaO with CO2 generally takes place in two stages, fast
reaction controlled and slow diffusion controlled. Dou et al. [69], Rout et al. [70] and Mohammadi et al. [71] performed kinetic studies on the carbonation reaction of different CaO-based synthetic sorbents and found that the carbonation reaction was controlled by both chemical reaction at the CaO-CaCO3 interface and carbonate layer diffusion. The fast
reaction controlled step will be continued until the carbonate layer surrounding the unreacted CaO core is completed. Then, the slow diffusion controlled step starts, during which the product layer restricts the access of CO2 molecules to reactive sites. Alvarez and Abanades
[72]reported that the gas diffusion and thus, the sorption rate, can be limited above a critical carbonate layer thickness of 50 nm. Mostafavi et al. [73] found that the CaO sorbent derived from limestone represented a higher initial carbonation reaction rate in comparison with that
10
derived from dolomite, because of excessive CaO active sites. On the other hand, the ultimate conversion was higher for dolomite-derived CaO sorbent at low temperature (550 °C), whereas this value was higher for limestone-derived CaO sorbent at high temperatures (600 and 675 °C). As a matter of fact, at the beginning of the reaction, the growth of CaCO3 layer
for limestone was faster than that of dolomite because of the higher rate of reaction. Since Knudsen diffusion is related to the square root of temperature, the diffusion through CaCO3
layer enhanced as the temperature increased above 550 °C. Therefore, limestone showed a higher ultimate conversion at 600 and 675 °C.
Although there are many advantages in using CaO as a CO2 sorbent, its industrial
application encounters with some critical issues such as the loss of sorption capacity in long-term operation and the loss of reactivity with sulfur containing gases due to the formation of CaSO4 [74-77]. The loss of CO2 sorption capacity during cyclic operation is mainly resulted
from the sintering phenomenon, which is referred to the agglomeration of small particles, the change of pore shapes, and the pore shrinkage. As can be seen in Figure 1.1, the amount of unreacted CaO increases along with the cycle number until a rigid interconnected CaO skeleton is formed after 50 cycles. Therefore, the carbonation reaction takes place only on the external surface of sample. The sintering phenomenon is mainly caused by the expansion of CaO particles during CO2 sorption. This expansion is strongly influenced by the
temperature and particle separation distance: the higher the carbonation temperature and the shorter the distance between two particles, the faster the sintering rate. Therefore, the capacity decay of CaO sorbents during multiple carbonation/calcination cycles mainly depends on the experimental temperature and the precursor type [78, 79].
Several studies have been conducted on the CO2 capture performance of limestone in
long-term cyclic operation. Grasa and Abanades [76] evaluated the CO2 capture performance
of limestone over 500 carbonation/calcination cycles and observed that the CO2 uptake
capacity significantly decreased during the first 20 cycles and then stabilized along with the cycle number around 0.075-0.08 residual conversion up to 500 cycles. Figure 1.2 shows the decrease of CO2 capture capacity along with the cycle number for Piaseck limestone. In
another study, Sun et al. [77] examined the CO2 sorption behavior of limestone through more
than 1000 carbonation/calcination cycles. They reported a calcium conversion between 4 and 17% (depending on the carbonation time) after 150 cycles.
Figure 1.2. Typical weight changes vs. time for a repeated number of calcination/carbonation cycles of Piaseck limestone [76].
The natural CaO-based sorbents containing MgO, such as dolomite and huntite, show a better stability during cyclic operations. Silaban et al. [74] observed that after six carbonation/calcination cycles, the capture capacity of limestone was decreased from 61% to 35% of its theoretical value. However, dolomite was already stable at a sorption capacity of 40%. The behavior of dolomite was ascribed to the presence of MgO in its structure, which provided a better structural stability. Bandi et al. [80] examined the CO2 capture performance
of calcite, dolomite and huntite for 47 carbonation/calcination cycles. The experimental results revealed that huntite could preserve around 84% of its initial capacity, while dolomite and calcite were able to maintain 55% and 38% of their initial capacity, respectively. However, huntite possessed less CO2 uptake capacity in comparison to dolomite and calcite
because of its higher content of inactive MgO. According to these studies, the deactivation of CaO-based sorbents derived from natural sources is unavoidable. Different strategies have
12
therefore been proposed to improve the CO2 sorption performance of CaO-based sorbents,
such as (i) application of various sources of calcium to produce CaO-based sorbents, (ii) incorporation of inert materials into CaO structure, and (iii) reactivation and treatment of CaO-based sorbents.
1.1.1.1. Application of various sources of calcium to produce CaO-based sorbents
Many research groups have focused on the production of CaO-based sorbents from various calcium precursors, including CaCO3 [81-83], Ca(OH)2 [83-85], organometallic
[86-92], and nano-sized CaO/CaCO3 [88, 91, 93-100], with the aim of synthesizing CaO-based
sorbents with meso- and macro-porous structure, high specific surface area, large pore volume, and small particle size (Table 1.1). These specifications must be met to have a sorbent with high and stable CO2 capture capacity. The performance of the CaO-based
sorbents derived from two most promising groups, organometallic and nano-sized CaO/CaCO3 precursors, are discussed further.
Table 1.1. Summary of investigations on CaO-based sorbents synthesized from different precursors.
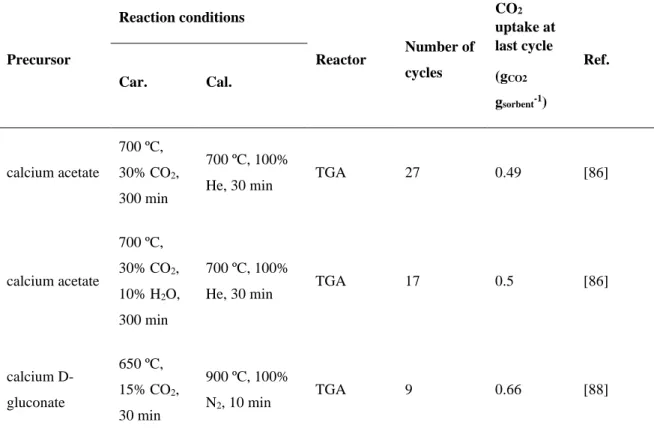
Precursor Reaction conditions Reactor Number of cycles CO2 uptake at last cycle (gCO2 gsorbent-1) Ref. Car. Cal. calcium acetate 700 ºC, 30% CO2, 300 min 700 ºC, 100%
He, 30 min TGA 27 0.49 [86]
calcium acetate 700 ºC, 30% CO2, 10% H2O, 300 min 700 ºC, 100%
He, 30 min TGA 17 0.5 [86]
calcium D-gluconate 650 ºC, 15% CO2, 30 min 900 ºC, 100% N2, 10 min TGA 9 0.66 [88]
calcium D-gluconate 650 ºC, 15% CO2, 30 min 920 ºC, 15% CO2, 2 min TGA 57 0.19 [88] nano-CaCO3 (40 nm) 650 ºC, 15% CO2, 20 min 850 ºC, 100% N2, 10 min TGA 100 0.17 [72] calcium naphthenate (FSP-made CaO) 700 ºC, 30% CO2, 300 min 700 ºC, 100%
He, 30 min TGA 60 0.39 [94]
calcium naphthenate (FSP-made CaO) 700 ºC, 30% CO2, 5 min nonisothermal to 900 °C, 100% He, 40 min TGA 20 0.39 [94] Calcium acetate 650 ºC, 15.30% CO2 850 ºC, 100% N2 FB 9 0.303 [89] CaCO3 650 ºC, 15.30% CO2 850 ºC, 100% N2 FB 9 0.285 [89] CaCO3 650 ºC, 15% CO2, 2 h 950 ºC, 100% N2 TGA 11 0.40 [82] Ca(NO3)2.4H2O 650 ºC, 15% CO2, 15 min 850 ºC, 100% N2, 10 min TGA 20 0.51 [95] Ca(NO3)2.4H2O 650 ºC, 15% CO2, 15 min 950 ºC, 100% CO2, 10 min TGA 20 0.2 [95]
14 CaO 750 ºC, 100% CO2, 40 min 750 ºC, 100% N2, 30 min TGA 9 0.32 [83] CaCO3 650 ºC, 100% CO2, 30 min 750 ºC, 100% N2, 30 min TGA 9 0.1 [83] Ca(OH)2 650 ºC, 100% CO2, 40 min 750 ºC, 100% N2, 40 min TGA 9 0.25 [83] 1.1.1.1.1. Organometallic precursors
Up to now, a number of studies have reported the synthesis of CaO-based sorbents from various organometallic precursors (OMPs) and the relation between the structural characteristics of OMPs-derived CaO-based sorbents with their CO2 uptake performance
[86-92]. The decomposition of OMPs leads to the formation of meso- and macro-porous structures with large surface area, which results in the enhancement of CO2 capture
performance [86, 88]. In an earlier work, Silaban et al. [92] found that the CaO-based sorbent synthesized from calcium acetate possessed a higher CO2 capture capacity than CaO
synthesized from calcium carbonate regardless of calcination temperature. Similarly, Lu et al. [86] examined the CO2 capture behaviour of the CaO-based sorbents derived from calcium
acetate monohydrate, calcium carbonate, calcium hydroxide, and calcium nitrate tetrahydrate. According to the experimental results, the CaO-based sorbent obtained from calcium acetate demonstrated the best performance with a CO2 sorption capacity of 0.49 gCO2
gsorbent-1 after 27 cycles (carbonation at 700 °C under 30% CO2 in He and calcination at 700
°C under He) because of its large BET surface area and pore volume. The SEM images revealed that this sorbent contained a fluffy structure, which contributed to its high surface area and large pore volume. In a further study, Liu et al. [88] prepared CaO-based sorbents using different precursors, including calcium acetate hydrate, calcium citrate tetrahydrate, calcium D-gluconate monohydrate, calcium formate, calcium L-lactate hydrate, calcium hydroxide, microsize calcium carbonate, nanosize (<70 nm) calcium carbonate, and nanosize (<160 nm) calcium oxide. Among the developed materials, the CaO-based sorbent derived
from calcium D-gluconate monohydrate exhibited the highest CO2 capture capacity of 0.66
gCO2 gsorbent-1 at the 9th cycle. Yang et al. [89] synthesized four types of CaO-based sorbents
from calcium acetate monohydrate, calcium carbonate, calcium hydroxide, and calcium oxide precursors by calcination and hydration reactions. The cyclic CO2 capture experiments
showed that the CaO-based sorbents derived from calcium acetate and calcium carbonate presented a higher CO2 sorption capacity (0.299 and 0.284 gCO2 gsorbent-1 at 650 C,
respectively) compared to the CaO-based sorbents derived from calcium oxide and calcium hydroxide due to the larger pore volume and higher specific surface area of the former ones. According to the cyclic carbonation/calcination experiments, the CO2 sorption capacity of
calcium acetate- and CaCO3-derived CaO-based sorbents increased over the initial cycles
and then decreased and reached to 0.303 and 0.285 gCO2 gsorbent-1 after 9 cycles. Grasa et al.
[87] studied the cyclic CO2 sorption performance of the CaO-based sorbents prepared from
calcium hydroxide, calcium acetate, and calcium oxalate under realistic calcination conditions (regeneration at temperatures around 900 C under CO2 atmosphere). Although
these synthetic sorbents performed well under mild reaction conditions, they showed a dramatic decay in CO2 capture capacity under realistic severe calcination conditions, the
behavior being similar to natural limestone. The best synthetic sorbent, which was derived from calcium acetate, exhibited a final CO2 capture capacity slightly higher than that of
limestone. This CO2 capture behavior did not justify the higher cost of sorbent production
from chemical precursors.
1.1.1.1.2. Nano-sized CaO/CaCO3 precursors
For porous CaO particles with critical particle size less than 44 nm or a large single CaO crystal with critical crystal size less than 220 nm, the carbonation reaction completes within the fast reaction controlled regime and the slow diffusion controlled regime does not exist [96, 101]. Therefore, different research groups have studied nano-sized CaO and CaCO3
materials as calcium precursors [88, 91, 93-100]. All of these studies have shown that nano-sized sorbents possess much better CO2 capture performance in comparison to micro-sized
based sorbents. However, there are two primary obstacles for using the nano-sized CaO-based sorbents in practical applications: (i) the nano-sized CaO-CaO-based sorbents synthesized from nano-sized particles are more susceptive to sintering, because of their higher surface area, when compared to micro-sized CaO-based sorbents [93] and (ii) the methods used for
![Figure 1.2. Typical weight changes vs. time for a repeated number of calcination/carbonation cycles of Piaseck limestone [76]](https://thumb-eu.123doks.com/thumbv2/123doknet/3174493.90588/37.918.197.752.391.630/figure-typical-changes-repeated-calcination-carbonation-piaseck-limestone.webp)
![Figure 1.8. Stability tests of nickel-based catalysts supported on Al 2 O 3 (▪), CeO 2 (▴), and SiC (•) [47]](https://thumb-eu.123doks.com/thumbv2/123doknet/3174493.90588/83.918.258.665.423.879/figure-stability-tests-nickel-based-catalysts-supported-ceo.webp)
![Figure 1.9. H 2 yield and selectivity of C-containing gas products of Ni/La 2 O 3 and LCx catalysts in SRG [53]](https://thumb-eu.123doks.com/thumbv2/123doknet/3174493.90588/86.918.282.676.432.688/figure-yield-selectivity-containing-products-lcx-catalysts-srg.webp)
![Figure 1.10. Product distribution for steam reforming of glycerol over Ni catalysts (T = 873 K, S/C=33, P = 1bar) [36]](https://thumb-eu.123doks.com/thumbv2/123doknet/3174493.90588/87.918.266.693.210.441/figure-product-distribution-steam-reforming-glycerol-ni-catalysts.webp)
![Figure 1.13. Schematic representation of the reaction mechanism over NiSn/MgO-Al 2 O 3 catalysts [50].](https://thumb-eu.123doks.com/thumbv2/123doknet/3174493.90588/91.918.283.675.105.566/figure-schematic-representation-reaction-mechanism-nisn-mgo-catalysts.webp)