Molecular simulations as test beds for bridging high throughput and high performance computing
151
0
0
Texte intégral
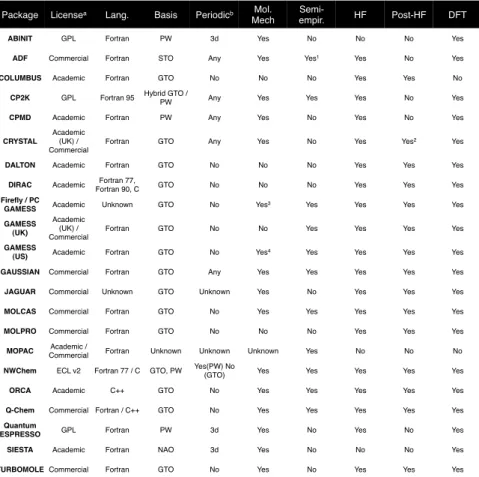
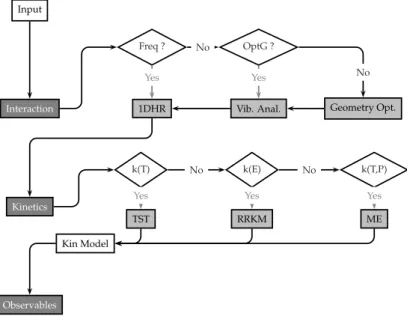
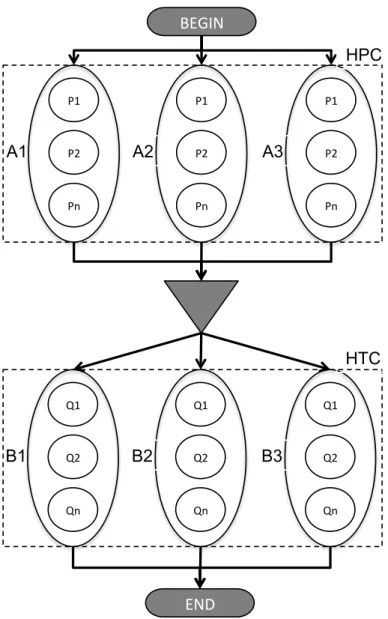
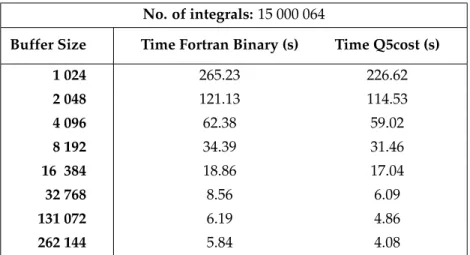
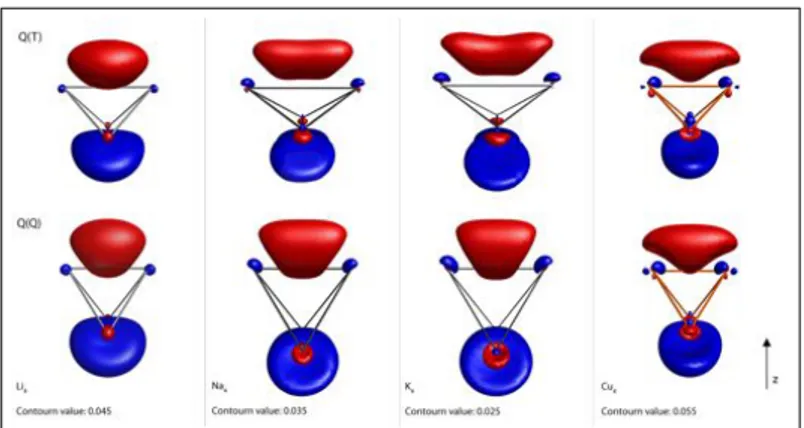
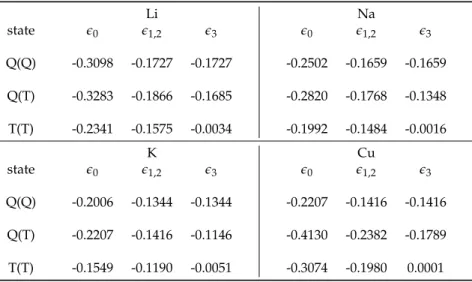
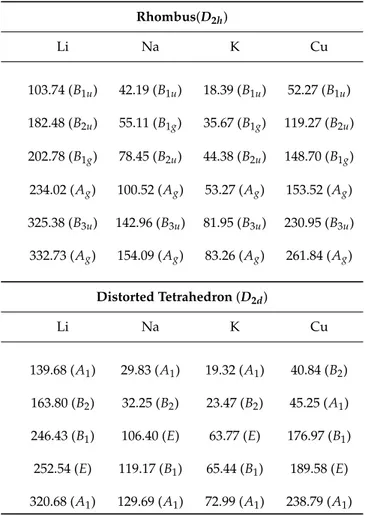
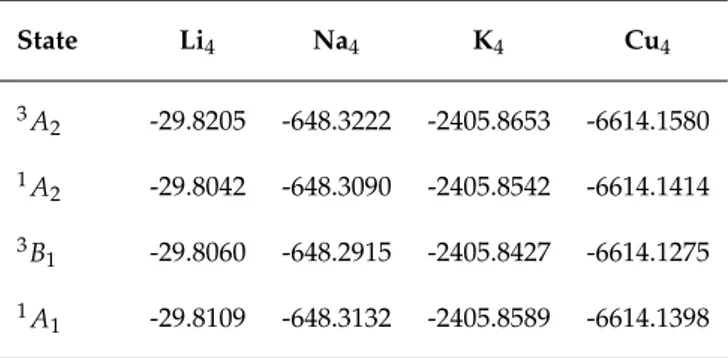
Figure

+7
Documents relatifs
