Étude de l'influence du repliement des chaînes de polyéthylène sur la température de fusion par simulation moléculaire
79
0
0
Texte intégral
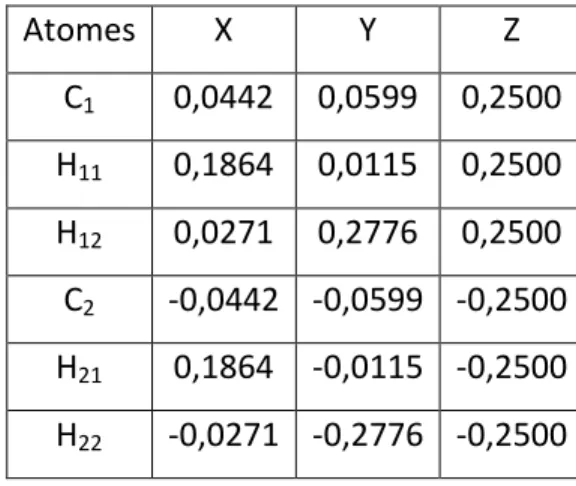
Figure
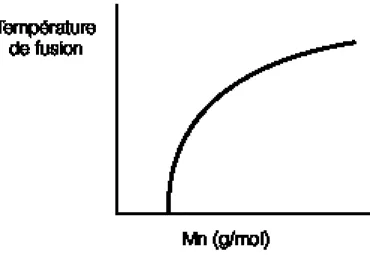
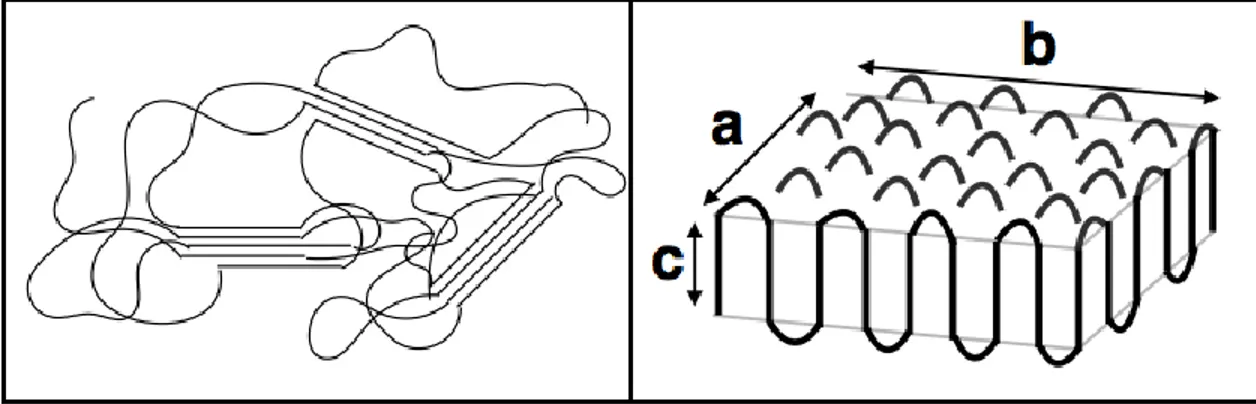
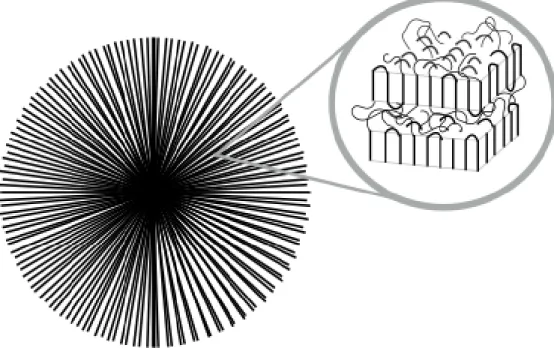

+7
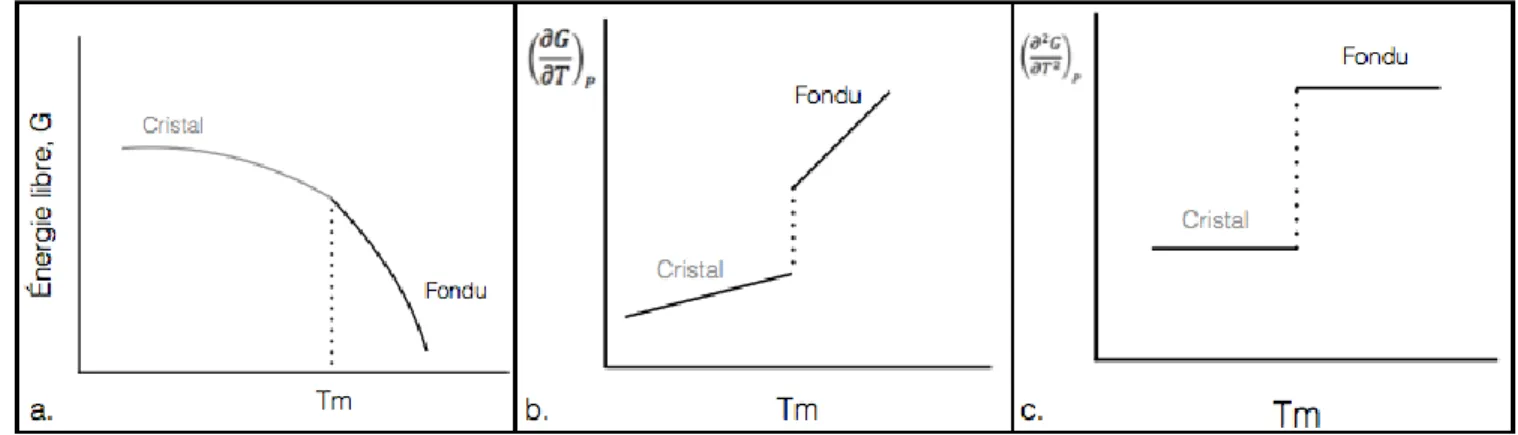



Documents relatifs