Utilisation du XtalFluor en synthèse organique
et développement de réactifs de fluoration électrophile
Thèse
Mathilde Vandamme
Doctorat en chimie
Philosophiæ Doctor (Ph. D.)
Québec, Canada
© Mathilde Vandamme, 2017
Utilisation du XtalFluor en synthèse organique
et développement de réactifs de fluoration électrophile
Thèse
Mathilde Vandamme
Sous la direction de :
Résumé
L’amélioration des méthodes de synthèse organique, que ce soit les réactions ou les réactifs, retient continuellement l’intérêt des chimistes. Ceci est particulièrement vrai pour les molécules fluorées, qui sont d’une grande utilité en chimie pharmaceutique, en agrochimie et en sciences des matériaux.
À cet effet, la compagnie OmegaChem a récemment développé deux réactifs de fluoration nucléophile capables de réaliser la déoxofluoration d’alcools, de cétones et d’acides carboxyliques en fluorures d’alkyles, difluorométhylènes et fluorures d’acyles respectivement. Il s’agit de tétrafluoroborate de diéthylaminodifluorosulfinium et de tétrafluoroborate de morpholinodifluorosulfinium, appelés XtalFluor-E et XtalFluor-M. Ceux-ci possèdent un pouvoir activant, mais une source externe de fluorure est nécessaire pour que la déoxofluoration s’opère. En raison de leur facilité de manipulation et leur faible coût, il s’est avéré intéressant d’utiliser cette nouvelle classe de réactifs dans d’autres réactions de fluoration, mais également dans des transformations nécessitant un agent activant. Dans le cadre des travaux de cette thèse, diverses réactions impliquant le XtalFluor-E ont été développées. Ainsi, des isonitriles ont pu être synthétisés à partir de formamides, puis impliqués dans des réactions multicomposantes. De la même manière, une méthode permettant de former des nitriles à partir d’amides primaires ou d’aldoximes a été développée. Des esters perfluorés ont également été synthétisés, à partir d’acides carboxyliques et d’alcools perfluorés variés. Enfin, une réaction de déoxofluoration éliminatrice a permis l’obtention de monofluoroalcènes cycliques.
La seconde partie du projet s’est focalisée sur la fluoration électrophile. Dans ce cas-là, le substrat joue le rôle de nucléophile tandis que l’atome de fluor est fourni sous forme électrophile. Au vu des limites des réactifs actuels (disponibilité commerciale, solubilité dans les solvants organiques, réactivité, etc.), l’objectif consiste à élaborer de nouveaux réactifs de fluoration électrophile comblant ces lacunes. Plus particulièrement, des dérivés de N-fluorosquaramides ont été brièvement étudiés.
Abstract
The improvement of synthetic methodologies, either reactions or reagents, continually retains the interest of chemists. This is particularly true for fluorinated molecules, which occupy a significant place in pharmaceutical chemistry, agrochemistry, and material sciences.
To this end, OmegaChem recently commercialized two nucleophilic fluorinating reagents allowing the deoxofluorination of alcohols, ketones and carboxylic acids into alkyl fluorides, difluoromethylenes and acyl fluorides, respectively. These are diethylaminodifluorosulfinium tetrafluoroborate and morpholinodifluorosulfinium tetrafluoroborate, named XtalFluor-E and XtalFluor-M. They possess an activating power, but an exogenous source of fluoride is required to perform deoxofluorination reactions. Because of their ease of handle and low cost, it was advantageous to use this new class of reagents in other fluorination reactions, but also in transformations requiring an activating agent. As part of this thesis, various reactions involving XtalFluor-E have been developed. Isocyanides could be synthesized from formamides and then involved in multi-component reactions. In the same way, a method allowing the formation of nitriles from primary amides or aldoximes has been developed. Perfluorinated esters have been also synthesized, from carboxylic acids and various perfluorinated alcohols. Finally, an eliminative deoxofluorination allowed the formation of cyclic monofluoroalkenes.
The second part of the project focused on electrophilic fluorination. In this case, the substrate behaves as the nucleophile whereas the fluorine atom is delivered as an electrophile. Given the limitations of the current reagents (commercial availability, solubility in organic solvents, reactivity, etc.), the objective is to develop new electrophilic fluorine sources that can address these issues. More particularly, N-fluorosquaramides derivatives were briefly investigated.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... viii
Liste des schémas ... ix
Liste des tableaux ... xi
Liste des abréviations ... xii
Remerciements ... xiv
Avant-propos ... xvi
CHAPITRE 1 Introduction ... 1
1.1 LE FLUOR EN CHIMIE ORGANIQUE ... 1
1.1.1 Propriétés physico-chimiques ... 1
1.1.2 Applications en chimie organique ... 3
1.2 MÉTHODES DE FLUORATION ... 6 1.2.1 Fluoration nucléophile ... 6 1.2.2 Fluoration électrophile ... 10 1.2.3 Fluoration radicalaire ... 13 1.3 LE XTALFLUOR ... 14 1.3.1 Découverte du XtalFluor ... 14 1.3.2 Méthodes de synthèse ... 16 1.3.3 Propriétés ... 18 1.3.4 Réactif de fluoration ... 20 1.3.5 Agent activant ... 26 1.4 OBJECTIFS DE LA THÈSE ... 35
1.4.1 Utilisation du XtalFluor-E en synthèse organique ... 35
1.4.2 Développement de nouveaux réactifs de fluoration électrophile ... 37
CHAPITRE 2 Synthèse d’isonitriles par déshydratation de formamides en utilisant le XtalFluor-E Synthesis of isocyanides through dehydration of formamides using XtalFluor-E 38 2.1 RÉSUMÉ ... 39
2.2 ABSTRACT ... 39
2.3 INTRODUCTION ... 39
2.4 RESULTS AND DISCUSSION ... 42
2.5 CONCLUSION ... 49
2.6 ACKNOWLEDGMENTS ... 49
2.7 SUPPORTING INFORMATION AVAILABLE ... 49
2.7.1 General information ... 49
2.7.2 Synthesis of the new formamides ... 50
2.7.3 Synthesis of isocyanides ... 54
2.7.4 Multi-component reactions ... 57
CHAPITRE 3 Synthèse de nitriles à partir d’aldoximes et d’amides primaires en utilisant le XtalFluor-E Synthesis of Nitriles from Aldoximes and Primary Amides Using XtalFluor-E ... 63
3.2 ABSTRACT ... 64
3.3 INTRODUCTION ... 65
3.4 RESULTS AND DISCUSSION ... 67
3.5 CONCLUSION ... 73
3.6 ACKNOWLEDGMENTS ... 73
3.7 SUPPORTING INFORMATION AVAILABLE ... 74
3.7.1 General information ... 74
3.7.2 Synthesis of aldoximes ... 74
3.7.3 Synthesis of amides ... 78
3.7.4 Synthesis of nitriles ... 80
CHAPITRE 4 Estérification directe d’acides carboxyliques avec des alcools perfluorés, effectuée par l’entremise de XtalFluor-E Direct Esterification of Carboxylic Acids with Perfluorinated Alcohols Mediated by XtalFluor-E ... 88
4.1 RÉSUMÉ ... 89
4.2 ABSTRACT ... 89
4.3 INTRODUCTION ... 90
4.4 RESULTS AND DISCUSSION ... 92
4.5 CONCLUSION ... 99
4.6 ACKNOWLEDGMENTS ... 100
4.7 ANNEXE ... 100
4.7.1 Estérification de l’acide 5-phénylvalérique avec des alcools non fluorés ... 100
4.7.2 Étude du mécanisme par chimie computationnelle ... 101
4.8 SUPPORTING INFORMATION AVAILABLE ... 102
4.8.1 General information ... 102
4.8.2 Esterification mediated by XtalFluor-E using perfluorinated alcohols ... 103
4.8.3 Control experiments ... 119
4.9 PARTIE EXPÉRIMENTALE DES RÉSULTATS NON PUBLIÉS (SECTION 4.7) ... 121
4.9.1 Estérification de l’acide 5-phénylvalérique avec des alcools non fluorés ... 121
4.9.2 Méthodes computationnelles ... 123
CHAPITRE 5 Déoxofluoration éliminatrice au moyen de XtalFluor-E : Synthèse de monofluoroalcènes en une étape à partir de dérivés de cyclohexanone Eliminative Deoxofluorination Using XtalFluor-E: A One-Step Synthesis of Monofluoroalkenes from Cyclohexanone Derivatives ... 124
5.1 RÉSUMÉ ... 125
5.2 ABSTRACT ... 125
5.3 INTRODUCTION ... 125
5.4 RESULTS AND DISCUSSION ... 129
5.5 CONCLUSION ... 135
5.6 ACKNOWLEDGMENTS ... 135
5.7 ANNEXE ... 135
5.7.1 Optimisation : Résultats complémentaires ... 135
5.7.2 Étendue de la réaction : discussion ... 138
5.8 SUPPORTING INFORMATION AVAILABLE ... 140
5.8.1 General information ... 140
5.8.2 Additional optimization results ... 142
CHAPITRE 6 Vers la synthèse de N-fluorosquaramides en tant que nouveaux
réactifs de fluoration électrophile ... 150
6.1 INTRODUCTION ... 150
6.1.1 Propriétés des squaramides ... 150
6.1.2 N-fluorosquaramides ... 151
6.1.3 Potentiel de fluoration électrophile des N-fluorosquaramides ... 152
6.1.4 Potentiel de fluoration radicalaire des N-fluorosquaramides ... 155
6.2 RÉSULTATS ET DISCUSSION ... 156
6.3 CONCLUSION ... 161
6.4 MÉTHODES COMPUTATIONNELLES ... 162
6.5 PARTIE EXPÉRIMENTALE ... 162
6.5.1 Informations générales ... 162
6.5.2 Synthèse des produits de départ ... 163
6.5.3 Tentatives de fluoration, chloration et bromation des squaramides ... 166
6.5.4 Méthylation des squaramides ... 167
6.5.5 Expériences de deutération ... 169
CHAPITRE 7 Conclusion et perspectives ... 170
7.1 RETOUR SUR LES OBJECTIFS ... 170
7.1.1 Utilisation du XtalFluor-E en synthèse organique ... 170
7.1.2 Développement de nouveaux réactifs de fluoration électrophile ... 171
7.2 PERSPECTIVES ... 171
7.2.1 Étendre la liste des réactions utilisant le XtalFluor-E comme agent activant 171 7.2.2 Réactifs de fluoration électrophile : concevoir de nouvelles structures ... 173
Liste des figures
Figure 1.1. Exemples de médicaments fluorés. ... 4
Figure 1.2. Exemples de produits agrochimiques fluorés. ... 5
Figure 1.3. Exemples de réactifs de fluoration nucléophile. ... 8
Figure 1.4. Réactifs de déoxofluoration. ... 10
Figure 1.5. Exemples de réactifs de fluoration électrophile. ... 12
Figure 1.6. Réactifs de fluoration électrophile récents. ... 13
Figure 1.7. Thermogrammes DSC du DAST, Deoxo-Fluor, XtalFuor-E et XtalFluor-M. Figure tirée de la référence 31b. ... 19
Figure 1.8. Structure des N-fluorosquaramides. ... 37
Figure 2.1. Activation of amides with [Et2NSF2]BF4 for the synthesis of 1,3,4-oxadiazoles and isocyanides. ... 41
Figure 2.2. Unproductive formamides. ... 45
Figure 2.3. Mechanistic proposal for the dehydration reaction. The BF4- counter-ion has been omitted for clarity. ... 46
Figure 3.1. Activation of amides and aldoximes with XtalFluor-E for the synthesis of nitriles. ... 67
Figure 5.1. Ensemble des cétones pour lesquelles la fluoration n’a pas été possible. ... 139
Figure 6.1. Structure des squaramides. ... 151
Figure 6.2. N-Fluorosquaramides et autres réactifs de fluoration électrophile. ... 152
Figure 6.3. Valeurs de FPD des principales classes de réactifs de fluoration électrophile dans le dichlorométhane. Figure tirée de la référence 241. ... 153
Figure 6.4. Comparaison des valeurs calculées de FPD (kcal/mol) des N-fluorosquaramides avec celles du NFSI et du Selectfluor dans le dichlorométhane et l’acétonitrile. ... 154
Figure 6.5. Valeurs de BDE des principales classes de réactifs de fluoration N–F dans l’acétonitrile. Figure tirée de la référence 243. ... 155
Figure 6.6. Comparaison des valeurs calculées de BDE (kcal/mol) des N-fluorosquaramides avec celles du NFSI et du Selectfluor dans l’acétonitrile. ... 156
Figure 6.7. Squaramides modèles. ... 157
Liste des schémas
Schéma 1.1. Fluoration nucléophile. ... 7
Schéma 1.2. Réaction de déoxofluoration. ... 9
Schéma 1.3. Fluoration électrophile. ... 11
Schéma 1.4. Synthèse de sels de dialkylaminodifluorosulfinium au moyen de BF3·Et2O. . 14
Schéma 1.5. Premier exemple d’utilisation d’un sel de dialkylaminodifluorosulfinium comme agent de déoxofluoration. ... 15
Schéma 1.6. Méthodes de synthèse du XtalFluor-E. ... 17
Schéma 1.7. Synthèse de sels de diéthylaminodifluorosulfinium à partir d’acides de Brønsted. ... 17
Schéma 1.8. Synthèse de fluorures d’alkyle par déoxofluoration d’alcools. ... 21
Schéma 1.9. Mécanismes de déoxofluoration d’un alcool avec le DAST et le XtalFluor-E. ... 22
Schéma 1.10. Formation de l’intermédiaire diéthylaminodifluorosulfane 1.3. ... 23
Schéma 1.11. Synthèse de difluorométhylènes par déoxofluoration de carbonyles. ... 24
Schéma 1.12. Synthèse de fluorures d’acyle par déoxofluoration d’acides carboxyliques. . 24
Schéma 1.13. Synthèse de fluorures de glycosyle. ... 25
Schéma 1.14. Synthèse d’un fluorure de sulfonyle. ... 26
Schéma 1.15. Utilisation du XtalFluor-E comme agent activant. ... 26
Schéma 1.16. Expansion de cycles dérivés de prolinol. ... 27
Schéma 1.17. Synthèse de 1,3,4-oxadiazoles à partir de 1,2-diacylhydrazines. ... 28
Schéma 1.18. Synthèse d’oxazolines à partir d’hydroxyamides. ... 29
Schéma 1.19. Synthèse d’oxazolines par désilylation in situ et cyclodéshydratation. ... 29
Schéma 1.20. Synthèse d’oxazolines par ouverture d’oxiranes. ... 30
Schéma 1.21. Halogénation d’alcools primaires. ... 30
Schéma 1.22. Amidation d’acides carboxyliques décrite par le groupe de Cossy. ... 31
Schéma 1.23. Amidation d’acides carboxyliques décrite par le groupe de Paquin. ... 31
Schéma 1.24. Aminofluoration intramoléculaire catalysée au fer. ... 32
Schéma 1.25. Aminofluoration intermoléculaire catalysée au fer. ... 33
Schéma 1.26. Synthèse de dérivés d’imidazolidinone par ouverture d’aziridines. ... 33
Schéma 1.27. Synthèse de diaryl- et triarylméthanes par benzylation de Friedel-Crafts. .... 34
Schéma 1.28. Allylation d’alcools benzyliques. ... 35
Schéma 1.29. Synthèse d’isonitriles à partir de formamides. ... 35
Schéma 1.30. Synthèse de nitriles à partir d’amides primaires ou d’aldoximes. ... 36
Schéma 1.31. Synthèse d’esters perfluorés à partir d’acides carboxyliques et d’alcools perfluorés. ... 36
Schéma 1.32. Synthèse de monofluoroalcènes à partir de cétones. ... 37
Scheme 2.1. Synthesis of N-formyl amides with crude isocyanides. ... 48
Scheme 2.2. Ugi-Smiles with a crude isocyanide. ... 48
Scheme 3.1. Initial results for the dehydration of 3.1 and 3.3 using XtalFluor-E. ... 68
Scheme 3.2. Synthesis of aromatic nitriles (3.6) from aldoximes (3.4) or primary amides (3.5). ... 69
Scheme 3.3. Synthesis of vinylic nitrile 3.9 from cinnamic acid derivatives 3.7 and 3.8. ... 70
Scheme 3.4. Synthesis of aliphatic nitriles from aldoximes (3.10) or primary amides (3.11). ... 71
Scheme 3.5. Synthesis of chiral nonracemic nitriles from primary amides derived from
protected amino acids. ... 72
Scheme 3.6. Synthesis of chiral nonracemic nitriles from L-mandelic acid and L-lactic acid and derivatives. ... 73
Scheme 4.1. Previous Work and the Current Method. ... 91
Scheme 4.2. Results for the Esterification of Various Carboxylic Acids with TFE Using XtalFluor-E.a,b ... 95
Scheme 4.3. Selected Results for the Esterification of Various Carboxylic Acids with Perfluorinated Alcohols Using XtalFluor-E.a,b ... 97
Scheme 4.4. Control Experiments and Mechanistic Hypothesis.a ... 99
Schéma 4.5. Estérification de l’acide 5-phénylvalérique (4.2) avec des alcools non fluorés. ... 101
Schéma 4.6. Voies mécanistiques possibles. ... 101
Schéma 4.7. Équilibre de l’intermédiaire 5.57 avec sa forme dissociée 5.58. ... 102
Scheme 5.1. Previous and Current Work. ... 127
Scheme 5.2. Eliminative Deoxofluorination of Various Cyclohexanone Derivatives Using XtalFluor-E.a,b ... 132
Scheme 5.3. Mechanistic Hypothesis.a ... 134
Schéma 5.4. Équilibre conformationnel entre les formes A et B de l’intermédiaire 5.6. ... 140
Schéma 6.1. Réactivité attendue des N-fluorosquaramides envers les énolates. ... 152
Schéma 6.2. Dissociation hétérolytique de réactifs de fluoration électrophile de type N–F. ... 153
Schéma 6.3. Dissociation homolytique de réactifs de fluoration électrophile de type N–F. ... 155
Schéma 6.4. Stratégie de synthèse des squaramides. ... 157
Schéma 6.5. Voies de fluoration des squaramides. ... 158
Schéma 6.6. Méthylation des squaramides. ... 160
Schéma 6.7. Chloration des squaramides. ... 160
Schéma 6.8. Bromation du squaramide 6.4. ... 161
Schéma 6.9. Deutération du squaramide 6.4. ... 161
Schéma 7.1. Utilisation du XtalFluor-E pour la synthèse d’isonitriles, nitriles, esters perfluorés et monofluoroalcènes cycliques. ... 171
Schéma 7.2. Synthèse de monofluoroalcènes à partir de cétones. ... 172
Schéma 7.3. Benzylation de Friedel-Crafts par activation de dérivés de phénylcyclopropanol. ... 172
Liste des tableaux
Tableau 1.1. Comparaison des propriétés atomiques de l’atome de fluor avec les atomes
d’hydrogène, chlore, brome et iode. ... 2
Tableau 1.2. Comparaison des caractéristiques des liaisons C–X. ... 3
Tableau 1.3. Comparaison des propriétés du DAST avec celles du XtalFluor-E. ... 18
Table 2.1. Selected optimization results for the dehydration of formamide 2.1. ... 42
Table 2.2. Scope of the dehydration of formamides with XtalFluor-E.a ... 44
Table 2.3. Synthesis of α-acyloxyamide using crude isocyanides.a,b ... 47
Table 4.1. Optimization Results for the Esterification of 5-Phenylvaleric acid (4.2) with TFE Using XtalFluor-E.a ... 93
Table 5.1. Key Optimization Results for the Eliminative Deoxofluorination of 5.1a Using XtalFluor-E.a ... 130
Tableau 5.2. Étude de l’influence de la température. ... 136
Tableau 5.3. Étude de l’influence du nombre d’équivalents de XtalFluor-E. ... 137
Tableau 5.4. Étude de l’influence de l’ordre et du temps d’ajout des réactifs. ... 138
Table 5.5. Additional Optimization Results for the Eliminative Deoxofluorination of 5.1a Using XtalFluor-E. ... 142
Liste des abréviations
Ac acétyle
Ar aryle
BDE bond dissociation enthalpy
Bn benzyle Boc tert-butoxycarbonyle Bu butyle Bz benzoyle Cbz carboxybenzyle Cys cystéine
DAST trifluorure de diéthylaminosulfure DBU 1,8-diazabicyclo[5.4.0]undéc-7-ène DCE 1,2-dichloroéthane
DIBAl-H hydrure de diisobutylaluminium DIPEA N,N-diisopropyléthylamine DMA diméthylacétamide DME 1,2-diméthoxyéthane DMF diméthylformamide DMPU 1,3-diméthyl-3,4,5,6-tétrahydro-2(1H)-pyrimidinone DMSO diméthylsulfoxyde
DSC calorimétrie différentielle à balayage
ee excès énantiomérique
équiv. équivalent
Et éthyle
FPD fluorine plus detachment
HFIP 1,1,1,3,3,3-hexafluoroisopropan-2-ol HMDS hexaméthyldisilamidure
HPLC chromatographie en phase liquide à haute pression
i-Pr iso-propyle Me méthyle MOM méthoxyméthyle MS tamis moléculaire NFSI N-fluorobenzènesulfonimide NMP N-méthyl-2-pyrrolidone
NMR résonance magnétique nucléaire (nuclear magnetic resonance)
Nu nucléophile
Ph phényle
PMB p-méthoxybenzyle
PTFE polytétrafluoroéthylène
RMN résonance magnétique nucléaire
rt température ambiante (room temperature)
Ser sérine
SMD modèle de solvatation basé sur la densité SN substitution nucléophile
t-Bu tert-butyle
TBAF fluorure de tétrabutylammonium
TBS tert-butyldiméthylsilyle
temp. température TES triéthylsilyle
Tf trifluorométhylsulfonyle TFA acide trifluoroacétique TFE 2,2,2-trifluoroéthanol
THF tétrahydrofurane
Tr trityle
TrisNHNH2 hydrazide de 2,4,6-triisopropylbenzènesulfonyle
Remerciements
Je tiens à remercier chaleureusement mon directeur de thèse, le professeur Jean-François Paquin, de m’avoir accueillie dans son laboratoire afin que je puisse y réaliser ma thèse, dans le domaine stimulant de la chimie des composés organofluorés. Je le remercie pour sa grande disponibilité, son soutien et ses conseils judicieux, mais aussi de m’avoir partagé son inestimable expérience dans ce domaine. Je souhaite également le remercier de m’avoir donné l’occasion d’écrire des articles dans des journaux renommés, et d’assister à de nombreux congrès.
Je remercie le professeur André Beauchemin d’avoir accepté d’évaluer cette thèse en tant qu’examinateur externe, ainsi que les professeurs Denis Giguère et Thierry Ollevier en tant qu’examinateurs internes.
Je souhaite ensuite remercier tous les membres du laboratoire, passés et présents, avec lesquels j’ai partagé de bons moments et des discussions enrichissantes au quotidien. De près ou de loin, ils m’ont tous aidée à devenir la chercheuse que je suis aujourd’hui. Un énorme merci à Massaba, qui a grandement contribué au projet relatif au XtalFluor, ainsi qu’à Eliane, Audrey et Léa, qui ont chacune apporté leur pierre à l’édifice. Un merci particulier à Elsa, qui m’a très bien accueillie dès mon arrivée à Québec et avec qui j’ai passé d’excellents moments, que ce soit au sein du labo ou en dehors. Merci aussi à Myriam et Audrey, avec qui j’ai eu beaucoup de fun lors des nombreuses soirées et sorties de lab, à PA et JD d’avoir toujours été disponibles lorsque j’avais des questions ou que je doutais de moi, à Justine d’avoir été une super voisine de hotte tout au long de mon doctorat, à Paul pour ses conseils en matière de calculs ab initio, à Majdouline pour sa gentillesse infinie, et à Marius, toujours partant pour une bière ou un barbecue.
Je remercie aussi tous les employés du département, qui ont toujours été disponibles et d’une aide précieuse. Je pense notamment à Pierre Audet, Christian Côté, Jean Laferrière, Mélanie Tremblay, Denyse Michaud et Marie Tremblay.
Cette thèse étant la raison de ma présence à Québec, je me dois également de remercier toutes les personnes qui m’ont entourée, encouragée, et ont formé ma « famille
québécoise ». Je pense ici à JF, mon compatriote belge, mais aussi à tous les membres de « Lundi BBQ », pour les très nombreuses soirées du vendredi, soirées film et sorties, ainsi que les multiples brunchs de lendemain de veille, appart’athons, week-ends en nature, et … barbecues du lundi. Merci donc à Alina, Laurence, Marjo, Arnaud, Fanny, Ben, Sean, Sinen, Romain, Steph, PJ, Julia, Anne-So, Julien, Nizar, Larbi, les quatre Marseillais et tous les autres. Je remercie également les nombreux colocs qui sont passés par le 524 Richelieu, et plus particulièrement Manon et Belén, qui ont toujours été là pour me remonter le moral dans les instants plus difficiles, et avec qui j’ai partagé énormément de moments inoubliables. J’espère sincèrement les revoir de l’autre côté de l’Atlantique : en Belgique, en France ou en Espagne.
Je tiens aussi à adresser mes remerciements à mes amis de Belgique, qui m’ont toujours soutenue à distance. Merci à Kala, Kiki et Pooka pour les nombreux skypes copines, et aux Quentin’s de s’être inquiétés de ma survie au Canada depuis le début…
Enfin, je remercie ma famille pour leurs encouragements et leurs visites à Québec. Un merci plus particulier à mes parents, qui m’ont constamment soutenue, que ce soit par e-mail, whatsapp, skype, par l’envoi de cartes ou de chocolat, et qui ont traversé l’Atlantique pour assister à la soutenance de ma thèse.
Avant-propos
Les travaux de recherche présentés ci-après font l’objet d’une collaboration avec la compagnie québécoise OmegaChem, spécialisée dans le développement de molécules de synthèse. Ces travaux sont financés par une subvention de recherche et développement coopérative (RDC) du CRSNG.
Cette thèse est divisée en 7 chapitres. Une introduction générale présente d’abord l’état de l’art de la chimie du fluor et du XtalFluor, et relie les différents chapitres qui vont suivre entre eux. Les chapitres 2 à 5 sont constitués d’articles scientifiques auxquels j’ai contribué de manière significative. Le texte et les figures de ces articles ont été recopiés sans modifications, excepté la numérotation des schémas, tableaux, figures et molécules. Le chapitre 6 décrit ensuite des résultats non publiés, qui nécessitent encore des recherches plus approfondies. Enfin, la thèse se conclut par un retour aux objectifs et la mise en place de perspectives.
Le chapitre 2 est tiré d’un article intitulé « Synthesis of isocyanides through dehydration of formamides using XtalFluor-E » paru dans Tetrahedron Letters et publié en ligne le 4 décembre 2014. L’optimisation de la réaction a été effectuée par Massaba Keita et Olivier Mahé, tous deux stagiaires postdoctoraux. J’ai réalisé une partie des expériences nécessaires à l’étendue de la réaction, en partenariat avec Massaba. Nous avons également toutes les deux contribué à la préparation du document d’informations supplémentaires. Le manuscrit a été rédigé principalement par mon directeur de thèse, le professeur Jean-François Paquin.
Le chapitre 3 provient d’un article intitulé « Synthesis of Nitriles from Aldoximes and Primary Amides Using XtalFluor-E » paru dans Synthesis et publié en ligne le 20 août 2015. Les premiers tests ont été réalisés par Massaba Keita, puis les expériences nécessaires à l’étendue de la réaction (synthèse des substrats de départ et formation des nitriles) ont été partagées entre Massaba et moi-même. Nous avons également écrit la partie expérimentale ensemble, tandis que le manuscrit a été rédigé principalement par mon directeur Jean-François Paquin.
Le chapitre 4 est issu d’un article intitulé « Direct Esterification of Carboxylic Acids with Perfluorinated Alcohols Mediated by XtalFluor-E » paru dans Organic Letters et publié en ligne le 7 décembre 2016. Massaba Keita a participé à l’optimisation de la réaction, tandis que Léa Bouchard et Audrey Gilbert, toutes deux stagiaires au baccalauréat, ont synthétisé quelques esters perfluorés. Quant à moi, j’ai orchestré le projet en complétant l’optimisation, et en effectuant la majeure partie des expériences nécessaires à l’étendue de la réaction et à l’étude du mécanisme. J’ai également préparé le document d’informations supplémentaires, avec la contribution de Léa et Audrey. Le manuscrit a été principalement rédigé par mon directeur Jean-François Paquin. La partie annexe comporte les résultats de Massaba et Audrey concernant les esters non fluorés, et l’étude du mécanisme de la réaction par chimie computationnelle a été réalisée par moi-même.
Quant au chapitre 5, il est tiré d’un article intitulé « Eliminative Deoxofluorination Using XtalFluor-E: A One-Step Synthesis of Monofluoroalkenes from Cyclohexanone Derivatives » paru dans Organic Letters et publié en ligne le 27 juin 2017. J’ai réalisé l’entièreté des expériences nécessaires à la publication de cet article, de même que la préparation du document d’informations supplémentaires. Le manuscrit a été rédigé principalement par mon directeur Jean-François Paquin. Les résultats annexes décrivent des expériences effectuées par moi-même.
Enfin, le chapitre 6 est composé de résultats non publiés issus d’expériences que j’ai réalisées seule, y compris les calculs ab initio. Même si ces expériences n’ont pas toutes abouti à des résultats concluants, cela constitue néanmoins des données intéressantes à prendre en compte lors d’une poursuite éventuelle du projet.
CHAPITRE 1
Introduction
1.1 L
E FLUOR EN CHIMIE ORGANIQUELorsque le fluor est évoqué, Monsieur Tout-le-Monde a tendance à penser au dentifrice, et plus particulièrement aux fluorures qui permettent de renforcer l’émail des dents et limiter l’apparition de caries. Selon certains, le fluor est même toxique, il est responsable de tous les maux et les industries pharmaceutiques nous empoisonnent !1 C’est dans de telles situations que le rôle du scientifique intervient : informer la population, discerner le vrai du faux, et faire la distinction entre les ions fluorures et les molécules organofluorées. Ces dernières comportent un atome de fluor lié de manière covalente au reste de la molécule et peuvent s’avérer précieuses pour de multiples applications, au vu des propriétés exceptionnelles de l’atome de fluor.
1.1.1 Propriétés physico-chimiques
L’intérêt porté par de nombreux chimistes sur l’atome de fluor s’explique par les propriétés extrêmes de cet atome.2,3 Le fluor est l’élément le plus léger de la série des halogènes, mais aussi le plus électronégatif du tableau périodique (3,98 sur l’échelle de Pauling) (Tableau
(1) Exemple typique d’information erronée circulant sur le web : Le fluor toxique règne dans la pharmacie. http://www.alterinfo.net/Le-fluor-toxique-regne-dans-la-pharmacie_a81311.html, consulté le 31 mai 2017. (2) Livres généraux sur la chimie des molécules organofluorées : (a) Kirsh, P. Introduction. Dans Modern
Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2013. (b)
Hiyama, T. Organofluorine Compounds; Chemistry and Applications; Yamamoto, H. (Éd.): Springer, 2000. (3) Revues : (a) Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. (b) O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319.
1.1).4 Selon les estimations de Bondi, son rayon de van der Waals est de 1,47Å, ce qui en fait le plus petit atome après l’hydrogène (1,2 Å).5 Il s’agit également de l’atome le moins polarisable, avec une polarisabilité qui s’élève à 0,557 Å3.6
Tableau 1.1. Comparaison des propriétés atomiques de l’atome de fluor avec les atomes
d’hydrogène, chlore, brome et iode.
Atome Électronégativité Rayon de
van der Waals (Å) Polarisabilité (Å 3) H 2,2 1,2 0,667 F 3,98 1,47 0,557 Cl 3,16 1,75 2,18 Br 2,96 1,85 3,05 I 2,66 1,98 4,7
Les liaisons C–F sont également dotées de propriétés exceptionnelles grâce au recouvrement presque optimal des orbitales 2s et 2p de l’atome de fluor avec celles du carbone.3 De ce fait, le lien C–F (1,38 Å) se classe en deuxième position, juste après le lien C–H (1,09 Å) en matière de longueur de liaison (Tableau 1.2). La liaison C–F comporte également une très grande énergie de liaison (115,7 kcal/mol), ce qui en fait la liaison simple la plus forte que peut faire le carbone avec n’importe quel autre atome. Ceci s’explique par la grande différence d’électronégativité entre ces deux atomes, conférant ainsi un caractère fortement ionique à la liaison. De la même façon, cela rend la liaison très polarisée, avec un moment dipolaire typique qui avoisine 1,41 D.
(4) Sen, K. D.; Jorgensen, C. K. Electronegativity, Springer-Verlag, New York, 1987. (5) Bondi, A. J. Phys. Chem. 1964, 68, 441–451.
Tableau 1.2. Comparaison des caractéristiques des liaisons C–X. Atome X liaison C–X (Å) Longueur de
Énergie de liaison C–X (kcal/mol) Moment dipolaire C–X (D) H 1,09 98,0 0,4 F 1,38 115,7 1,41 Cl 1,77 77,2 1,46 Br 1,94 64,3 1,38 I 2,13 50,7 1,19
Les propriétés physiques extrêmes de l’atome de fluor énumérées ci-dessus mènent également à des propriétés chimiques intéressantes.2,3 À cause de son effet inductif électroattracteur non négligeable, l’introduction d’un atome de fluor dans une molécule augmente de manière considérable l’acidité des groupements ionisables à proximité, tout en diminuant la basicité des groupements fonctionnels voisins. On remarque aussi une influence sur la lipophilie puisque d’une manière générale, les molécules fluorées auront tendance à être plus lipophiles que leur analogue non fluoré. Enfin, l’atome de fluor peut être impliqué dans des ponts hydrogène en tant qu’accepteur de liaisons hydrogène.7 Cependant, ces liaisons sont de faible énergie étant donné la petite polarisabilité de la liaison C–F et des doublets du fluor, qui ne contribue ainsi que moindrement au transfert d’électrons.
1.1.2 Applications en chimie organique
1.1.2.1 Chimie médicinale
Les molécules fluorées sont omniprésentes en chimie médicinale puisque environ 25 % des médicaments actuellement sur le marché comportent au moins un atome de fluor (Figure 1.1).8 L’insertion d’un atome de fluor dans une molécule altère de manière significative ses
(7) Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306–322.
(8) Revues : (a) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. (b) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. (c) Kirk, K. L. Org. Process. Res. Dev. 2008, 12, 305–321. (d) Wang, J.; Sánchez-Roselló, M.; Luis Aceña, J.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.;
propriétés chimiques et peut ainsi augmenter son affinité avec sa cible biologique (par des interactions dipôle-dipôle, ponts H, etc.), mais également améliorer ses propriétés pharmacocinétiques. Les molécules fluorées étant bien souvent plus lipophiles que leur analogue non fluoré, le passage à travers les bicouches lipidiques sera facilité et le médicament aura une meilleure biodisponibilité. D’autre part, la force de la liaison C–F ainsi que l’effet électroattracteur de l’atome de fluor permettent à la molécule de mieux résister aux divers processus métaboliques impliqués (métabolismes oxydatif et hydrolytique).
Figure 1.1. Exemples de médicaments fluorés.
En outre, les molécules fluorées sont utilisées comme sondes biochimiques pour l’étude de divers processus biologiques, et le fait que les noyaux 19F soient actifs en RMN permet l’utilisation de l’imagerie par résonance magnétique (IRM) in vivo.9
Enfin, la tomographie par émission de positons (TEP) utilisant des molécules marquées au fluor radioactif 18F permet de diagnostiquer, localiser et détecter la récurrence et la progression de diverses maladies telles que le cancer.10
Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. (e) Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359.
(9) Revues: (a) Bartusik, D.; Tomanek, B. Adv. Drug Deliv. Rev. 2013, 65, 1056–1064. (b) Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. Chem. Soc. Rev. 2013, 42, 7971–7982.
N CO2H OH OH Ph PhHNO2C i-Pr F Atorvastatine Lipitor®
régulateur du métabolisme lipidique
MeHN O Ph CF3 N N O CO2H F HN Ciprofloxacine Ciprobay® antibactérien Fluoxétine Prozac® antidépresseur
1.1.2.2 Agrochimie
L’effet des substituants fluorés sur l’activité biologique des systèmes organiques a également été largement utilisé dans le domaine de l’agrochimie.11 Ainsi, de nombreux herbicides, insecticides et fongicides comportent au moins un atome de fluor au sein de leur structure, bien souvent sous forme de fluorure d’aryle ou d’aryltrifluorométhyle (Figure 1.2).
Figure 1.2. Exemples de produits agrochimiques fluorés.
1.1.2.3 Chimie des matériaux
Depuis la découverte du polytétrafluoroéthylène (PTFE) en 1930, de nombreux polymères fluorés ont été exploités dans le domaine des matériaux, grâce à leur grande stabilité chimique et thermique.11e,12 Ceux-ci se retrouvent par exemple utilisés pour des
(10) Fletcher, J. W.; Djulbegovic, B.; Soares, H. P.; Siegel, B. A.; Lowe, V. J.; Lyman, G. H.; Coleman, R. E.; Wahl, R.; Paschold, J. C.; Avril, N.; Einhorn, L. H.; Suh, W. W.; Samson, D.; Delbeke, D.; Gorman, M.; Shields, A. F. J. Nucl. Med. 2008, 49, 480–508.
(11) (a) Jeschke, P. ChemBioChem 2004, 5, 570–589. (b) Jeschke, P. Pest. Manag. Sci. 2010, 66, 10–27. (c) Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.-P.; Leroux, F. R. J. Fluorine Chem. 2013, 152, 2–11. (d) Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. (e) Harsanyi, A.; Sandford, G. Green
Chem. 2015, 17, 2081–2086.
(12) Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508.
NO2 F3C NO2 N F F N H O N H O Cl F Cl F N OMe O MeO O N CF3 Teflubenzuron Nomolt® insecticide Trifloxystrobine Flint® fongicide Benfluraline Balan® herbicide
revêtements de poêles (PTFE, Teflon®), des vêtements imperméables (PTFE, Goretex®), des réfrigérants (chlorofluorocarbones) et dans des cellules photovoltaïques.13
1.2 M
ÉTHODES DE FLUORATIONLes molécules organofluorées sont très peu abondantes naturellement : seuls 19 composés ont été identifiés à ce jour.14 Ceci incite donc fortement les chimistes à consacrer leurs recherches au développement de nouvelles méthodes de fluoration, qui sont maintenant largement décrites dans la littérature.15
L’introduction d’un atome de fluor peut être réalisée par trois approches différentes, qui varient selon la nature électronique du fluor (nucléophile, électrophile ou radicalaire). Les réactions de fluoration électrophile et nucléophile sont les méthodes les plus courantes, bien que la fluoration radicalaire émerge comme une alternative viable.16 Nous nous consacrerons ici essentiellement aux fluorations nucléophile et électrophile.
1.2.1 Fluoration nucléophile
En fluoration nucléophile, l’atome de fluor est fourni sous forme de fluorure, tandis que le substrat joue le rôle de l’électrophile.15,17 Il s’agit bien souvent d’une simple substitution
(13) Xu, X.-P.; Li, Y.; Luo, M.-M.; Peng, Q. Chin. Chem. Lett. 2016, 27, 1241–1249. (14) Walker, M. C.; Chang, M. C. Y. Chem. Soc. Rev. 2014, 43, 6527–6536.
(15) Revues récentes : (a) Prakash, C. K. S.; Wang, F.; O’Hagan, D.; Hu, J.; Ding, K.; Dai, L.-X. Flourishing Frontiers in Organofluorine Chemistry. Dans Organic Chemistry – Breakthroughs and Perspectives; Ding, K., Dai, L.-X., Eds.; Wiley-VCH: Weinheim, Germany, 2012. (b) Liang, T.; Neumann, C. N.; Ritter, T.
Angew. Chem., Int. Ed. 2013, 52, 8214–8264. (c) Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev.
2015, 115, 826–870. (d) Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612–633. (e) Champagne, P. A.;
Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174.
(16) (a) Rueda-Becerril, M. ; Chatalova Sazepin, C. ; Leung, J. C. T. ; Okbinoglu, T. ; Kennepohl, P. ; Paquin, J.-F. ; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. (b) Sibi, M. P.; Landais, Y. Angew. Chem.,
Int. Ed. 2013, 52, 3570–3572. (c) Chatalova Sazepin, C.; Hemelaere, R.; Paquin, J.-F.; Sammis, G. Synthesis
2015, 47, 2554–2569.
(17) Revues récentes consacrées à la fluoration nucléophile: (a) Hollingworth, C.; Gouverneur, V. Chem.
nucléophile au cours de laquelle une chaîne alkyle ou un cycle aromatique comportant un groupe partant réagit avec une source de fluorure (Schéma 1.1).
Schéma 1.1. Fluoration nucléophile.
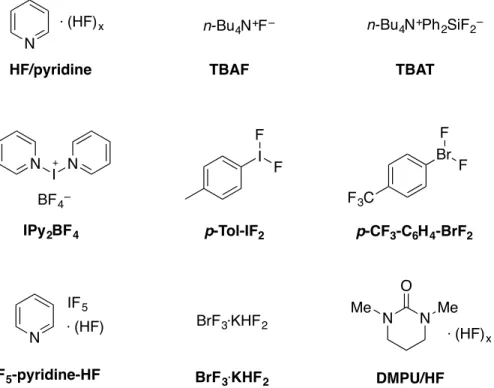
De nombreuses sources de fluorure ont été utilisées au fil des années. Ainsi, les réactifs de fluoration traditionnels sont essentiellement des fluorures de métaux alcalins (KF, CsF), des réactifs à base de HF (HF/pyridine, Et3N·3HF), des silicates et des stannates hypervalents fluorés et des fluorures de tétraalkylammonium (fluorure de tétrabutylammonium, abrégé TBAF) (Figure 1.3). Les dérivés d’halogène hypervalent constituent également une classe importante de réactifs de fluoration nucléophile, comme le tétrafluoroborate de bis(pyridine)iodonium(I) (IPy2BF4),18 le difluorure de para-iodotoluène (p-Tol-IF2)19 ou le difluorure de para-trifluorométhylphénylbromonium (p-CF3-C6H4-BrF2).20 Enfin, plus récemment, de nouveaux réactifs ont été développés de manière à étendre encore davantage les possibilités de fluoration nucléophile de manière plus efficace et pour un large éventail de substrats. Parmi ces nouveaux réactifs, on retrouve deux dérivés du TBAF (une version anhydre du TBAF et le TBAF(t-BuOH)4),21,22 le IF5-pyridine-HF,23 le BrF3-KHF224 et le DMPU/HF (complexe de 1,3-diméthyl-3,4,5,6-tétrahydro-2(1H)-pyrimidinone avec HF).25
(18) Barluenga, J.; Gonzalez, J. M.; Campos, P. J.; Asensio, G. Angew. Chem., Int. Ed. Engl. 1985, 24, 319– 320.
(19) Tsushima, T.; Kawada, K.; Tsuji, T. Tetrahedron Lett. 1982, 23, 1165–1168. (20) Frohn, H. J.; Giesen, M. J. Fluorine Chem. 1998, 89, 59–63.
(21) Sun, H.; DiMagno, S. G. J. Am. Chem. Soc. 2005, 127, 2050–2051.
R2 R1 X X R source de F– R2 R1 F F R X = halogénure ou sulfonate
Figure 1.3. Exemples de réactifs de fluoration nucléophile.
La réaction de déoxofluoration est un cas particulier de fluoration nucléophile et est une des stratégies les plus intéressantes pour l’introduction d’un atome de fluor au sein d’une molécule organique. Ceci s’explique par l’abondance et l’accessibilité de précurseurs comportant des fonctions alcools.26 Les produits fluorés sont obtenus en une synthèse monotope au cours de laquelle l’alcool est d’abord activé, puis subit une substitution nucléophile par un ion fluorure (Schéma 1.2).
(22) Kim, D. W.; Jeong, H.-J.; Lim, S. T.; Sohn, M.-H. Angew. Chem., Int. Ed. 2008, 47, 8404–8406.
(23) Hara, S.; Monoi, M.; Umemura, R.; Fuse, C. Tetrahedron 2012, 68, 10145–10150. (24) Shishimi, T.; Hara, S. J. Fluorine Chem. 2014, 168, 55–60.
(25) Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. (26) (a) Dax, K. Synthesis by Substitution of Hydroxy Groups in Alcohols. Dans Science of Synthesis; Percy, J. M., Éd.; Thieme Chemistry; Stuttgart, Germany, 2006; pp 71–148. (b) Al-Maharik, N.; O’Hagan, D.
Aldrichimica Acta 2011, 44, 65–75. (c) Vandamme, M.; Paquin, J.-F. Synthesis by Substitution of Hydroxy
Groups in Alcohols. Dans Science of Synthesis Knowledge Updates 2016/1; Paquin, J.-F., Éd.; Thieme Chemistry: Stuttgart, Germany, 2016; pp 395–408.
N N N ⋅ (HF)x n-Bu4N+F– n-Bu 4N+Ph2SiF2– I N I F F F3C Br F F BF4– IF5 ⋅ (HF) BrF3⋅KHF2 N N Me Me O ⋅ (HF)x
HF/pyridine TBAF TBAT
DMPU/HF BrF3⋅KHF2
IF5-pyridine-HF
Schéma 1.2. Réaction de déoxofluoration.
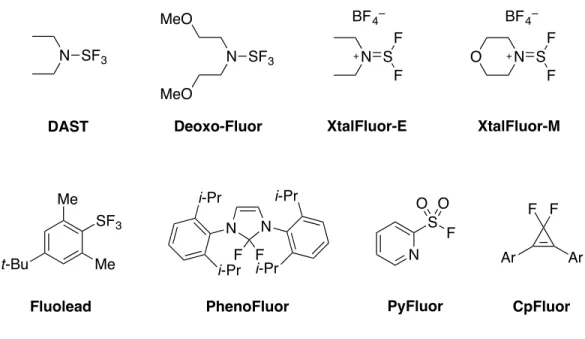
Parmi les nombreux réactifs capables d’effectuer cette transformation, le DAST27 (trifluorure de diéthylaminosulfure) et le Deoxo-Fluor28 (trifluorure de bis(2-méthoxyéthyl)aminosulfure) sont les deux réactifs commerciaux les plus couramment utilisés (Figure 1.4).29 Cependant, des inconvénients sont associés à leur utilisation. Il s’agit notamment de leur instabilité thermique, leur mauvaise chimiosélectivité et la génération de produits secondaires d’élimination. Ceci a mené ainsi au développement récent de nouveaux réactifs de déoxofluoration (Figure 1.4), tels que le Fluolead30 (trifluorure de
4-tert-butyl-2,6-diméthyl phénylsoufre), les XtalFluor-E et -M31 (sels de tétrafluoroborate d’aminodifluorosulfinium), le PhenoFluor (1,3-bis(2,6-diisopropylphényl)-2,2-difluoro-2,3-dihydro-1H-imidazole) utilisé pour les phénols32 et les alcools33, le PyFluor34 (fluorure de 2-pyridinesulfonyle) et le CpFluor35 (1,2-diaryl-3,3-difluorocyclopropène).
(27) Middleton, W. J. J. Org. Chem. 1975, 40, 574–578.
(28) Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M. Chem. Commun. 1999, 215–216. (29) Revue: Singh, R. P. ; Shreeve, J. M. Synthesis 2002, 2561–2578.
(30) Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205.
(31) (a) Beaulieu, F.; Beauregard, L.-P.; Courchesne, G.; Couturier, M.; Laflamme, F.; L’Heureux, A. Org.
Lett. 2009, 11, 5050–5053. (b) L’Heureux, A.; Beaulieu, F.; Bennet, C.; Bill, D. R.; Clayton, S.; Laflamme,
F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M. J. Org. Chem. 2010, 75, 3401–3411. (c) Mahé, O.; L'Heureux, A.; Couturier, M.; Bennett, C.; Clayton, S.; Tovell, D.; Beaulieu, F.; Paquin, J.-F. J. Fluorine
Chem. 2013, 153, 57–60.
(32) Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133, 11482–11484.
(33) Sladojevich, F.; Arlow, S. I.; Tang, P.; Ritter, T. J. Am. Chem. Soc. 2013, 135, 2470–2473. (34) Nielsen, M. K.; Ugaz, C. R.; Li, W. P.; Doyle, A. G. J. Am. Chem. Soc. 2015, 137, 9571–9574. (35) Li, L.; Ni, C.; Wang, F.; Hu, J. Nature Commun. 2016, 7, 13320.
R2 R1 OH R2 R1 F F– R2 R1 O [X] [X]–F alcool activé
Figure 1.4. Réactifs de déoxofluoration.
1.2.2 Fluoration électrophile
En fluoration électrophile, les rôles sont inversés puisque le substrat se comporte comme le nucléophile tandis que l’atome de fluor est fourni sous forme d’un équivalent de « F+ ».15,36 Le nucléophile peut aussi bien se présenter sous la forme d’un carbanion (ex : réactifs de Grignard), d’une insaturation riche en électrons (arène, alcène ou alcyne), ou d’un substrat comportant un groupement labile lié à un nucléophile (Schéma 1.3). Bien que les sources de fluor électrophile soient souvent considérées comme des sources de « F+ », elles ne génèrent véritablement aucune espèce de cette sorte puisque cela serait très défavorable d’un point de vue énergétique.
(36) Revue consacrée à la fluoration électrophile : Taylor, S. D.; Kotoris, C. C.; Hum, G. Tetrahedron 1999,
55, 12431–12477. N SF3 N SF3 MeO MeO N S F F BF4– N S F F BF4– O SF3 Me Me t-Bu N N F F i-Pr i-Pr i-Pr i-Pr
DAST Deoxo-Fluor XtalFluor-E XtalFluor-M
Fluolead PhenoFluor F F Ar Ar CpFluor N S F O O PyFluor
Schéma 1.3. Fluoration électrophile.
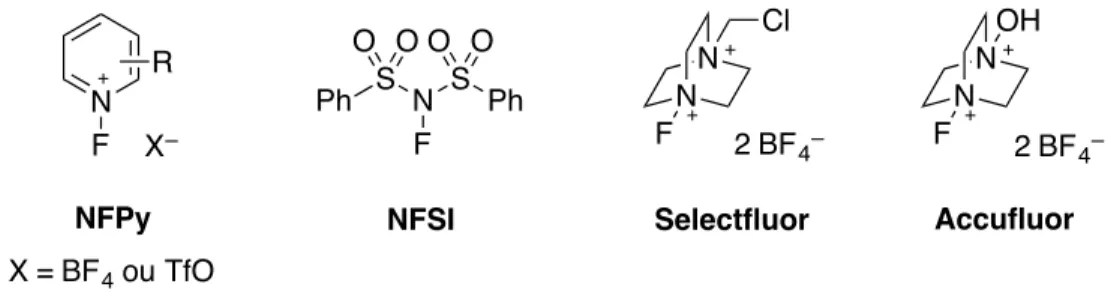
Les premiers réactifs de fluoration électrophile comportaient des liaisons O–F (ex : CH3OF, HOF, CsSO4F), Xe–F (XeF2) ou F–F (F2), mais ces composés montraient une très grande réactivité et une faible sélectivité. La plupart des sources de « F+ » utilisées de nos jours sont des réactifs dans lesquels l’atome de fluor est lié à un atome d’azote, ce qui les rend plus stables et dont la commercialisation est ainsi possible.37 Les sels de N-fluoropyridinium (NFPy)38, le NFSI (N-fluorobenzènesulfonimide),39 le Selectfluor (bis(tétrafluoroborate) de 1-chlorométhyl-4-fluoro-1,4-diazoniabicyclo[2,2,2]octane)40 et son dérivé l’Accufluor (bis(tétrafluoroborate) de 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2,2,2]octane)41 constituent les réactifs utilisés le plus couramment (Figure 1.5).
(37) Revues : (a) Lal, G. S.; Pez, G. P.; Syvret, R. G. Chem. Rev. 1996, 96, 1737–1756. (b) Baudoux, J.; Cahard, D. Org. React. 2007, 69, 1–326.
(38) Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. (39) Differding, E.; Ofner, H. Synlett 1991, 187–189.
(40) Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, L.; Syvret, R. G. J. Chem. Soc., Chem.
Commun. 1992, 595–596.
(41) Stavber, S.; Zupan, M.; Poss, A. J.; Shia, G. A. Tetrahedron Lett. 1995, 36, 6769–6772.
R source de " F + " F R X = SiR3, SnR3, BR2 ou BR3– X F R3C F R3C
Figure 1.5. Exemples de réactifs de fluoration électrophile.
Bien que ces réactifs aient démontré leur efficacité pour toutes sortes de transformations, il subsiste toujours des réactions pour lesquelles ces sources de « F+ » ne se comportent pas comme attendu (ou espéré). Ainsi, il a été observé dans certains cas un transfert de la phényle sulfone du NFSI,42 et dans d’autres cas, la réactivité des quatre molécules présentées ci-dessus diffère beaucoup. De hauts taux de conversion obtenus avec le PhSO2N(F)t-Bu peuvent donner de mauvaises conversions et sélectivités en utilisant une source commerciale de « F+ » telle que le NFSI.43 De subtiles différences dans la structure du réactif de fluoration peuvent donc mener à des résultats très différents. Enfin, un problème récurrent reste la question de la solubilité de la source de « F+ » dans le milieu réactionnel. Le Selectfluor nécessite par exemple des solvants très polaires tels que l’acétonitrile, l’eau ou le diméthylformamide (DMF), qui ne sont pas toujours compatibles avec les réactions souhaitées.
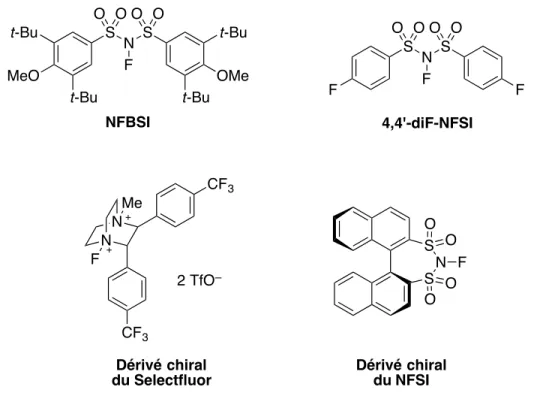
La recherche de nouveaux réactifs de fluoration électrophile doit dès lors être poursuivie, afin que ceux-ci possèdent une réactivité appropriée envers les substrats désirés et une bonne solubilité dans les solvants organiques couramment utilisés (THF, toluène, etc.). De plus, le développement de réactifs chiraux permettrait une fluoration énantiosélective. On a ainsi assisté ces dernières années à l’apparition de nouvelles sources de « F+ », comme de
(42) Snieckus, V.; Beaulieu, F.; Mohri, K.; Han, W.; Murphy, C. K.; Davies, F. A. Tetrahedron Lett. 1994,
35, 3465–3468.
(43) (a) Lee, S.-H.; Riediker, M.; Schwartz, J. Bull. Korean Chem. Soc. 1998, 19, 760–766. (b) Marterer, J.; Paquin, J.-F. Résultats non publiés.
N F R N F S S Ph Ph O O O O N N Cl F N N OH F 2 BF4– 2 BF 4– X– X = BF4 ou TfO
nouveaux dérivés de NFSI,44 mais aussi des dérivés chiraux de Selectfluor45 et de NFSI46 (Figure 1.6).
Figure 1.6. Réactifs de fluoration électrophile récents.
1.2.3 Fluoration radicalaire
En fluoration radicalaire, la liaison C–F est formée par réaction d’un carbone radicalaire (généré in situ de diverses manières) avec une source de fluor atomique.16 Les premiers réactifs utilisés pour cette approche ont été le XeF2, l’hypofluorite et le fluor moléculaire.47
(44) NFBSI : (a) Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. J. Fluorine Chem. 2011, 132, 222–225. 4,4'-diF-NFSI : (b) Wang, F.; Li, J.; Hu, Q.; Yang, X.; Wu, X. Y.; He, H. Eur. J. Org. Chem. 2014, 3607–3613.
(45) Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V. Angew. Chem., Int. Ed. 2013, 52, 9796–9800.
(46) Zhu, C.-L.; Maeno, M.; Zhang, F.-G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.-A.; Cahard, D. Eur. J. Org. Chem. 2013, 6501–6505.
(47) (a) Grakauskas, V. J. Org. Chem. 1969, 34, 2446–2450. (b) Rozen, S. Acc. Chem. Res. 1988, 21, 307– 312. (c) Patrick, T. B.; Khazaeli, S.; Nadji, S.; Hering-Smith, K.; Reif, D. J. Org. Chem. 1993, 58, 705–708.
N F S S O O O O N N Me F 2 TfO– Dérivé chiral du NFSI NFBSI 4,4'-diF-NFSI Dérivé chiral du Selectfluor CF3 CF3 F F N F S S O O O O OMe MeO t-Bu t-Bu t-Bu t-Bu S S N O O F O O
De plus récentes contributions dans le domaine ont montré que des réactifs de fluoration électrophile de type N–F16a ainsi que certains solvants fluorés48 pouvaient se comporter comme des agents de transfert de fluor atomique.
1.3 L
EX
TALF
LUORParmi les réactifs de fluoration nucléophile, nous nous sommes intéressés plus particulièrement au XtalFluor. Celui-ci possède un double rôle puisqu’il peut se comporter à la fois comme agent de fluoration et comme agent activant.
1.3.1 Découverte du XtalFluor
Les premiers exemples de sels de dialkylaminodifluorosulfinium ont été rapportés dans la littérature en 1977 par le groupe de Markovskii.49 Les auteurs décrivent que le DAST et ses dérivés réagissent avec l’éthérate de trifluorure de bore (BF3·Et2O) pour donner les sels d’aminodifluorosulfinium correspondants (Schéma 1.4). Dans ce cas-ci, le trifluorure de bore n’est pas utilisé comme acide de Lewis, mais plutôt comme accepteur irréversible d’ions fluorures pour former le tétrafluoroborate de manière irréversible.50
Schéma 1.4. Synthèse de sels de dialkylaminodifluorosulfinium au moyen de BF3·Et2O.
(48) Döbele, M.; Vanderheiden, S.; Jung, N.; Bräse, S. Angew. Chem., Int. Ed. 2010, 49, 5986–5988. (49) Markovskii, L. N.; Pashinnik, V. E.; Saenko, E. P. Zh. Org. Khim. 1977, 13, 1116–1117. (50) Minkwitz, R.; Molsbeck, W.; Oberhammer, H.; Weiss, I. Inorg. Chem. 1992, 31, 2104–2107.
N S R R F F F BF3⋅Et2O N S R R F F BF4 -R = Me, Et, –(CH2)5– –(CH2)2O(CH2)2–
D’autres sels de dialkylaminodifluorosulfinium ont ensuite été rapportés par différents groupes, qui ont fait réagir un dérivé du DAST avec un accepteur d’ions fluorures, tel que BF3, PF5, SeF4, SbF5 et AsF5.51,52,53,54
En 1996, la substitution d’un hydroxyle allylique par un fluorure dans des prostaglandines représente le premier exemple de déoxofluoration qui emploie un sel de dialkylaminodifluorosulfinium (Schéma 1.5).55 Plus précisément, le tétrafluoroborate de morpholinodifluorosulfinium est utilisé en adjonction avec un dérivé de triméthyldifluorosilicate de tris(morpholine)sulfonium comme source de fluorure pour donner un mélange des deux épimères.
Schéma 1.5. Premier exemple d’utilisation d’un sel de dialkylaminodifluorosulfinium
comme agent de déoxofluoration.
(51) Cowley, A. H.; Pagel, D. J.; Walker, M. L. J. Am. Chem. Soc. 1978, 100, 7065–7066. (52) Mews, R.; Henle, H. J. Fluorine Chem. 1979, 14, 495–510.
(53) Pauer, F.; Erhart, M.; Mews, R.; Stalke, D. Z. Zeitschrift fuer Naturforsch., B: Chem. Sci. 1990, 45, 271– 276.
(54) Pashinnik, V. E.; Martynyuk, E. G.; Shermolovich, Y. G. Ukr. Khim. Zh. (Russ. Ed.) 2002, 68, 83–87. (55) Bezuglov, V. V.; Pashinnik, V. E.; Tovstenko, V. I.; Markovskii, L. N.; Freimanis, Y. A.; Serkov, I. V.
Russ. J. Bioorg. Chem. 1996, 22, 814–822.
O O OMe OH O O OMe F O N S F F BF4 CH3CN source de F -O N S N N O O Me3SiF2 -source de F -85 %
Enfin, c’est au cours de ces huit dernières années que la compagnie québécoise OmegaChem a exploité le potentiel de déoxofluoration de ces composés en présence d’une source externe de fluorure. OmegaChem a ainsi mis sur le marché deux nouveaux réactifs : le tétrafluoroborate de diéthylaminodifluorosulfinium et le tétrafluoroborate de morpholinodifluorosulfinium, appelés respectivement XtalFluor-E et XtalFluor-M (Figure 1.4).31
1.3.2 Méthodes de synthèse
La première méthode de synthèse a été décrite en 1977 par le groupe de Markovskii (Schéma 1.6).49 Le XtalFluor-E est synthétisé par réaction du DAST avec BF3·Et2O dans l’éther, puis recristallisé à chaud dans le 1,2-dichloroéthane (DCE) pour mener à la formation de cristaux en forme d’aiguilles après un rapide refroidissement. Ces cristaux ont un point de fusion compris entre 74 et 76 °C et sont sensibles à l’humidité.
En 2010, des chimistes de chez OmegaChem ont effectué une recristallisation à une température plus élevée. Celle-ci ne conduit pas à des cristaux de même morphologie puisque des flocons ont été obtenus à la place des aiguilles.31b Ce produit est plus dense, plus propre, plus stable et moins hygroscopique. Son point de fusion s’élève à 83-85 °C. Une diffraction par rayons X a pu démontrer la génération des deux polymorphes différents. Le XtalFluor-E sous forme d’aiguilles est appelé polymorphe de type I, tandis que celui sous forme de flocons correspond au polymorphe de type II.
Cette méthode de synthèse comporte un gros inconvénient, puisque le DAST est un réactif qui nécessite d’être préalablement distillé. Cette distillation est dangereuse et requiert d’importantes mesures de sécurité en raison du caractère très explosif du DAST à température élevée. Une voie de synthèse alternative a ainsi été développée, dans laquelle le DAST est formé in situ (Schéma 1.6). La diéthyltriméthylsilylamine réagit tout d’abord avec le SF4 dans le dichlorométhane, puis le BF3·THF est ajouté directement sur le DAST brut. Après filtration, le XtalFluor-E est obtenu avec un rendement de 90 % sous forme de cristaux de type II (polymorphe désiré).
Schéma 1.6. Méthodes de synthèse du XtalFluor-E.
Enfin, les chimistes de chez OmegaChem ont également mis au point une méthode pour accéder à la synthèse de triflates de dialkylaminodifluorosulfinium (Schéma 1.7). Il a en effet été montré que le DAST réagit de manière très exothermique avec HBF4 pour former le XtalFluor-E, avec élimination concomitante de HF. Ainsi, cette méthode par échange d’acide de Brønsted donne accès à des sels de dialkylaminodifluorosulfinium comportant d’autres types de contre-anions. Ces nouveaux sels peuvent posséder des propriétés intéressantes, comme une augmentation de la température de fusion (97-101 °C pour le dérivé triflate).
Schéma 1.7. Synthèse de sels de diéthylaminodifluorosulfinium à partir d’acides de
Brønsted. N S F F F BF3⋅Et2O Et2O N S F F BF4 N SiMe3 1) SF4, CH2Cl2 2) BF3⋅THF N S F F BF4 N S F F F 3) filtration Synthèse décrite en 1977 Synthèse décrite en 2010 90 % 82 % N S F F F HBF4⋅Et2O Et2O N S F F BF4 96 % 81 % N S F F OTf Et2O HOTf
1.3.3 Propriétés
Le XtalFluor-E a été conçu comme alternative au DAST, dont l’utilisation comporte de nombreux inconvénients. Parmi ceux-ci, nous pouvons citer une purification dangereuse, une grande instabilité, la libération de HF, la génération de produits secondaires d’élimination, un coût élevé et une manipulation difficile. En comparaison, le XtalFluor-E se présente sous la forme d’un solide cristallin relativement stable et moins coûteux, qui ne libère pas de HF libre et dont la quantité de produits secondaires d’élimination est relativement limitée (Tableau 1.3).31
Tableau 1.3. Comparaison des propriétés du DAST avec celles du XtalFluor-E.
DAST XtalFluor-E
Purification Distillation sous vide Recristallisation Stabilité Décomposition soudaine et très
exothermique à 155 °C
Décomposition progressive aux alentours de 205 °C
Libération de HF Oui Non
Produits secondaires
(élimination) Fréquents Peu fréquents
Coût56 1,97 $CAD/mmol 0,824 $CAD/mmol
Apparence Liquide fumant Solide cristallin
La stabilité du XtalFluor-E a été étudiée par des analyses de calorimétrie différentielle à balayage (DSC), afin d’établir si celui-ci est sécuritaire d’un point de vue thermique (Figure 1.7). Le DAST possède un pic très étroit à 155 °C et dégage 1641 J/g, indiquant une décomposition soudaine et très exothermique. Le Deoxo-Fluor montre une température de décomposition similaire (158 °C), mais avec un pic plus large et un dégagement de 1031 J/g. Le XtalFluor-E, quant à lui, se décompose aux alentours de 205 °C avec un dégagement exothermique de 1260 J/g. Une température de décomposition élevée et un
(56) Les prix ont été calculés à partir de la plus grande quantité disponible chez Sigma-Aldrich (juillet 2017). Le DAST est sous forme de solution 1,0 M dans CH2Cl2.
faible dégagement d’énergie sont significatifs d’un composé plus stable, et donc plus sécuritaire. Ainsi, le XtalFluor-E est plus stable que le DAST et le Deoxo-Fluor puisque sa température de décomposition est supérieure d’environ 50 °C. De meilleurs résultats ont encore été observés avec le XtalFluor-M, qui se décompose aux alentours de 243 °C et dégage 773 J/g.
Figure 1.7. Thermogrammes DSC du DAST, Deoxo-Fluor, XtalFuor-E et XtalFluor-M.
Figure tirée de la référence 31b.
En plus de cela, lorsque la température est fixée à 90 °C, le XtalFluor-E ne subit aucune dégradation visible endéans la période de temps consacrée à l’expérience (5000 minutes), contrairement au DAST et au Deoxo-Fluor qui se dégradent en moins de 300 et 1800 minutes respectivement.
1.3.4 Réactif de fluoration
Le XtalFluor a été développé initialement en tant que réactif de déoxofluoration pour des transformations variées, telles que la formation de fluorures d’alkyle, de difluorométhylènes ou de fluorures d’acyle.
1.3.4.1 Déoxofluoration d’alcools
La déoxofluoration d’alcools, par réaction avec le XtalFluor et un promoteur, permet d’obtenir les fluorures d’alkyle correspondants (Schéma 1.8).31 Dans cette transformation, le promoteur peut être une source de fluorures (Et3N·3HF ou Et3N·2HF), mais également une base forte telle que le 1,8-diazabicyclo[5.4.0]undéc-7-ène (DBU) dans certains cas. Le XtalFluor-E et le XtalFluor-M ont généralement une réactivité similaire, tandis que les autres sels d’aminodifluorosulfinium étudiés n’ont pas fourni de meilleurs résultats.31c Toutes sortes d’alcools ont été fluorés de cette manière, incluant des alcools primaires, secondaires, tertiaires, allyliques et anomériques. Dans le cas d’alcools allyliques, la réaction s’effectue cependant uniquement via un mécanisme SN2’. En outre, la formation de produits secondaires d’élimination a été rapportée pour certains substrats. Les deux produits ayant des propriétés physiques similaires, leur séparation demeure quelques fois difficile, voire impossible.
Schéma 1.8. Synthèse de fluorures d’alkyle par déoxofluoration d’alcools.
Depuis cette découverte et avec la commercialisation des XtalFluor-E et -M, de nombreux autres exemples ont été décrits dans la littérature.57
(57) (a) Srinivasarao, M.; Park, T.; Chen, Y.; Fuchs, P. L. Chem. Commun. 2011, 47, 5858–5860. (b) Lueg, C.; Schepmann, D.; Günther, R.; Brust, P.; Wünsch, B. Bioorg. Med. Chem. 2013, 21, 7481–7498. (c) Juncosa Jr.; J. I.; Groves, A. P.; Xia, G.; Silverman, R. B. Bioorg. Med. Chem. 2013, 21, 903–911. (d) Huchet, Q. A.; Kuhn, B.; Wagner, B.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Fluorine
Chem. 2013, 152, 119–128. (e) Kardivel, M.; Fairclough, M.; Cawthorne, C.; Rowling, E. J.; Babur, M.;
McMahon, A.; Birkket, P.; Smigova, A.; Freeman, S.; Williams, K. J.; Brown, G. Bioorg. Med. Chem. 2014,
22, 341–349. (f) Kardivel, M.; Fanimarvasti, F.; Forbes, S.; McBain, A.; Gardiner, J. M.; Brown, G. D.;
Freeman, S. Chem. Commun. 2014, 50, 5000–5002. (g) Gagnon, M.-C.; Turgeon, B.; Savoie, D.; Parent, J.-F.; Auger, M.; Paquin, J.-F. Org. Biomol. Chem. 2014, 12, 5126–5135. (h) Holl, K.; Schepmann, D.; Fischer, S.; Ludwig, F.-A.; Hiller, A.; Donat, C. K.; Deuther-Conrad, W.; Brust, P.; Wünsch, B. Pharmaceuticals
2014, 7, 78–112. (i) Khazaei, K.; Yeung, J. H. F.; Moore, M. M.; Bennet, A. J. Can. J. Chem. 2015, 93,
1207–1213. (j) Carpentier, C.; Godbout, R.; Otis, F.; Voyer, N. Tetrahedron Lett. 2015, 56, 1244–1246. (k) Davies, S. G.; Fletcher, A. M.; Frost, A. B.; Roberts, P. M.; Thomson, J. E. Org. Lett. 2015, 17, 2254–2257.
OH R2 R1 F R2 R1 XtalFluor promoteur promoteur = Et3N⋅3HF, Et3N⋅2HF ou DBU Exemples représentatifs OH F SN2' 90 % N HO Cbz N F Cbz 86 % ee 98 % fluoro/alcène = 6,9:1 N Cbz + OH F 93 %
Le mécanisme de cette transformation est similaire à celui de la déoxofluoration d’un alcool par le DAST, puisque ces deux réactions impliquent la formation d’un intermédiaire diéthylaminodifluorosulfane (Schéma 1.9).27,31b Les différences notoires entre les deux mécanismes sont, d’une part, la libération de HF pour la déoxofluoration au moyen de DAST, et d’autre part, la nécessité d’une source externe de fluorure pour la réaction utilisant le XtalFluor-E.
Schéma 1.9. Mécanismes de déoxofluoration d’un alcool avec le DAST et le XtalFluor-E.
Un intermédiaire diéthylaminodifluorosulfane a pu être isolé par le groupe de John Vederas en 1999 lors de la déoxofluoration de l’alcool 1.1 par le DAST (Schéma 1.10).58 Cet intermédiaire a été obtenu avec un rendement de 12 %, en plus du produit d’élimination (7 %) et du produit fluoré (52 %).
(58) Sutherland, A.; Vederas, J. C. Chem. Commun. 1999, 1739–1740. N S F F F HO R - HF N S F F O F F R
Déoxofluoration d'un alcool avec le DAST
R N S O F N S F F HF + +
Déoxofluoration d'un alcool avec le XtalFluor-E
BF4 HO R - HBF4 N S F F O F F R R N S O F + source externe de fluorure F +
Schéma 1.10. Formation de l’intermédiaire diéthylaminodifluorosulfane 1.3.
1.3.4.2 Déoxofluoration d’aldéhydes et cétones
La déoxofluoration d’aldéhydes et de cétones par le XtalFluor a été développée parallèlement à la déoxofluoration des alcools (Schéma 1.11).31 Les conditions réactionnelles sont semblables et une source de fluorure est également nécessaire (Et3N·3HF ou Et3N·2HF). Ceci a conduit à la synthèse de difluorométhylènes variés, décrits par OmegaChem31b ou plus tard par d’autres groupes.59 Tout comme pour la déoxofluoration d’alcools, des produits secondaires d’élimination sont observés, dans ce cas-ci des fluoroalcènes.
(59) En plus de la réf. 57d, voir : (a) Yu, L.-F.; Eaton, J. B.; Fedolak, A.; Zhang, H.-K.; Hanania, T.; Brunner, D.; Lukas, R. J.; Kozikowski, A. P. J. Med. Chem. 2012, 55, 9998–10009. (b) Nemoto, H.; Takubo, K.; Shimizu, K.; Akai, S. Synlett 2012, 23, 1978–1984. (c) Chernykh, A. V.; Feskov, I. O.; Chernykh, A. V.; Daniliuc, C. G.; Tolmachova, N. A.; Volochnyuk, D. M.; Radchenko, D. S. Tetrahedron 2016, 72, 1036– 1041. N OMe OMe i-Pr CO2Me O NBoc2 SF2 NEt2 N OMe OMe i-Pr CO2Me NBoc2 N OMe OMe i-Pr CO2Me F NBoc2 + + 1.2, 7 % 1.3, 12 % 1.4, 52 % N OMe OMe i-Pr CO2Me OH NBoc2 DAST CH2Cl2, -78 °C 1.1





![Figure 2.1. Activation of amides with [Et 2 NSF 2 ]BF 4 for the synthesis of 1,3,4-oxadiazoles and isocyanides](https://thumb-eu.123doks.com/thumbv2/123doknet/5540884.132522/58.918.207.708.326.663/figure-activation-amides-et-nsf-synthesis-oxadiazoles-isocyanides.webp)
