Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines
Texte intégral
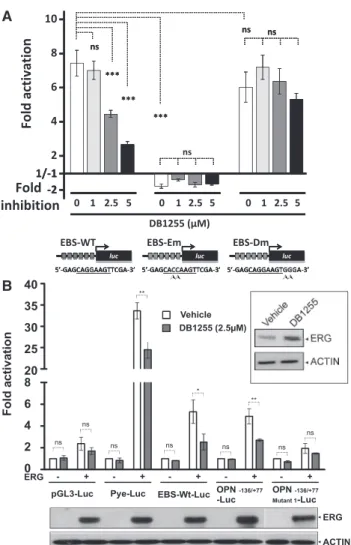
Figure

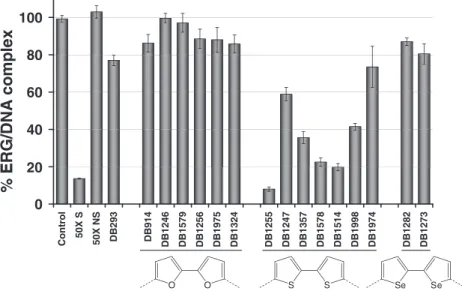
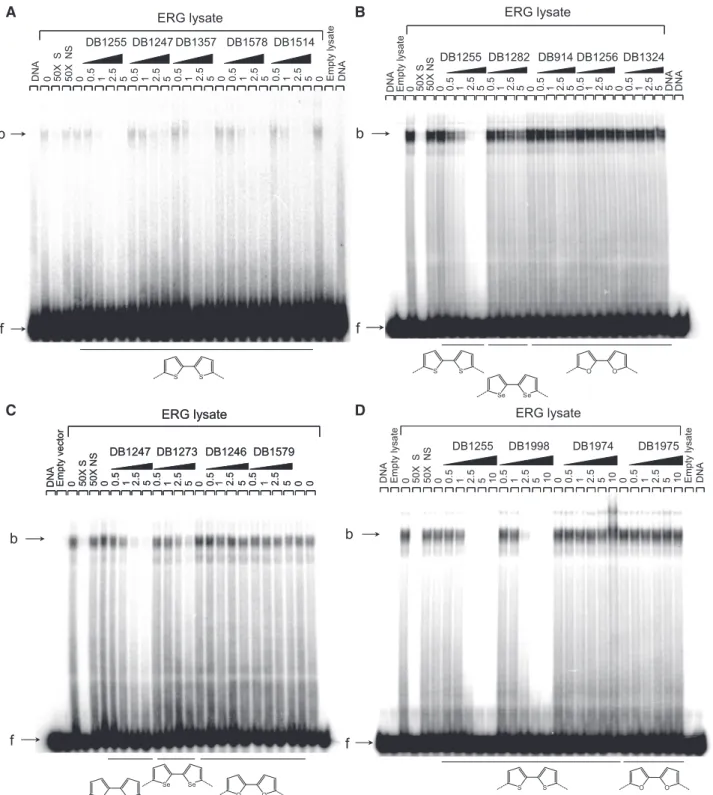
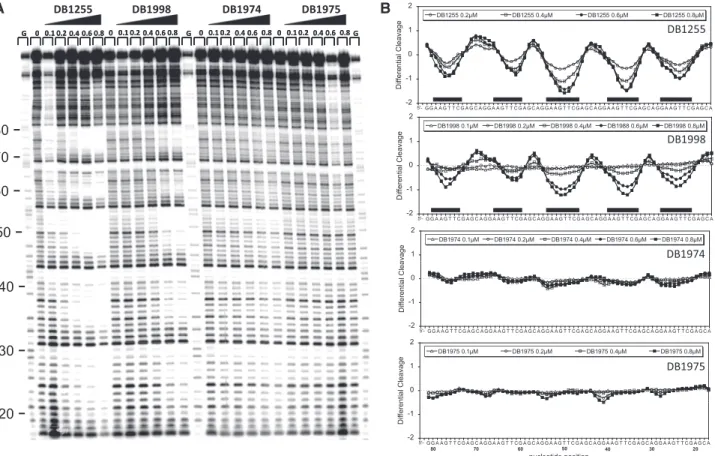
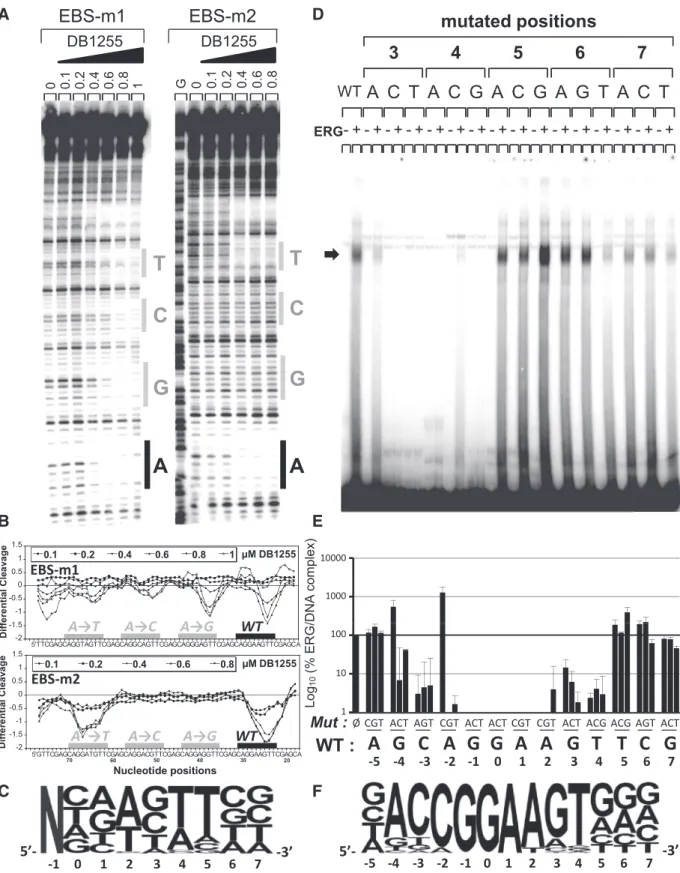

Documents relatifs
Thereafter, Krivine also proves the converse, which is that every arithmetical for- mula which is true in the ground model is realized by the term which implements the trivial
Keywords: dependent types, sequent calculus, classical logic, control opera- tors, call-by-value, delimited continuations, continuation-passing style transla- tion, value
Scott que, ao mesmo tempo que afirma que “aqueles que se propõem a codificar o sentido das palavras lutam por uma causa perdida porque as palavras, como as ideias e as coisas que
Par contre, la classe Politique Congolaise manque en générale des visionnaires; comme à l’époque MOBUTU, toute la classe Politique de l’Opposition se focalisée seulement sur
Cette recherche a permis de découvrir plusieurs éléments au sujet des complications pos- topératoires d’une opération suite à une fracture de la hanche, plus
The absence of pure Feynman’s cascade and suppressed efficiency of the pure Kelvin-wave cascade create specific circumstances under which there exists a range in the kelvon
Consigne: « Par groupes de 4 ou 5 vous allez devoir schématiser le trajet du sang dans le corps en tenant compte des connaissances acquises lors des dernières
The idea of shuttling a small amount of liquid between different temperature zones by means of a pneumatic pump offers decisive advantages over methods published so far: (a) a
