Remerciements
Je tiens avant tout à remercier le Docteur Anne Nègre-Salvayre pour m’avoir accueillie au sein de son équipe, pour avoir cru en mes capacités scientifiques, et pour m’avoir fait confiance lors des demandes de bourses auprès du GRRC. Vous avez toujours été très disponible et m’avez constamment soutenue durant ces 3 années de thèse, notamment durant les derniers mois, lorsque la rédaction et la soutenance de thèse devaient s’accomplir en un temps très réduit. Je vous suis très reconnaissante de votre investissement et de votre soutien lors de cette période. Je vous souhaite de tout cœur une excellente continuation à la direction de l’équipe, qui par ailleurs est de plus en plus reconnue pour sa qualité scientifique.
Je remercie chaleureusement le Professeur Robert Salvayre pour son accueil ainsi que pour son soutien et sa confiance lorsqu’il a été question que j’intègre l’équipe en tant que thésarde. Vous m’aviez alors dit que les étudiants mal classés en DEA peuvent néanmoins faire de brillants chercheurs, s’ils sont motivés. Croyez-moi que je compte bien vous donner raison ! Je vous remercie également pour le partage de votre grande expérience de la recherche, qui se traduit notamment par votre recul sur les résultats, votre exigence du détail, sans compter votre savoir faire rédactionnel, ainsi que votre talent de mise en page des figures.
Merci au Professeur Alain Tedgui d’avoir accepté de présider cette soutenance. J’ai eu l’honneur de vous avoir dans mon jury de thèse et je vous en suis très reconnaissante étant donné votre stature internationale dans le monde de la recherche médicale et de l’athérosclérose. Par ailleurs, je tiens à vous remercier vivement pour votre aide précieuse lors de ma recherche de post doc.
Je remercie Mme Michèle Garlatti d’avoir participé à mon jury de thèse. J’ai beaucoup apprécié d’avoir pu échanger avec vous sur le stress du reticulum endoplasmique : après trois années passées sur cette voie de signalisation, avoir l’opportunité d’en discuter avec une spécialiste le jour de la soutenance m’est apparue comme un bel achèvement. Merci à vous.
Je remercie enfin Mr Eric Ogier-Denis d’avoir accepté de participer à mon jury de thèse et d’avoir évalué mon manuscrit. Malheureusement, nous n’avons finalement pas pu nous rencontrer, mais j’espère avoir une future occasion d’échanger avec vous sur la thématique de nos travaux qui est pour le moins assez proche.
Je souhaite remercier l’association du GRRC (Groupe de Réflexion et de Recherche en Cardiovasculaire) pour l’aide qu’elle fournit aux jeunes chercheurs, et par la même, je les remercie de m’avoir financé deux années de thèse. Merci et longue vie au GRRC ainsi qu’au club jeune !
Nathalie, tu auras marqué ma thèse, comme d’autres avant moi, et d’autres après. Au-delà de la science, des protocoles et de l’emplacement de chaque produit au labo, j’ai tellement apprécié toutes ces conversations sur à peu prés tout de la vie. Avec toutes ces discussions, tu imagines combien de fois tu me reviendras en tête au cours de ma vie ! Je te remercie donc pour tous ces bons moments, souvent drôles, tes conseils scientifiques et professionnel, ton humour et ainsi que ton franc-parler qui fait également avancer. Bonne continuation dans ta carrière de chercheur et surtout beaucoup de bonheur avec Manon, Chloé et Eric, à Saint Léon ou ailleurs.
Merci beaucoup à vous Jean-Claude pour toute l’immunohisto et cyto. Et lorsqu’avec votre œil critique, vous approuvez qu’un résultat est positif : quel bonheur !!! Merci pour votre efficacité dans le travail, votre savoir faire et surtout votre gentillesse. Bientôt vous pourrez être à plein temps dans votre jardin, et je suis certaine que vous installerez un épouvantail fait de lames et lamelles tellement elles vous manqueront !
Merci Cécile pour tes conseils techniques avec ta rigueur légendaire qui est tellement précieuse lorsque l’on apprend le métier. J’ai apprécié les discussions scientifiques que l’on a pu avoir pour cause de sujets assez proches, ainsi que les discussions cinématographiques ou simplement vestimentaires… Je te souhaite également bonne continuation dans la recherche et beaucoup de bonheur à toi et ta famille.
Merci à toi Françoise : même si nous n’avons pas travaillé ensemble, j’ai apprécié tes conseils et ta disponibilité pour mes questions cliniques. Bon courage pour continuer à mener de front tout ce que tu gères, sans oublier tes 3 hommes.
Merci à toutes nos aides techniques, notamment Marie-Hélène pour m’avoir aidée pour de nombreux western. Merci à Stéphanie, Corinne et Christelle pour tout ce que vous faites au quotidien : commandes, entretiens, réactifs. Comme souvent, on se rend compte à quel point vous êtes indispensables quand vous n’êtes plus là…Une petite pensée aussi pour Béatrice et Christian Mora pour toutes ces LDL préparées qui m’auront servi durant mes deux premières années de thèse. J’aurais eu peu de temps pour travailler avec toi Elodie, et c’est avec regrets car je suis certaine que j’aurais beaucoup appris de ton savoir en biologie moléculaire. Bonne continuation dans le labo, je suis persuadée que tu seras d’une aide précieuse pour les futurs gros papiers.
Merci aux secrétaires, Eve et Danièle pour leur compagnie et leurs services, ainsi qu’un grand merci à Chantal, pour son professionnalisme, son efficacité et sa gentillesse.
J’en arrive à remercier tous mes collègues étudiants du labo : Sylvain avant tout. Bonne chance à toi et que le meilleur pour la suite (Je mets déjà de côté toutes les friandises américaines qu’il te faut goûter). Plein de bons souvenirs avec Edwige, Cindy, gardienne du labo à veiller soir et week end, avec Aurélie, Cécile I, Raphaël, Christelle, Carole, Aurélien. Bonne chance à vous tous, concernant la science bien sûr, mais concernant tout le reste surtout.
Parce que le manuscrit de thèse donne l’opportunité d’écrire publiquement un petit mot, alors j’en profite pour citer les amis qui me tiennent à cœur et les remercier d’être là, en tant qu’amis : Caro, Karine, Léa, Camille, Julien D, Coralie (pour la science aussi), Edwige, Marjorie, Lise et Guillaume.
Enfin je terminerai par remercier ma famille, et avant tout mes Parents. Que de chemin parcourut depuis la ferme en Normandie jusqu’à ce diplôme de Docteur…et c’est à vous que je le dois. Déjà parce que la vie d’aventuriers que vous avez choisit nous à ouvert l’esprit et nous a permis de choisir des voies auxquelles nous n’aurions pas été confrontées autrement, ensuite parce que vous avez toujours répondu à mes besoins pendant ces longues années d’étude, même lors de périodes pas faciles pour vous. Votre audace nous a enseigné la persévérance (voir l’entêtement), et même si Dakar était un peu trop loin pour venir le jour de ma soutenance, mes pensées et mes remerciements allaient tout droit vers vous. Merci pour tout cela, pour nous avoir mené là où nous sommes maintenant.
Merci à mon frère, Pierre, ainsi qu’à Caro d’avoir étés présents pour ma soutenance : étant donné la distance et l’incompréhensibilité de l’exposé pour les non-initiés, cela me touche beaucoup. J’ai été heureuse aussi qu’une part de ma famille ait pu être présente, donc encore Merci Pierre. Merci à Eliza et Maurice, mes ‘parents adoptifs’ lors de mes débuts à Toulouse. Je suis heureuse que vous ayez pu venir pour ce moment qui a marqué une étape de ma vie. Un grand Merci également à mes Beaux-Parents, Philippe et Maïté, pour tout ce que vous faites pour nous, pour votre soutien moral constant, ainsi que pour votre soutien matériel qui a rendu nos thèses confortables et nous a permis de réaliser d’autres projets.
Les gens les plus importants figurants à la fin en sciences, je remercie du fond du cœur mon mari. Tout récemment, je te remercie pour m’avoir grandement facilité cette fin de thèse à toute allure en assurant la maintenance domestique ainsi qu’en préparant notre départ à New York. Au-delà de ça, je te remercie car j’ai pu observer un tournant au milieu de ma thèse, tournant où je me suis beaucoup plus investie et où je me suis vraiment épanouie dans mon travail. Je sais que cela s’est opéré en partie grâce à toi, en me communiquant ta passion pour la science, ton investissement au travail, ta persévérance dans les moments difficiles, ta notion d’efficacité aussi. Nous avons cette chance d’avancer ensemble dans notre vie de chercheur et cela nous entraîne vers le haut, ainsi que vers des contrées lointaines… Vivement la suite !
Résumé
L’athérosclérose est une pathologie inflammatoire chronique, caractérisée par la formation d’une lésion dans la paroi des moyennes et grosses artères. L’évolution des lésions est lente, liée au vieillissement vasculaire, et reste longtemps silencieuse. Les complications majeures de l’athérosclérose surviennent lors de sa rupture et des phénomènes de thrombose qui sont à l’origine des infarctus et des accidents cardio ou neurovasculaires, premières causes de mortalité dans les pays développés. La rupture de la plaque d’athérosclérose est due à l’apoptose des cellules vasculaires. Les lipoprotéines de basse densité (LDL) jouent un rôle majeur dans la formation de la plaque de par leur oxydation dans l’intima. Les LDL oxydées sont impliquées dans la formation des lésions primaires (stries graisseuses), mais pourraient jouer un rôle dans l’évolution des plaques vers des stades plus avancés en raison de leurs propriétés inflammatoires et proapoptotiques.
L’objectif de ce travail a été d’étudier les voies impliquées dans l’apoptose des cellules vasculaires induites par les LDL oxydées, et plus précisément l’implication du stress du reticulum endoplasmique (stress du RE), et de la signalisation spécifique de ’Réponse aux protéines mal repliées’ (UPR). En effet, de nombreux facteurs peuvent venir perturber l’homéostasie du RE et induire l’UPR. L’UPR est une réponse d’adaptation au stress, qui dans un premier temps favorise la survie, mais peut orienter la cellule vers l’apoptose lorsque le stress est intense ou se prolonge. Nos travaux de recherche ont été orientés vers le mécanisme de protection d’une protéine chaperonne du RE, ORP150, et d’un agent pharmacologique inhibiteur du stress du RE, le Salubrinal, contre l’apoptose induite par les LDL oxydées dans les cellules vasculaires.
Nous montrons dans un premier travail que ORP150 est induite dans les cellules endothéliales et les cellules musculaires lisses traitées avec des LDL oxydées. Des siRNA pour ORP150 potentialisent l’apoptose, alors que la transfection d’un vecteur codant pour ORP150 protège des effets toxiques des LDL oxydées. La mort cellulaire induite par les LDL oxydées dépend d’une dérégulation de l’homéostasie calcique qui active les voies pro-apoptotiques calcium-dépendantes. La surexpression de ORP150 bloque l’apoptose des cellules endothéliales en inhibant la dérégulation du calcium. Dans la deuxième partie de ce travail, nous mettons en évidence que les LDL oxydées et les produits d’oxydation lipidiques (7-cétocholestérol et 4-hydroxynonénal) induisent un stress prolongé du RE, caractérisé par l’activation des ‘sensors ‘ PERK, IRE-1α et ATF6, et impliqué dans l’apoptose via la phosphorylation de JNK. La surexpression de ORP150 inhibe le stress du RE en se liant aux sensors via un effet ‘BiP-like’. L’inhibition du stress du RE par ORP150 contribue à son effet anti-apoptotique. Dans la dernière partie de ce travail, nous avons étudié l’effet d’un inhibiteur du stress du RE, le salubrinal, sur la toxicité des LDL oxydées. Cette molécule induit la phosphorylation du facteur eIF2α et active le facteur de transcription ATF4, deux protéines impliquées dans la survie cellulaire.
Ces résultats mettent en évidence (i) l’importance du stress du RE comme nouvelle voie de signalisation pro-apoptotique induite par les LDL oxydées dans les cellules vasculaires, (ii) le rôle protecteur majeur de ORP150 contre ce stress et l’apoptose induits par les LDL oxydées, et permettent par ailleurs (iii) de proposer une approche pharmacologique (Salubrinal) contre l’apoptose et l’instabilité de la plaque.
Summary
Atherosclerosis is a chronic inflammatory pathology characterized by the formation of a lesion in the arterial wall of large and medium arteries. It is an age-linked, slowly evolutive pathology, with acute events due to plaque rupture, thrombosis and cardiovascular events such as heart attack and stroke, wich are the most prevalent causes of mortality in western countries. Vascular cell apoptosis, including endothelial cell, smooth muscle cell or macrophage apoptosis, participate to plaque weakening and to its rupture. Atherosclerosis is a multifactorial pathology, in which lipids and low density lipoproteins (LDL) play a major role because of their oxidation within the subendothelial space. Oxidized LDL (oxLDL), are involved in the formation of early lesions (fatty streaks), but could also participate to the evolution of the lesions towards more advanced states, because of their inflammatory and proapoptotic properties.
The aim of this work was to evaluate the apoptotic signaling evoked by oxLDL in vascular cells, and more precisely the implication of the endoplasmic reticulum stress (ER stress) and the subsequent Unfolded Protein Response (UPR). The UPR is induced as an adaptative defence system in response to various stresses, but switches toward apoptosis as function of stress intensity. We focused this study on the protective antiapoptotic role of an ER stress-associated chaperone protein, ORP150, and of a molecule, Salubrinal, which inhibits ER stress and apoptosis induced by oxLDL in vascular cells.
The first part of this work is focused on the antiapoptotic effect of ORP150 against oxLDL. ORP150 silencing by siRNA potentiates oxLDL-induced apoptosis, while the transfection of a vector coding for ORP150 overexpression was protective. Apoptosis evoked by oxLDL included a deregulation of calcium homeostasis, and the activation of calcium-dependent apoptotic signaling which were completely inhibited by ORP150-forced expression. In the second part of this work, we show that oxLDL induce a prolonged ER stress in endothelial cells, characterized by the lasting activation of ER stress sensors, PERK, IRE-1α and ATF6. ER stress induction was inhibited by ORP150, which binds the sensors, and inhibits their activation via a ‘Bip-Like’ mechanism. The last part of this work is focused on the antiapoptotic effect of a small molecule, Salubrinal, known to inhibit ER stress by promoting eIF2α phosphorylation and thus inhibiting protein translation. We report that Salubrinal inhibits oxLDL-induced apoptosis by triggering eIF2α phosphorylation and the activation of the survival transcription factor ATF4.
These results shed light on ER stress as a new signaling pathway evoked by oxLDL in vascular cells and involved in apoptosis, and on the protective role of ORP150 overexpression, which could modulate vascular cell apoptosis within the arterial wall. Moreover we identify salubrinal as a potent protective agent against oxLDL toxicity, and thus with potential therapeutical interest against (ER stress-dependent) apoptosis in atherosclerosis.
Sommaire
Liste des abréviations ……….. p5
Liste des figures ……… p7
Introduction bibliographique ……….... p9
A. L’athérosclérose ………. P9
A.1 : Définition
………p9
A.2 : Facteurs de Risque ………. p11
A.3 : Mécanismes de l’Athérogénése ……… p11
A.3.1 : La dysfonction endothéliale ………. p15 A.3.2 : L’oxydation des LDL dans l’intima ……….. p15 A.3.3 : Recrutement des Monocytes et des Lymphocytes T ………... p15 A.3.4 : Formation des cellules spumeuses ………. p17 A.3.5 : Migration et Prolifération des cellules musculaires lisses …… p19 A.3.6 : Evolution de la plaque ……… p19B. Les LDL oxydées ……….. p23
B.1 : Les lipoprotéines ……… p23
B.1.1 : Origine des LDL ……….. p23 B.1.2 : Captation des LDL ……….. p25B.2 : Oxydation des LDL ……….. p25
B.2.1 : Le stress oxydant ……… p27 B.2.2 : La peroxydation lipidique ………... p29 B.2.3 : Les récepteurs scavenger ………. p33B.3 : Effets cellulaires des LDL oxydées ………. p34
C. Apoptose et Athérosclérose ……….. p37
C.1 : L’Apoptose ……….. p37
C.2 : L’Apoptose dans l’athérosclérose ……… p37
C.3 : Mécanismes d’Apoptose ………... p39
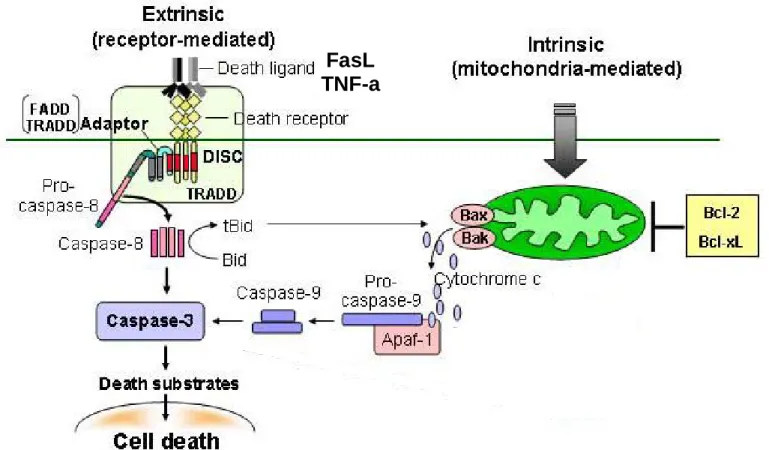
C.3.1 : La Voie Extrinsèque ……… p39 C.3.2 : La Voie Intrinsèque ………. p41 C.3.3 : Rôle du Calcium ……….. p42C.4 : Mécanismes d’apoptose induits par les LDL oxydées ……… p43
D. Le Stress du Reticulum Endoplasmique ……… p47
D.1 : Description du RE ……….. p47
D.2 : Fonction du RE ……….. p48
D.2.1 : Synthèse des lipides et detoxification ……… p48
D.2.2 : Homéostasie calcique ……….. p49
D.2.3 : Synthèse et maturation des protéines ………... p57
D.3 : Le Stress du Reticulum Endoplasmique ………... p63
D.3.1 : L’Unfolded Protein Response ………. p67
D.3.2 : Transition vers l’apoptose ……… p75
D.4 : Stress du RE et athérosclérose ………... p81
E. Oxygen Regulated Protein 150kDa ………... p85
E.1 : Caractéristiques ……….. p85
E.1.1 : Structure de ORP150 ……… p85
E.1.2 : Induction et propriétés de ORP150 ………. p87
E.2 : Régulation transcriptionnelle de ORP150 ……….. p87
E.3 : Mécanismes d’action ………. p90
E.3.1 : ORP150 comme protéine chaperonne ………... p90
E.3.2 : ORP150 et ATP ……….. p91
E.3.3 : ORP150 et calcium ……… p91
E.3.4 : ORP150 et stress du RE ……….. p92
E.4 : ORP150 et maladies cardiovasculaires ………. p92
Objectif de l’étude ………... p95
Matériel et Méthodes ……….. p97
Résultats expérimentaux ………... p103
Introduction ………. p103
I : ORP150 inhibe la dérégulation du calcium cytosolique et
La signalisation pro-apoptotique calcium-dépendante, impliquée
dans l’apoptose
II : Implication du stress du reticulum endoplasmique dans
l’apoptose induite par les LDL oxydées et effet protecteur de
ORP150
Discussion ……… p111
III : La voie PERK/eIF2α/ATF4 prévient l’apoptose induite par
les LDL oxydées. Effet du Salubrinal……….. p113
Conclusion Générale ……….. p127
Références bibliographiques ………. p131
Annexes ……… p149
Liste des abréviations
AVC : Arrêt Vasculaire CérébralATF (ATF4, ATF6) : Activating Transcription Factor BiP : Binding immunoglobulin Protein
CE : Cellules Endothéliales
CHOP : CCAAT Homologous Protein CML : Cellules Musculaires Lisses COX : Cycloxygenase
DAG : Diacylglycérol
eIF : eucaryotic Initiation Factor
ERAD : Endoplasmic Reticulum Associated Degradation ERO : Espèces réactives de l’Oxygène
ERSE : Endoplasmic Reticulum Stress response Element FT : Facteur Tissulaire
GADD : Growth Arrest and DNA Damage inducible protein GRP : Glucose Regulated Protein
HDL : High Density Lipoprotein 4-HNE : 4-Hydroxynonénal Hsp : Heat shock protein IFN : Interferon
IDL : Intermediate Density Protein IL : Interlekine
IP3 : Inositol triphopshate (inositol 1,4,5 triphosphate) IP3R : Récepteur à l’inositol triphosphate
Ire1 : Inositol Requiring enzyme 1 JNK : Jun Kinase
LDL : Low Density Lipoprotein LDL-R : Récepteur aux LDL
M-CSF : Macrophages Colony Stimulating Factor MDA : Malondialdéhyde
NAC : N-acetyl cystéine
NF-κB : Nuclear Factor- kappa B NO : Monoxyde d’azote
NOS (eNOS, iNOS) : Nitric Oxide Synthetase, endogène ou inductible O2-° : anion superoxyde
ONOO- : peroxynitrite
ORP150 : Oxygen Regulated Protein 150 kDa PERK : PKR like endoplasmic Reticulum Kinase PDI : Protein Disulfide Isomerase
PIP3 : Phosphatidyl Inositol 3,4,5 triphosphate PIP2 : Phosphatidyl Inositol diphosphate
PKR : Protein Kinase double strand RNA activated PLC : Phospholipase C
PP1 : Protein Phosphatase 1
PUFA : Poly unsaturated Fatty Acids RE : Reticulum Endoplasmique
SERCA : Sarco-Endoplasmic Reticulum Calcium ATPase SOC : Store Operated Channel
SR (SR-A, SR-B) : Scavenger Receptor (Classe A ou Classe B) TGFβ : Transforming Growth Factor
TNF : Tumor Necrosis Factor
TRPC : Transient Receptor Potential Canonical channel UPR : Unfolded Protein Response
UPRE : Unfolded Protein Response Element VLDL : Very Low Density Lipoprotein
Liste des figures
Figure 1 : Structure de la paroi d’une artère. Evolution de la plaque d’athérome ……… p8 Figure 2 : Forces de frottement et athérogenèse ……….. p14 Figure 3 : Etapes de la formation de la plaque d’athérome ………. p18 Figure 4 : Schéma d’une lipoprotéine ………. p22 Figure 5 : Captation des LDL natives par le récepteur ApoB/E ……….. p24 Figure 6 : Oxydation des LDL ………... p28 Figure 7 : Les récepteurs ‘scavenger’……….. p32 Figure 8 : Voie extrinsèque et voie intrinsèque de l’apoptose ………. p40 Figure 9 : Principaux membres de la famille Bcl-2 ……… p40 Figure 10 : Le Reticulum Endoplasmique ………... p46 Figure 11 : Régulation de l’homéostasie calcique ………. p50 Figure 12 : Contrôle qualité des protéines au sein du RE ……… p56 Figure 13 : Calnexine et Calreticuline ……….. p58 Figure 14 : Structure des chaperonnes de la famille Hsp ………. p60 Figure 15 : Les senseurs du stress du RE ……….. p66 Figure 16 : Activation de PERK et Ire1 ………. p68 Figure 17 : Membres eIF et initiation de la traduction ………... p70 Figure 18 : Phosphorylation de la sous-unité alpha de eIF2 ……… p70 Figure 19 : Activation de la voie PERK/eIF2α ………. p70 Figure 20 : Activation des voies ATF6/XBP-1 et Ire1/XBP-1 ……… p72 Figure 21 : Schéma récapitulatif de l’UPR ………... p74 Figure 22 : Activation de l’apoptose UPR-dépendante ……….. p74 Figure 23 : Voies de survie et apoptose au sein de l’UPR ……… p78 Figure 24 : UPR et adaptation au stress ……….. p80 Figure 25 : structure du gène de ORP150 ………... p96 Figure 26 : Séquence ‘ERSE-like’ de ORP150 ………... p88 Figure 27 : Structure Chimique du Salubrinal ………... p115
Tableau 1 : Concentrations sanguines en LDL et HDL ………. p10 Tableau 2 : Classification de Stary ……… p16 Tableau 3 : Classification des caspases ……….. p40
Revue Générale
A. L’Athérosclérose
A.1 : Définition
L’athérosclérose apparaît lors du vieillissement des vaisseaux et est caractérisée par la formation de plaques au niveau de la paroi des artères de gros et moyen calibre. Ces plaques sont composées de dépôts graisseux riches en cholestérol (athéro provient du grec ‘athéré’ pour ‘bouillie’), enchâssés dans une gangue fibreuse solide (‘scléros’ signifiant ‘dur’ en grec)1. L’évolution de la plaque d’athérome reste longtemps cliniquement silencieuse.
Les premières complications de l’athérosclérose sont liées à l’épaississement de la paroi qui accompagne la progression de la plaque (cf. : Figure 1). Le diamètre artériel est localement réduit, ce qui a pour conséquence une diminution du débit sanguin en aval de la lésion. Cet événement est à l’origine de douleurs (angines de poitrine) correspondant à une souffrance du muscle cardiaque en manque de nutriments et d’oxygène. Les angines de poitrine sont souvent un avertissement à l’apparition d’un phénomène plus grave qu’est la survenue d’un infarctus provoqué lors de l’oblitération d’un vaisseau par un caillot sanguin, le thrombus, qui se forme suite à l’érosion ou à la rupture de la plaque. L’infarctus du myocarde va provoquer la nécrose d'une partie plus ou moins importante du muscle cardiaque. L’autre complication majeure de l’athérosclérose est l’accident vasculaire cérébral (AVC) qui est provoqué par la rupture d’un vaisseau ou une oblitération de la circulation sanguine dans le territoire cérébral (ischémie cérébrale). Les AVC représentent en France la troisième cause de mortalité et la première cause d'invalidité en raison des déficits moteurs et cognitifs qu’ils entraînent (atteinte du langage, de la compréhension, de la mémoire...).
En 2005, le rapport de l’OMS estimait à 17,5 millions le nombre d’individus décédés de maladies cardiovasculaires, ce qui représente 30% de l’ensemble des décès. De ces 17,5 millions, 45% succombent à un AVC et 30% à un infarctus. Depuis l’année dernière, les données ont été modifiées en France car la mortalité d'origine cardiovasculaire a baissé de 50 % en vingt-cinq ans, alors que celle liée au cancer n'a que peu diminué: 30% des décès sont attribués au cancer contre 29% pour les maladies cardiovasculaires.
A.2 : Facteurs de risque
L’athérosclérose est une pathologie multifactorielle qui résulte de la combinaison entre des facteurs de risque non-modifiables que sont l’âge, le sexe et les antécédents familiaux, et des facteurs environnementaux tels qu’une alimentation riche en graisses en en sucres, l’inactivité et le tabagisme. De telles conditions de vie favorisent l’installation d’une hypertension, d’une hyperglycémie (diabète), de dyslépidémies et de l’obésité, pathologies qui accélèrent la formation des plaques et leurs complications2. La teneur du sang en lipides est une composante majeure de l’incidence des maladies cardiovasculaires et notamment la concentration en « mauvais cholestérol », les LDL, par opposition au « bon cholestérol » représenté par les HDL. Ainsi le rapport LDL sur HDL est proportionnel au risque d’apparition de ces maladies (cf. : Tableau 1).
Des régions du monde jusqu’à présent épargnées des maladies cardiovasculaires s’y trouvent aujourd’hui confrontées en raison de l’évolution de leur style de vie qui se rapproche du modèle occidental. C’est le cas en Afrique, Inde et Asie3. Ces nouvelles données ont amené à prendre en compte les déterminants sociologiques et économiques comme facteurs de risque des maladies cardiovasculaires (80% d’entre elles concernent les populations au revenu faible ou moyen).
A.3 : Mécanismes de l’athérogenèse.
La paroi d’une artère normale est constituée de trois couches cellulaires séparées par du tissu élastique (cf. : Figure1). De la lumière du vaisseau vers l’extérieur, se trouvent :
- L’intima formée d’une monocouche de cellules endothéliales (CE) et de l’espace sous-endothélial riche en collagène et protéoglycanes.
- La media constituée de cellules musculaires lisses (CML). Cette couche est séparée des deux autres par la limitante élastique interne (côté intima) et la limitante élastique externe (côté adventice).
- L’adventice formée par du tissu conjonctif comprenant la vascularisation propre du vaisseau.
• Théorie inflammatoire de l’athérogenèse
Il y a une trentaine d’années, la théorie de l’athérogenèse était dominée par le rôle des lipides étant donné les fortes corrélations qui existaient entre l’hypercholestérolémie et l’apparition des plaques d’athérome4. L’implication des facteurs de croissance et la prolifération des CML à l’origine de la sténose ont ensuite pris le relais dans l’intérêt porté aux mécanismes de l’athérogenèse5. À la fin des années 80, la plaque était donc visualisée comme un amas de débris lipidiques enrobés dans une capsule de CML en prolifération. La meilleure connaissance des cellules immunitaires depuis ces 20 dernières années ont laissé petit à petit la place à une théorie inflammatoire de l’athérogenèse6. La participation active du système immunitaire serait indispensable à la formation de la plaque mais également à sa progression.
• Rôle physiologique de l’endothélium
L’endothélium de l’intima est la partie du vaisseau directement en contact avec la circulation sanguine, d’où un rôle important de régulation, notamment celui du maintien du tonus vasculaire. L’endothélium sécrète de nombreuses substances vasoactives dont la principale est l’oxyde nitrique (NO), dérivé réactif de l’oxygène produit de façon constitutive par la eNOS (endothelial NO synthase)7. Le NO est indispensable à l’homéostasie vasculaire et ses principaux rôles sont :
- Le maintien d’une surface anti-thrombotique
- Favoriser la prolifération des CE permettant ainsi la réparation de l’endothélium. - Un fort pouvoir anti-oxydant éliminant les espèces réactives de l’oxygène (ERO), dont
l’anion superoxyde (O2°– ) qui endommage fortement l’endothélium.
- Une action sur les CML de la media d’une part en inhibant leur prolifération, et d’autre part en ayant un effet relaxant sur ces cellules.
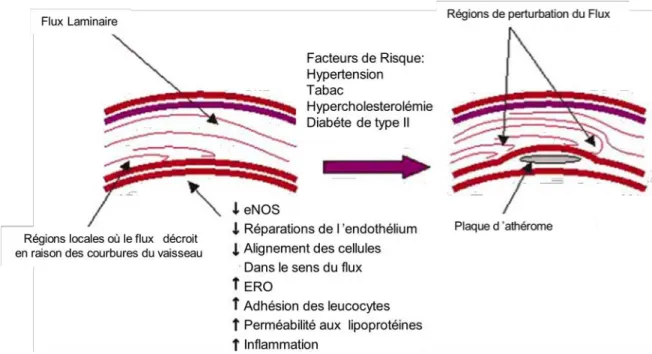
L’effet vasodilatateur du NO est équilibré par la sécrétion de peptides vasoconstricteurs, tels que l’endothéline et l’angiotensine II. La balance des effets de ces substances dicte le tonus du vaisseau qui lui-même conditionne l’écoulement du flux sanguin. Lors de son passage, le flux sanguin applique des forces de frottement contre la paroi vasculaire. Lorsque ces forces de frottement sont constantes, cela confère à l’endothélium un statut anti-inflammatoire, anti-oxydant et anti-thrombotique8.
Figure 2: Importance des forces de frottement dans la dysfonction endothéliale
et dans l’apparition de la plaque d’athérome.D’après KS. Cunningham et AI.
Gotlieb, 2005 (ref:8)
A.3.1 : La dysfonction endothéliale
Certaines zones de l’arbre circulatoire telles que les courbures artérielles, imposent une contrainte physique à l’écoulement sanguin, rendant les forces de frottement diminuées, voire négatives à ce niveau. Cet événement perturbe l’homéostasie de l’endothélium vasculaire et est à l’origine de la dysfonction endothéliale9 (cf. : Figure 2). Ce dysfonctionnement est aggravé par la présence des facteurs de risques précédemment décrits : hypercholestérolémie, diabète, hypertension, tabagisme, ainsi que certains agents infectieux ou toxines, qui vont modifier le métabolisme du NO, diminuer sa biodisponibilité et provoquer une augmentation de la perméabilité endothéliale. Les LDL pro-athérogènes, présentes en plus ou moins grande quantité, voient leur passage dans le sous-endothelium facilité par ces conditions10. Ces événements sont l’explication au développement préférentiel des plaques d’athérome au niveau des bifurcations artérielles11.
A.3.2 : Oxydation des LDL dans l’intima
Le passage des LDL à travers l’endothélium est également rendu possible par la petite taille des LDL : plus elles sont petites, plus elles sont athérogènes12.Une fois l’endothélium traversé, ces LDL se retrouvent dans l’espace sous-endothélial où elles restent piégées en raison d’interactions qui s’établissent entre des constituants de l’ApoB100 et les protéoglycanes de la matrice13. La survenue préalable d’une dysfonction endothéliale conduit à une production d’ERO par ces cellules, radicaux qui vont attaquer la partie lipidique et protéique des LDL emprisonnées dans l’intima. Ce processus aboutit à l’oxydation des LDL et à la formation de produits dérivés (cf. : Chapitre LDL oxydées) qui induisent l’expression de molécules d’adhésion par les CE permettant le recrutement des leucocytes sur le site de lésion14.
A.3.3 : Recrutement des monocytes et lymphocytes T
15Les protéines d’adhésion exprimées à la surface des CE sont principalement représentées par VCAM-1 (Vascular Cell Adhesion Molecule-1), ainsi que par la P et la E selectine. VCAM-1 n’est pas uniquement induite par les LDL oxydées, mais elle est également exprimée lorsque le flux sanguin perd son caractère laminaire9. D’ailleurs, les cellules endothéliales expriment VCAM-1 en réponse au cholestérol uniquement dans les régions où le flux sanguin est perturbé, soit dans les zones propices au développement des lésions16. VCAM-1, P et E selectine reconnaissent la β2-intégrine et le ligand VLA-4
Tableau 2: Les premières lésions lipidiques se produisent dés les premières
années de vie mais peuvent régresser. Le développement des plaques d’athérome
de produit dans la troisième décennie de vie. Adapté de la classification de
Herbert.C Stary.
exprimés à la surface des monocytes et des lymphocytes T. Alors que les P et E selectines permettent le roulement des cellules immunitaires à la surface de l’endothélium, la fixation à VCAM-1 induit leur extravasation par diapédèse entre les jonctions serrées des CE17 18. La migration de ces leucocytes dans l’intima est également rendue possible grâce à un gradient de chemokines établit par les CE, comprenant le facteur MCP-1 (Monocyte Chemoattractant Protein-1) capté par le récepteur CCR2 des leucocytes 19. Une fois dans l’intima, les monocytes se multiplient et acquièrent un phénotype macrophagique sous l’effet du M-CSF (Macrophages Colony Stimulating Factor) d’abord sécrété par les CE puis par les macrophages eux-mêmes. Les lymphocytes T passent la barrière endothéliale suivant le même principe que les monocytes, et sont ensuite stimulés par les antigènes présents dans l’intima, dont les LDL oxydées.
A.3.4 : Formation des cellules spumeuses
La sollicitation des monocytes a pour but initial d’épurer l’intima du cholestérol accumulé. Les macrophages vont ainsi internaliser les LDL oxydées par l’intermédiaire de leurs récepteurs éboueurs ou ‘scavenger receptor’20, dont l’expression n’est pas régulée. De ce fait les macrophages se surchargent en cholestérol et se transforment en cellules spumeuses qui s’accumulent dans la paroi21 pour former les stries graisseuses caractéristiques des lésions précoces de l’athérosclérose. Les stries graisseuses sont déjà présentes en grand nombre chez les jeunes adultes où elles sont asymptomatiques, et peuvent évoluer en plaque d’athérome ou disparaître (cf. : Tableau 2).
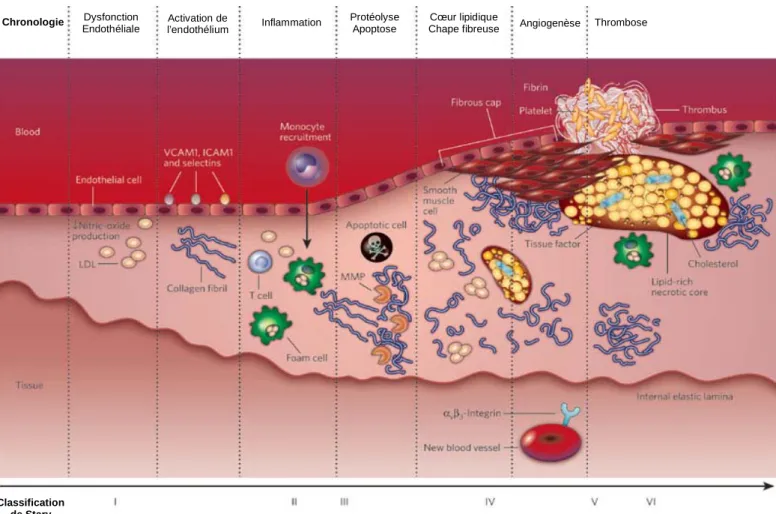
Sous l’influence du M-CSF (mais aussi des lymphocytes T), les cellules spumeuses produisent des cytokines pro-inflammatoires (IL-1, IL-6, TNFα, IFN-γ). Les lymphocytes T sécrètent des cytokines pro-inflammatoires, mais aussi anti-inflammatoires (IL-10, IL-4)22. Il en résulte néanmoins la présence d’une réaction inflammatoire locale qui induit la progression de la lésion, notamment par l’arrivée continue de nouveaux monocytes qui se transforment en cellules spumeuses. Par ailleurs l’IL-1 et le TNF-α induisent l’expression de VCAM-1 dans les CE23. Les lipides de la plaque sont d’abord essentiellement intracellulaires puisqu’ils sont digérés par les macrophages, puis ils deviennent extracellulaires lorsque la capacité épuratrice de ces cellules est dépassée. À ce stade, les lipides se regroupent au centre de la plaque pour former un amas appelé cœur lipidique composé de cholestérol et d’esters de cholestérol, mais également de débris apoptotiques et de cellules nécrosées, sources inflammatoires pour la plaque (cf. : Chapitre Apoptose).
Chronologie Dysfonction Endothéliale Activation de l’endothélium Inflammation Protéolyse Apoptose Cœur lipidique
Chape fibreuse Angiogenèse Thrombose
Classification de Stary
Figure 3: Etapes de la formation de la plaque d’athérome. Les LDL natives passent dans le
sous-endothélium où elles sont piégées et oxydées. Cette oxydation provoque l’arrivée des
monocytes sur la lésion qui vont phagocyter les LDL oxydées et se transformer en cellules
spumeuses. Ces dernières s’accumulent, forment le cœur lipidique et induisent également la
migration et la prolifération des CML. Les CML recouvrent le cœur lipidique pour former la
chape fibreuse. D’après Javier Sanz et Zahi A. Fayad, Nature 2008 (ref: 334).
A.3.5 : Migration et prolifération des CML
En parallèle de la formation des stries graisseuses et du cœur lipidique, les CML de la media migrent vers l’intima où elles prolifèrent. Cet événement se produit en réponse à :
- la levée d’inhibition de prolifération des CE sur les CML
- La sécrétion du CD40 ligand par les lymphocytes T qui participe à la migration des CML24.
- la production d’ERO25 et de cytokines (TNF-α)26 par les cellules de la paroi vasculaire.
- la production de facteurs de croissance, dont le PDGF (Platelet Derived Growth Factor) et le TGFβ (Transforming Growth Factor) libérés par les macrophages, les CML et les CE27.
Une fois dans l’intima, les CML synthétisent de la matrice extracellulaire et du collagène qui permettent la formation d’une chape fibreuse autour du cœur lipidique afin d’isoler le centre de la plaque de la circulation générale28 (cf. : Figure 3 ).
A.3.6 : Evolution de la plaque
La plaque se développe longtemps sans altérer le calibre vasculaire, car le vaisseau s'adapte par un élargissement compensatoire appelé remodelage vasculaire. Lorsque la masse intimale excède 40 % de la surface totale de la paroi, le remodelage excentrique de la paroi n'est plus suffisant pour contenir la plaque, et son développement progresse vers la lumière du vaisseau. Une plaque est dite stable lorsque le coeur lipidique est peu étendu et calcifié, et que la chape fibreuse est épaisse sans entamer le diamètre du vaisseau. De telles plaques restent cliniquement silencieuses.
• Complications cliniques
Les manifestations cliniques graves de la maladie athéromateuse ne sont pas en rapport avec la taille mais avec son instabilité29. Une plaque instable est caractérisée par un endothélium fragilisé et une chape fibreuse fine30. En effet, l'intégralité fonctionnelle de l’endothélium maintient les propriétés thrombo-résistantes du vaisseau, de même que l’intégrité de la chape fibreuse protége de la mise en contact avec le cœur lipidique pro-thrombogène.
Lorsque le coeur lipidique occupe plus de 40 % du volume de la plaque, les cytokines pro-inflammatoires largement présentes vont stimuler l’activité des metalloprotéases matricielles et l’apoptose des CML, tandis que d’autres comme l’IFN-γ inhibent la production de collagène, événements aboutissant à l’amincissement de la chape fibreuse31. Par ailleurs, lorsque la plaque entame le diamètre du vaisseau, les forces de frottement qui s'exercent sur la circonférence de la paroi agressent l’endothélium8. Les phénomènes d’apoptose des cellules vasculaires sont donc très importants dans la survenue de rupture de la plaque, puisque l’apoptose des CE contribue à l’érosion de l’endothelium, alors que celle des CML fragilise la chape fibreuse (cf. : chapitre Apoptose). Par ailleurs, les cellules vasculaires en apoptose libèrent des microparticules directement responsables d’une activité pro-coagulante en raison d’une forte expression de facteur tissulaire (FT) à leur surface32.
La rupture de plaque expose au sang le matériel lipidique fortement thrombogène. Il se produit une réaction de coagulation initiée par le facteur tissulaire qui induit la formation d’un thrombus33. L’érosion de l’endothélium sans rupture de plaque est également à l’origine de la formation de micro-thrombi en raison des propriétés pro-adhésives et pro–coagulantes des cellules endothéliales en apoptose34. Le thrombus est à l’origine des complications majeures de l’athérosclérose (AVC et infarctus). Cependant, toute rupture de plaque n’aboutit pas systématiquement à une obstruction de la circulation car un système de fibrinolyse permet de dissoudre les caillots formés. Cette fibrinolyse est une source importante de PDGF qui va stimuler la prolifération des CML. Il est ainsi courant d’observer des sténoses consécutives aux ruptures de plaque. Ces dernières sont la plupart du temps asymptomatique car leur évolution étant lente, la diminution du débit sanguin est naturellement compensée par une néo-angiogénèse. Chaque étape d’évolution de la plaque a été caractérisée et a permis d’établir une classification des lésions (cf. : Tableau 2, p20).
Bien que la théorie actuelle de l’athérogenèse soit inflammatoire, le rôle des LDL reste primordial dans la formation et l’évolution de la lésion. Nous allons voir dans le chapitre suivant la structure et les fonctions de ces molécules dont les effets in vitro sur les cellules vasculaires sont très nombreux.
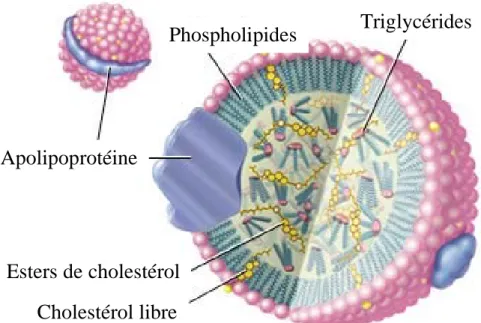
Phospholipides
Triglycérides
Apolipoprotéine
Esters de cholestérol
Cholestérol libre
Figure 4: Schéma d’une lipoprotéine. Si on considère la LDL, son
cœur contient en moyenne 1600 molécules d’esters de
cholestérol, 100 molécules de triglycérides et 1 molécule de
carotène. Sur la couche externe, on trouve 600 molécules de
cholestérol (dont les acides gras polyinsaturés représentent 25 à
50%), 700 molécules de phospholipides et 6 molécules de
tocophérol. D’après R. Salvayre, 2002 (ref: 50)
B. Les LDL oxydées
L’importance du cholestérol et plus particulièrement des LDL dans l’athérosclérose est connue depuis plus de 30 ans4. Le rôle des LDL dans l’initiation et la progression de la plaque d’athérome dépend des transformations biochimiques qu'elles subissent au sein de l'intima, notamment des transformations oxydatives. Des auto-anticorps dirigés contre les LDL oxydées sont présents au sein des plaques d’athérome humaines, attestant de leur présence35.
B.1 : Les lipoprotéines
Les principaux composés lipidiques, comme le cholestérol libre, les esters de cholestérol, les triglycérides ou les phospholipides, ne peuvent circuler sous forme libre dans la circulation sanguine du fait de leur hydrophobicité. Ces éléments vont donc s’associer et former des micelles dans lesquelles s’enchâsse une composante protéique, d’où leur nom de lipoprotéines. Les triglycérides et les esters de cholestérol forment le cœur des lipoprotéines, tandis que les phospholipides et le cholestérol libre forment la couche extérieure dans laquelle s’insèrent les apolipoprotéines. Cette monocouche périphérique permet la solubilisation des lipoprotéines dans le plasma grâce aux propriétés amphiphiles des phospholipides (cf. : Figure 4).
Il existe plusieurs types de lipoprotéines mais seules certaines sont athérogènes: les VLDL, les IDL, et les LDL (notamment les LDL petites et denses) dont les taux sont étroitement liés au risque cardiovasculaire36. Les lipoprotéines assurent le transport des lipides d’un tissu à un autre. Durant ce trajet, elles subissent des modifications qui affectent leur composition, leur structure et leur fonction. Arrivées à destination, les lipoprotéines sont captées par des récepteurs spécifiques afin de délivrer leur contenu aux cellules. Le cholestérol entrant est utilisé pour le stockage et la production d’énergie, le maintien des membranes cellulaires et la fabrication des hormones stéroïdiennes et des acides biliaires.
B.1.1 : Origine des LDL
Les lipoprotéines sont classées en fonction de leur taille et de leur contenu en lipides, leur densité étant inversement proportionnelle à leur contenu en lipides. Les principales apoprotéines sont l’Apo A, E, C et B, chacune d’elles possédant différents sous-types. Les premières lipoprotéines engagées dans l’assimilation des lipides sont les chylomicrons,
Figure 5: Captation des LDL natives par le
lipoprotéines les plus grosses. Ils se forment dans la muqueuse de l’intestin durant l’absorption des graisses et permettent l’apport de cholestérol au foie. Les VLDL (Very Low Density Lipoprotein, ApoE, C et B) se forment dans le foie et transportent les triglycérides vers les tissus extra-hépatiques. Après avoir perdu l’ApoE et une partie de leurs triglycérides, les VLDL deviennent des IDL (Intermediate Density Lipoprotein, ApoE et B). Certaines IDL sont captées par le foie tandis que les autres perdent encore plus de triglycérides et deviennent des LDL (Low Density Lipoprotein) auxquelles il ne reste plus que l’ApoB. Enfin, les HDL (High Density Lipoprotein, ApoA) sont synthétisées par le foie et l’intestin et permettent d’absorber le cholestérol qui quitte les cellules, notamment au niveau de la paroi artérielle. Une partie de ce cholestérol est ramenée au foie (transport reverse du cholestérol) où il est excrété dans la bile et éliminé dans les fèces. Les HDL permettent donc d’abaisser le taux de cholestérol plasmatique et cette propriété leur confère un rôle protecteur vis-à-vis du développement de l’athérosclérose, d’autant plus qu’ils possèdent un fort pouvoir antioxydant apporté par l’enzyme paraoxanase37.
B.1.2 : Captation des LDL
Dans les conditions normales, les LDL sont captées par un récepteur spécifique ubiquitaire à toutes les cellules, le récepteur ApoB/E (ou LDL-R)38. La voie cellulaire d’endocytose des LDL couplées à leur récepteur a été initialement décrite par Brown et Goldstein, ce qui leur a valu le prix Nobel de médecine en 198539. Les LDL circulantes se fixent sur leur récepteur au niveau des puits recouverts de clathrine. Les LDL sont ensuite internalisées par endocytose dans les vésicules lysosomiales où tous les constituants lipidiques et protéiques sont dégradés (cf. :Figure 5). Ce mécanisme assure à la cellule un approvisionnement en cholestérol dont le stockage réprime l’expression de novo des récepteurs aux LDL et limite ainsi leur captation supplémentaire 39. Les récepteurs ApoB/E ne reconnaissent pas les LDL modifiées ou oxydées40.
B.2 : Oxydation des LDL
L’oxydation des LDL intervient dans l’intima des artères après leur passage à travers l’endothélium. Elle se produit en réponse au stress oxydant généré par les CE qui participe activement à la formation de la plaque et à sa progression.
B.2.1 : Le stress oxydant (Beaudeux, JL, 2006)
Un stress oxydant est provoqué par un déséquilibre entre la production d’espèces réactives de l’oxygène (ERO) et les systèmes antioxydants. Les ERO regroupent les radicaux libres oxygénés (espèces chimiques possédant un électron célibataire non apparié) dont le chef de file est l'anion superoxyde (O2°–), et les dérivés non-radicalaires (ne
possédant pas d’électron célibataire), auquel appartient le peroxyde d’hydrogène H2O2. La production d’ERO au sein des différentes cellules de la paroi artérielle est physiologique et continue41. Elle est assurée par différents systèmes tels que la NAD(P)H oxydase, les lipoxygénases, la cyclo-oxygénase 1 (COX1), l’eNOS, le cytochrome p450 et la xanthine oxidase (XO), qui sont actives aussi bien au niveau des CE que des CML42. La chaîne respiratoire mitochondriale produit également l’anion superoxyde qui est convertit ensuite en H2O2 par la superoxyde dismutase (SOD). La présence concomitante d’antioxydants endogènes permet de neutraliser les ERO en les transformant en molécules stables et non réactives. Il s’agit de la SOD, de la catalase, de la glutathion peroxydase et de la thioredoxine réductase. Une seconde ligne de défense est représentée par les piégeurs de radicaux libres, pour la plupart apportés par l'alimentation, et dont le rôle essentiel est de neutraliser les effets toxiques des ERO, limitant ainsi l’atteinte de l'intégrité cellulaire. Parmi ceux-ci, on retrouve la vitamine E (α-tocophérol), la vitamine C (ascorbate), les caroténoïdes (vitamine A et β-carotène)
L’équilibre entre éléments oxydants et anti-oxydants est rompu en présence des facteurs de risque de l’athérosclérose (hypertension, diabète, hyperlipidémie, tabagisme). Ces conditions induisent l’expression accrue de la NAD(P)H oxydase, de la COX-2, de la thromboxane synthetase, des lipoxygénases et de l’iNOS (inductible NO Synthase)42. L’activation de la iNOS conduit à une production massive de NO toxique pour la cellule7. Le métabolisme de la chaîne respiratoire est également activé en présence de fortes concentrations de glucose43, et conduit à une surproduction d’anion superoxyde44. L’O2°- en excès réagit très rapidement avec le NO pour former l’ion peroxynitrite ONOO- très toxique45. Il résulte de l’ensemble de ces événements à la fois une diminution de la biodisponibilité du NO protecteur (issu de la eNOS), et un fort stress oxydant participant à la dysfonction endothéliale et à l’origine de l’oxydation des LDL. Des souris déficientes pour la 15-lipoxygénase présentent moins de lésions que des souris sauvages, mettant en évidence l’importance de cette enzyme dans l’oxydation des LDL46.
Les cellules immunitaires présentes sur le site de lésion participent également à l’oxydation des LDL. La NAD(P)H oxydase, l’iNOS ainsi que la myeloperoxidase des macrophages et cellules T sont largement impliquées dans l’oxydation des LDL20 46. Les LDL deviennent fortement oxydées avec atteinte de leur apolipoprotéine ce qui empêche leur reconnaissance par les récepteurs classiques de captation des LDL. Le stress oxydant joue un rôle important tout au long du processus athéromateux 4748.
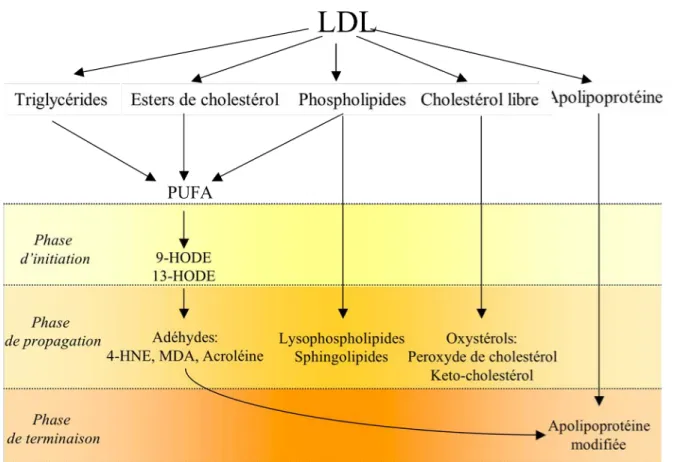
B.2.2 : La peroxydation lipidique
L’oxydation des LDL, ou peroxydation lipidique, implique une cascade de dégradation en chaîne des acides gras, conduisant à la formation d'hydroperoxydes instables qui vont également attaquer la composante protéique de la lipoprotéine49. L’oxydabilité des LDL varie en fonction de leur composition : elle augmente avec la teneur en acides gras polyinsaturés, de leur structure, de leur teneur en antioxydants, et des conditions pathologiques50. L’oxydation des LDL se déroule selon plusieurs étapes (cf. : Figure 6) :
- La phase de latence permet la consommation des antioxydants par les ERO51
- La phase d'initiation correspond à l’attaque des acides gras polyinsaturés (les PUFA pour Poly Unsaturated Fatty Acids). Ils sont particulièrement vulnérables du fait de leurs doubles liaisons. Ces premières modifications des LDL n’affectent pas l’apolipoprotéine.
- La phase de propagation correspond à la formation d’aldéhydes depuis les PUFA. Ils sont représentés, entre autres, par le 4-hydroxynonénal (4HNE), le malondialdéhyde (MDA) et l’acroléine. Les autres lipides sont également modifiés durant cette phase : ainsi le cholestérol est oxydé en oxystérols (cholestérol peroxyde, kéto-cholestérol…). Les aldéhydes libérés, 4-HNE et MDA, peuvent se lier à l’Apo B100 des LDL, modifiant dans un premier temps son activité physiologique, puis activant ensuite sa dégradation52. Ces molécules de LDL fortement modifiées ne sont plus reconnues par les récepteurs classiques mais par les récepteurs scavenger des macrophages.
- La terminaison de la réaction d’oxydation survient soit par épuisement du substrat, soit par formation de dérivés stables.
• Les Produits d’Oxydation Lipidique
L’oxydation des LDL aboutit à la production de plusieurs dérivés lipidiques dont les effets sont résumés ici.
Les Acides Gras oxydés
Au sein de la particule LDL, Les PUFA sont présents sous forme d’esters de phospholipides en surface de la LDL, et sous forme d’esters de cholestérol dans le cœur. La péroxidation des PUFA donne des composés très bioréactifs, tels que le 13-hydroxyperoxide (13-HPODE) ou le 9-HODE, dont le rôle principal est celui d’intermédiaires indispensables à la phase de propagation 53. Néanmoins, les hydroperoxides participent aussi à l’effet toxique des LDL oxydées 54, 55.
Les Aldéhydes
Le MDA, le 4-HNE et l’acroléine sont les plus abondants. Ces dérivés sont très réactifs vis-à-vis des groupements thiols et amines 56 et forment des adduits sur les protéines, altérant l’homéostasie cellulaire et induisant l’apoptose 57. Ces composés sont peu impliqués dans les phénomènes d’inflammation, mais interviennent plutôt dans la prolifération et l’apoptose des cellules vasculaires en fonction de leur concentration 58.
Les Oxystérols
Les oxystérols les plus largement présents dans les LDL oxydées sont le 7-kétocholestérol et le 7/25 hydroperoxycholestérol (7-0H et 25-OH) 59, qui seraient responsables de nombreux effets biologiques des LDL oxydées (inflammation, régulation du métabolisme du cholestérol), et de l’apoptose des cellules vasculaires en culture 60, 61. Le 7-kétocholestérol induit l’apoptose des CML par le biais d’un stress oxydatif produit suite à l’activation d’une NADPH oxydase 62. D’ailleurs, des antioxydants comme le NAC ou le glutathion protégent les macrophages de l’apoptose induite par le 7-kétocholestérol 63. Alors que le 7-OH induit la synthèse du facteur tissulaire, cette propriété n’est pas retrouvée pour le 7-kétocholestérol 64.
Les Phospholipides oxydés et les Sphingolipides
Les phospholipides oxydés peuvent êtres dégradés en lysophosphatidylcholine (LPC) et en acide lysophosphatidique (LPA). La LPC induit la prolifération des CML65, des CE 66, et des macrophages 67, mais a cependant également été décrite comme provoquant l’apoptose des CML68. Les sphingolipides regroupent le céramide, la sphingosine et la sphingosine-1-phosphate. Ils peuvent êtres formés durant l’oxydation des LDL ou via la signalisation dépendante des LDL oxydées et des cytokines pro-athérogènes, et sont impliqués dans la prolifération69 ou l’apoptose cellulaire70.
La toxicité des LDL oxydées est due à la présence de ces lipides modifiés 71. Leurs effets variant selon leur concentration52 et le degré d’oxydation des LDL, il est difficile
Figure 7: Principaux récepteurs scavenger impliqués dans la captation des
d’établir la part respective de chaque dérivé dans les effets observés in vitro des LDL oxydées. Les LDL minimalement oxydées, bien que leur oxydation ne soit que partielle, participent également aux effets toxiques de ces molécules et à la formation de la plaque 55
49.
B.2.3 : Les Récepteurs Scavenger
72Les modifications subies par les lipides composant les LDL oxydées empêchent leur reconnaissance par le LDL-R 40. Les lipides oxydés sont alors reconnus par les récepteurs éboueurs, ou récepteurs scavenger, des cellules, principalement les macrophages. Plusieurs types de récepteurs scavengers sont impliqués dans le développement des lésions d’athérosclérose (cf. : Figure 7).
Classe A
SR-AI (Scavenger Receptor class AI) et SR-A2 sont surtout présents au niveau des macrophages 73, mais également au niveau de CE et de CML de la plaque 74. Ces récepteurs sont responsables de la captation d’environ 80% des LDL oxydées par les macrophages 75. Les SR-AI et AII reconnaissent également les cellules apoptotiques 76.
Classe B
Le CD36 et le SR-BI, bien qu’appartenant à la même famille, ont des fonctions différentes dans le métabolisme des lipides. Le CD36 a une distribution assez large, dont les macrophages et les CE 76. Il reconnaît les LDL oxydées avec une assez faible affinité 77, mais fixe également les LDL natives et minimalement oxydées 78. Au contraire, le SR-BI assure le transport reverse du cholestérol puisqu’il fixe les HDL. Il est fortement exprimé au niveau du foie et des macrophages 79. Des souris KO pour SR-BI développent des lésions d’athérosclérose plus importantes que des souris contrôles 80.
Autres
LOX-1 (Lectin-like OXidized low density receptor) est exprimé par les macrophages, les CE et les CML et fixe les LDL oxydées. Des souris surexprimant cette protéine présentent une accélération du développement des lésions 81. Le CD68, appelé macrosialine chez la souris, est exprimé dans les lysosomes et les endosomes des macrophages. Ce récepteur ne semble pas impliqué dans la captation des LDL oxydées, mais plutôt dans leur digestion lysosomale 82. La relevance de ces récepteurs dans le processus athéromateux reste à approfondir.
Les LDL minimalement oxydées ont été nommées « cheval de Troie » par l’équipe d’Hajjar DP 83, car leur ressemblance avec les LDL natives leur permet d’être en partie captées par le LDL-R 71, alors qu’elles sont chargées en éléments bioactifs. LOX-1 et CD36 sont également impliqués dans la captation des LDL minimalement oxydées 40.
• Dégradation des LDL oxydées
Les LDL oxydées sont captées puis internalisées dans le compartiment endo-lysosomal où elles sont dégradées. Les composés toxiques libérés dans le cytosol vont induire leurs effets toxiques. Cependant, la dégradation des LDL oxydées est plus lente que celle des LDL natives 84, et les dérivés oxydés peuvent s’accumuler dans les lysosomes, inactiver ces derniers, induire leur rupture puis la mort cellulaire 85. Contrairement à la captation des LDL par le récepteur Apo B/E, l’ingestion de lipides par les récepteurs scavenger n’est soumise à aucun mécanisme de rétrocontrôle : l’expression des récepteurs scavenger à la surface de la cellule est même amplifiée en présence de ces lipides 21. Enfin, les oxystérols sont également susceptibles de s’intercaler dans les membranes cellulaires altérant ainsi leur structure et leur fonction 60.
B.3 : Effets cellulaires des LDL oxydées
Les LDL oxydées captées et dégradées activent diverses voies de signalisation impliquées dans la réponse inflammatoire, la prolifération ou l’apoptose des cellules. Leurs principaux effets sont résumés ici :
- La surexpression par l’endothélium de protéines d’adhésion. Les composants oxydés des LDL, dont les phospholipides et les aldéhydes, induisent la transcription du gène VCAM-1 par l’intermédiaire du facteur nucléaire NF-κB 23.
- La surexpression des récepteurs scavenger 21
- La surexpression de facteurs de croissance et de cytokines impliqués dans le recrutement des monocytes 23, dans la prolifération des CML86 ainsi que dans la réponse inflammatoire 87, 88. La prolifération des CML en culture est induite pour de faibles concentrations en LDL oxydées et implique notamment la voie des sphingolipides 8990.
- L’augmentation de sécrétion de matrice extracellulaire permettant la rétention des LDL dans la plaque et assurant la solidité de la chape fibreuse 2.
- La surexpression de métalloprotéases qui stimulent la migration cellulaire, le remodelage mais induisent également la fragilisation de la plaque 92.
- L’altération de l’hémostase dans un sens pro-thrombotique : au niveau de l’endothélium, les LDL oxydées augmentent la production de FT, et au niveau des plaquettes, elles stimulent la production de thromboxane A2 93.
La signalisation induite par les LDL oxydées varie en fonction de leur composition, de leur concentration et de leur temps d’action 94. De nombreux travaux ont montré que les LDL oxydées étaient toxiques pour les cellules vasculaires en culture, et induisaient leur apoptose. L’importance de cette mort cellulaire dans la progression de la plaque et les mécanismes d’apoptose activés par les LDL oxydées font l’objet du chapitre suivant.
C. Apoptose et Athérosclérose
C.1 : L’Apoptose
L'apoptose a été mise en évidence en 1972 par Andrew H. Wyllie lors de l'étude de tissu par microscopie électronique 95. Il désigna ce phénomène sous le terme de mort cellulaire naturelle ou ‘apoptosis’, terme provenant d'un ancien poète grec désignant la chute des feuilles en automne (apo : au loin et ptosis : chute). On nomme apoptose le processus par lequel des cellules déclenchent leur auto destruction en réponse à un signal. C'est une mort cellulaire physiologique, génétiquement programmée, nécessaire à la survie des organismes pluricellulaires. Elle est en équilibre constant avec la prolifération cellulaire.
L’apoptose se caractérise par un rétrécissement de la cellule, la condensation de la chromatine et la fragmentation de l’ADN entre chaque nucléosome 95. Elle induit également une redistribution des phospholipides membranaires : la phosphatidylsérine, normalement constitutive du feuillet interne, est exposée sur le feuillet externe. Ce phénomène permet la reconnaissance des cellules en apoptose par les macrophages et leur phagocytose, évitant ainsi l’apparition d’une réaction inflammatoire, caractéristique de la mort par nécrose 96. Les cellules en apoptose génèrent également des ERO qui oxydent les lipides membranaires, dont les phospholipides 97. La cellule expose donc en surface des phospholipides oxydés, de structure semblable à celle des LDL oxydées, et qui sont reconnus par les récepteurs scavenger des macrophages, majoritairement le CD36 98.
La mort par nécrose survient lors d’une atteinte des fonctions cellulaires et se caractérise par des altérations de la membrane et une lyse de la cellule. Le contenu cytoplasmique de la cellule, dont les protéases lysosomiales, est déversé dans le territoire environnant, ce qui provoque une réaction inflammatoire importante.
C.2 : L’apoptose dans l’athérosclérose
99 30L’apoptose est un processus qui survient dans la plaque depuis sa formation jusqu’à la rupture et qui concerne tous les types cellulaires. Cette mort cellulaire devient néfaste à partir du moment où elle dépasse la prolifération cellulaire et surtout lorsque l’épuration des corps apoptotiques ne se produit plus. Les cellules meurent alors par nécrose et viennent alimenter le cœur nécrotique, amplifiant la réaction inflammatoire. L’apoptose au sein des
plaques d’athérome a été mise en évidence au niveau de plaques instables et serait associée à une érosion ou à une rupture de la plaque, ainsi qu’aux phénomènes thrombogènes 100.
• Apoptose des cellules endothéliales
Dans les zones vasculaires naturellement protégées de l’apparition de plaques en raison de l’existence d’un flux laminaire, les CE ne se divisent que très peu et ont une durée de vie très longue (jusqu’à 20 ans). A l’opposé, l’endothélium présent au niveau des courbures artérielles est caractérisé par un plus grand turn-over des cellules, signifiant que l’apoptose des CE est plus importante dans ces régions 101. L’apoptose des CE expose les éléments thrombogènes du sous-endothélium en surface de la plaque 102, sans compter que les CE en apoptose sont elles-mêmes pro-coagulantes 103 104. L’ensemble de ces éléments met en évidence le rôle important de l’apoptose de CE dans la dysfonction endothéliale et la rupture de plaque. L’ensemble des facteurs impliqués dans l’apparition de l’athérosclérose favorise l’apoptose des cellules endothéliales vasculaires, en particulier des concentrations élevées en glucose comparables à celles retrouvées chez les patients diabétiques 105, les ERO 106, l’angiotensine 107 et le lipopolysaccharide bactérien 108. De nombreuses études in vitro ont montré que les LDL oxydées étaient toxiques pour les cellules endothéliales et provoquaient leur apoptose ou leur nécrose 109110, 111.
• Apoptose des cellules musculaires lisses
L’apoptose des CML participe à l’amincissement de la chape fibreuse, affaiblit et déstabilise fortement les plaques dont le cœur lipidique est important 112. Les produits dérivés de l’oxydation des LDL tels que le 4-HNE, induisent la mort cellulaire des CML 57. La synthèse de cytokines pro-inflammatoires par les cellules immunitaires augmente la sensibilité des CML à l’apoptose 113,114. Par ailleurs, les CML en apoptose sont pro-coagulantes et contribuent au risque de formation de thrombus 103, 104. Un fort taux d’apoptose des CML n’est pas associé à une diminution de la plaque car la mort provoquée étant souvent massive, son intensité dépasse la capacité épuratrice des macrophages. Les corps apoptotiques non-phagocytés et les cellules mourant par nécrose provoquent une réaction inflammatoire ainsi qu’une réponse auto-immune qui aggravent la lésion 115116.