Rôle de SHP-1 et de son activation pharmacologique
dans la réponse à la fumée de cigarette de tabac
Mémoire
Marie Pineault
Maîtrise en sciences cliniques et biomédicales - avec mémoire
Maître ès sciences (M. Sc.)
Rôle de SHP-1 et de son activation pharmacologique
dans la réponse à la fumée de cigarette de tabac
Mémoire
Marie Pineault
Sous la direction de :
Mathieu Morissette, directeur de recherche
François Maltais, codirecteur de recherche
Résumé
PROBLÉMATIQUE. Plusieurs membres d’une famille porteurs d’une mutation
hétérozygote du gène PTPN6 sont affectés par une forme d’emphysème précoce et sévère. Cette mutation (PTPN6Ala455Thr) cause une perte de fonction partielle de la
phosphatase SHP-1. OBJECTIFS. 1) Étudier les effets de l’activation de SHP-1 sur la réponse inflammatoire pulmonaire en contexte tabagique. 2) Caractériser les impacts pulmonaires et systémiques causés par la mutation ptpn6Ala457Thr dans un
modèle murin en contexte tabagique. MÉTHODES. Des souris BALB/c sauvages exposées à la fumée de cigarette durant quatre jours ont reçu des agonistes de SHP-1, soit le SC-60 ou le SC-43. Des souris ptpn6Ala457Thr ont été exposées à la
fumée de cigarette durant huit semaines pour évaluer l’effet de la mutation sur l’inflammation et les fonctions pulmonaires. Le rôle de SHP-1 dans l’activation des macrophages a été évalué en stimulant des macrophages dérivés de la moelle osseuse des souris ptpn6Ala457Thr avec divers agonistes des toll like receptor (TLR).
Des souris ptpn6Ala457Thr ont été immunisées avec du NP-Ficoll afin d’observer les
effets de la mutation ptpn6Ala457Thr sur les lymphocytes B et la production d’anticorps.
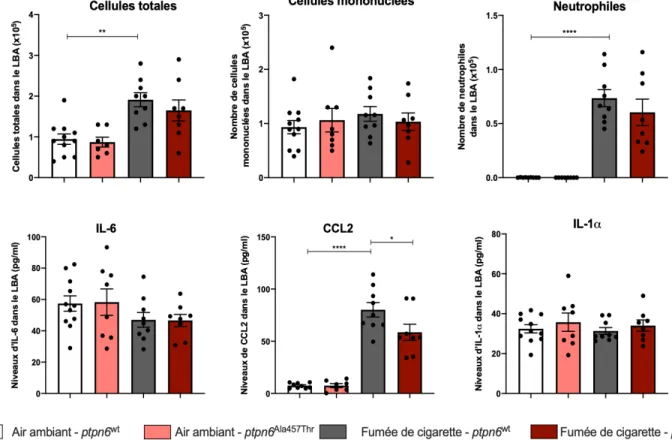
RÉSULTATS. Les agonistes de SHP-1 limitent l’augmentation du nombre de
cellules totales dans les lavages broncho-alvéolaires en contexte tabagique. La mutation ptpn6Ala457Thr n’influence pas la réponse inflammatoire pulmonaire ni les
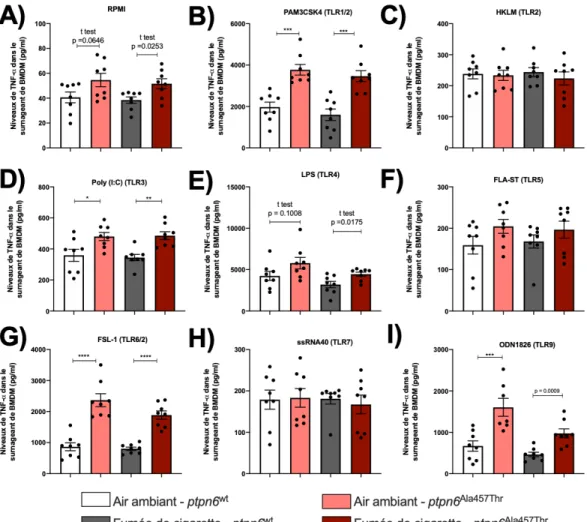
fonctions pulmonaires suite à une exposition de huit semaines à la fumée de cigarette. En réponse aux agonistes des TLR, les macrophages dérivés de la moelle osseuse de souris ptpn6Ala457Thr sécrètent davantage de TNF-a et d’IL-6 que ceux
de souris sauvages. Les souris ptpn6Ala457Thr produisent moins d’anticorps IgG1
comparés aux souris contrôles. CONCLUSION. SHP-1 est un régulateur important de l’inflammation induite par la fumée de cigarette et par l’activation des voies des TLR. La mutation ptpn6Ala457Thr affecte la réponse immune à un antigène, indiquant
Abstract
BACKGROUND. A new mutation (PTPN6Ala455Thr) was recently discovered in a
Quebecoise family in which many members develop a severe and early form of emphysema. The mutation is located in the PTPN6 gene and causes a diminution of the activity of the phosphatase SHP-1. OBJECTIVES. 1) Investigate the effects of SHP-1 activation in the context of cigarette smoke. 2) Characterize the pulmonary and systemic impacts of the ptpn6Ala457Thr mutation in a mouse model of smoking.
METHODS. BALB/c mice exposed to cigarette smoke were administered SHP-1
agonists, either SC-60 or SC-43, daily for 4 days. Mutant ptpn6Ala457Thr mice were
exposed to cigarette smoke for 8 weeks in order to assess the effect of the mutation on pulmonary inflammation and function. The role of SHP-1 in macrophages differentiation and function was assessed by stimulating bone marrow-derived macrophages of ptpn6Ala457Thr mice with various TLR agonists. In addition, production
of specific antibodies in response to NP-Ficoll immunization of ptpn6Ala457Thr mice
was performed to determine the effect of the mutation on B cells functions.
RESULTS. Administration of SHP-1 agonists SC-60 or SC-43 mitigated the increase
in total cells number in broncho-alveolar lavage of mice exposed to cigarette smoke. However, the ptpn6Ala457Thr mutation did not affect the pulmonary inflammation and
lung function changes induced by 8 weeks exposure to cigarette smoke. Nevertheless, bone marrow-derived macrophages from ptpn6Ala457Thr mice secreted
more TNF-a et d’ IL-6 in response to TLR agonists compared to those from wild-type mice. Finally, ptpn6Ala457Thr exhibited reduced levels of IgG1 antibodies following
NP-Ficoll immunization compared to wild-type mice. CONCLUSION. SHP-1 seems to be an important regulator of inflammatory response to cigarette smoke and to TLR stimuli. The ptpn6Ala457Thr mutation seems to affect processes of the adaptive
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... viii
Liste des abréviations, sigles, acronymes ... x
Remerciements ... xiii
Introduction ... 1
Le tabagisme ... 1
Impacts du tabagisme sur le poumon ... 1
Réponse inflammatoire à la fumée de cigarette ... 2
Altérations de l’immunité pulmonaire causées par la fumée de cigarette ... 3
Immunité innée ... 4 Le macrophage alvéolaire ... 4 Le neutrophile ... 7 Immunité adaptative ... 8 Le lymphocyte B ... 9 Le lymphocyte T ... 10
La maladie pulmonaire obstructive chronique ... 12
Pathophysiologie ... 13
L’importance de la composante génétique de la MPOC ... 15
Nouvelle mutation associée à l’emphysème ... 16
Src homology region 2 domain-containing phosphatase-1 (SHP-1) ... 18
La famille des tyrosines phosphatases ... 18
Description de SHP-1 ... 19
Modèles murins de déficience en SHP-1 ... 21
Pathologies associées à SHP-1 ... 22
Rôle de SHP-1 dans l’immunité ... 23
Cellules lymphocytaires ... 23
Cellules myéloïdes ... 26 Macrophages ... 26 Neutrophiles ... 29 Chapitre 1 : Hypothèse ... 31 1.1 Pertinence du projet ... 31 1.2. Hypothèse ... 31 1.3 Objectifs spécifiques ... 32
Chapitre 2 : Matériel et Méthodes ... 33
2.1 Traitement in vitro avec les agonistes de SHP-1 sur les cellules J774A.1 .... 33
2.2 Dérivation de macrophages à partir de la moelle osseuse ... 34
2.2.1 Stimulation des macrophages dérivés de la moelle osseuse ... 34
2.3 Immunobuvardage ... 35
2.4 Protocoles d’exposition à la fumée de cigarette in vivo ... 36
2.4.1 Souris et exposition à la fumée de cigarette ... 36
2.4.2 Activation de SHP-1 à l’aide d’agonistes ... 37
2.5 Euthanasie et prélèvements des échantillons ... 38
2.6 Dosage des médiateurs inflammatoires ... 39
2.7 Histologie pulmonaire ... 39
2.8 Protocole d’immunisation avec le NP-Ficoll ... 40
2.9 Dosages des anticorps spécifiques au NP-Ficoll ... 40
2.10 Analyses statistiques ... 41
Chapitre 3 : Résultats ... 43
3.1 Caractérisation des effets des agonistes de SHP-1 in vitro ... 43
3.1.1. Le SC-60 et le SC-43 modulent les niveaux de phosphorylation de STAT3 dans une lignée de macrophages murins. ... 43
3.1.2 Le SC-43 limite la réponse de macrophages dérivés de la moelle osseuse suite à une stimulation au LPS ... 44
3.2 Caractérisation des impacts des agonistes de SHP-1 sur l’inflammation pulmonaire en contexte tabagique ... 46
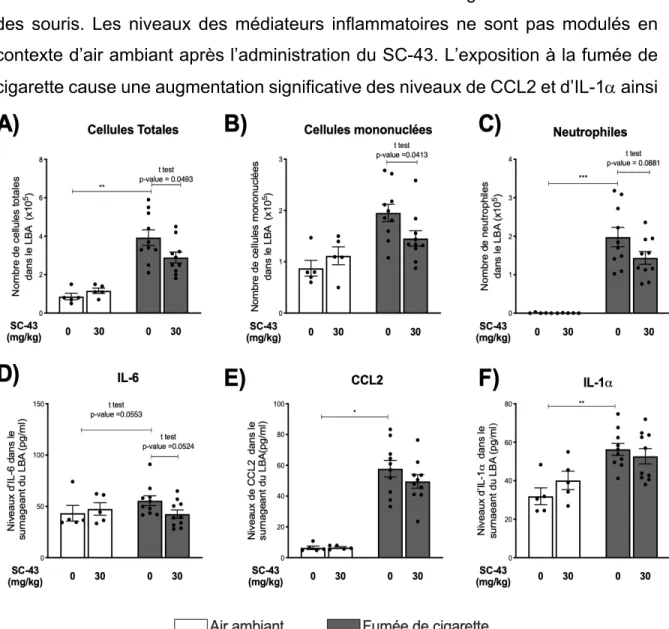
3.2.1 L’administration du SC-43 limite l’inflammation pulmonaire induite par une exposition à la fumée de cigarette ... 46
3.2.2 L’inflammation pulmonaire induite par la fumée de cigarette est limitée par l’administration du SC-60 ... 48
3.3 Caractérisation des effets pulmonaires de la mutation hétérozygote
ptpn6Ala457Thr dans un modèle murin ... 50
3.3.1 En contexte d’exposition chronique à la fumée de cigarette, la mutation ptpn6Ala457Thr ne module pas la réponse inflammatoire pulmonaire ... 50
3.3.2 Les fonctions pulmonaires ne sont pas affectées par la mutation ptpn6Ala457Thr suite à une exposition à la fumée de cigarette de 8 semaines ... 52
3.4 Investigation des effets de la mutation hétérozygote ptpn6Ala457Thr sur les progéniteurs des macrophages ... 53
3.4.1 La réponse des macrophages dérivés de la moelle osseuse des souris ptpn6Ala457Thr diffère de celle de souris sauvage suite à une stimulation avec des agonistes des TLR. ... 53
3.5 Caractérisation des anomalies immunitaires causées par la mutation ptpn6Ala457Thr ... 57
3.5.1. Présence d’amas cellulaires dans les poumons de souris ptpn6Ala457Thr ... 57
3.5.2 La réponse immune des souris ptpn6Ala457Thr à un antigène est altérée comparativement aux souris sauvages ... 58
Chapitre 4 : Discussion ... 61
4.1 Activation de SHP-1 dans l’inflammation pulmonaire ... 62
4.2 Effets pulmonaires de la mutation ptpn6Ala457Thr en contexte tabagique ... 65
4.3 Impacts systématiques de la mutation ptpn6Ala457Thr ... 69
4.4 Forces et limites ... 72
4.5 Perspectives ... 74
Conclusion ... 78
Liste des figures
Introduction
Figure 1. Schéma de la réponse pulmonaire à la fumée de cigarette. …………12
Figure 2. Schéma du mécanisme d’auto-inhibition de la phosphatase SHP-1. ………..20
Figure 3. Schéma de l’implication de SHP-1 dans certaines voies de signalisation dans les macrophages. ………26
Résultats
Figure 4. Modulation des niveaux de la phosphorylation de STAT3 suite à un traitement avec des activateurs de SHP-1. ………44
Figure 5. Le SC-43 module la réponse in vitro des macrophages dérivés de la moelle osseuse. ……….………45
Figure 6. Le SC-43 limite l’inflammation pulmonaire induite par une exposition à la fumée de cigarette. ………..47
Figure 7. Le SC-60 limite l’infiltration cellulaire pulmonaire induite par une exposition à la fumée de cigarette. ……….49
Figure 8. La réponse inflammatoire pulmonaire de souris
femelles ptpn6Ala457Thr induite par une exposition de huit semaines à la fumée
Figure 9. Les fonctions pulmonaires de souris femelles ptpn6Ala457Thr sont
similaires à celles de souris sauvages suite à une exposition de huit semaines à la fumée de cigarette. ………..52
Figure 10. Les niveaux de TNF-⍺ sécrétés par des macrophages dérivés de la
moelle osseuse de souris ptpn6Ala457Thr exposées à la fumée de cigarette
durant huit semaines en réponse à divers agonistes des TLR diffèrent de ceux de souris sauvages. ………54
Figure 11. Les niveaux d’IL-6 sécrétés par des macrophages dérivés de la
moelle osseuse de souris ptpn6Ala457Thr exposées à la fumée de cigarette
durant huit semaines en réponse à divers agonistes des TLR diffèrent de ceux de souris sauvages. ………56
Figure 12. Les poumons de souris mâles ptpn6Ala457Thr âgés de neuf mois
présentent des structures anormales et de la destruction du parenchyme pulmonaire………58
Figure 13. Schéma expérimental du protocole d’immunisation au NP-Ficoll.
………...…..59
Figure 14. Les souris ptpn6Ala457Thr ont une réponse humorale différente aux
Liste des abréviations, sigles, acronymes
A1AT : alpha-1-antitrypsine
BAFF: « B cells activation factor »
BMDM : « Bone marow-derived macrophages » ou macrophages dérivés de la moelle osseuse »
C : Celsius
CCL2 : « chemiokin ligand 2 » CD : Cluster de différenciation
CPAUL : Comité de protection des animaux de l’Université Laval ELISA: « enzyme -linked immunoabsorbant assay »
FLA-ST: Standard de Flagellin S. typhimurium
FSL1: Pam2CGDPKHPKSF Lipopeptide synthétique dérivé de Mycoplasma
fermentan
GLOD: « Global Initiative for Chronic Obstructive Lung Disease » GM-CSF: « Granulocyte-macrophages colony-stimulating factor » GSTM1 : « Glutathione S-transferase Mu-1 »
GSTP1: « Glutathione S-transferase P-1 » GSTT1: «Glutathione S-transferase theta-1 » GWAS: « Genome wide association study » HKLM: Heat-Kill Listeria Monocytogene Ig: Immunoglobuline
IgA: Immunoglobuline A IgG1: Immunoglobuline G1 IgG3: Immunoglobuline G3 IgM: Immunoglobuline M IL-1a: Interleukine 1 alpha
IL-4 : Interleukine 4 IL-6: Interleukine 6 IL-10: Interleukine 10 IL-12: Interleukine 12 IL-13 : Interleukine 13
JAK: « Janus kinase-Signal Transducer » LPS: Lipopolysaccharide
LTA : « Lymphoctoxin alpha » NE: « neutrophil elastase »
MAPK: « Mitogene-activated protein kinase » MMPs : Métalloprotéinases
MMP-9 : métalloprotéinase 9 MMP-12 : métalloprotéinase 12
MPOC : Maladie pulmonaire obstructive chronique ODN1826: « immunostimulatory oligonucleotide 1826 »
PAM3CSK4: PAM3CysSerLys4 Synthetic triacylated lipopeptide PBS: Phosphate-buffered saline
Poly (I:C): « Polyinosinic–polycytidylic acid sodium salt » PP2A: protéine phosphatase 2
PTPB1: « protein-tyrosine phosphatase 1B »
PTPN6: « protein tyrosine phosphatase non-receptor type 6 » ROS: « Reative oxygene species »
SERPINA1: « serpin family A member 1 » SERPINA3: « serpin family A member 3 »
SHP-1: « Src homology region 2 domain-containing phosphatase-1 » SHP-2: « Src homology region 2 domain-containing phosphatase-2 » SIRPα: « Signal regulatory protein alpha »
SOD2: « superoxide dismutase 2 » SOD2: « superoxide dismutase 3 »
STAT3: « Signal transducer and activator of transcription 3 » TBS: « Tris-buffered saline »
TIMP2 : « Tissue inhibitor metalloproteinase 2 » TGFB1 : « Transformaing growth factor 1 » TLT: Tissus lymphoïdes tertitaires
TLR: « Toll like receptor »
TNF-a : « Tumor necrosis factor alpha » V: Volt
VEMS: Volume expiratoire maximal par seconde a : alpha
b: bêta l: gamma k: kappa
Remerciements
Je tiens d’abord à remercier mes parents qui depuis toujours m’ont appuyée et supportée dans mes études ! Je serais éternellement reconnaissante pour tout ce que vous avez fait pour moi.
J’aimerais également remercier Mathieu Morissette, mon directeur de recherche, sans qui cette maitrise n’aurait pas été aussi enrichissante et motivante. Ton soutient, ton dévouement pour tes étudiants et tes conseils ont fortement contribué à la réussite de ma maitrise. Bien hâte de continuer au doctorat dans ton labo !
J’aimerais également prendre le temps de remercier les professionnelles de recherche exceptionnelles qui m’ont aidée durant mes deux années de maitrise. Merci Marie-Ève, Marie-Josée, Sophie et plus récemment Joanie ! Merci, Marie-Jo pour tes leçons de tricot, mais surtout merci de m’avoir permis d’apprendre énormément. Merci à toi Sophie pour tes belles chansons et tes jeux de mots parfois douteux, mais sérieusement, merci pour les nombreuses fois où tu m’as grandement aidée. Finalement, merci, Joanie pour ces fous rires, ces discussions ! Merci de m’avoir prise sous ton aile en tant que stagiaire ! Ce fut un immense plaisir de travailler avec vous toutes.
Sans oublier les étudiants du labo qui m’ont grandement aidée dans mon projet, mais qui ont également rendu ces deux années de maitrise formidables. Merci Nadia Milad, Gabrielle Bouffard, Éric Jubinville, Ariane Lechasseur, Mélanie Hamel-Auger et Maude Talbot. Je vous remercie pour la folie, les fous rires, mais grandement pour avoir joué au hacky quotidiennement ! Finalement, un merci tout spécial à mon amoureux qui m’a encouragée, motivée et écoutée durant ces quatre dernières années. Merci pour tout !
Introduction
Le tabagisme
Actuellement plus de 1,1 milliard d’individus sont des consommateurs de la cigarette de tabac mondialement, malgré les risques qui y sont associés (OMS, 2019). La moitié des consommateurs décéderont des maladies causées par l’exposition à la fumée de cigarette et ces derniers décéderont en moyenne 10 ans plus tôt que l’espérance de vie moyenne (CDC, 2019). La cigarette est d’ailleurs la cause de près de 80 % des décès de la maladie pulmonaire obstructive chronique (MPOC) (CDC, 2020). En plus des effets pulmonaires, l’exposition à la fumée de cigarette est un facteur de risque pour plusieurs maladies, dont la maladie coronarienne, les accidents vasculaires cérébraux et plusieurs types de cancers non pulmonaires (CDC, 2020). La cigarette est aussi un fardeau économique important pour notre société. En effet, aux États-Unis, près de 170 milliards de dollars sont dépensés annuellement dans les soins médicaux portés aux individus consommant la cigarette (CDC, 2019). Toutefois, malgré les nombreuses connaissances sur les effets du tabagisme sur la santé, aucun traitement curatif n’est disponible pour traiter la MPOC.
Impacts du tabagisme sur le poumon
Les effets nocifs de la cigarette sur la santé sont documentés et les connaissances sur les composés de la cigarette ont évoluées. En effet, dans les années 90, déjà plus de 3500 des composés étaient connus (Hoffmann & Hoffmann, 1997). Plus de 7000 composés chimiques sont identifiés (Talhout et al., 2011). Parmi ces composés, 68 seraient cancérigènes (National Center for Chronic Disease, Health Promotion Office on, & Health, 2014).
Une des caractéristiques principales de la réponse pulmonaire dans la réponse aigüe et chronique à la fumée de cigarette est l’induction d’une inflammation pulmonaire. De manière aigüe, l’immunité innée, principalement les macrophages et les neutrophiles, répond aux dommages causés par l’exposition à la fumée de cigarette. Lorsque l’inhalation de la fumée de cigarette est répétée de façon chronique, le système de l’immunité adaptative, principalement les lymphocytes, est recruté. Une augmentation des médiateurs inflammatoires dans l’environnement pulmonaire menant ainsi à une infiltration cellulaire et à l’activation des cellules immunitaires est caractéristique de l’inflammation induite par la fumée de cigarette (Morissette, Shen, Thayaparan, & Stampfli, 2015).
Réponse inflammatoire à la fumée de cigarette
Le système respiratoire est le premier affecté par l’exposition à des substances nuisibles dans l’air, notamment par la fumée de cigarette. La réponse inflammatoire est initiée par les dommages causés par la fumée de cigarette au surfactant pulmonaire et aux cellules structurales du poumon, telles les cellules épithéliales. Le surfactant pulmonaire est une structure essentielle au poumon. Il s’agit d’une monocouche de phospholipides qui tapisse l’interface air-liquide des alvéoles pulmonaires. En plus des phospholipides qui constituent 90 % du surfactant, celui-ci contient aussi des lipides neutres et des protéines. Cette structure joue plusieurs rôles essentiels, dont la réduction de la tension de surface, ce qui empêche les alvéoles de collapser lors de l’expiration (Han & Mallampalli, 2015). L’exposition à la fumée de cigarette altère la structure du surfactant, notamment en oxydant les lipides (M. Oulton, Moores, Scott, Janigan, & Hajela, 1991; M. R. Oulton, Janigan, MacDonald, Faulkner, & Scott, 1994; Rodriguez-Capote, Manzanares, Haines, & Possmayer, 2006). Une fois endommagé, le surfactant doit être internalisé et dégradé par les macrophages alvéolaires (J. R. Wright & Dobbs, 1991). Les macrophages internalisent le surfactant par les récepteurs éboueurs (Postlethwait,
l’homéostasie du surfactant. Morissette et ses collaborateurs ont proposé un mécanisme d’initiation de la réponse inflammatoire à la fumée de cigarette. Suite à la recapture des lipides oxydés par les macrophages alvéolaires, ces derniers libèrent de l’IL-1a, ce qui induit une production du GM-CSF et le recrutement d’autres cellules inflammatoires, notamment les neutrophiles (Morissette et al., 2015). Bien que ce processus inflammatoire soit initialement nécessaire, il engendre plus de dommages tissulaires et devient délétère avec une exposition chronique à la fumée de cigarette. Néanmoins, les mécanismes impliqués dans le développement des maladies chroniques liées à cette réponse demeurent mal connus.
Altérations de l’immunité pulmonaire causées par la fumée de
cigarette
En plus d’induire une réponse inflammatoire pulmonaire, le tabagisme altère considérablement l’homéostasie pulmonaire. En effet, tant les effecteurs de l’immunité innée que de l’immunité adaptative jouent un rôle important dans la réponse à la fumée de cigarette. Une fois inhalée, la fumée de cigarette cause d’abord des dommages aux cellules épithéliales qui tapissent le tractus respiratoire. Par exemple, la fumée de cigarette altère la réponse épithéliale à des pathogènes. Van der Toorn et ses collaborateurs ont montré qu’un extrait de cigarette diminue la capacité des cellules humaines gingivales à produire la b-défensive 2, un peptide antimicrobien (van der Toorn et al., 2007). Au poumon, la fumée de cigarette induit l’apoptose des cellules épithéliales et réduit la mobilité des cellules ciliées en plus d’induire l’hyperproduction de mucus (Lee, Taneja, & Vassallo, 2012). Puis, les dommages causés par la fumée de cigarette engendrent la libération de médiateurs inflammatoires, comme CCL2, TNF-a, IL-1a et IL-8 (Lee et al., 2012). Ces médiateurs sont nécessaires au recrutement de cellules immunitaires tels les macrophages et les neutrophiles. La fumée de cigarette active également plusieurs voies de signalisation impliquées dans l’activation cellulaire, comme les voies des
MAP kinases et celles associées au facteur de transcription NF-kB, un facteur important dans la régulation de la production de médiateurs inflammatoire (Lee et al., 2012).
Immunité innée
Les processus de l’immunité innée sont effectués principalement par les macrophages, les granulocytes, les monocytes, les cellules dendritiques et les cellules « natural killer ». Ces effecteurs sont les premiers à bâtir une réponse inflammatoire à la fumée de cigarette.
Le macrophage alvéolaire
Au plan physiologique, les macrophages alvéolaires jouent un rôle essentiel en tant que sentinelles. Ces cellules immunitaires sont d’ailleurs les premiers effecteurs de la réponse pulmonaire à un agent pathogène ou aux toxines. Leur rôle premier est de phagocyter les particules étrangères, les agents pathogènes ou bien les débris dans les poumons. Un groupe a montré que la délétion des macrophages avant une infection à un virus augmente la quantité de virus mesurée au poumon en plus de limiter la production de médiateurs inflammatoire et de diminuer la survie chez la souris (Tumpey et al., 2005). Des observations similaires ont été faites chez des souris infectées avec Brucella abortus, une bactérie à Gram négatif. La charge bactérienne était plus importante lorsqu’il y avait une déplétion des macrophages par des liposomes de clodronate dans les poumons, la rate et le foie (Archambaud et al., 2010). Afin d’éliminer les agents pathogènes, les macrophages produisent des espèces réactives de l’oxygène et de grandes quantités d’oxyde nitrique (Boukhenouna, Wilson, Bahmed, & Kosmider, 2018). Outre leurs rôles dans la défense contre les pathogènes, les macrophages alvéolaires sont également
endommagé (J. R. Wright & Dobbs, 1991). De plus, une fois activés les macrophages pulmonaires peuvent relâcher plusieurs métalloprotéinases (mmp-2,
mmp-9, mmp-12) afin de dégrader la matrice extracellulaire et permettre la migration
cellulaire dans le tissu. Ces protéases sont essentielles afin de garder un équilibre entre les protéases et les antiprotéases, un équilibre important pour l’homéostasie du parenchyme pulmonaire.
Toutefois, en contexte tabagique, la morphologie et les fonctions des macrophages sont altérées. En effet, il est connu depuis la fin des années 60 que la fumée de cigarette augmente le nombre et la taille des macrophages au niveau du poumon chez l’humain (Pratt, Finley, Smith, & Ladman, 1969). Pratt et ses collaborateurs ont également observé des agrégats intracellulaires dans les macrophages de sujets fumeurs (Pratt et al., 1969). Ces accumulations seraient des lipides neutres (Morissette et al., 2015). Il a également été montré que l’expression des récepteurs éboueurs, impliqués dans la gestion de l’export lipidique, est augmentée en contexte tabagique (Jubinville et al., 2017). Une augmentation de la taille des macrophages est d’ailleurs une caractéristique importante de la réponse à la fumée de cigarette. L’augmentation de la capture d’une plus grande quantité de lipides oxydés induit un phénotype de macrophages spumeux (Martin, 1973). Cette augmentation de la taille des macrophages causée par une exposition à la fumée de cigarette persiste même après une cessation tabagique (Morissette et al., 2015).
La fumée de cigarette affecte également l’activation des macrophages alvéolaires. Les macrophages alvéolaires de souris exposées à la fumée de cigarette sécrètent davantage d’IL-1a que ceux de souris non exposées (Morissette et al., 2015). L’exposition à la fumée de cigarette cause une augmentation de la libération de médiateurs inflammatoire comme le TNF-a et l’IL-8 (Rom, Avezov, Aizenbud, & Reznick, 2013). De plus, une fois activés, les macrophages sécrètent des métalloprotéinases qui participent grandement à la destruction du tissu
pulmonaire. Chez l’humain, une augmentation de mmp-12 est observée dans les poumons de patients atteints de MPOC (Molet et al., 2005). Lim et ses collaborateurs ont montré qu’une déplétion des macrophages à l’aide de liposome de clodronate limitait la réponse inflammatoire à une exposition à la fumée de cigarette dans un modèle murin (Lim, Kim, Lee, Bae, & Kim, 2018). Lorsqu’il y avait une déplétion des macrophages, moins de cellules inflammatoires et de neutrophiles étaient présents dans les lavages broncho-alvéolaires et moins de médiateurs inflammatoires étaient relâchés suite à une exposition à la fumée de cigarette. Toutefois, une déplétion complète des macrophages pulmonaires induit un déséquilibre du système immunitaire et donc pourrait altérer la réponse homéostatique à une insulte pulmonaire.
Une autre fonction importante des macrophages est leur capacité à phagocyter et éliminer les agents pathogènes. Une étude a montré que la capacité à phagocyter des cellules épithéliales apoptotiques par les macrophages alvéolaires de patients atteints de MPOC est diminuée en comparaison à celle de sujets sains (Hodge et al., 2007). Certains récepteurs associés à la reconnaissance de pathogènes tels que le CD91, le CD31 et le CD44 sont diminués dans les macrophages alvéolaires de patients atteints de MPOC en comparaison à des individus sains. Ces résultats suggèrent que le tabagisme pourrait affecter leur rôle en tant que phagocyte principal au niveau du poumon (Hodge et al., 2007). La capacité des macrophages alvéolaires à phagocyter Listeria monocytogenes est également diminuée chez les individus fumeurs (King, Savici, & Campbell, 1988). Chez la souris, une étude a montré qu’une exposition à la fumée de cigarette de six à huit semaines affecte la réponse à une inoculation de bactérie Pseudomonas
aeruginosa. Lorsque les souris sont exposées à la fumée de cigarette, la charge
bactérienne au poumon est beaucoup plus importante que lorsque les souris ne sont pas exposées. Une infiltration cellulaire plus importante et une augmentation des médiateurs inflammatoires dans le poumon sont observées chez les souris
effets négatifs sur la défense de l’hôte induit par une exposition chronique à la fumée de cigarette corrèlent avec la prévalence d’infections respiratoires observées chez les fumeurs et les patients MPOC. En somme, la fumée de cigarette altère considérablement plusieurs fonctions essentielles des macrophages alvéolaires.
Le neutrophile
Les neutrophiles sont des leucocytes qui sont également impliqués dans la réponse initiale à un agent pathogène et la défense de l’hôte. Les neutrophiles sont recrutés rapidement dans une réponse inflammatoire. Ces leucocytes sont attitrés par de très faibles quantités de signaux chimioattractant (Wang, Artemenko, Cai, Iglesias, & Devreotes, 2014). Alors qu’ils sont beaucoup plus petits que les macrophages, les neutrophiles sont aussi en mesure de phagocyter et d’éliminer les agents infectieux. De plus, les neutrophiles contiennent des granules cytotoxiques remplies d’enzymes hydrolytiques qui sont normalement sécrétées en contexte pro-inflammatoire (Dale, Boxer, & Liles, 2008). Dès la fin des années 60, Klebanoff a postulé que les myéloperoxydases sont impliquées dans la génération de peroxyde d’hydrogène et que ces enzymes seraient en partie responsables des propriétés antimicrobiennes des neutrophiles (Klebanoff, 1968). D’autres études ont ensuite montré que le NADPH oxydase est également importante dans la génération d’espèce réactive de l’oxygène (Babior, Curnutte, & McMurrich, 1976). En plus de la génération des espèces réactives de l’oxygène, les neutrophiles sécrètent des protéases comme la neutrophil elastase (NE) et les cathepsines qui permettent de détruire les micro-organismes (Demkow & van Overveld, 2010).
Toutefois, l’implication du neutrophile dans la réponse à la fumée de cigarette demeure peu étudiée et comprise. Il a été observé chez des patients atteints de MPOC que les neutrophiles s’accumulent au niveau du poumon (Hogg et al., 2004). En étudiant la réponse à la fumée de cigarette dans le temps chez la souris, D’Hulst et son équipe ont remarqué tant une augmentation de neutrophiles que de
macrophages dans les lavages broncho-alvéolaires de souris (D'Hulst A, Vermaelen, Brusselle, Joos, & Pauwels, 2005). Ce recrutement de neutrophiles observés semble être dépendant de l’IL-1a. La déplétion de l’IL-1a diminue la neutrophilie induite par une exposition à la fumée de cigarette (Botelho et al., 2011). Cette étude a aussi montré un rôle important du neutrophile et de l’IL-1a dans la réponse inflammatoire lors d’exacerbation virale. En effet, des souris exposées à la fumée de cigarette et infectées avec le virus influenza A H1N1 ont une plus grande infiltration cellulaire au niveau des lavages broncho-alvéolaires. Toutefois, en bloquant l’IL-1a par des anticorps, cette exacerbation de la réponse à un pathogène est limitée (Botelho et al., 2011). De plus, la capacité des neutrophiles à phagocyter a été étudiée in vitro à l’aide d’extrait de cigarette. Un traitement de deux heures avec un extrait de cigarette de 4 % a diminué la capacité des neutrophiles à phagocyter la bactérie E.Coli de 35 % (Stringer, Tobias, O'Neill, & Franklin, 2007).
Une étude s’est intéressée à l’implication de la protéase NE dans la destruction du parenchyme pulmonaire, notamment en générant un modèle murin de délétion de cette protéine. Une diminution du nombre de neutrophiles dans le poumon est observée en plus d’une diminution de la destruction du parenchyme pulmonaire. Cette étude suggère un rôle du neutrophile dans le développement de ces pathologies pulmonaires (Shapiro et al., 2003). Ainsi, le neutrophile et ses fonctions semblent être altérés par la fumée de cigarette. Par contre, le rôle et l’implication du neutrophile dans la physiopathologie des maladies associées à la fumée de cigarette demeurent à investiguer.
Immunité adaptative
L’immunité adaptative quant à elle est composée des lymphocytes B et T. La réponse à la fumée de cigarette est d’abord médiée par l’immunité innée, mais
Un recrutement au poumon de lymphocytes T et B est observé chez les personnes atteintes de MPOC (Donovan et al., 2019). Toutefois, le rôle de l’immunité adaptative dans le développement et la persistance de l’inflammation chronique en contexte tabagique demeure peu étudié.
Le lymphocyte B
Le lymphocyte B est un leucocyte impliqué dans la réponse à un antigène, notamment en sécrétant des anticorps qui mènent à la neutralisation et à la clairance des pathogènes ou en établissant une mémoire qui accélère la réponse à une deuxième exposition au même antigène. Quelques études ont montré un rôle des lymphocytes B dans la pathogenèse des maladies associées à la fumée de cigarette.
Dans les voies respiratoires des patients atteints de MPOC, une infiltration de lymphocyte B corrèle avec la sévérité de la maladie (Hogg et al., 2004). Chez l’humain, la présence de tissus lymphoïdes tertiaire (TLT) a été documentée. Ces structures sont composées entres autres de lymphocytes T et B (Hogg et al., 2004). Une étude a été réalisée dans un modèle murin d’exposition à la fumée de cigarette. La présence de TLT a été observée dans les poumons de souris exposées durant 24 semaines, mais aucune présence de ces structures suite à une exposition de quatre jours ou de huit semaines. Suite à une cessation tabagique, les TLT étaient toujours présents malgré une diminution de l’inflammation pulmonaire (Morissette, Jobse, et al., 2014). De plus, l’apparition d’auto-anticorps a été observée dans la pathogenèse de la MPOC (Wen, Krauss-Etschmann, Petersen, & Yu, 2018). Dans les expectorations de patients atteints de MPOC, une augmentation de ces auto-anticorps a été montrée. Cette même étude a montré une augmentation de la quantité d’auto-anticorps dans les lavages de souris exposées à la fumée de cigarette durant huit et 24 semaines qui n’était pas renversée par une cessation tabagique (Morissette, Jobse, et al., 2014). D’ailleurs, une augmentation du B-cell
activating factor (BAFF), au niveau transcriptionnel dans le poumon et protéique
dans les lavages broncho-alvéolaires est observée chez les patients fumeurs. De plus, une augmentation des niveaux de BAFF dans les lavages broncho-alvéolaires chez la souris suite à une exposition d’une semaine a déjà été observée. Lorsque BAFF est bloqué par des anticorps, la présence d’auto-anticorps et de TLR est diminuée en contexte tabagique (Morissette et al., 2016).
Le rôle des lymphocytes B a également pu être étudié grâce à la déplétion de ce type cellulaire dans un modèle murin d’exposition à la fumée de cigarette. Dans une étude utilisant des souris déficientes en lymphocytes B, la destruction du parenchyme pulmonaire causée par une exposition à la fumée de cigarette de quatre et six mois est diminuée chez ces souris (John-Schuster et al., 2014). En somme, même si l’immunité innée a été beaucoup plus étudiée dans les maladies associées au tabagisme, le rôle des lymphocytes B n’est pas à négliger.
Le lymphocyte T
Le lymphocyte T est aussi impliqué dans la défense contre un antigène. Il existe deux types principaux de lymphocytes T, soit les CD4+ et les CD8+. Les lymphocytes T CD4+ sont plutôt des cellules qui orchestrent la réponse immunitaire, notamment en recrutant les cellules de l’immunité innée et activant les lymphocytes B. Les lymphocytes T CD8+ sont des cellules cytotoxiques, induisant la mort des cellules ciblées via la sécrétion d’enzyme (David Male, Brostoff, Roth, & Roitt, 2012). Tout comme pour le lymphocyte B, les études s’intéressant au rôle du lymphocyte T dans les maladies associées au tabagisme sont beaucoup moins nombreuses que celles portant sur les cellules de l’immunité innée.
MPOC, des lymphocytes T CD4+ et CD8+ ont été retrouvés. La présence de lymphocytes T dans les TLT, des structures qui se développent suite à une exposition chronique à la fumée de cigarette, a été documentée (Hogg et al., 2004). Une augmentation des cellules CD8+ est observée dans les poumons de souris, alors que le nombre de cellules CD4+ n’est pas changé en réponse à la fumée de cigarette. Cette même étude a montré que la réponse inflammatoire induite par une exposition de 6 mois à la fumée de cigarette est limitée lorsque les lymphocytes T CD8+ sont absents. La destruction du parenchyme pulmonaire induite par une exposition chronique à la fumée n’est pas observée chez les souris déficientes en lymphocytes CD8+ (Maeno et al., 2007). Dans un modèle d’exposition pulmonaire à l’acroléine, un irritant présent dans la fumée de cigarette, une augmentation de lymphocytes CD8+ est observée dès 2 semaines d’exposition. Une augmentation du nombre de macrophages est observée dans les lavages broncho-alvéolaires des souris exposées, mais dans des souris déficientes en cellules CD8+ le recrutement de macrophages est limité, voir presque qu’absent (Borchers et al., 2007). Ces études témoignent de l’implication de lymphocytes T dans la réponse à un irritant pulmonaire.
En résumé, la fumée de cigarette altère tant les effecteurs de l’immunité innée que de l’immunité adaptative. La réponse inflammatoire est premièrement médiée par les macrophages pulmonaires via la sécrétion de médiateurs inflammatoires et le recrutement d’autres cellules inflammatoires comme les neutrophiles. Puis, en contexte d’inflammation chronique, les lymphocytes B et T sont recrutés au poumon et participent à la formation de TLT (Figure 1). Tous ces processus cellulaires participent activement au développement de la MPOC à long terme.
La maladie pulmonaire obstructive chronique
Malgré l’importante capacité d’adaptation et de réparation du poumon aux insultes constantes induites par la fumée de cigarette, à long terme le tabagisme est le principal facteur de risques de plusieurs maladies, dont la maladie pulmonaire obstructive chronique (MPOC). La prévalence de MPOC modérée à sévère chez les individus de plus de quarante ans est d’environ 10 % mondialement (Decramer, Janssens, & Miravitlles, 2012). La MPOC sera la troisième cause de décès mondialement d’ici 2030 (OMS, 2020).
Figure 1. Schéma de la réponse pulmonaire à la fumée de cigarette. 1. La fumée
de cigarette engendre d’abord une réponse inflammatoire suite à l’oxydation du surfactant pulmonaire, 2. ce qui induit l’augmentation de la capture des lipides endommagés par les macrophages. Ces derniers libèrent des médiateurs inflammatoires (IL-1a, CCL2, GM-CSF) afin de recruter des cellules inflammatoires telles les neutrophiles. 3. Lorsque l’insulte pulmonaire est soutenue et chronique, les cellules de l’immunité adaptative, principalement les lymphocytes B et T, sont impliquées et peuvent mener au développement des tissus lymphoïdes tertiaires.
1. 2. Lymphocytes B Lymphocytes T IL-1a CCL2 GM-CSF Macrophage Cellules épithéliales Neutrophile Circu latio nsa ngui ne Immunité adaptative Immunité innée 3.
Tissus lymphoïdes tertiaires Dommages au surfactant
Pathophysiologie
Une des caractéristiques de la MPOC est une limitation irréversible du débit respiratoire. La MPOC comprend deux entités, soit l’emphysème et la bronchite chronique. La bronchite chronique est caractérisée par une inflammation des bronches et est associée à une hyperproduction de mucus et une production d’expectoration durant au moins trois mois de deux années consécutives (Pauwels & Rabe, 2004). L’emphysème se définit par une destruction du parenchyme pulmonaire menant à une diminution des surfaces d’échange des gaz. Une perte de l’élasticité du poumon est également observée et peut être causée par une activation incontrôlée des protéases, qui favorisent la destruction du tissu conjonctif (Decramer et al., 2012).
D’un point de vue clinique, plusieurs symptômes sont associés à la MPOC. La toux est généralement le premier symptôme à apparaitre. Cette toux peut s’accompagner d’expectorations, qui font partie de la définition de la bronchite chronique. La dyspnée est également un des symptômes clés dans la MPOC. L’évaluation de la limitation du débit respiratoire par la spirométrie est le test standard dans le diagnostic de la MPOC. Le coefficient de Tiffeneau correspond à un ratio du volume maximal expiré lors de la première seconde d’une expiration forcée (VEMS) sur la capacité vitale forcée (CVF). Normalement, ce ratio se situe autour de 0,8 et une valeur en dessous de 0,7 suggère un syndrome obstructif, telle la MPOC (Pauwels & Rabe, 2004).
La sévérité spirométrique de la MPOC est classifiée par les stades établis par la Global Initiative for Chronic Obstructive Lung disease (GOLD). Globalement, la classification se base sur la valeur de VEMS mesurée qui est comparée à la valeur prédite à partir de la taille, le sexe et l’âge des individus. Cette classification est importante dans la prise en charge des patients. Le stade léger se définit par un
VEMS supérieur à 80 % de la prédite (GOLD 1), le stade GOLD 2 est déterminé par une VEMS entre 50-80 % de la valeur prédite. Lorsque le VEMS se situe entre 30-50 % de la valeur prédite, il s’agit d’un stade sévère (GOLD 3). Finalement, lorsqu’on parle d’un stade très sévère de la maladie (GOLD 4), le VEMS est inférieur à 30 % de la valeur prédite (Decramer et al., 2012; GOLD, 2020). La progression de cette maladie est souvent affectée par la fréquence élevée d’exacerbation. Ces exacerbations mènent souvent à l’hospitalisation et se manifestent par une aggravation de la toux, de la dyspnée et de la production d’expectoration. Les infections respiratoires bactériennes et virales demeurent les causes les plus importantes de ces exacerbations (Decramer et al., 2012).
Actuellement, seule une prise en charge des symptômes est possible et aucun traitement curatif à la MPOC n’est disponible. Cette prise en charge est déterminée en fonction de la sévérité de la maladie. Les bronchodilatateurs inhalés constituent le traitement principal pour la MPOC. Les glucocorticostéroïdes en inhalation sont souvent prescrits afin de prévenir les exacerbations. Finalement, lorsque l’hopoxémie s’installe dans les stades les plus avancés, en plus des premiers traitements, l’oxygène est recommandé (Decramer et al., 2012). Dans tous les cas, l’arrêt tabagique est fortement recommandé.
Alors que la pollution de l’air est également responsable de l’apparition de la MPOC, le tabagisme en est le principal facteur de risque modifiable. Au plan pathophysiologique, l’exposition à la fumée de cigarette entraine de nombreux impacts sur la réponse immune et mène au développement de cette pathologie pulmonaire.
L’importance de la composante génétique dans la MPOC
La MPOC est étudiée depuis plusieurs décennies, mais la cause exacte de son apparition demeure inconnue. Le principal facteur de risque pour cette maladie est le tabagisme. Toutefois, étant donné que la majorité des fumeurs ne développeront pas la MPOC dans leur vie et que le déclin des fonctions pulmonaires est très hétérogène chez ces patients, des susceptibilités génétiques sont en cause pour cette maladie complexe.
Plusieurs études génétiques ont été effectuées afin d’identifier des gènes potentiellement associés à la MPOC. Essentiellement, des voies de signalisations reliées à l’inflammation (IL-4, IL-6, IL-13, IL-1b, IL-1RN, LTA, TNF et TGFB1), aux protéases et antiprotéases (MMP-9, TIMP2 et SERPINA3) et au stress oxydatif (GSTM1, GSTP1, GSTT1, SOD2 et SOD3) ont été identifiées à l’aide de méta-analyses (Bosse, 2012). Ces trois voies de signalisations sont grandement impliquées dans la réponse à la fumée de cigarette et dans le développement de la MPOC. Plus spécifiquement des études de type genome-wide association study (GWAS) ont également permis de déterminer certains loci associés à la MPOC (Bosse, 2012). Une association identifiée sur le locus du récepteur a-nicotinique acétylcholine sur le chromosome 15q25 et une sur le locus de la hedgehog
interacting protein (HHIP) ont été identifiées (Pillai et al., 2009). Ces deux
associations ont ensuite été confirmées par des GWAS subséquentes dans diverses cohortes. Ces études ont également identifié deux autres loci associés à la MPOC soit le 4q22.1 sur le gène FAM13A et le locus 19q13 contenant les gènes RAB4B,
EGLN2, MIA et CYP2A6 (Cho et al., 2010; Cho et al., 2012). D’ailleurs les loci 15q25
et 19q13 avaient déjà été associés au tabagisme, qui est le principal facteur de risque pour la MPOC (Thorgeirsson et al., 2010; Tobacco & Genetics, 2010). Ces études de type GWAS ont permis de découvrir plusieurs loci susceptibles de participer à la pathogenèse de la MPOC (Bosse, 2012).
Toutefois, jusqu’à tout récemment la seule mutation associée à une transmission héréditaire de l’emphysème est celle retrouvée dans le gène SERPINA1, codant pour l’antitrypsine A1AT (Bosse, 2012). Ces mutations seraient responsables de 1 à 2 % des cas d’emphysème (Decramer et al., 2012). Plusieurs variants de l’allèle de SERPINA1 ont été documenté et causent chacun un certain degré de sévérité. D’ailleurs le variant le plus susceptible de causer l’emphysème sévère est le variant Z. Chez les patients homozygotes pour ce variant, les niveaux circulants de l’alpha-1-antitrypsin sont 10 à 15 % des niveaux normaux (Gooptu, Ekeowa, & Lomas, 2009). La protéine alpha-1-antitrypsine est un inhibiteur de protéase, qui joue un rôle important dans la protection du parenchyme pulmonaire. L’antitrypsine inhibe l’activité de certaines protéases et diminue donc la destruction du parenchyme pulmonaire. En contexte tabagique où la sécrétion de protéases est accrue, une déficience en cet inhibiteur peut entrainer l’apparition précoce de l’emphysème pulmonaire. Un traitement pour l’emphysème causé par la déficience en alpha-1-antitrypsine consiste à supplémenter les patients avec un inhibiteur de alpha-1-protéase (AIP1) (Strange, 2018).
Nouvelle mutation associée à l’emphysème
Récemment, une nouvelle mutation a été associée à l’emphysème pulmonaire. Il s’agit en fait de la 2e mutation héréditaire associée à cette maladie.
La mutation a été découverte par des chercheurs de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) au sein d’une famille québécoise. Les membres de cette famille sont affectés par une forme d’emphysème précoce et sévère qui est situé dans les lobes inférieurs. La transmission de cette mutation suit un patron autosomal- dominant et affecte donc les membres porteurs hétérozygotes (Bosse et al., 2019).
La présence d’une mutation du gène SERPINA1 a d’abord été exclue par le séquençage de ce gène spécifiquement. Puis un séquençage du génome complet a été effectué dans un échantillon de la famille afin d’identifier la mutation. Finalement, le génotype des 55 membres de la famille a été testé à l’aide du séquençage Sanger. Parmi les 55 participants, 29 sont porteurs d’une mutation hétérozygote située sur le gène PTPN6, un gène qui code pour la protéine SHP-1. La présence d’emphysème a été confirmée par tomodensitométrie chez 20 des 27 porteurs de la mutation (pour deux patients les tomodensitométries n’étaient pas disponibles), soit 74 % des hétérozygotes. Les individus porteurs de cette mutation et ayant un historique de tabagisme avaient tous un diagnostic d’emphysème (sauf 1 patient). Ces observations reflètent donc une pénétrance quasi complète de la mutation en contexte tabagique. En comparaison, seulement un patient non porteur de la mutation avait un diagnostic d’emphysème sur les 19 individus qui ont été testés. Ce patient porteur de l’allèle sauvage était un ancien fumeur et présentait une forme d’emphysème typiquement causée par le tabagisme (Bosse et al., 2019). Les individus porteurs de la mutation PTPN6 ont un score qualitatif d’emphysème évalué par tomodensitométrie plus élevé que les individus sains. La capacité de diffusion au monoxyde de carbone est diminuée chez les individus PTPN6Ala455Thr
comparativement aux individus non porteurs. Le coefficient de Tiffeneau (VEMS/CVL) des patients porteurs de la mutation est diminué significativement comparé à celui de patients sains (Bosse et al., 2019).
Plus précisément, la mutation se situe sur le chromosome 12 et cause un changement d’une guanine pour une adénine à la position 1363 dans l’exon 12 du gène. Cette mutation résulte en un changement de l’acide aminé alanine qui est non polaire pour l’acide aminé thréonine qui est polaire à la position 455 de la protéine SHP-1. Ce changement d’acide aminé est localisé très près du site actif de SHP-1, ce qui cause une diminution de l’activité de cette protéine (Bosse et al., 2019). Toutefois, le rôle de SHP-1 dans le maintien de l’homéostasie pulmonaire tant en contexte physiologique que tabagique nécessite une étude plus approfondie.
Src
homology
region 2
domain-containing
phosphatase-1 (SHP-1)
La famille des tyrosines phosphatases
Globalement, les phosphatases sont des protéines régulatrices qui modulent l’activité de leur substrat en retirant des groupements phosphates. La phosphorylation et la déphosphorylation sont des mécanismes de régulation bien conservés qui ont un rôle critique dans le développement, la différenciation ainsi que la régulation de plusieurs types cellulaires.
Ces phosphatases sont impliquées dans plusieurs maladies pulmonaires, comme la fibrose pulmonaire idiopathique. En effet, « Src homology region 2 domain-containing phosphatase-2 » (SHP-2), une tyrosine phosphatase, aurait des effets antifibrotiques au niveau du poumon (Verma & Sharma, 2018). Dans le syndrome aigu de détresse respiratoire qui est caractérisé par un œdème pulmonaire, l’inhibition de la « protein-tyrosine phosphatase 1B » (PTP1B) augmente l’œdème pulmonaire observé dans un modèle murin (Grinnell, Chichger, Braza, Duong, & Harrington, 2012) .
De plus, quelques études ont montré l’implication des tyrosines phosphatases dans la réponse à la fumée de cigarette. Par exemple, la protéine phosphatase 2A (PP2A) semble avoir un effet protecteur sur la réponse à la fumée de cigarette (Wallace et al., 2012). En effet, chez la souris lorsqu’on administre la protéine PP2A par instillation intratrachéale, l’infiltration cellulaire observée suite à l’exposition à la fumée de cigarette semble être diminuée. À l’inverse, lorsqu’on inhibe PP2A à l’aide de l’acide okadaïque, l’inflammation pulmonaire induite par l’exposition à la fumée
l’activation de PP2A permet de diminuer le nombre de cellules totales des lavages broncho-alvéolaires induit par l’exposition à la fumée de cigarette et on émit l’hypothèse que la phosphatase PP2A diminuerait l’activité de la cathepsine S, une protéine qui pourrait avoir un lien avec la MPOC (Doherty et al., 2019). Par ailleurs, la phosphatase PTP1B a également été étudiée dans le contexte de la MPOC. En effet, dans un modèle murin la perte de PTP1B aggrave la réponse pulmonaire suite à une infection au virus respiratoire syncytial (Foronjy et al., 2016). La protéine SHP-2 a également été étudiée en contexte tabagique. L’absence de SHP-SHP-2 limite l’infiltration cellulaire observée suite à une exposition à la fumée de cigarette et cet effet pourrait être médié par la sécrétion de l’IL-8. De plus, lorsque des souris sont exposées à la fumée de cigarette durant quatre jours, une augmentation des niveaux protéiques de SHP-2 est observée dans le poumon (F. F. Li et al., 2012). Ainsi, le rôle des phosphatases dans diverses maladies pulmonaires est de plus en plus étudié, mais beaucoup reste à découvrir.
Description de SHP-1
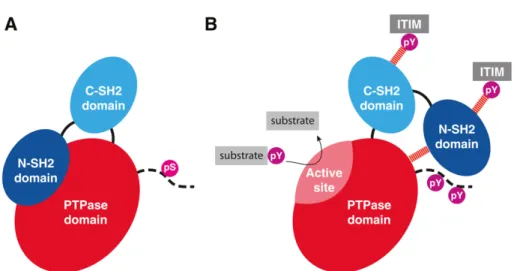
La protéine SHP-1 est composée de deux domaines SH2, d’un domaine tyrosine phosphatase ainsi que d’une queue C terminale contenant plusieurs sites de phosphorylation. Des analyses par cristallographie ont montré qu’il y a une liaison intramoléculaire entre le domaine N-SH2 et le site actif de la protéine (Figure 2). Ainsi, sans phosphorylation, la phosphatase demeure inactive (Yang et al., 2003).
Plus précisément, cette phosphatase est principalement exprimée par les cellules hématopoïétiques, mais également par d’autres types cellulaires, comme les cellules épithéliales ou neuronales (Plutzky, Neel, & Rosenberg, 1992). Cette protéine est localisée dans le cytoplasme des cellules hématopoïétiques, mais elle se retrouve dans le noyau dans les cellules épithéliales (Tsui, Hasselblatt, Martin, Mok, & Tsui, 2002).
Quelques études se sont intéressées au rôle de SHP-1 au niveau des cellules épithéliales. Les souris ptpn6H-KO, dans lesquelles une délétion spécifique de
SHP-1 dans les hépatocytes a été réalisée, ont une réponse exacerbée à une instillation au LPS comparé aux souris sauvages. Il y a une augmentation de TNF-a, d’IL-1b et de l’IL-6 dans le sang. D’ailleurs, suite à l’instillation de LPS, une augmentation du recrutement de cellules, principalement des neutrophiles, est observée dans la cavité péritonéale des souris ptpn6H-KO. Cette délétion spécifique de SHP-1 au
niveau des hépatocytes a eu également des impacts au niveau de la réponse fonctionnelle des hépatocytes. Suite à une stimulation in vitro au LPS, les hépatocytes de souris ptpn6H-KO sécrètent davantage de médiateurs inflammatoires
comparativement à ceux de souris sauvages. Toutefois, la réponse des macrophages péritonéaux, qui n’ont pas la délétion de SHP-1, demeure normale (Adhikari, Martel, Marette, & Olivier, 2017).
Figure 2. Schéma du mécanisme d’auto-inhibition de la phosphatase SHP-1.
A) Le domaine N-SH2 cache le site actif de la phosphatase et B) lorsqu’il y a phosphorylation de certains résidus tyrosine de la protéine, le domaine N-SH2 dégage le site actif. Tiré de (Abram & Lowell, 2017).
Dans les cellules endothéliales, SHP-1 est un régulateur de la sécrétion de plusieurs facteurs essentiels pour la différenciation, la prolifération et de la mobilisation des progéniteurs des cellules endothéliales, notamment, le VEGF, c-Kit, G-CSF (Trop, Tremblay, & Bourdeau, 2008). Par ailleurs, au niveau des neurones, SHP-1 est connu pour avoir un rôle d’inhibition du réarrangement neuronal. Dans un modèle de dommage cortical, l’expression et l’activité de SHP-1 sont augmentées suite à une lésion, cependant cette augmentation n’est pas observée chez les souris déficientes en SHP-1. De plus, l’absence de SHP-1 améliore l’élongation des fibres neurales en plus d’améliorer les fonctions motrices suite à la lésion (Tanaka, Fujita, Ueno, Shultz, & Yamashita, 2013).
Modèles murins de déficience en SHP-1
Les modèles murins de mutation de SHP-1 ont permis de découvrir les nombreux rôles de SHP-1 dans la régulation de la différenciation et de l’activation de plusieurs types cellulaires. Le premier modèle murin d’une mutation de SHP-1 à avoir été découvert est la souris « Motheaten » ou me/me en 1975. Green et collaborateur ont décrit des souris ayant entre autres des lésions cutanées, d’où l’origine du nom du modèle (Green & Shultz, 1975). Ces souris développent des problèmes inflammatoires et d’auto-immunité. Cette mutation homozygote est située dans le gène ptpn6 codant la protéine SHP-1. La mutation se situe dans le domaine N-SH2 et induit une protéine entièrement non fonctionnelle. Dès l’âge de trois semaines, les souris « motheaten » développent des lésions cutanées et aux pattes. L’importance de SHP-1 est montrée par le fait que ces souris ne survivent pas plus de huit semaines. Une seconde mutation très similaire a permis de mieux étudier le rôle de SHP-1 étant donné que les souris survivent un maximum de 25 semaines (Shultz, Coman, Bailey, Beamer, & Sidman, 1984). En effet, les souris « motheaten viable » (mev/mev) ont une mutation moins sévère dans laquelle
SHP-1 fonctionne à SHP-10-20 % de son activité normale. Ces souris présentent également une accumulation de macrophages au niveau de poumon (Shultz et al., 1984).
Un autre modèle murin souvent utilisé afin d’étudier l’importance de SHP-1 est la souris ptpn6spin. Cette mutation est située dans le domaine C-SH2 de SHP-1
et cause des altérations de la signalisation des voies associées à SHP-1. Cette mutation homozygote induite par l’agent mutagène N-ethyl-N- nitrosourea induit un phénotype inflammatoire. En effet, ces souris développent des lésions enflammées et purulentes au niveau des pattes (Croker et al., 2008).
Pathologies associées à SHP-1
Récemment, il a été montré que le métabolisme du glucose est augmenté chez les souris « motheaten viable » (Dubois et al., 2006). Cette étude ouvre la porte à l’étude de SHP-1 dans le développement de nouvelle cible thérapeutique pour le diabète de type 2. Certaines études génétiques ont également montré une association entre des variants du gène PTPN6 et des dermatites neutrophiliques chez l’humain (Nesterovitch et al., 2011).
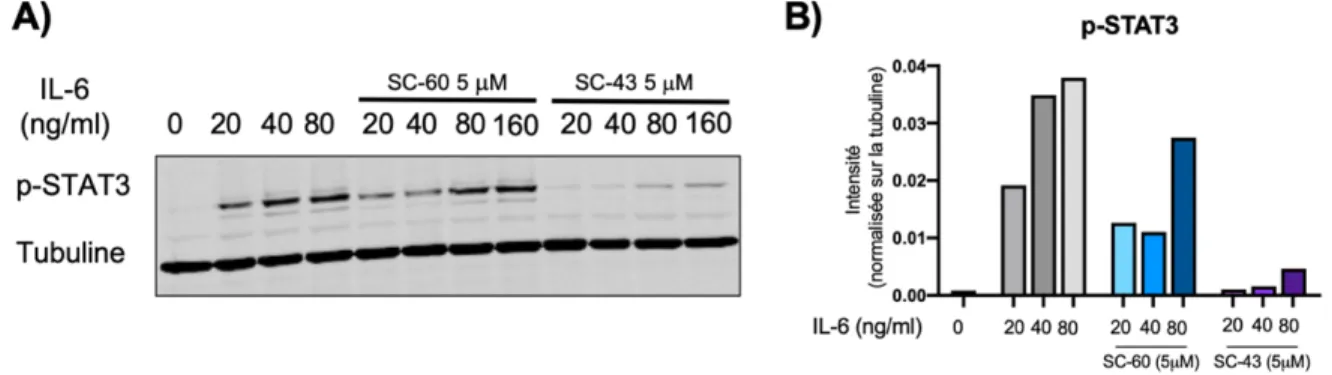
Actuellement l’étude de SHP-1 dans le cancer est une avenue intéressante pour plusieurs types de cancers, comme les carcinomes hépatocellulaires. En effet, SHP-1 est un régaleur important de la phosphorylation de STAT3, un facteur de transcription (Huang, Su, Liu, Shiau, & Chen, 2017). La voie de STAT3 est grandement étudiée étant donné qu’il s’agit d’un facteur de transcription impliqué dans la régulation de la prolifération. Des agonistes de SHP-1 ont été dérivés de la sorafénib, un médicament actuellement utilisé dans le traitement de cette maladie. La sorafénib est un inhibiteur multikinases (Edginton, Zimmerman, Vasilyeva, Baker, & Panetta, 2016). Le SC-60 et le SC-43 sont deux dérivés de la sorafénib plus spécifiques. Le SC-60 est un des dérivés qui montrent un potentiel d’inhiber la voie de signalisation de STAT3 sans affecter les voies des kinases (Liu et al., 2017). Les effets observés du SC-60 et du SC-43 montrent que ces molécules augmentent
l’activité de SHP-1 (Hu et al., 2017; Tai et al., 2014). Le SC-43 et le SC-60 agissent en liant le domaine N-SH2, ce qui empêche l’auto-inhibition de SHP-1 (Figure 2).
Dans un modèle murin de cancer hépatocellulaire, l’administration du SC-60 diminue significativement la taille de la tumeur et augmente l’activité de SHP-1 (Tai et al., 2014). Cette phosphatase régule plusieurs voies de signalisation liées à l’inflammation. De plus, étant donné la récente découverte de la mutation liant 1 à l’emphysème pulmonaire, il est pertinent d’étudier les effets d’agonistes de SHP-1 dans la réponse inflammatoire à la fumée de cigarette. Toutefois, les études de cette protéine régulatrice dans les maladies associées au tabagisme sont peu nombreuses.
Rôle de SHP-1 dans l’immunité
SHP-1 est fortement exprimée dans les cellules myéloïdes et lymphocytaires, notamment par les macrophages, les neutrophiles et les lymphocytes. Ces types cellulaires sont également tous impliqués dans la réponse pulmonaire à la fumée de cigarette.
Cellules lymphocytaires Lymphocytes T
Les phénotypes observés chez les souris « motheaten » ne semblent pas être médiés par les lymphocytes T et B. L’ablation de ces deux types cellulaires ne restaure pas les phénotypes causés par la mutation. Le croisement entre des souris
mev/mev et des souris RAG-/-, des souris n’ayant pas de lymphocytes B et T matures,
résultent en des souris qui développent le même phénotype que celui des souris « motheaten ». Ces résultats indiquent donc que les lymphocytes B et T ne sont pas
nécessaires au développement des problèmes inflammatoires et d’auto-immunité de ces souris (C. C. Yu et al., 1996). Toutefois, l’absence de SHP-1 cause des anomalies lymphocytaires assez importantes.
Plus spécifiquement, plusieurs études ont montré que SHP-1 est impliqué dans le développement et la sélection positive des lymphocytes T. L’introduction d’une forme inactive de SHP-1 dans des souris entraîne une augmentation des cellules simplement CD4 positive dans le thymus en comparaison à des souris sauvages (Plas et al., 1999). La même observation a été démontrée chez les souris « motheaten » (Zhang et al., 1999). De plus, les souris « motheaten » ont un niveau plus élevé de lymphocytes T régulateurs et ces lymphocytes sont plus efficaces dans leur rôle de suppression (Iype, Sankarshanan, Mauldin, Mullins, & Lorenz, 2010). Ainsi, SHP-1 semble un important régulateur du développement des lymphocytes T. De plus, SHP-1 pourrait avoir des effets sur la polarisation des lymphocytes T en Th1. En effet, les lymphocytes CD4+ de la rate et des ganglions de souris « motheaten » sécrètent davantage d’INF-g in vitro (W. M. Yu, Wang, Keegan, Williams, & Qu, 2005).
Lymphocytes B
Au niveau des lymphocytes B, l’absence de SHP-1 cause également des problèmes dans leur développement et leur différenciation. En effet, les souris « motheaten » ont une réponse immune déficiente et montrent des niveaux élevés des immunoglobulines sériques IgM, IgG et IgA (Shultz & Green, 1976). Les souris « motheaten » produisent également des autoanticorps (Shultz, 1988).
Chez la souris, il existe plusieurs types de lymphocytes B, dont les B-2 qui sont les lymphocytes B développant une réponse immune et produisant des
chez la souris contrairement à l’humain. Ce sous-type de lymphocytes induit plutôt une réponse non spécifique, en sécrétant des immunoglobulines (IgM principalement), à un antigène. Afin d’étudier plus spécifiquement le rôle de SHP-1 dans les lymphocytes B, Pao et ses collaborateurs ont généré un modèle murin de délétion spécifique de SHP-1 dans les lymphocytes B (ptpn6fl/fl cre-CD19). Ces
travaux ont montré que l’absence de SHP-1 module les proportions des populations lymphocytaires sans affecter les niveaux de lymphocytes B. Chez les souris n’ayant pas SHP-1, une diminution des lymphocytes B-2, mais une augmentation des B-1a sont observées dans la rate, la moelle osseuse, les ganglions lymphatiques ainsi que dans la cavité péritonéale (Pao et al., 2007). Ainsi, SHP-1 semble important dans le développement des sous-types de lymphocytes B et son absence pour avoir également un impact sur la fonctionnalité de ces cellules.
Effectivement, l’absence de SHP-1 a affecté la réponse immune à un antigène spécifique aux lymphocytes B, soit le NP-Ficoll. Les souris n’ayant pas SHP-1 ne sécrètent aucun IgG3 et IgG1 spécifiques au NP-Ficoll comparativement à la réponse des souris sauvages. Toutefois, les différences observées entre les souris ptpn6fl/fl cre-CD19 et sauvages dans la réponse à un antigène T dépendant
soit le NP-CGG sont beaucoup moins importantes (Pao et al., 2007). Une seconde étude s’est également intéressée au rôle de SHP-1, mais dans les lymphocytes B activés. Les auteurs de cette étude n’ont pas observé de débalancement dans les populations de lymphocytes B comparativement à ce qui a été observé avec une délétion spécifique de SHP-1 dans les lymphocytes B. Toutefois, une réponse immune diminuée en IgG1 est observée chez les souris ayant une délétion de SHP-1 dans les lymphocytes B activés (Y. F. Li, Xu, Ou, & Lam, 20SHP-14). Ainsi, SHP-SHP-1 pourrait avoir un plus grand impact dans le fonctionnement des lymphocytes B.
Cellules myéloïdes
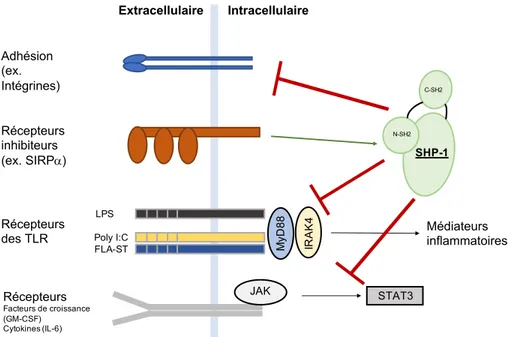
L’étude des impacts des mutations dans le gène ptpn6 dans les cellules myéloïdes a également permis de comprendre le rôle de SHP-1. En effet, cette protéine est importante dans la régulation de la prolifération, de la différenciation, de l’adhésion, de la régulation par les récepteurs inhibiteurs comme SIRPa et les fonctions de plusieurs types cellulaires, comme les macrophages (Figure 3).
Macrophages
Au niveau pulmonaire, les souris « motheaten » développent une pneumonie pouvant affecter tous les lobes du poumon. Les lésions pulmonaires sont caractérisées par la présence de macrophages, particulièrement au niveau des alvéoles (Green & Shultz, 1975). Il a été bien caractérisé par Nakayama et ses
Extracellulaire Intracellulaire N-SH2 C-SH2 SHP-1 Adhésion (ex. Intégrines) Récepteurs inhibiteurs (ex. SIRPa) Récepteurs des TLR LPS Poly I:C FLA-ST Récepteurs Facteurs de croissance (GM-CSF) Cytokines (IL-6) JAK STAT3 IR A K 4 M yD 8 8 Médiateurs inflammatoires
Figure 3. Schéma de l’implication de SHP-1 dans certaines voies de signalisation dans les macrophages. SHP-1 est une phosphatase qui
régule plusieurs voies de signalisation dont celle de l’adhésion via l’inhibition de la signalisation des récepteurs des intégrines. SHP-1 limite la signalisation des récepteurs des TLR et la signalisation via les voies JAK/STAT. Adapté de (Abram & Lowell, 2017)
macrophages F4/80+ au niveau du poumon qui s’aggrave dans le temps (Nakayama, Takahashi, Shultz, Miyakawa, & Tomita, 1997). Cette accumulation de macrophages au niveau pulmonaire pourrait être la cause de décès des souris. De plus, une étude a montré l’accumulation de cristaux éosinophiliques dans le poumon de souris me/me. Cette molécule est produite par les macrophages activés, indiquant ainsi que ces derniers pourraient être suractivés (Guo, Johnson, & Schuh, 2000).
SHP-1 est également un régulateur de l’adhésion, principalement un inhibiteur. Une étude a montré que les macrophages dérivés de la moelle osseuse de souris « motheaten » sont plus adhérents que ceux de souris sauvages (Roach et al., 1998). Une seconde étude a démontré que l’inactivation SHP-1 dans la lignée myélo-monocytaire U937 augmente l’adhérence de ces cellules (Q. Dong, Siminovitch, Fialkow, Fukushima, & Downey, 1999).
D’autres études se sont intéressées à l’implication de SHP-1 dans la capacité fonctionnelle des macrophages à phagocyter. Plus précisément, lorsque les macrophages alvéolaires de souris sauvages sont en présence de sodium stibogluconate, un inhibiteur de SHP-1, ils ont un index de phagocytose plus élevé. De plus, cette même étude a confirmé ces résultats en testant la capacité des macrophages alvéolaires de souris « motheaten » à phagocyter. Les macrophages alvéolaires de souris me/me ont un indice de phagocytose plus élevée que ceux de souris sauvages (Janssen et al., 2008). Ces résultats suggèrent donc que SHP-1 régule les mécanismes de phagocytose.
L’étude de la réponse des macrophages de souris ayant une mutation de SHP-1 à divers stimuli est bien documentée. Toutefois, étant donné la différence entre ces types cellulaires, des résultats divergents ont été obtenus, limitant ainsi