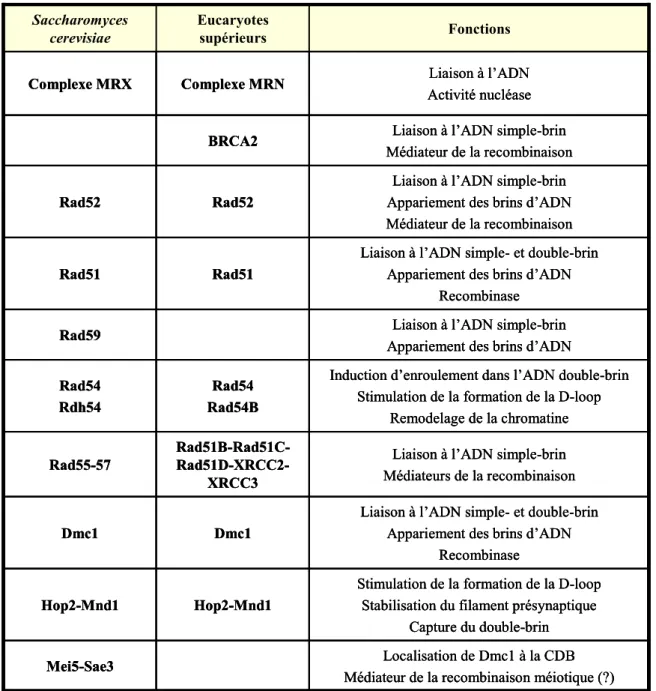
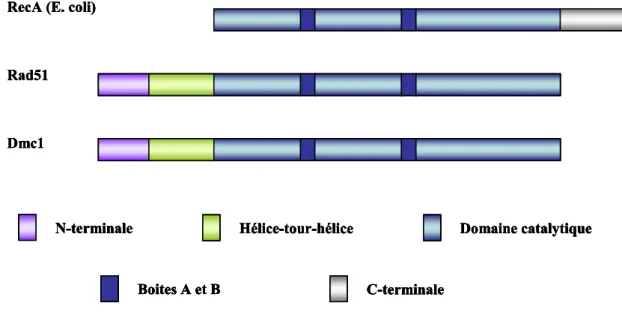
THESE
En vue de l’obtention duDOCTORAT DE L’UNIVERSITE DE TOULOUSE
Discipline : GénotoxicologiePrésentée et soutenue par
Giuseppina BLANCA
Le 26 septembre 2008
RAD51 ET LA TUBULINE-GAMMA : CORRELATION ENTRE
REPARATION PAR RECOMBINAISON HOMOLOGUE ET
ETABLISSEMENT DU FUSEAU MITOTIQUE.
Directeur de thèse Dr. Martine DEFAIS
JURY
Pr. Christophe CAZAUX Professeur, Université Paul Sabatier, Toulouse Président Dr. Serge BOITEUX Directeur de Recherche CNRS, Fontenay aux Roses Rapporteur Dr. Evelyne SAGE Directeur de Recherche CNRS, Orsay Rapporteur Dr. Martine DEFAIS Directeur de Recherche CNRS, Toulouse Directeur de thèse
Institut de Pharmacologie et Biologie Structurale
Evelyne Sage pour avoir accepté d’être les rapporteurs de cette thèse.
Je tiens à remercier tout particulièrement le Docteur Martine Defais, ma directrice de thèse, pour m’avoir accueillie au sein de son laboratoire. Je vous remercie pour m’avoir toujours soutenue pendant ces quatre années et pour la confiance que vous m’avez témoignée. Vous avez cru en moi et sans cela, je ne serais pas arrivée au bout de ce chemin. Sans compter votre enthousiasme contagieux qui m’a permis de surmonter les moments de désespoir. En un seul mot : merci !
Je souhaite remercier Giada et Maryse pour leur participation à ce travail, ce qui m’a permis d’accélérer dans cette course.
Un grand merci à Claire qui, avant son départ, m’a appris la rigueur et la précision que nécessite ce travail et qui m’a également transmis toutes ses connaissances dans le domaine de la biologie moléculaire et cellulaire.
Je remercie les autres membres de l’équipe de Génotoxicologie : Uli, Nicolas, Neil et Giuseppe. Vos diverses spécialités ainsi que vos compétences m’ont apporté de nombreuses suggestions et de précieux conseils.
Je souhaite également remercier l’ensemble des personnes des différentes équipes et services de l’IPBS qui m’ont aidée au cours de ce travail de thèse, en particulier la dernière arrivée des italiennes : Lorena. De plus, je remercie les personnes avec lesquelles j’ai eu l’occasion de collaborer, en particulier l’équipe du Docteur Michel Wright à l’ISTMT de Toulouse.
J’aurais une pensée particulière pour le Docteur Giovanni Maga sans qui je n’aurais jamais réalisé cette expérience en France. Merci pour avoir toujours pris le temps de répondre à mes appels et pour l’amitié que tu m’as manifestée.
Merci à tous ceux qui ont supporté mes plaintes, qui m’ont permis de penser à autre chose, qui ont aimé discuter avec moi et qui m’ont aidée à me connaître un peu mieux. C’est principalement grâce à ces personnes là que je suis arrivée à réaliser ce projet. Merci à Elisa, Sophie, Gabriele et la « coloc de Balma ».
Enfin merci à tous ceux qui, au cours de ces cinq années passées à Toulouse, ont ajouté des pièces à cet énorme puzzle qui représente ma vie : Donata, Séverine ainsi que tous les italiens.
J’espère pouvoir vous rendre un jour ce que vous tous m’avez donné jusqu’à aujourd’hui.
Pour finir, je souhaite enfin dédier ce travail à ceux qui m’ont épaulée jour après jour tout au long de ces huit dernières années et qui sont toujours restés à mes côtés, même sans comprendre : ma mère, mon père et mon frère.
SOMMAIRE
Liste des abréviations……….…page 4 Avant-propos………...………..……..page 7 INTRODUCTION………...……….….…….page 8 I. LES CASSURES DOUBLE-BRIN DE L’ADN (CDBs)………page 9 II. MECANISMES DE REPARATION DES CDBs DE L’ADN CHEZ LES MAMMIFERES………...………...page 12 La signalisation d’une CDB sur l’ADN………page 17 1° : La modification de la chromatine...page 18
L’histone
γ
H2AX………...………..page 19La protéine MDC1………...…………...………page 20 La protéine 53BP1………...………page 21 2° : Le complexe MRN………...……….………page 22 La famille des PIKKs………...………...…page 23 3° : La résection des extrémités endommagées……...……….…..page 25 La protéine RPA………...………...………page 26 4° : Résumé………...………….……….page 27 III. LA REPARATION PAR JONCTION DES EXTREMITES NON-HOMOLOGUES (NHEJ)………...…page 29 IV. LA REPARATION PAR RECOMBINAISON HOMOLOGUE (HR)…...…..page 32 A. Rôle biologique de la Recombinaison Homologue……….……….page 32 B. Le cœur catalytique de la Recombinaison Homologue……….…………..page 33 1° : La recombinase RecA...page 35 2° : La recombinase Rad51………..……..page 37 La protéine Rad52……….…………..page 40 Les paralogues de Rad51………page 42 Les protéines Rad54 et Rad54B……….………….page 45 3° : La protéine suppresseur des tumeurs BRCA2………..………...page 46
Rôle de BRCA2 dans la recombinaison mitotique……….……….page 46 Rôle de BRCA2 dans la recombinaison méiotique……….…………page 51 Rôle de BRCA2 dans la progression du cycle cellulaire………page 52 La protéine Dss1………..………...page 52 La protéine PALB2……….page 53 4° : La recombinase Dmc1………...………..…page 54 C. La phase postsynaptique de la Recombinaison Homologue………..…..page 56 1° : La famille des hélicases RecQ………...page 58 La protéine BLM……….………page 61 La protéine WRN……….………page 62 2° : La famille XPF/MUS81………...……….page 64 Le complexe MUS81-EME1……….………...page 65
V. LA REPARATION PAR RECOMBINAISON HOMOLOGUE : LES
MECANISMES………...page 67 A. La réparation des cassures double-brin………...page 67 B. Le rétablissement des fourches de réplication bloquées………....………..page 69 C. La réparation des pontages interbrins………..…..………..page 71 VI. LA REGULATION DE LA REPONSE AUX CASSURES DOUBLE-BRIN DE L’ADN………..……..page 74 1° : La protéine suppresseur des tumeurs BRCA1……….………...…….page 79 BRCA1 dans la réparation des CDBs de l’ADN……….……page 79 Rôle de BRCA1 dans la signalisation des dommages sur l’ADN………...…page 80 Rôle de BRCA1 dans l’activation du checkpoint………..………..…page 81 Le complexe BRCA1/BARD1………..………page 81 2° : L’Anémie de Fanconi………..….page 83 VII. LES CENTROSOMES ET LA REGULATION DU CYCLE CELLULAIRE CHEZ LES MAMMIFERES………..……....….page 87 La tubuline-
γγγγ
et l’organisation du centrosome……….………page 88 RESULTATS………...…..………….……...page 91 PRESENTATION DU THEME DE RECHERCHE………...………..page 92 I. ETUDE DE LA CINETIQUE DE FORMATION DES CASSURES DOUBLE-BRIN DE L’ADN……….………page 95 INTRODUCTION………...………..…….page 95 RESULTATS………..………page 96Cinétique d’apparition de
γγγγ
H2AX après différents traitements………...……page 96 II. ETUDE DU ROLE FONCTIONNEL DE L’ASSOCIATION ENTRE RAD51 ET LATUBULINE-γγγγ………...……….…...page 104
INTRODUCTION………..………...……….…….….page 104 RESULTATS………...……….……page 105 A. Mesure de la Recombinaison Homologue in vivo : les vecteurs pBHPR, pBHRF et pBHRR………..……….……..page 105 B. Mesure de la Recombinaison Homologue in vivo : le vecteur pDR-GFP……….page 112 III. ETUDE DE LA LOCALISATION PHYSIQUE DE LA TUBULINE-γγγγ AU NIVEAU NUCLEAIRE………..………....page 121 INTRODUCTION………..……….…...………..page 121 RESULTATS………...…..………...page 122 A. Distribution de la tubuline-
γγγγ
dans les compartiments nucléaires……….page 122 B. Identification d’une forme modifiée de la tubuline-γγγγ
………..…..……....page 123 C. Nouvelles interactions nucléaires de la tubuline-γγγγ
………...………….page 127 D. Localisation physique de la tubuline-γγγγ
au noyau……….…….……page 129 IV. ETUDE DU ROLE DE RAD51 DANS LE RECRUTEMENT DE LA TUBULINE-γγγγ AU NOYAU………...……….……….page 133 INTRODUCTION……….………...………page 133 RESULTATS………...……….page 134 A. Effet de la fonctionnalité de Rad51 sur le recrutement de la tubuline-γγγγ
au noyau……….………...………page 134 B. Effet de la présence de Rad51 sur le recrutement de la tubuline-γγγγ
au noyau……….………...page 136 DISCUSSION ET PERSPECTIVES………...……….page 139 MATERIELS ET METHODES………...…………...………..page 146 REFERENCES BIBLIOGRAPHIQUES………...………..page 163LISTE DES ABREVIATIONS
53BP1 p53 Binding Protein 1
aa aminoacides
ATCC American Type Culture Collection ATM Ataxia Telangiectasia Mutated protein
ATP Adenosine Triphosphate
ATR Ataxia Telangiectasia mutated and Rad3-related protein ATRIP ATR Interacting Protein
BARD1 BRCA1-Associated Ring Domain 1
BASC BRCA1-Associated genome Surveillance Complex
Bcl B-cell Leukemia/Lymphoma protein
BER Base Excision Repair
BFP Blue Fluorescent Protein
BIR Break-Induced Replication
BLAP75 BLM-Associated Protein, 75 kDa BLM Bloom syndrome mutated protein
bp paire de bases
BRAF35 BRCA2 Associated Factor, 35 kDa BRCA1/BRCA2 Breast Cancer susceptibility protein 1 / 2
BRCC BRCA1-BRCA2-Containing Complex
BRCT BRCA1 C-Terminus domain
BS Syndrome de Bloom
BUBR1 Budding Uninhibited by Benzimidazoles R1 protein CDB Cassure Double-Brin de l’ADN
Cdk Cycline-Dependent Kinase
Chk1/Chk2 Checkpoint Kinase 1 / 2
CHO cellules issues d’Ovaires de Hamster Chinois DNA-PKcs DNA-dependant Protein Kinase - catalytic subunit
DSBR Double-Strand Break Repair
DT40 lignée de lymphocytes B immortalisés de poulet
EBV Epstein-Barr Virus
EGFP Enhanced Green Fluorescent Protein
ERCC Excision Repair Cross-Complementing domain
ES Embryonic Stem cells
FA Anémie de Fanconi
FANC Gène ou Protéine Fanconi FAAP24 FA-Associated Protein, 24 kDa FITC Fluoresceine-Iso-ThioCyanate
FRAP Fluorescence Recovery After Photobleaching
γ-TuSC γ-tubulin Small Complex
γ-TuRC γ-tubulin Ring Complex GCP γ-tubulin Complex Protein GFP Green Fluorescent Protein
H2AX variante X de la famille des histones H2A
HeLa cellules humaines épithéliales provenant d’un adénocarcinome de l’ovaire
HR Homologous Recombination
HU Hydroxyurea
IRIF Ionizing Radiation Induced Foci
kDa kilo Dalton
LOH Loss of Heterozygosity
MDC1 Mediator of DNA Damage Checkpoint protein 1 MEF Mouse Embryonic Fibroblast
MMC Mitomycine C
MMR Mismatch Repair
MMS Méthyle Méthane Sulfonate Mre11 Meiotic Recombination protein 11
MRN MRE11/RAD50/NBS1 Complexe
MTOC Microtubules Organization Center Nbs1 Nijmegen Breakage Syndrome protein 1 NER Nucleotide Excision Repair
NHEJ Non-Homologous End Joining NLS Nuclear Localization Signal OB Oligosaccharide Binding domain PARP-1 Poly (ADP-Ribose) Polymerase 1 PCM Pericentriolar Matrix
PCNA Proliferating Cell Nuclear Antigen PCR Polymerase Chain Reaction
PIKK Phosphatydil-Inositol-3-Kinase related Kinase
pol DNA polymerase
RF-C Replication Factor C
RAD Radiation sensitivity abnormal
RI Radiations Ionisantes
ROS Reactive Oxygen Species
RPA Replication Protein A
SDSA Synthesis-Dependent Strand Annealing siRNA small interference RNA
SMC1 Structural Maintenance of Chromosome protein 1 SSA Single-Strand Annealing
SSB Single-Strand Binding protein
TdT Terminal-deoxynucleotidyl Transferase
TLS Translesion Synthesis
ToBP1 Topoisomerase II Binding Protein 1
TR Texas-Red
U2OS cellules humaines épithéliales provenant d’un ostéosarcome
UV Ultraviolet Radiation
WRN Werner syndrome mutated protein
WS Syndrome de Werner
XLF XRCC4-like Factor
XPF Xeroderma Pigmentosum, groupe de complémentation F XPV Xeroderma Pigmentosum, groupe de complémentation V XRCC X-ray Repair Cross-Complementing protein
AVANT-PROPOS
Dans l’introduction de ce manuscrit j’ai présenté les réponses cellulaires impliquées dans la réparation des cassures double-brin de l’ADN, considérées comme les lésions les plus toxiques pour la cellule.
En particulier, dans une première partie nous avons décrit les étapes de la réponse aux dommages de l’ADN communes à tous les mécanismes impliqués dans la réparation de ce type de lésions, dans les cellules de mammifères. Ensuite, nous avons décrit de façon générale un des deux mécanismes de réparation de cassures double-brin principalement impliqués, la réparation par Jonction des Extrémités Non-Homologues, pour successivement focalisé notre attention sur la réparation par Recombinaison Homologue. Nous avons décrit les protéines impliquées, les mécanismes par lesquels elles agissent et leur régulation vis-à-vis de la progression du cycle cellulaire. Dans la dernière partie de cette introduction nous avons examiné les caractéristiques du centrosome et de son composant principal, la tubuline-γ qui est impliquée dans la régulation de la réponse cellulaire aux lésions.
J’ai presenté ensuite le thème central de mon travail de thèse, à savoir le rôle fonctionnel et la régulation de l’association entre Rad51 et la tubuline-γ au niveau d’un même complexe nucléaire, qui est traité dans la section des résultats. Ceux-ci sont ensuite discutés. La section « matériels et méthodes » se trouve à la fin du manuscrit.
I. LES CASSURES DOUBLE-BRIN DE L’ADN (CDBs).
Les cassures double-brin de l’ADN (CDBs) sont considérées comme les lésions les plus toxiques pour la cellule, puisqu’elles menacent directement l’intégrité génétique. La faillite de la réparation de ce type de lésion peut être à l’origine d’une perte des chromosomes, d’un réarrangement du matériel génétique, de la mort cellulaire ou de la carcinogénèse (1).
Les CDBs peuvent avoir différents origines. Des processus comme les stress mécaniques (2), les transpositions (3), les mécanismes de diversification des immunoglobulines dans les lymphocytes (4), la méiose responsable de la génération de la diversité génétique (5) et la réplication de l’ADN, sont des exemples de facteurs endogènes qui peuvent être à l’origine de la formation d’une CDB. Par exemple, au cours de la phase S d’une cellule humaine, une cinquantaine de CDBs apparaît (6).
Les CDBs peuvent également être le résultat de l’action directe ou indirecte d’agents exogènes, comme les radiations ultraviolettes émises par le soleil (UV), les radiations ionisantes et certains agents thérapeutiques. De même les espèces réactives du métabolisme cellulaire peuvent entrainer l’apparition des CDBs (Figure 1).
La formation des CDBs après un traitement aux radiations ionisantes peut être un effet direct, alors que dans le cas des radiations UV il s’agit toujours d’un effet indirect. En effet, les cibles de la radiation solaire sont l’ADN et les chromophores (molécules cellulaires photosensibles). A cause des structures aromatiques des bases, l’ADN absorbe très efficacement ce type de radiation (7) ; les types de dommages majoritairement produits sont les dimères de pyrimidines et les photoproduits (6-4). Ces lésions créent des distorsions de la double hélice et peuvent inhiber la réplication et la transcription (8). Les radiations UV peuvent aussi induire la formation des espèces réactives de l’oxygène (ROS) (voir ci-dessous) qui peuvent menacer pas seulement l’ADN mais aussi les lipides et les protéines (9).
CAUSES Exogènes • radiations ionisantes • agents chimiques Endogènes • radicaux libres • cassures simple-brin pendant la réplication • fourches de réplication bloquées Spécialisées • recombinaison V(D)J • commutation de classe • hypermutation somatique • méiose CONSEQUENCES Mort cellulaire Contrôle du cycle cellulaire Réparation Incorrecte Instabilité génomique Carcinogénèse CDB
Figure 1. Causes, réponse cellulaire et conséquences des cassures double-brin de l’ADN. Les CDBs de l’ADN peuvent avoir une origine endogène ou exogène ou elles peuvent être produites par certains mécanismes de recombinaison spécialisées. La réponse aux dommages de l’ADN est très complexe et dépend de la coordination entre les mécanismes de réparation et l’activation des checkpoints. Si les dommages sont excessifs, la cellule se dirige vers la mort cellulaire. La fidélité de la réparation est extrêmement importante puisque une réparation incorrecte entraine une instabilité génétique qui peut contribuer à la carcinogénèse. (D’après Van Gent DC et al., 2001)
CAUSES Exogènes • radiations ionisantes • agents chimiques Endogènes • radicaux libres • cassures simple-brin pendant la réplication • fourches de réplication bloquées Spécialisées • recombinaison V(D)J • commutation de classe • hypermutation somatique • méiose CONSEQUENCES Mort cellulaire Contrôle du cycle cellulaire Réparation Incorrecte Instabilité génomique Carcinogénèse CDB CAUSES Exogènes • radiations ionisantes • agents chimiques Endogènes • radicaux libres • cassures simple-brin pendant la réplication • fourches de réplication bloquées Spécialisées • recombinaison V(D)J • commutation de classe • hypermutation somatique • méiose CAUSES CAUSES Exogènes • radiations ionisantes • agents chimiques Exogènes • radiations ionisantes • agents chimiques Endogènes • radicaux libres • cassures simple-brin pendant la réplication • fourches de réplication bloquées Endogènes • radicaux libres • cassures simple-brin pendant la réplication • fourches de réplication bloquées Spécialisées • recombinaison V(D)J • commutation de classe • hypermutation somatique • méiose Spécialisées • recombinaison V(D)J • commutation de classe • hypermutation somatique • méiose CONSEQUENCES Mort cellulaire Contrôle du cycle cellulaire Réparation Incorrecte Instabilité génomique Carcinogénèse CONSEQUENCES CONSEQUENCES Mort cellulaire Contrôle du cycle cellulaire Réparation Incorrecte Instabilité génomique Carcinogénèse CDB
Figure 1. Causes, réponse cellulaire et conséquences des cassures double-brin de l’ADN. Les CDBs de l’ADN peuvent avoir une origine endogène ou exogène ou elles peuvent être produites par certains mécanismes de recombinaison spécialisées. La réponse aux dommages de l’ADN est très complexe et dépend de la coordination entre les mécanismes de réparation et l’activation des checkpoints. Si les dommages sont excessifs, la cellule se dirige vers la mort cellulaire. La fidélité de la réparation est extrêmement importante puisque une réparation incorrecte entraine une instabilité génétique qui peut contribuer à la carcinogénèse. (D’après Van Gent DC et al., 2001)
Les CDBs d’origine exogènes sont très utilisées dans le contexte d’une thérapie anticancéreuse. Leur accumulation au cours des traitements tue les cellules cancéreuses. C’est le cas des traitements par des radiomimétiques tels que la bléomycine et la néocarcinostatine, qui produisent des CDBs de façon directe (10), des inhibiteurs des topoisomérases (11) et des radiations ionisantes (12).
Les topoisomérases sont des enzymes qui contrôlent la superhélicité de l’ADN au cours de différents processus cellulaires ; elles sont caractérisées par la formation d’une liaison covalente avec l’ADN, pendant leur cycle catalytique (13). Cette étape de transition est stabilisée par les inhibiteurs des topoisomérases, comme l’étoposide et la camptothécine, qui induisent de cette façon des cassures à simple- ou à double-brin sur l’ADN.
Les radiations ionisantes induisent différents types de lésions en même temps : cassures simple-brin, modifications des bases, dommages au niveau du groupe sucre-phosphate et CDBs. Quand un rayon ou une particule ionisante à haut niveau énergétique pénètre dans la cellule, il heurte les molécules cellulaires, y compris l’eau ; ces collisions déterminent la formation des ROS et si ces espèces se forment à proximité de l’ADN, les radicaux libres diffusent dans la chromatine et génèrent des dommages de façon casuelle (12).
L’action délétère des CDBs représente une sérieuse menace pour la viabilité des cellules et leur stabilité génomique. Ces lésions touchant simultanément les deux brins d’ADN doivent donc être prise en charge rapidement par la machinerie cellulaire.
II. MECANISMES DE REPARATION DES CDBs DE L’ADN CHEZ LES
MAMMIFERES.
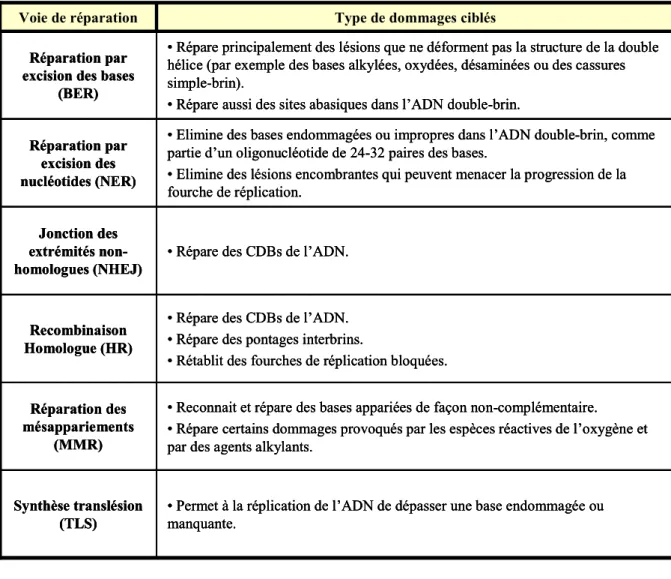
Les cellules des eucaryotes ont développé des voies multiples de réponse aux dommages de l’ADN, pour restaurer l’intégrité génétique, la séquence normale de l’ADN et sa structure. Le Tableau 1 résume les mécanismes de réparation principaux dans les cellules de mammifères et le type de dommage envers lequel ils agissent.
• Permet à la réplication de l’ADN de dépasser une base endommagée ou manquante.
Synthèse translésion (TLS)
• Reconnait et répare des bases appariées de façon non-complémentaire.
• Répare certains dommages provoqués par les espèces réactives de l’oxygène et par des agents alkylants.
Réparation des mésappariements
(MMR)
• Répare des CDBs de l’ADN. • Répare des pontages interbrins.
• Rétablit des fourches de réplication bloquées. Recombinaison
Homologue (HR)
• Répare des CDBs de l’ADN. Jonction des
extrémités non-homologues (NHEJ)
• Elimine des bases endommagées ou impropres dans l’ADN double-brin, comme partie d’un oligonucléotide de 24-32 paires des bases.
• Elimine des lésions encombrantes qui peuvent menacer la progression de la fourche de réplication.
Réparation par excision des nucléotides (NER)
• Répare principalement des lésions que ne déforment pas la structure de la double hélice (par exemple des bases alkylées, oxydées, désaminées ou des cassures simple-brin).
• Répare aussi des sites abasiques dans l’ADN double-brin. Réparation par
excision des bases (BER)
Type de dommages ciblés Voie de réparation
Tableau 1. Les voies de réparation des lésions de l’ADN chez les mammifères.
• Permet à la réplication de l’ADN de dépasser une base endommagée ou manquante.
Synthèse translésion (TLS)
• Reconnait et répare des bases appariées de façon non-complémentaire.
• Répare certains dommages provoqués par les espèces réactives de l’oxygène et par des agents alkylants.
Réparation des mésappariements
(MMR)
• Répare des CDBs de l’ADN. • Répare des pontages interbrins.
• Rétablit des fourches de réplication bloquées. Recombinaison
Homologue (HR)
• Répare des CDBs de l’ADN. Jonction des
extrémités non-homologues (NHEJ)
• Elimine des bases endommagées ou impropres dans l’ADN double-brin, comme partie d’un oligonucléotide de 24-32 paires des bases.
• Elimine des lésions encombrantes qui peuvent menacer la progression de la fourche de réplication.
Réparation par excision des nucléotides (NER)
• Répare principalement des lésions que ne déforment pas la structure de la double hélice (par exemple des bases alkylées, oxydées, désaminées ou des cassures simple-brin).
• Répare aussi des sites abasiques dans l’ADN double-brin. Réparation par
excision des bases (BER)
Type de dommages ciblés Voie de réparation
• Permet à la réplication de l’ADN de dépasser une base endommagée ou manquante.
Synthèse translésion (TLS)
• Reconnait et répare des bases appariées de façon non-complémentaire.
• Répare certains dommages provoqués par les espèces réactives de l’oxygène et par des agents alkylants.
Réparation des mésappariements
(MMR)
• Répare des CDBs de l’ADN. • Répare des pontages interbrins.
• Rétablit des fourches de réplication bloquées. Recombinaison
Homologue (HR)
• Répare des CDBs de l’ADN. Jonction des
extrémités non-homologues (NHEJ)
• Elimine des bases endommagées ou impropres dans l’ADN double-brin, comme partie d’un oligonucléotide de 24-32 paires des bases.
• Elimine des lésions encombrantes qui peuvent menacer la progression de la fourche de réplication.
Réparation par excision des nucléotides (NER)
• Répare principalement des lésions que ne déforment pas la structure de la double hélice (par exemple des bases alkylées, oxydées, désaminées ou des cassures simple-brin).
• Répare aussi des sites abasiques dans l’ADN double-brin. Réparation par
excision des bases (BER)
Type de dommages ciblés Voie de réparation
Dans cette introduction nous avons choisi de focaliser l’attention sur les principales voies de réparation des CDBs dans les cellules de mammifères ; en effet, la plupart des protéines impliquées dans tous ces processus, participent à plusieurs voies de réparation et certaines lésions peuvent être réparées par plusieurs voies.
Tous les eucaryotes ont développé plusieurs mécanismes pour faire face aux CDBs de l’ADN, ce qui montre l’importance et la difficulté de leur réparation. Ces systèmes entrainent la participation d’une quantité imposante de protéines qui fonctionnent dans un réseau de signalisation impliqué dans la reconnaissance du dommage, l’arrêt du cycle cellulaire et l’activation des voies de réparation.
La réponse cellulaire à une CDB peut survenir à différentes phases du cycle cellulaire et quand la cellule subit un nombre important de dommages, plusieurs voies de signalisation peuvent se chevaucher ; ces circonstances peuvent conduire à la mort cellulaire pour éviter la propagation d’une cellule avec un génome hautement instable (14). Cela rend la réponse cellulaire aux CDBs extrêmement critique, puisque si les mécanismes de réparation ne sont pas efficaces, l’alternative pour la cellule à la mort cellulaire peut être la carcinogenèse ou d’autres effets biologiques délétères.
Dans les cellules de mammifères, les CDBs de l’ADN sont réparées principalement par deux mécanismes : la jonction des extrémités non-homologues (NHEJ) et la recombinaison homologue (HR). Le premier mécanisme permet une réparation « error-prone » dans le sens où une partie de l’information génétique peut être perdue après la réparation de la lésion ; le deuxième est par contre très fidèle, parce que la réparation met en jeu des séquences d’ADN homologues à la séquence endommagée, présentes sur la chromatide sœur ou sur le chromosome homologue.
Chez les mammifères, il existe un troisième mécanisme que permet la réparation des CDBs en utilisant des séquences homologues comme substrat pour la réparation : l’appariement entre simple-brins (SSA) (Figure 2). Ce mécanisme est actif quand les séquences homologues se trouvent à proximité de la cassure sur la même molécule d’ADN ; dans ce cas, l’appariement entre les séquences complémentaires détermine la perte du matériel génétique entre elles (15). Ce mécanisme est indépendant de Rad51 et il est favori quand des mutations désactivent une des protéines du cœur de la HR. En général, puisque le SSA détermine toujours la perte de matériel génétique, la cellule cherche à contrôler son activation et à basculer l’équilibre vers les deux autres mécanismes.
Figure 2. Mécanisme de réparation par appariement entre simple-brins (SSA). Après la résection des extrémités de la cassure, la protéine Rad52 promeut l’appariement entre les séquences complémentaires. Une endonucléase élimine en suite les extrémités non homologues qui ne sont pas appariées (probablement XPF-ERCC1).
Figure 2. Mécanisme de réparation par appariement entre simple-brins (SSA). Après la résection des extrémités de la cassure, la protéine Rad52 promeut l’appariement entre les séquences complémentaires. Une endonucléase élimine en suite les extrémités non homologues qui ne sont pas appariées (probablement XPF-ERCC1).
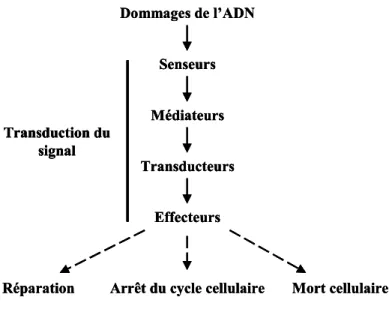
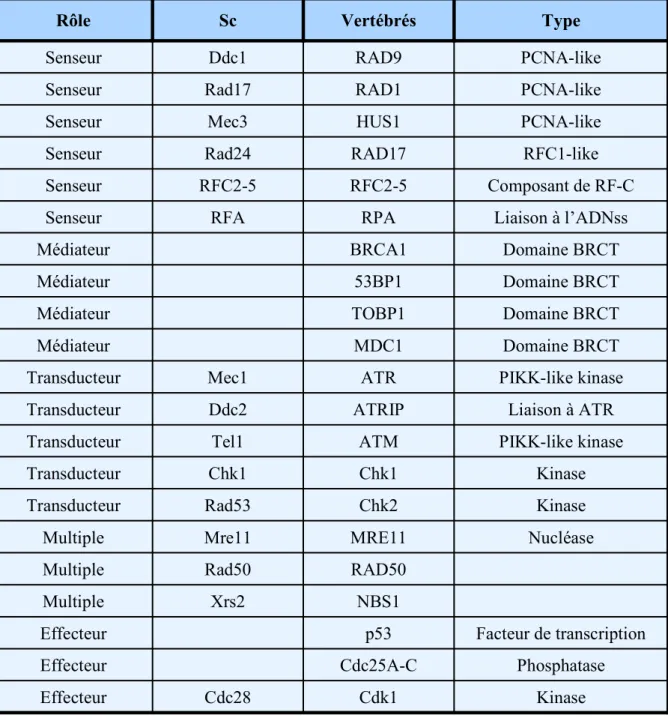
Ces voies de réponse aux dommages de l’ADN impliquent la participation de plusieurs composants qui peuvent être classés dans les catégories suivantes : les senseurs du signal, les transducteurs du signal, les médiateurs et les effecteurs (Figure 3). Le Tableau 2 donne une vue d’ensemble sur les acteurs principaux et le rôle qu’ils effectuent.
Réparation Arrêt du cycle cellulaire Mort cellulaire Transduction du signal Dommages de l’ADN Senseurs Médiateurs Transducteurs Effecteurs
Figure 3. Organisation de la réponse aux dommages de l’ADN.
Réparation Arrêt du cycle cellulaire Mort cellulaire Transduction du signal Dommages de l’ADN Senseurs Médiateurs Transducteurs Effecteurs
Réparation Arrêt du cycle cellulaire Mort cellulaire Transduction du signal Transduction du signal Dommages de l’ADN Senseurs Médiateurs Transducteurs Effecteurs
Domaine BRCT 53BP1 Médiateur Kinase Cdk1 Cdc28 Effecteur Phosphatase Cdc25A-C Effecteur Facteur de transcription p53 Effecteur NBS1 Xrs2 Multiple RAD50 Rad50 Multiple Nucléase MRE11 Mre11 Multiple Kinase Chk2 Rad53 Transducteur Kinase Chk1 Chk1 Transducteur PIKK-like kinase ATM Tel1 Transducteur Liaison à ATR ATRIP Ddc2 Transducteur PIKK-like kinase ATR Mec1 Transducteur Domaine BRCT MDC1 Médiateur Domaine BRCT TOBP1 Médiateur Domaine BRCT BRCA1 Médiateur Liaison à l’ADNss RPA RFA Senseur Composant de RF-C RFC2-5 RFC2-5 Senseur RFC1-like RAD17 Rad24 Senseur PCNA-like HUS1 Mec3 Senseur PCNA-like RAD1 Rad17 Senseur PCNA-like RAD9 Ddc1 Senseur Type Vertébrés Sc Rôle
Tableau 2. Classification des gènes impliqués dans la réponse aux dommages de l’ADN dans Saccharomyces cerevisiae (Sc) et chez les vertébrés.
Domaine BRCT 53BP1 Médiateur Kinase Cdk1 Cdc28 Effecteur Phosphatase Cdc25A-C Effecteur Facteur de transcription p53 Effecteur NBS1 Xrs2 Multiple RAD50 Rad50 Multiple Nucléase MRE11 Mre11 Multiple Kinase Chk2 Rad53 Transducteur Kinase Chk1 Chk1 Transducteur PIKK-like kinase ATM Tel1 Transducteur Liaison à ATR ATRIP Ddc2 Transducteur PIKK-like kinase ATR Mec1 Transducteur Domaine BRCT MDC1 Médiateur Domaine BRCT TOBP1 Médiateur Domaine BRCT BRCA1 Médiateur Liaison à l’ADNss RPA RFA Senseur Composant de RF-C RFC2-5 RFC2-5 Senseur RFC1-like RAD17 Rad24 Senseur PCNA-like HUS1 Mec3 Senseur PCNA-like RAD1 Rad17 Senseur PCNA-like RAD9 Ddc1 Senseur Type Vertébrés Sc Rôle Domaine BRCT 53BP1 Médiateur Kinase Cdk1 Cdc28 Effecteur Phosphatase Cdc25A-C Effecteur Facteur de transcription p53 Effecteur NBS1 Xrs2 Multiple RAD50 Rad50 Multiple Nucléase MRE11 Mre11 Multiple Kinase Chk2 Rad53 Transducteur Kinase Chk1 Chk1 Transducteur PIKK-like kinase ATM Tel1 Transducteur Liaison à ATR ATRIP Ddc2 Transducteur PIKK-like kinase ATR Mec1 Transducteur Domaine BRCT MDC1 Médiateur Domaine BRCT TOBP1 Médiateur Domaine BRCT BRCA1 Médiateur Liaison à l’ADNss RPA RFA Senseur Composant de RF-C RFC2-5 RFC2-5 Senseur RFC1-like RAD17 Rad24 Senseur PCNA-like HUS1 Mec3 Senseur PCNA-like RAD1 Rad17 Senseur PCNA-like RAD9 Ddc1 Senseur Type Vertébrés Sc Rôle
Tableau 2. Classification des gènes impliqués dans la réponse aux dommages de l’ADN dans Saccharomyces cerevisiae (Sc) et chez les vertébrés.
Les senseurs du signal sont évidemment les premiers participants à la réponse cellulaire ; seuls les transducteurs peuvent transmettre, aux autres composants de la voie, les indications nécessaires pour que le problème soit résolu le plus rapidement et le plus correctement possible. La plupart des senseurs et transducteurs a été identifié par des études génétiques dans la levure. Ceux-ci incluent, entre autres, le complexe formé par Rad9p, Hus1p, et Rad1p (appelé complexe 9-1-1) et un complexe de Rad17p avec les quatre sous-unités plus petites de RF-C (Replication Factor C), RFC2-5 (16) ; le complexe 9-1-1 est un hétérotrimère qui
ressemble à PCNA (Proliferating Cell Nuclear Antigen) et il est chargé sur l’ADN par RF-C. Senseurs/transducteurs incluent aussi quatre protéines kinases qui sont conservées de la levure aux mammifères (17) : deux membres de la famille des protéines kinases PIKK (Phosphatydil-Inositol-3-Kinase related Kinase), ATM (Ataxia Telangiectasia Mutated protein) et ATR (Ataxia Telangiectasia mutated and Rad3-related protein), et les deux Serine/Thréonine kinases Chk1 et Chk2 (Checkpoint Kinase 1 et 2). Toutes ces protéines citées ici, seront décrites en détail en dessous.
La classe des médiateurs a été, par contre, récemment clarifiée et il s’agit d’une classe de molécules qui n’ont pas d’activité catalytique. La protéine Rad9p de la levure, est le premier médiateur qui a été génétiquement défini ; les orthologues dans les cellules des vertébrés incluent 53BP1 (p53 Binding Protein 1), BRCA1 (Breast Cancer susceptibility protein 1), MDC1 (Mediator of DNA Damage Checkpoint protein 1) et ToBP1 (Topoisomerase II Binding Protein 1). Les médiateurs ont en commun un domaine BRCT (BRCA1 C-Terminus domain) qui permet la liaison aux protéines phosphorylées, la capacité de former foyers nucléaires et la capacité de promouvoir les interactions protéine-protéine.
Enfin, les effecteurs ont été identifiés grâce aux connaissances sur les transitions du cycle cellulaire et sur l’activation des checkpoints. Le concept de checkpoint naît des études réalisées dans la levure où la progression du cycle cellulaire est visible de l’extérieur ; l’activation du checkpoint se manifeste comme un arrêt dans une phase particulière du cycle cellulaire. Les kinases dépendant des cyclines (Cdk) promeuvent les transitions du cycle cellulaire et la modulation de leur activité est l’objectif ultime de l’activation du checkpoint. Dans certains cas, la modulation d’un régulateur peut influencer les effets de l’activation du checkpoint, même si le régulateur n’est pas la cible du checkpoint. Un effecteur, donc, est celui qui influence la signalisation du checkpoint mais qui aussi montre des changements dans ses caractéristiques suite à l’activation du checkpoint (par exemple son expression, son état de phosphorylation, sa localisation, etc.). Les homologues de Cdc25 et la protéine suppresseur des tumeurs p53, sont des exemples d’effecteurs (17). Les premiers sont phosphorylés et inhibés après activation du checkpoint chez les mammifères, Drosophila melanogaster,
Xenopus laevis et la levure ; alors que le dernier est phosphorylé après induction des CDBs (dans les mammifères et la Drosophila melanogaster) et augmente en concentration (dans les mammifères).
La HR et le NHEJ ont en commun les premières étapes de la réponse aux dommages de l’ADN, qui permettent de signaler la présence d’une lésion. Les étapes suivantes de la réparation sont réalisées par des protéines spécifiques de l’un ou de l’autre mécanisme.
Le choix du mécanisme de réparation à activer qui s’occupera de l’élimination de la lésion, dépend de plusieurs facteurs. Un de ceux-ci est par exemple la nature chimique de la lésion et la structure de ses extrémités qui sont dépendant du mécanisme qui l’a induite. Par exemple, un substrat typique pour le mécanisme du NHEJ est une CDB qui présente deux extrémités contondantes.
Mais le choix est surtout strictement corrélé au cycle cellulaire. L’existence de tous ces mécanismes de réparation qui peuvent être dirigés contre le même type de dommage sur l’ADN, implique que tous ces mécanismes soient fortement régulés. En effet, l’équilibre entre eux change selon l’espèce, selon les lignées cellulaires de la même espèce et pendant les différentes phases du cycle dans la même cellule. Par exemple, dans la levure, le mécanisme dominant pour la réparation des CDBs de l’ADN est la HR ; le NHEJ est très limité et imprécis, dans le sens où après le déroulement de ce mécanisme une partie de l’information génique est perdue dans la plupart des cas. Une explication peut se trouver dans le fait que les cellules des eucaryotes supérieurs possèdent des protéines qui sont absentes dans la levure, comme par exemple la DNA-PKcs (DNA-dependant Protein Kinase - catalytic subunit), BRCA1 et Artemis. Le NHEJ dans les eucaryotes supérieurs a un rôle extrêmement important ; il est actif pendant tout le cycle même s’il est prédominant dans la phase G1 et dans la transition G1-S. Par contre la HR est particulièrement active pendant les phases S et G2 du cycle, puisque l’accessibilité du substrat est un facteur limitant pour son efficacité (18, 19).
Nous donnerons quelques détails de la régulation de ces voies de réparation au cours du cycle cellulaire après avoir décrit leurs mécanismes, puisque ceux-ci sont fortement corrélés et, de plus, ils partagent un nombre important des protéines.
Avant de décrire les mécanismes à travers lesquels se déroule la réparation des CDBs de l’ADN, nous donnerons une description des étapes précoces de la réponse aux dommages qui sont communes entre les deux mécanismes.
La signalisation d’une CDB sur l’ADN.
Dans les cellules de mammifères, comme dans la levure, une CDB sur l’ADN a trois conséquences immédiates : la modification de la chromatine ; la liaison à l’ADN du complexe MRN (MRX dans la levure) qui est composé par les protéines Mre11 (Meiotic Recombination
protein 11), Rad50 et Nbs1 (Nijmegen Breakage Syndrome protein 1 ; Xrs2p dans la levure) ; la résection du double-brin pour exposer des simple-brins d’ADN.
1° : La modification de la chromatine.
La modification de la chromatine, comme il a été montré dans des fibroblastes embryonnaires de souris (MEFs), est un mécanisme dépendant d’énergie qui implique une réduction, localisée au site de la lésion, de la densité de la chromatine (20). Par des études en microscopie à fluorescence et électronique, le laboratoire de M. J. Kruhlak a réussi à caractériser les propriétés biophysiques de la chromatine endommagée. Il a été montré que la chromatine s’ouvre dans l’espace de quelques secondes après la formation de la CDB et que dans les mêmes temps, les premiers senseurs sont recrutés sur le site. Toutefois, en absence d’ATP, les senseurs sont recrutés sur le site de la coupure mais ils ne s’associent pas stablement et par conséquent, ils ne s’accumulent pas sur le site mais ils diffusent dans le noyau. Cela veut dire que soit la modification de la chromatine, soit le recrutement des senseurs sont des mécanismes dépendant de l’énergie.
Les changements de la chromatine sont de grande importance fonctionnelle, parce que les traitements qui empêchent ces changements, interférent avec toutes les phases successives de réparation du site endommagé.
Notamment, des travaux récents indiquent que l’ouverture de la chromatine est nécessaire pour le recrutement des transducteurs et des médiateurs. Par exemple, dans les cellules de mammifères, la formation des foyers de Rad51 dépend strictement de la décondensation de la chromatine (21). De plus, le déplacement des nucléosomes à proximité de la cassure, permet le chargement de Rad51 sur la lésion (22). Donc, cet événement est sûrement indispensable pour rendre la lésion plus accessible aux protéines de la réparation.
Egalement, la décondensation de la chromatine favorise probablement l’exposition des modifications des histones, comme par exemple la méthylation de l’histone H3 sur la Lys79 chez les mammifères. Cette méthylation augmente in vitro l’affinité pour le médiateur 53BP1 et la déplétion de la méthyltransférase qui modifie la Lys79, interfère avec la formation des foyers nucléaires de 53BP1 après l’induction de CDBs dans la lignée U2OS (cellules humaines épithéliales provenant d’un ostéosarcome) (23). Il faut remarquer que le niveau de méthylation de la Lys79, après l’induction des dommages sur l’ADN, ne change pas et, peut-être pour cette raison, l’augmentation de l’accessibilité aux modifications, obtenue avec la
décondensation de la chromatine, est impliquée dans la réponse aux dommages. Il existe d’autres modifications des histones impliquées dans les mêmes mécanismes, comme par exemple les acétylations (24) et les phosphorylations (25).
L’histone
γγγγ
H2AX.La plus caractérisée des modifications induites par les dommages sur l’ADN est sans doute la phosphorylation de la variante H2AX de l’histone H2A (γH2AX) (26-28). Cette histone est modifiée sur un résidu serine conservé à l’extrémité C-terminale (la Ser139 chez les mammifères et la Ser129 chez la levure) ; la modification est réalisée par les kinases appartenant à la famille des PIKKs : ATM, ATR et, dans quelque cas particulier, par la DNA-PKcs (29).
La structure physique et la composition de la chromatine contenant γH2AX ne sont pas connues, mais la phosphorylation d’H2AX a été liée à des phénomènes différents, à la fois opposés ; par exemple, elle a été liée à la décondensation de la chromatine dans la levure
Saccharomyces cerevisiae (30) et, au contraire, à la condensation de la chromatine et au « silencing » transcriptionnel du chromosome sexuel du mâle pendant la spermatogenèse, dans la souris (31). De plus, des nombreux complexes qui remodèlent la chromatine (comme par exemple, INO80 et NuA4/Tip60) et des composants structuraux associés à la chromatine (comme par exemple, les cohésines), sont assemblés à l’ADN de façon dépendante de γH2AX (25, 32).
Les souris invalidées pour H2AX sont viables, mais montrent une haute sensibilité aux radiations et une augmentation dans la fréquence des cancers. Les cellules souches embryonnaires (ES) dérivantes de ces animaux, montrent une diminution dans l’efficacité de la HR, lorsque la recombinaison V(D)J dans les lymphocytes, qui est réalisé par le NHEJ, n’est pas affectée (33).
L’histone γH2AX est responsable de l’assemblage des complexes multi-protéiques dans des foyers nucléaires (34, 35). La phosphorylation de la Ser139 a lieu dans l’espace de quelques minutes après l’induction de la CDB par des radiations ionisantes (36) et γH2AX ne se distribue pas de façon homogène dans le noyau, mais il forme des foyers (IRIF) dont le nombre, dans les cellules de mammifères, correspond au nombre des CDBs.
Ces foyers de γH2AX colocalisent, entre autres, avec les foyers de 53BP1 qui se forment aussi très rapidement au site de la cassure, avec les foyers de BRCA1 et avec les protéines du complexe MRN qui se forment plusieurs heures après l’irradiation.
Mais il est maintenant établi que la présence de γH2AX n’est pas nécessaire pour reconnaître la CDB. En effet, les fibroblastes et les cellules B provenant de souris invalidées pour H2AX, ne sont pas capables de former des foyers de 53BP1 ou de BRCA1 ou des protéines MRN après exposition aux radiations ionisantes (33). Néanmoins, il faut remarquer que, dans ces cellules, BRCA1, 53BP1 et Nbs1 sont encore recrutées au site de la cassure (37). Donc, γH2AX joue probablement le rôle de maintenir et d’accumuler les protéines de la réparation au site de la CDB.
L’imagerie en temps réel, appliquée à des cellules de mammifères, a permis de recueillir d’autres informations sur les IRIF, en tant que sites d’amplification du signal. L’induction de CDBs localisée, en utilisant des faisceaux laser qui traversent le noyau au long d’une ligne définie, ont comme résultat l’induction localisée de γH2AX, mais le signal ne génère pas la formation de foyers, même après plusieurs heures (37). Ces données suggèrent que les IRIFs ne sont pas le résultat de la réorganisation des facteurs de réparation et que la localisation des CDBs, marqués par γH2AX, reste fixe à l’intérieur du noyau. Au contraire, l’augmentation des dimensions de l’IRIF, peut être simplement le reflet d’une extension de γH2AX sur plusieurs megabases de la chromatine qui entoure la lésion (36).
La protéine MDC1.
Concomitant à l’extension de γH2AX pourrait être le recrutement des protéines qui se lient à γH2AX. Entre celles-ci, la protéine MDC1 est indispensable pour la formation des IRIF (38). Il a été montré que : dans des extraits de cellules non endommagées, MDC1 fait partie d’un complexe contenant MRN (39) ; MDC1 interagit directement et spécifiquement avec γH2AX à travers son domaine BRCT (40) ; de plus, un peptide phosphorylé dérivant de l’extrémité C-terminale d’H2AX peut interagir avec MRN seulement en présence de MDC1 (41). Les données le plus récentes suggèrent que MDC1 s’interpose entre γH2AX et Nbs1 (42) (Figure 4). Ainsi, l’extension des protéines de la réparation sur la chromatine, pourrait venir du recrutement séquentiel sur γH2AX de MDC1, MRN et ATM, qui en suite génèrent d’autre γH2AX en position distale à la lésion et un recrutement ultérieur des mêmes et d’autres facteurs. Des mutants d’H2AX qui sont phosphorylés, mais qui ne peuvent pas lier MDC1, échouent dans la formation des IRIF de 53BP1, de Nbs1 et d’ATM activée (43). De cette façon MDC1 pourrait être l’élément clé pour que γH2AX puisse former les IRIF.
Avant la lésion Après la lésion
foyer nucléaire
Figure 4. Modèle représentant le mécanisme d’interaction entre MDC1 et le complexe MRN avant et après la lésion. Pour les détails suivre le texte. (D’après Spycher C et al., 2008)
Avant la lésion Après la lésion
foyer nucléaire
Avant la lésion
Avant la lésion Après la lésion
foyer nucléaire
Après la lésion
foyer nucléaire
Figure 4. Modèle représentant le mécanisme d’interaction entre MDC1 et le complexe MRN avant et après la lésion. Pour les détails suivre le texte. (D’après Spycher C et al., 2008)
Un autre rôle important de MDC1 a été révélé dans des cellules U2OS déficientes en MDC1 où l’histone H2AX est phosphorylée mais ne se maintient pas au site de la lésion. Probablement, la liaison de MDC1 protège γH2AX de l’action de phosphatases ou sert à recruter et maintenir ATM activée en proximité (40).
Ainsi, même si les protéines des IRIF sont recrutées aux sites des CDBs de façon indépendante de γH2AX, leur organisation dans des foyers semble nécessiter l’amplification de γH2AX sur la chromatine et des interactions protéine-protéine qui renforcent les complexes. Cette amplification est probablement nécessaire pour potentialiser la réponse aux dommages dans les cas où le signal est bas, comme après l’exposition à des basses doses de radiations ionisantes (44).
La protéine 53BP1.
La protéine 53BP1 s’accumule aux sites des cassures quelques secondes après leur formation même en absence de γH2AX mais, dans ces conditions, elle ne forme pas des foyers mais diffuse dans le noyau. Cette protéine, suppresseur des tumeurs, est un homologue de la protéine Rad9p de la levure qui, dans cette espèce, est un important régulateur du checkpoint.
Certaines données de la littérature indiquent qu’un signal pour le recrutement de 53BP1 aux sites de dommage pourrait être la méthylation des histones (45). Mais des données récentes assurent aussi que le recrutement de 53BP1 à la CDB est dépendante de MDC1 qui en détermine la phosphorylation ; les deux protéines agiraient dans la même voie et MDC1 serait un régulateur en amont de 53BP1, en déterminant sa localisation et sa phosphorylation (46). Il semblerait donc que la localisation de 53BP1 au site de la lésion soit régulée par deux mécanismes différents, mais il n’est pas encore clair pourquoi et si les deux mécanismes sont corrélés entre eux. Probablement la modalité de recrutement de 53BP1 est spécifique de sa fonction.
2° : Le complexe MRN.
La protéine MDC1 est responsable du recrutement du complexe MRN à travers son interaction avec la protéine Nbs1. Ce complexe est conservé entre les espèces et il est essentiel puisqu’il participe à plusieurs mécanismes cellulaires impliquant des dommages sur l’ADN, comme la réparation des cassures, la signalisation des cassures, la réplication de l’ADN, la recombinaison méiotique et l’induction de la mort cellulaire (47).
Le complexe MRN est constitué de trois sous-unités. La première est la nucléase Mre11 qui probablement est impliquée dans la modification des extrémités d’ADN pendant la réparation par HR (48). La deuxième est l’ATPase et adenylate kinase Rad50 qui, avec Mre11, faciliterait l’interaction entre les molécules d’ADN pendant la réparation (49). La troisième sous-unité, Nbs1, ne possède pas d’activité catalytique, mais favorise les interactions protéine-protéine (50) ; elle serait responsable, comme décrit ci-dessus, du recrutement du complexe MRN au niveau des foyers de γH2AX à travers son interaction avec MDC1.
Néanmoins, il faut remarquer que MDC1 n’est pas indispensable pour toutes les fonctions du complexe MRN ; son activité de modification des extrémités d’ADN dépend de MDC1, selon le type de CDB : l’accumulation du complexe MRN sur l’ADN à simple-brin n’est pas dépendante de MDC1 ; par contre, son accumulation et rétention sur de la chromatine contenant γH2AX, est fortement dépendante de MDC1 (51).
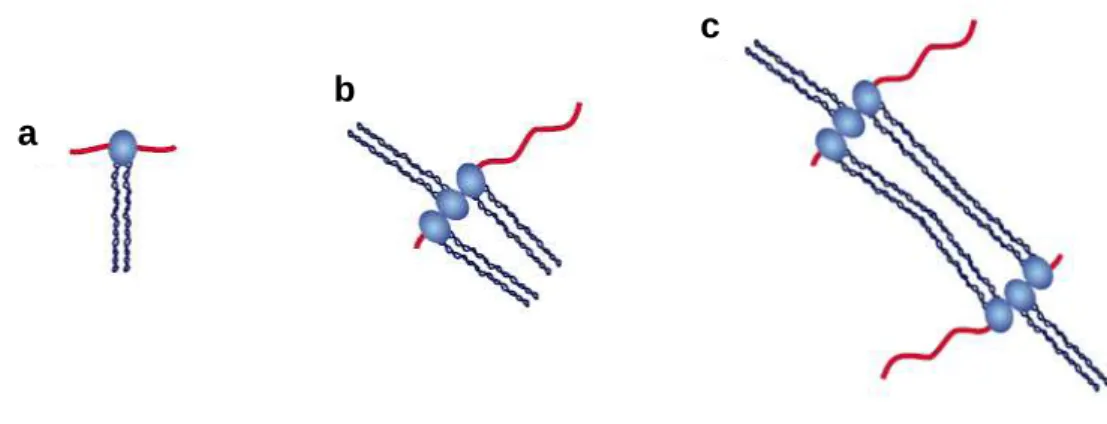
Des études biochimiques et biophysiques ont montré que le complexe formé par Rad50 et Mre11 forme des oligomères sur de l’ADN linéaire. Le complexe Rad50/Mre11 contient deux molécules de Rad50 qui forment un dimère par leurs domaines globulaires ; à ces domaines se lient aussi deux sous-unités de la nucléase Mre11, qui ne sont pas globulaires mais en
serpentin. Nbs1 est lié aussi au domaine globulaire mais sa stœchiométrie n’est pas connue. Le complexe Rad50/Mre11 ainsi formé, se lie à l’ADN à travers les domaines globulaires et les extrémités à serpentin promeuvent les interactions entre différents complexes. De cette façon, les oligomères de Rad50/Mre11 attachent plusieurs molécules d’ADN, en favorisant la réparation (49, 52) (Figure 5).
Figure 5. Attachement des molécules d’ADN endommagées par le complexe Rad50/Mre11. (a) Le complexe Rad50/Mre11 se lie à l’ADN (lignée rouge) à travers ses domaines globulaires. (b) Sur un ADN linéaire, le complexe forme des oligomères à proximité des extrémités de la cassure. (c) Les oligomères attachent les molécules d’ADN en favorisant la réparation de la lésion. (D’après Wyman C and Kanaar R, 2006)
a
b
c
Figure 5. Attachement des molécules d’ADN endommagées par le complexe Rad50/Mre11. (a) Le complexe Rad50/Mre11 se lie à l’ADN (lignée rouge) à travers ses domaines globulaires. (b) Sur un ADN linéaire, le complexe forme des oligomères à proximité des extrémités de la cassure. (c) Les oligomères attachent les molécules d’ADN en favorisant la réparation de la lésion. (D’après Wyman C and Kanaar R, 2006)
a b c a a b b c c
La famille des PIKKs.
Plusieurs études ont montré que le complexe MRN est nécessaire pour l’activation de certains membres de la famille des protéines kinases PIKK : ATM, ATR et la DNA-PKcs. Ces composants de la famille PIKK sont activés en réponse à différents types de dommage (53), activent les checkpoints du cycle cellulaire et régulent la réparation à travers la phosphorylation des protéines cibles. D’autres membres de la famille PIKK sont impliqués dans le contrôle de la croissance cellulaire (mTor ; (54)) et dans la régulation de la structure de la chromatine (Trrap ; (55)).
Les défauts dans l’activité de ces enzymes sont associés à une dérégulation du checkpoint, à une hypersensibilité aux agents génotoxiques, à la mort cellulaire et à une prédisposition au cancer. En effet les phosphorylations effectuées par les PIKKs sont impliquées dans plusieurs mécanismes cellulaires, de la signalisation du dommage à la régulation de la formation des foyers.
L’activité kinase d’ATM est stimulée rapidement quand les cellules sont exposées à des radiations ionisantes (56, 57). ATM se trouve dans la cellule sous forme de dimères et d’oligomères qui sont inactifs ; après l’induction des dommages, les formes inactives deviennent des monomères et les monomères s’autophosphorylent sur la Ser1981 en se localisant au site de la lésion (58).
Il a été montré in vitro que la monomérisation d’ATM est réalisé par l’action du complexe MRN (59). En effet, un rôle du complexe MRN serait celui de rapprocher plusieurs molécules d’ADN en augmentant la concentration locale de CDBs (voir ci-dessus) et ainsi induire la monomérisation d’ATM (60, 61).
De plus, selon la concentration de CDBs, l’autophosphorylation d’ATM sur la Ser1981 nécessite, in vitro, de l’interaction avec Nbs1 (62). Ainsi le deuxième rôle de MRN pourrait être de promouvoir l’activité kinase d’ATM à travers l’interaction d’ATM avec Nbs1. Toutefois, à hautes concentrations de CDBs, ATM peut se transformer en monomère même en absence du complexe MRN.
Après autophosphorylation, ATM phosphoryle plusieurs protéines impliquées dans la réponse aux dommages, comme par exemple p53 (56, 57), BRCA1 (63), Nbs1 (64), SMC1 (Structural Maintenance of Chromosome protein 1) (65), Rad17 (66) et 53BP1 (67).
La phosphorylation d’ATM détermine aussi l’activation du checkpoint par les protéines Chk1 et Chk2, qui peuvent être aussi des substrats d’ATR (68) et qui pourraient accomplir la tache de propager le signal de la lésion.
Plusieurs cibles d’ATM sont de protéines impliquées directement ou indirectement dans la réparation par HR et quelques unes de ces cibles peuvent aussi être phosphorylé par la DNA-PKcs.
L’histone H2AX est une des cibles immédiates des PIKKs (69) ; en particulier, après des radiations ionisantes, elle est une cible d’ATM (70). Les inhibiteurs des PIKKs, comme la wortmannine, ont comme effet l’élimination de γH2AX et, par conséquent, l’impossibilité pour les protéines associées à γH2AX, de s’accumuler dans des agrégats (34). Aussi la DNA-PKcs peut phosphoryler H2AX après induction de dommages, mais seulement en absence d’ATM ; cela suggère que ATM bloque l’accès d’H2AX à la DNA-PKcs (29).
ATM phosphoryle aussi des protéines du NHEJ, comme Artemis, et elle joue un rôle important dans la stabilisation des extrémités cassées pendant la recombinaison V(D)J opérée par le NHEJ. Néanmoins, il parait que ATM promeut la réparation des CDBs par HR ; en effet, des inhibiteurs spécifiques de son activité kinase (comme KU55933 (71)) réduisent la
HR induite par des CDBs et inhibent la formation de foyers Rad51 induit par les radiations ionisantes (72).
ATR, au contraire, ne montre pas des changements dans la structure ou dans l’activité après traitements génotoxiques. Il a été suggéré que la voie d’activation d’ATR passe par la formation du complexe RPA/ADN simple-brin (voir ci-dessous). ATR semble agir principalement dans le checkpoint qui répond aux radiations UV et à un arrêt de la mitose dû à un problème dans la synthèse d’ADN (73). Les souris invalidées pour ATR meurent précocement au stade embryonnaire, effet dérivant d’un manque de coordination entre la phase S et la mitose pendant la croissance cellulaire normale.
ATR se trouve sous forme de complexe avec la protéine ATRIP (ATR Interacting Protein) qui est nécessaire pour ses fonctions. Une étude récente in vitro a montré que ToBP1, un médiateur, se lie et stimule l’activité kinase d’ATR, humaine et de Xenopus laevis (74). Puisque ToBP1 est un composant des IRIF dans des cellules U2OS, la voie d’activation d’ATR par ToBP1 pourrait avoir un rôle dans la réponse aux dommages (75).
3° : La résection des extrémités endommagées.
L’étape suivante dans la réparation par HR d’une CDB, est le traitement des extrémités de l’ADN pour la génération de simple-brins avec extrémité 3’-OH sortante. Ce mécanisme a été bien clarifié dans les bactéries mais la machinerie équivalente dans les eucaryotes n’a pas encore été identifiée. Le processus est réalisé dans les bactéries par le complexe hélicase/nucléase RecBCD qui s’occupe aussi de charger sur l’ADN la recombinase RecA (voir ci-dessous) (76). La structure cristalline du complexe lié à l’ADN a montré que les deux extrémités d’ADN sont ouvertes et chaque simple-brin est guidé à l’intérieur d’un canal par action de l’hélicase (77). La nucléase responsable du traitement des extrémités se trouve en aval de l’hélicase et, de cette façon, elle a accès à un brin ou à l’autre.
Des expériences génétiques dans la levure ont montré que le traitement des extrémités est réalisé par le complexe Rad50/Mre11, mais il n’a pas été prouvé qu’il exerce aussi le rôle de nucléase (78). L’activité de Rad50/Mre11 dans les eucaryotes pourrait être légèrement modifiée par l’interaction avec d’autres facteurs ou ce complexe pourrait être moins impliqué dans la résection des extrémités mais, plutôt, pourrait agir comme cofacteur pour une autre nucléase, pour laquelle plusieurs candidats existent.
En tout cas le traitement des extrémités pour exposer des simple-brins d’ADN est exigé pour tous les types de cassures à réparer. Selon le traitement appliqué pour générer la CDB, les extrémités peuvent ne pas être réparées (comme après l’action d’une endonucléase) ou peuvent être transformées pour éliminer les protéines liées de façon covalente (comme après le traitement avec des inhibiteurs de topoisomérases) ou peuvent nécessiter l’élimination des groupes chimiques quand la coupure a lieu au niveau de l’anneau de ribose (comme après des radiations ionisantes). La structure des extrémités est donc déterminante du type de modification apporté, de façon à faciliter la réparation ; en effet, l’extrémité simple-brin de l’ADN peut être utilisée soit comme primer pour la synthèse, soit comme substrat pour une ligation.
La protéine RPA.
Les extrémités d’ADN ainsi formées, sont ensuite recouverte par une protéine qui possède une haute affinité pour l’ADN simple-brin, RPA (Replication Protein A) (79). RPA est un hétérotrimère qui peut éliminer de structures secondaires dans l’ADN simple-brin et qui, pour cette raison, est impliqué dans tous les processus métaboliques qui impliquent de l’ADN simple-brin. D’un point de vue stœchiométrique, un hétérotrimère de RPA recouvre 25 nucléotides d’ADN simple-brin.
Le complexe formé par RPA et l’ADN simple-brin est reconnu en tant que signal d’ADN endommagé, sur la base d’expériences in vivo et in vitro réalisés dans la levure, chez l’homme et dans Xenopus laevis (80, 81) ; il a été montré que RPA est impliqué dans le recrutement, sur l’ADN simple-brin, d’ATR/ATRIP, de Rad17 et du complexe 9-1-1, et stimule l’activité kinase d’ATR à travers Rad17. En effet, la déplétion d’une sous-unité de RPA, détermine la perte des foyers d’ATR/ATRIP induits par des radiations ionisantes, dans des cellules HeLa (cellules humaines épithéliales provenant d’un adénocarcinome de l’ovaire) ; et de plus, les mutants de levure déficients en RPA ne peuvent pas former foyers d’ATRIP à la CDB. Ces résultats amènent à imaginer l’existence d’un mécanisme conservé dans la levure et dans les vertébrés, dans lequel le complexe RPA/ADN simple-brin au site de la cassure recrute Rad17/9-1-1 et ATR/ATRIP pour faciliter la phosphorylation de ce dernier par l’autre.
Il existe quand même des théories discordantes, selon lesquelles le remodelage de la chromatine serait responsable du recrutement d’ATM et de la nucléase Mre11 qui seraient impliqués dans la résection des extrémités d’ADN, dans le recrutement d’ATR et dans l’activation de Chk1 dans des cellules humaines (48).
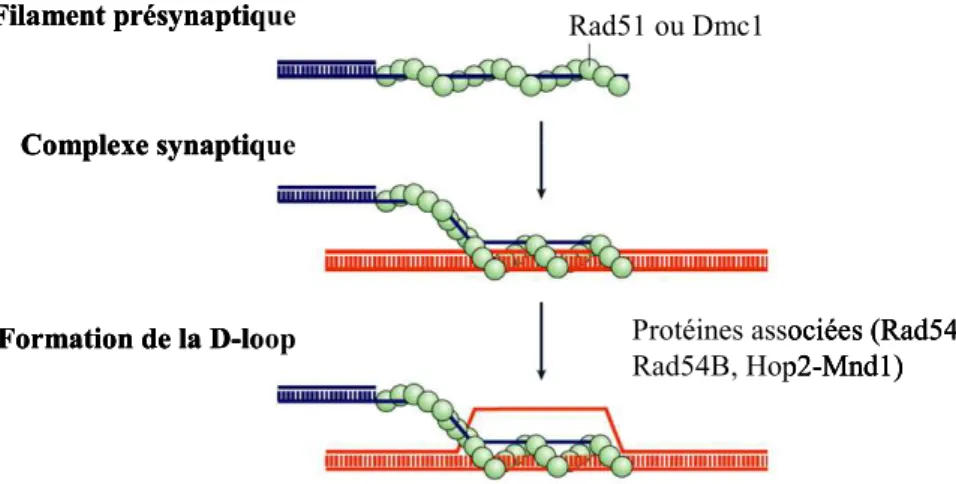
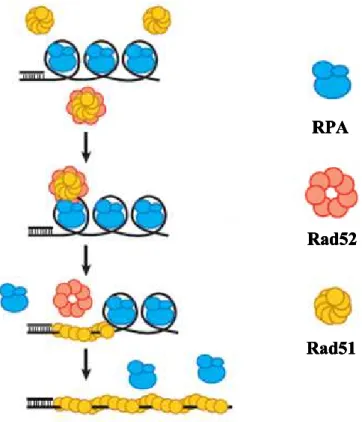
La fonction de RPA dans la HR est extrêmement complexe, puisque selon les circonstances, elle peut avoir un rôle de stimulateur ou d’inhibiteur dans l’assemblage du filament présynaptique (voir ci-dessous) (82). L’action stimulatrice de RPA a été remarquée en 1994, quand a été reportée pour la première fois, l’activité recombinase de Rad51 dans la levure
Saccharomyces cerevisiae (83). De plus, des mutations dans la sous-unité majeure de RPA sont associés à des défauts dans la synapse ; donc, RPA joue probablement un rôle dans l’invasion du filament complémentaire pendant la HR (84).
Néanmoins, une quantité de RPA suffisante à saturer l’ADN simple-brin disponible, supprime les activités ATPase et recombinase de Rad51 et de Dmc1 (85, 86). Mais ils existent plusieurs médiateurs de la HR que s’opposent à l’activité inhibitrice de RPA ; une quantité catalytique de médiateurs est suffisante pour déplacer RPA de l’ADN simple-brin et permettre au filament présynaptique de se former (87).
4° : Résumé.
Pour résumer, trois signaux sont générés par une CDB et ces signaux guident les événements en aval (Figure 6). Un de ceux-ci est la décondensation de la chromatine, qui détermine l’activation d’ATM, l’association à la chromatine de 53BP1 et le recrutement du complexe de réparation.
Un autre est l’association du complexe MRN qui est indépendante d’ATM/ATR et détermine le recrutement et l’activation d’ATM. BRCA1 peut interagir avec MRN et participer à cette activité, puisqu’elle peut s’associer à l’ADN endommagé de façon indépendante de l’activation d’ATM et peut recruter ATM activé.
Le troisième signal est la génération d’ADN simple-brin qui détermine le recrutement de RPA et successivement, d’ATR, du complexe 9-1-1 et de Rad17.
Ces mécanismes peuvent être dissociés ; par exemple la génération de γH2AX ne nécessite pas Rad17. D’un autre côté, ils partagent des composants ; par exemple, le complexe MRN peut aider dans la génération d’ADN simple-brin.
Les événements suivant l’interaction des senseurs avec la CDB, déterminent le recrutement d’autres protéines au site de la cassure et la propagation du complexe nucléoprotéique tout au long du chromosome. Ces événements incluent la génération de γH2AX par ATM, ATR ou les deux, qui recrute MDC1. MDC1 stabilise γH2AX, solidifie l’interaction entre ATM, MRN, 53BP1 et BRCA1 et induit la propagation de γH2AX sur le chromosome.
Les médiateurs peuvent aussi recruter et aider dans l’activation des substrats d’ATM/ATR, comme par exemple Chk1 et Chk2, pour amplifier et propager le signal de la lésion.
Décondensation de la chromatine Le complexe MRN
se lie à la CDB
Résection des extrémités
RPA recouvre l’ADN simple-brin
53BP1 se lie aux histones méthylées MRN recrute et active ATM
ATM génère γΗ2ΑX
Rad51 se lie et forme des foyers
de façon indépendante de γH2AX MDC1 se lie àγH2AX, recrute MRN et ATM et détermine la propagation de γH2AX γH2AX recrute MDC1 RPA recrute ATR/ATRIP , le complexe 9-1-1 et Rad17
Figure 6. Modèle de la signalisation d’une CDB de l’ADN. Pour les détails suivre le texte. (D’après Su TT, 2006).
Décondensation de la chromatine Le complexe MRN
se lie à la CDB
Résection des extrémités
RPA recouvre l’ADN simple-brin
53BP1 se lie aux histones méthylées MRN recrute et active ATM
ATM génère γΗ2ΑX
Rad51 se lie et forme des foyers
de façon indépendante de γH2AX MDC1 se lie àγH2AX, recrute MRN et ATM et détermine la propagation de γH2AX γH2AX recrute MDC1 RPA recrute ATR/ATRIP , le complexe 9-1-1 et Rad17 Décondensation de la chromatine Le complexe MRN se lie à la CDB Décondensation de la chromatine Le complexe MRN se lie à la CDB
Résection des extrémités
RPA recouvre l’ADN simple-brin Résection des extrémités
RPA recouvre l’ADN simple-brin
53BP1 se lie aux histones méthylées MRN recrute et active ATM
ATM génère γΗ2ΑX
53BP1 se lie aux histones méthylées MRN recrute et active ATM
ATM génère γΗ2ΑX
Rad51 se lie et forme des foyers
de façon indépendante de γH2AX MDC1 se lie àγH2AX, recrute MRN et ATM et détermine la propagation de γH2AX MDC1 se lie àγH2AX, recrute MRN et ATM et détermine la propagation de γH2AX γH2AX recrute MDC1 RPA recrute ATR/ATRIP , le complexe 9-1-1 et Rad17
Figure 6. Modèle de la signalisation d’une CDB de l’ADN. Pour les détails suivre le texte. (D’après Su TT, 2006).
III
. LA REPARATION PAR JONCTION DES EXTREMITES
NON-HOMOLOGUES (NHEJ).
La structure des extrémités d’une CDB est déterminante du mécanisme impliqué dans sa réparation. Le NHEJ répare principalement un CDB avec les deux extrémités contondantes, mais il peut aussi réparer des CDBs avec des groups 5’-P et 3’-OH qui présentent des microrégions d’homologies (comme celles qui sont produites par les nucléases) ; dans ce cas, la réparation implique l’alignement d’une ou de quelques bases complémentaires et provoque des petites délétions ou des petites insertions (88). Dans les cellules de mammifères, le mécanisme commence avec une transformation limitée des extrémités par intervention du complexe MRN qui interagit avec d’autres facteurs faisant partie du cœur du NHEJ : Ku70, Ku80 et la DNA-PKcs (Figure 7). Les protéines Ku70 et Ku80 forment un hétérodimère caractérisé par une haute affinité pour les extrémités d’ADN. La cristallographie a montré que l’hétérodimère Ku70/80 forme un anneau avec un trou que s’adapte à l’ADN, ce qui explique sa préférence pour la liaison des extrémités de l’ADN (89). Les données actuelles suggèrent que l’hétérodimère Ku70/80 recrute la DNA-PKcs. Une fois liée aux extrémités cassées, la DNA-PKcs est activée et elle se phosphoryle elle-même et d’autres cibles impliquées dans des mécanismes de réparation, comme par exemple les protéines RPA, WRN (Werner syndrome mutated protein) et la nucléase Artemis ; la DNA-PKcs peut aussi phosphoryler l’histone H2AX dans des cellules déficientes en ATM (voir ci-dessus) (90). La liaison de la DNA-PKcs à l’ADN détermine le déplacement de Ku70/80 du côté opposé aux extrémités (91), probablement pour faciliter l’accès à d’autres protéines aux extrémités de la cassure. En effet, le cœur du NHEJ comprend aussi les composants suivants, impliqués dans les phases finales du processus : XRCC4, l’ADN-ligase IV et XLF. En particulier, la dernière étape de jonction est obtenue par médiation de l’ADN-ligase IV qui est associée à un dimère de XRCC4 (92) (Figure 7).
XRCC4 peut se lier à l’ADN, est nécessaire pour la stabilité de l’ADN-ligase IV in vivo et stimule son activité adénylase et ligase (93, 94). Des études récentes ont permis l’identification d’une protéine XRCC4-like, XLF (aussi connue comme Cernunnos), qui interagit avec le complexe ADN-ligase IV/XRCC4 (95, 96). Sa fonction n’a pas été encore clarifiée mais, les patients qui possèdent des mutations dans le gène codant XLF, montrent une extrême radiosensibilité et des défauts dans la réparation des CDBs ; ces patients sont aussi immunodéficients parce que la résolution des CDBs intermédiaires est nécessaire pour l’assemblage de gènes des immunoglobulines, un processus dans lequel le NHEJ joue un rôle fondamental. Dans ce cadre, par exemple, l’endonucléase Artemis est importante pour l’ouverture des structure en épingle à cheveux qui se forment pendant la recombinaison V(D)J (97).
Le résultat du NHEJ peut être aussi modifié par la Terminal-deoxynucleotidyl Transferase (TdT) et par deux membres de la famille X des ADN polymérases (pol µ et pol λ) par addition « non-templated » de nucléotides aux extrémités ou par extension de l’ADN simple-brin au 3’-OH qui peut s’apparier de façon transitoire par micro-homologie à l’autre extrémité cassée (98).