Comparison of normalization methods for differential gene expression analysis in RNA-Seq experiments: A matter of relative size of studied transcriptomes
10
0
0
Texte intégral
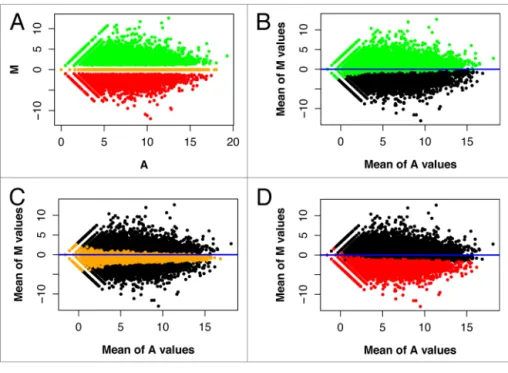
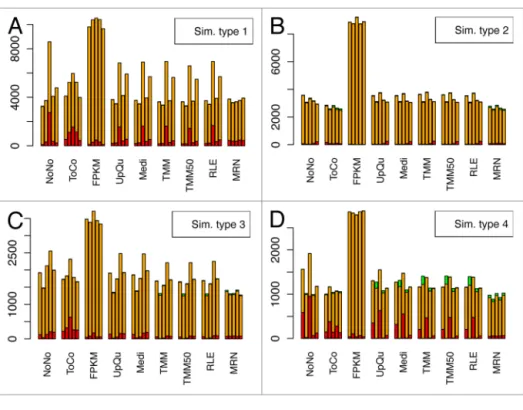
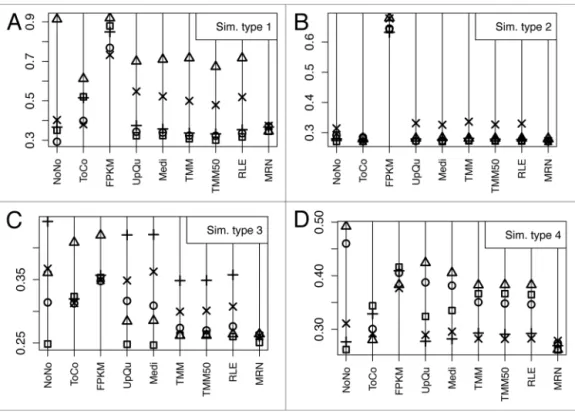
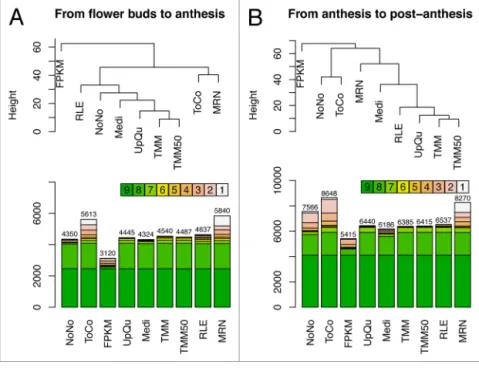
Figure
Documents relatifs
