Université de Sherbrooke
Étude des implications biochimiques et moléculaires sous-jacentes à la pharmacothérapie ciblée contre la proprotéine convertase PACE4 dans le cancer de la prostate.
Par Frédéric Couture
Programmes de Doctorat en biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph.D.)
en biochimie
Sherbrooke, Québec, Canada Janvier, 2018
Membres du jury d’évaluation
Robert Day, Départment de chirurgie – service d’urologie Guylain Boissonneault, Département de biochimie Fernand Gobeil Jr, Département de Pharmacologie Chantal Guillemette, Faculté de Pharmacie, Université Laval
“Science is a way of thinking much more than it is a body of knowledge.” ― Carl Sagan
R
ÉSUMÉ
Étude des implications biochimiques et moléculaires sous-jacentes à la pharmacothérapie ciblée contre la proprotéine convertase PACE4 dans le cancer de la prostate.
Par Frédéric Couture
Programmes de Doctorat en biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de philosophiae doctor (Ph.D.) en biochimie, Faculté de médecine et des sciences de la santé, Université de
Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Le cancer de la prostate est le cancer le plus fréquent chez les hommes et la capacité des tumeurs à développer une résistance face aux thérapies anti-androgéniques vient souvent compromettre le pronostic des patients. Le développement de nouvelles approches thérapeutiques afin de circonvenir à la progression de ces tumeurs représente un besoin important la gestion de ce type de cancer. Plusieurs démonstrations récentes établissent l’implication de la famille des proprotéines convertases dans la progression tumorale. Ces enzymes ont pour fonctions biologiques de cliver une variété de précurseurs protéiques jouant des rôles importants dans la tumorigénèse. Dans le cancer de la prostate, la proprotéine convertase PACE4 est fortement surexprimée dans les cellules cancéreuses et joue un rôle dans la prolifération et la capacité à former des tumeurs, ce qui en fait une cible thérapeutique d’intérêt. En ce sens, des inhibiteurs peptidomimétiques ont été développés dans l’optique de la thérapie ciblée contre la PACE4. Toutefois, dans le but de développer une approche thérapeutique optimale, il convient néanmoins de comprendre le niveau de redondance fonctionnelle entre les différents membres de la famille des convertases, qui sont connus pour partager plusieurs de leurs substrats, ainsi que les mécanismes moléculaires régissant l’activité de la PACE4 et de ses substrats sous-jacents. L’utilisation d’une approche de répression génique stable envers les différentes convertases a permis de mettre en lumière les fonctions uniques de la PACE4 dans la progression tumorale. De plus, grâce à une approche de protéomique comparative, le premier substrat de la PACE4 dans le cancer de la prostate; le growth differenciation factor 15, a été découvert. Ce substrat permet de commencer à dresser l’implication de PACE4 dans le paysage moléculaire du cancer de la prostate. Grâce à des modalités d’imagerie moléculaire, l’emploi de versions radiomarquées des inhibiteurs peptidiques a également permis de démontrer que les composés s’accumulent dans les cellules cancéreuses en fonction des niveaux de PACE4 présents, et ce, tant in cellulo qu’in vivo. Ces données suggèrent un potentiel pour le développement d’un examen théranostique pour prédire la réponse tumorale à la pharmacothérapie anti-PACE4. Finalement, l’analyse de l’épissage alternatif de l’ARNm de PACE4 a permis l’élucidation des caractéristiques biochimiques et des fonctions spécifiques d’une nouvelle isoforme; la PACE4-altCT, qui est exprimée chez les cellules cancéreuses de la prostate, mais aussi d’autres types de cancer. Cette découverte a permis de redéfinir le modèle de travail en intégrant le concept de la rétention intracellulaire de cette isoforme qui semble médier la plupart de l’activité pro-proliférative reliée à l’activité PACE4, ce qui en fait la cible pharmacologique principale des inhibiteurs peptidiques dans le cancer de la prostate, mais aussi un biomarqueur potentiel.
Mots clés : PACE4, thérapie ciblée, cancer de la prostate, imagerie moléculaire, inhibiteur peptidique, épissage alternatif, pharmacologie.
S
UMMARY
Biochemical and molecular implications downstream of PACE4-targeted therapy in prostate cancer By
Frédéric Couture Biochemistry Doctoral Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Doctor degree diploma philosophiae doctor (Ph.D.) in biochemistry, Faculty of medicine and health sciences, Université de
Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Prostate cancer is the most common cancer among men. The capabilities of tumors to adapt and overcome antiandrogenic therapy is persistently worsening patients prognostic and the development of novel therapeutic approaches to circumvent tumor progression therefore represents an unmet need. Many reports now demonstrate the implication of the enzymes from the proprotein convertase family in the progression of tumor from many cancer types. These enzymes are responsible for the processing of various protein precursors playing important roles in tumorigenesis. In prostate cancer, the proprotein convertase PACE4 is strongly overexpressed in cancer cells and plays a role in cell proliferation and tumor formation thus making a strong case for its use as a pharmacological target. For this reason, PACE4 peptidomimetic inhibitors were generated to develop PACE4-targeted therapies. However, to develop an optimal therapeutic approach regarding the inhibition of this enzyme, a complete understanding of the level of functional redundancy between the different convertases in prostate cancer is needed. Moreover, understanding the molecular mechanisms both upstream and downstream of PACE4 in prostate cancer cells would allow a better understanding of the considerations underneath such a therapeutic strategy. Using a stable gene silencing approach to knockdown all co-expressed member of the convertase family in prostate cancer cells, the roles of PACE4 in tumor progression were found to be unique and non-redundant among the other family member. Through a comparative proteomic approach, the first PACE4-specific substrate in prostate cancer; growth and differentiation factor 15, was identified. With this substrate growth factor, it is now possible to initiate the dissection of PACE4 biochemical functions in the prostate cancer molecular landscape. Using a radiolabelled version of the PACE4 peptide inhibitors, it was possible to demonstrate using molecular imaging that when applied in cellulo and in vivo, the compound is uptaken by cancer cells as well as by tissues according to their PACE4 expression levels. These data suggest that such PACE4 molecular imaging with pharmacological inhibitor could be developed as a theranostic assay to predict which tumor could be treated by PACE4-targetted therapy. Lastly, PACE4 mRNA alternative splicing analysis permitted the discovery of a new PACE4 isoform; named PACE4-altCT, which is strongly overexpressed by prostate cancer cells as well as other cancer types. As this isoform displays specific biochemical features and functions, notably being intracellularly retained and mediating most of the PACE4-associated cell growth capabilities, this discovery further redefined our working model, pointing to PACE4-altCT as the pharmacological target of inhibitory peptides in prostate cancer as well as a potential biomarker.
Keywords : PACE4, targeted therapy, prostate cancer, molecular imaging, peptide inhibitor, alternative splicing, pharmacology.
T
ABLE DES MATIERES
Résumé ... i
Summary ... ii
Table des matières ... iii
Liste des figures ... vii
Liste des tableaux ... ix
Liste des abréviations... x
Introduction ... 1
Les origines et bases du cancer ... 2
Les statistiques du cancer ... 4
Le cancer de la prostate ... 6
Statistiques du cancer de la prostate ... 6
La prostate ... 7
Anatomie ... 7
Structures et fonctions ... 8
Origines et évolution du cancer de la prostate ... 9
Étiologie du cancer de la prostate ... 9
Causes du cancer de la prostate ... 12
Gestion clinique du cancer de la prostate ... 15
Dépistage ... 15
Diagnostic ... 18
Approches thérapeutiques ... 21
Besoins actuels pour le cancer de la prostate ... 23
Les proprotéines convertases ... 26
Fonctions biologiques des proprotéines convertases ... 26
La redondance chez les proprotéines convertases ... 31
Les proprotéines convertases dans le cancer ... 33
Expression et fonctions des PCs dans le cancer ... 34
Les proprotéines convertases comme cibles pharmacologiques dans le cancer ... 36
La PACE4 et le cancer de la prostate ... 40
Fonction de la PACE4 dans le cancer de la prostate ... 41
Les inhibiteurs peptidiques de la PACE4 ... 43
L’inhibiteur peptidique de la PACE4; le Multi-Leucine ... 44
Optimisation des propriétés pharmacologiques des inhibiteurs peptidiques de PACE4 ... 47
La PACE4 comme cible thérapeutique et marqueur du cancer de la prostate ... 50
Hypothèse/problématique ... 52
Objectifs ... 52
Article 1- Role of Proprotein Convertases in Prostate Cancer Progression ... 54
Résumé : ... 54
Abstract ... 55
INTRODUCTION ... 56
MATERIAL AND METHODS ... 57
RESULTS ... 62
Knockdown of endogenous PCs in prostate cancer model cell lines ... 62
Effect of PC knockdowns on cell proliferation ... 64
Secreted growth factors are implicated in PCs-mediated proliferation effects ... 65
Only PACE4 downregulation prevents prostate tumor growth in vivo and reduces neovascularization .. 66
The silencing of PACE4 in prostate cancer cell lines enhances quiescence in tumor xenografts ... 70
DISCUSSION ... 73
ACKNOWLEDGMENTS ... 76
Article 2- PACE4-based molecular targeting of prostate cancer using an engineered 64Cu-radiolabeled peptide inhibitor ... 77
Résumé : ... 77
Abstract ... 78
INTRODUCTION ... 79
MATERIAL AND METHODS ... 81
RESULTS ... 85
Peptide synthesis ... 85
NOTA-coupled ML peptide retains original PACE4 affinity and is retained in prostate cancer cells expressing PACE4. ... 85
64Cu-NOTA-ML in-vivo stability ... 87
64Cu-NOTA-ML distribution is specific to PACE4-expressing tissues in-vivo ... 88
64Cu-NOTA-ML targets LNCaP prostate cancer xenografts and ML does not accumulate in PACE4-negative xenografts ... 90
DISCUSSION ... 94
CONCLUSION ... 97
Article 3 - Multi-Leu PACE4 inhibitor retention within cells is PACE4-dependent and a prerequisite for
antiproliferative activity. ... 98
Résumé : ... 98
Abstract : ... 99
INTRODUCTION ... 100
MATERIAL AND METHODS ... 102
RESULTS ... 104
DISCUSSION ... 111
CONCLUSION ... 112
ACKNOWLEDGMENTS ... 112
Article 4 - PACE4 Undergoes an Oncogenic Alternative Splicing Switch in Cancer ... 113
Résumé : ... 113
Abstract : ... 114
INTRODUCTION ... 115
MATERIAL AND METHODS ... 117
RESULTS ... 128
PACE4 mRNA levels correlate with tumor aggressiveness in PCa tissue specimens ... 128
PCa exploits a PACE4 mRNA terminal exon splicing event that is specific to tumor cells. ... 131
PACE4-altCT is generated by a stabilized mRNA variant that leads to intracellular retention, enhanced stability and increased auto-activation ... 136
PACE4 splicing is regulated by intra-exonic DNA methylation ... 141
The expression of PACE4-altCT in normal tissues and various cancer types suggests a common tumor mechanism ... 145
PACE4-altCT is the main isoform responsible for PCa cell-sustained growth capabilities ... 149
Identification of GDF-15 as a specific PACE4 substrate and pharmacological target engagement marker ... 153
DISCUSSION ... 162
ACKNOWLEDGMENTS: ... 166
SUPPLEMENTARY METHODS: ... 168
Discussion ... 179
PACE4; une proprotéine convertase aux fonctions uniques ... 181
GDF-15 ; premier indice moléculaire de l’action de PACE4 ... 182
PACE4-altCT ; fonctions distinctes et redondantes d’une isoforme ... 185
Thérapie ciblée pour la PACE4; avantages et limitations ... 190
Marqueur d’engagement de la cible ... 194
Imagerie moléculaire théranostique ... 195
Conclusion ... 197
Liste des références... 199
Remerciements ... 219
Annexe 1 - Tableau des numéros d’autorisations des emprunts de figures ... 220
Annexe 2 – Liste des communications scientifiques ... 221
Publications ... 221
Présentations par affiche ... 223
L
ISTE DES FIGURES
Figure 1 – Évènements marquants dans le développement de thérapies ciblées ... 2
Figure 2 – Statistiques relatives au cancer aux États-Unis depuis 1975 ... 5
Figure 3 – Anatomie uro-génitale ... 8
Figure 4 – Histologie de la prostate ... 9
Figure 5 – Histologie des adénocarcinomes prostate ... 11
Figure 6 – Étapes précoces de la carcinogenèse de l’épithélium prostatique ... 12
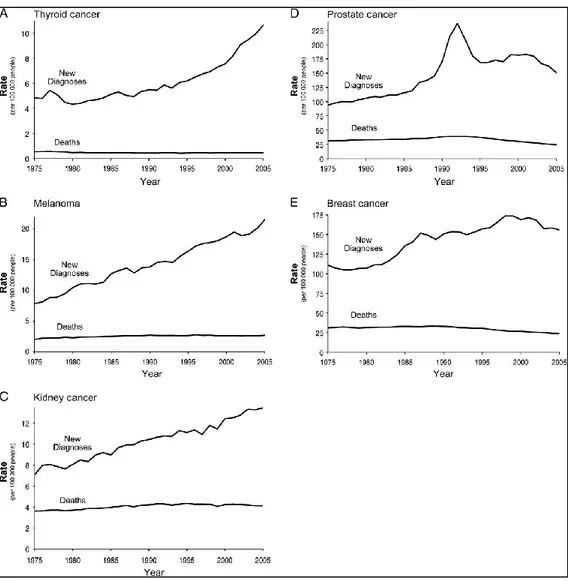
Figure 7 – Taux de diagnostic et de mortalité du SEER “Surveillance, Epidemiology, and End Results ” de 1975 to 2005 aux États-Unis ... 17
Figure 8 – Exemple d’échantillonnage permis par les biopsies transrectales ... 18
Figure 9 – Voies de synthèse des androgènes et cibles pharmacologiques pour la cancer de la prostate. ... 22
Figure 10 – Biosynthèse de l’insuline ... 26
Figure 11 – Nomenclature et organisation structurales des proprotéines convertases ... 27
Figure 12 – Processus d’auto-activation des PCs ... 28
Figure 13 – Localisation cellulaire des PCs... 29
Figure 14 – Clivage du précurseur POMC par les PCs PC1/3 et PC2. ... 31
Figure 15 – Homologie structurale du domaine catalytique des PCs ... 32
Figure 16 – Expression des PCs par dans les différents types de cancers de la cohorte du The Cancer Genome Atlas (TCGA). ... 35
Figure 17 – Implications des PCs dans les différentes étapes de la progression tumorale ... 36
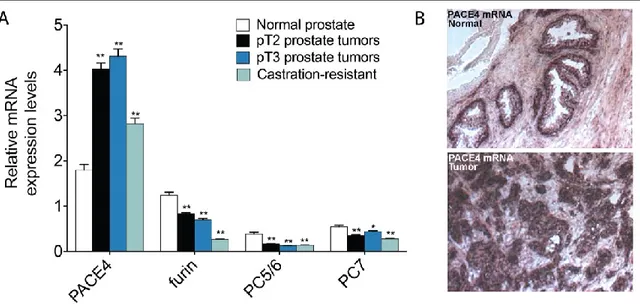
Figure 18 – Surexpression de la proprotéine convertase PACE4 dans le cancer de la prostate ... 41
Figure 19 – Impact de PACE4 sur la progression tumorale. ... 42
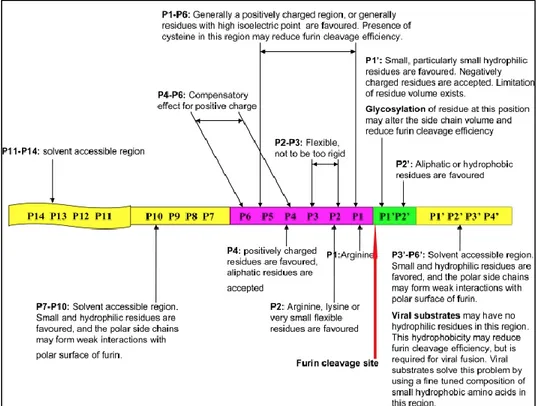
Figure 20 – Préférences en matière de résidus pour les positions P14 à P4’ chez les substrats de furine ... 44
Figure 21 – Nécessité de la pénétration cellulaire du peptide ML pour le maintien de son efficacité anti- proliférative sur les cellules de cancer de la prostate ... 45
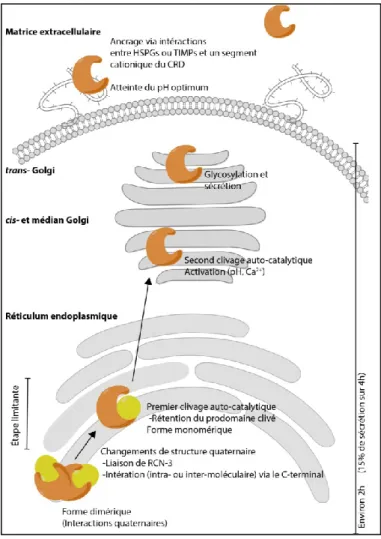
Figure 22 – Routage cellulaire et modifications post-traductionnelles de la PACE4 ... 47
Figure 23 – Structures des mimétiques de l’arginine ... 48
Figure 24 – Efficacité pharmacologique in vivo du C23 sur la progression tumorale ... 50
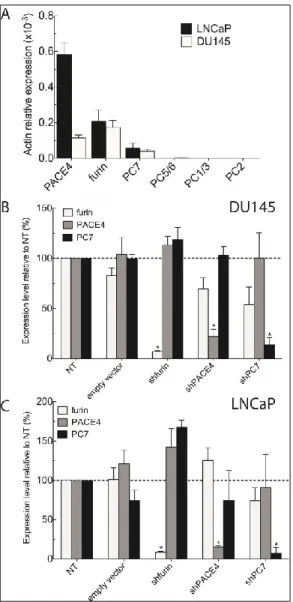
Figure 25 – shRNA efficiency screening on DU145 cells. ... 63
Figure 26 – Stable knockdown of endogenous PCs in DU145 and LNCaP prostate cancer cells ... 64
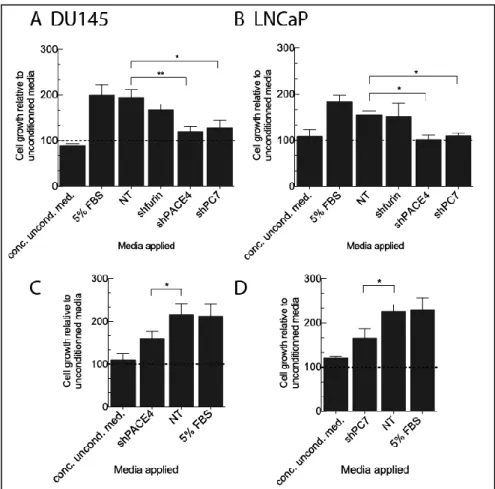
Figure 27 – Cellular proliferation upon PCs knockdown. ... 65
Figure 28 – Mitogenic properties of secreted factors derived from PCs knockdown cell lines. ... 66
Figure 29 – In vivo tumorigenicity assay of PC knockdown prostate cancer cell lines. ... 67
Figure 30 – H&E staining on LNCaP tumors. ... 68
Figure 31 – Xenograft microvessels density of tumors derived from LNCaP cell lines. ... 69
Figure 32 – Immunohistochemistry against cell-cycle markers and EGFR protein for DU145 and LNCaP tumor xenografts. ... 70
Figure 33 – EGFR Western blots on DU145-derived tumors ... 72
Figure 34 – NOTA-LLLLRVKR-NH2 in vitro affinity toward PACE4 ... 86
Figure 35 – Peptide uptake within prostate cancer cells ... 87
Figure 36 – Radiolabeled peptide integrity analysis in vivo ... 88
Figure 37 – Radiopeptide biodistribution ... 89
Figure 38 – PET imaging of LNCaP tumor-bearing athymic nude mice ... 92
Figure 39 – PACE4-lacking PC3 xenografts PET imaging ... 93
Figure 40 – PACE4 expression levels in the studied cell lines ... 104
Figure 41 – Knockdown cell proliferation rates... 106
Figure 43 – 64Cu/NOTA-ML uptake and retention correlation with PACE4 levels in cells ... 109
Figure 44 – Anti-proliferative properties of ML peptide on the tested cell lines ... 110
Figure 45 (partie 1) – Alternative splicing of PACE4 terminal exon is strongly enhanced in prostate cancer specimens and correlates with tumor aggressiveness. ... 129
Figure 46 – Antibody validation panel and distinct intracellular localization by IHC ... 131
Figure 47 (partie 1) – PACE4 mRNA splicing analysis and primer design ... 133
Figure 48 – PACE4 alternative splicing results in 3’UTR shortening and in the generation of an isoform differentially retained by cells with enhanced stability and increased auto-activation ... 137
Figure 49 – PACE4 isoforms secretion, stability and activity. ... 139
Figure 50 – PACE4 alternative splicing and polyadenylation is dependent on CTCF-mediated exon inclusion and regulated by intra-exonic DNA methylation. ... 142
Figure 51 – 5-aza-dC efficiency at hypomethylating CpG dinucleotides of interest in PCSK6 gene and ChIP in response to 5-aza-dC-induced hypomethylation in the LNCaP cell line. ... 144
Figure 52 – Mapping of PACE4-altCT across human tissues and different cancer types reveals a common tumor molecular switch mechanism... 146
Figure 53 – PACE4-altCT expression across human tissues and different cancer types reveals a common tumor molecular switch mechanism... 149
Figure 54 – PACE4-altCT is responsible of PACE4-associated sustained growth capabilities in prostate cancer cells. ... 150
Figure 55 – PACE4 expression and proliferation-related phenotypes in stable lentiviral-transduced overexpressing and siRNA-silenced cells. ... 152
Figure 56 (partie 1) – Secretome analysis heatmap representation and PC substrates analysis by western blots ... 154
Figure 57 – Identification and validation of GDF-15 as a PACE4-specific substrate in prostate cancer. ... 157
Figure 58 – GDF-15 as a PACE4-activity marker in vivo. ... 160
Figure 59 – Summary and working model. ... 163
Figure 60 – Sucession d’événements cellulaires et moléculaires menant à la carcinogenèse de l’épithélium prostatique. ... 179
Figure 61 – Statistiques relatives à la progression sur 19 ans des patients atteints du cancer de la prostate aux États-Unis ... 180
Figure 62 – Structure, expression et usage des niveaux sériques de GDF-15 ... 184
Figure 63 – Implications multiples de GDF-15 dans la progression tumorale du cancer de la prostate. ... 185
Figure 64 – Mécanismes suggérés pour réguler l’activité des PCs et le clivage de leurs substrats... 187
Figure 65 – Cinétique de captation tumorale et d’accumulation vésicale du 64Cu/NOTA-C23. ... 192
Figure 66 – Gamme dynamique des protéines plasmatiques ... 194
Figure 67 – Résumé des applications potentielles découlant des découvertes relatives à la PACE4 et à ses inhibiteurs dans le cancer de la prostate. ... 198
L
ISTE DES TABLEAUX
Tableau 1 – Classification TNM du cancer de la prostate ... 19
Tableau 2 – Scénarios de traitements du cancer de la prostate ... 20
Tableau 3 – Principaux agents thérapeutiques utilisés pour le traitement du CRPC. ... 23
Tableau 4 – Nouvelles thérapies expérimentales en essais cliniques pour le cancer de la prostate ... 25
Tableau 5 – Distribution tissulaire, phénotypes associés aux souris knockout et exemples de substrats des PCs ... 30
Tableau 6 – Inhibiteurs des proprotéines convertases utilisés in vivo ... 39
Tableau 7 – Propriétés des peptides en tant qu’agents thérapeutiques ... 48
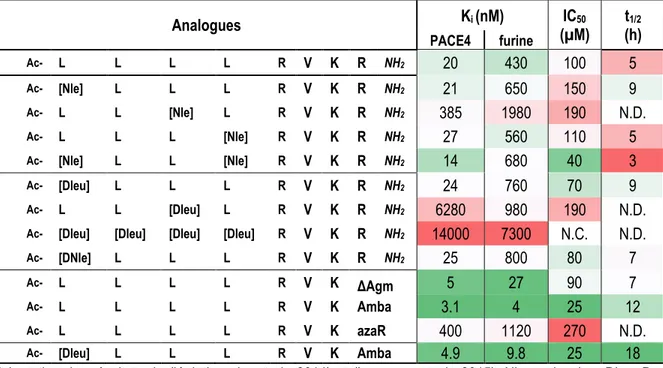
Tableau 8 – Meilleurs candidats des variants peptidomimétiques du ML de 2e génération. ... 49
Tableau 9 – Specific shRNA silencing sequences ... 58
Tableau 10 – qPCR primer sequences ... 58
Tableau 11 – Physicochemical properties of the presented peptide. ... 85
Tableau 12 – Comparison between 64Cu-NOTA-ML target-specific distribution and PACE4 reported expression pattern... 90
Tableau 13 – Primers used for PCR reactions ... 117
Tableau 14 – Primers used for the alternative splicing analysis (see Figure 47) ... 119
Tableau 15 – Assay design for pyrosequencing analyses of the PCSK6 DNA methylation level ... 123
L
ISTE DES ABRÉVIATIONS
17β‑HSD 17-β hydroxystéroide déshydrogénase [18F]-FDG [18F]-fluoro-2-deoxy-D-glucose
3’UTR 3’ untranslated region 5-aza-dC 5-aza-deoxy-cytidine
64Cu Cuivre-64 / copper-64
1-PDX 1-antitrypsin Portland
Ac- Acétyl- / acetyl-
ACN Acetonitrile
AD Androsténedione
ADAM A Disintegrin and metalloproteinase domain-containing proteins
Amba 4-amidinobenzylamide
ANCT Adjacent non-cancerous tissues
APS / PSA Antigène prostate spécifique / prostate specific antigen AR Récepteur aux androgènes / Androgen receptor
C23 Peptide [Dleu]LLLRVK-Amba
C8 Acide octanoïque
ChIP Chromatin immunoprecipitation
CHX Cycloheximide
CLU Clusterine / clusterin
CMK décanoyl-Arg-Val-Lys-Arg-chlorométhylcétone/decanoyl-Arg-Val-Lys-Arg-chloromethylketone
CRD Domaine riche en cystéines
CRPC Cancer de la prostate résistant à la castration / castrate resistant prostate cancer
CTCF CCCTC-binding factor DCM Dichloromethane DHEA(-S) Déhydroépiandrostérone(-sulfaté) DHT Dihydrotestostérone DMF N,N'-dimethylformamide DNMT DNA methyl-transferase Eβ+ Positron energy
EGF(R) Epidermal growth factor (receptor) EST Expressed sequence tag
FBS Fetal bovine serum
GDF-15 Growth differentiation factor 15
GnRH Gonadolibérine / gonadotropin-releasing hormone HGFR Hepatocyte growth factor receptor
HIF Hypoxia-inducible factor-1 HPC1 Hereditary prostate cancer 1 HR-MS High-resolution mass spectrometry HSPG heparan sulfate proteoglycans IGF-R Insulin-like growth factor (-receptor) IHC Immunohistochimie / immunohistochemistry
IP Immunoprecipitation
IR Récepteur à l’insuline / insulin receptor IRM Imagerie par résonance magnétique
ITGA6 Integrin alpha-6
Ki Constante d’inhibition
LH Hormone lutéinizante
LRP1 Low-density lipoprotein receptor-related protein 1 MIC1 Macrophage inhibitory cytokine-1
miRNA Micro-ARN / micro-RNA
ML Multi-leucine / multi-leucine
(MT)-MMP (Membrane-type) matrix metalloproteinases
MS Mass spectrometry
MTT 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide MVD Microvessels density
NNS Nombre requis à dépister / Number needed to screen NOTA 1,4,7-triazacyclononane-1,4,7-triacetic acid
NT Non-target
PACE4-altCT PACE4 avec le C-terminal alternatif / PACE4-alternative C-Terminal PACE4-FL PACE4 pleine longueur / PACE4-full length
PARP Poly-ADP ribose polymérase / poly-ADP ribose polymerase PC Proprotéine convertase / proprotein convertase
PCa Prostate cancer
PEG Poly-éthylène glycol / poly-ethylene glycol
PIN Néoplasie prostatique intraépithéliale / Prostate intraepithelial neoplasia
POMC Pro-opiomélanocortine
PPAR Peroxisome proliferator-activated receptor-
PTEN Phosphatase and tensin homolog gene
r- Recombinant
RAD51 Double-strand-break repair protein rad21 homolog
RE Réticulum endoplasmique
RFX5 Regulatory factor X5
RPMI Roswell Park Memorial Institute medium RUNX3 Runt-related transcription factor 3
ROS Espèces réactives de l’oxygène / Reactive oxygen species
RT-qPCR Transcription inverse suivie de PCR quantitatif / reverse transcription-quantitative PCR
S2 Schneider 2
SEER Surveillance, Epidemiology, and End Results SILAC Stable isotope labeling by amino acids in cell culture
shRNA Short hairpin RNA
SWATH-MS Sequential window acquisition of all theoretical mass spectrometry
TCGA The Cancer Genome Atlas
TEP / PET Positron emission tomography TGF- Transforming growth factors
TGN Réseau transgolgien / trans golgi network TIMP Tissue inhibitors of metaloproteinase
TP-PCR Three primers PCR
VEGF Vascular endothelial growth factor
XTT 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide ZNF143 Zinc Finger Protein 143
I
NTRODUCTION
Au cours du dernier siècle, l’espérance de vie humaine a augmenté de plus de trois décennies, passant d’environ 30 ans à plus de 75 ans dans les pays les plus développés (Global Burden of Disease Study, 2016; Riley, 2009). Ce changement graduel au niveau démographique s’est également accompagné d’une transition importante quant aux causes de décès, résultant en un changement majeur quant au rôle de la médecine. Les maladies infectieuses souvent fatales comme la tuberculose désormais quasiment disparues où devenues maîtrisables ont laissé place à de nouvelles causes principales de mortalité telles que les maladies cardiovasculaires et le cancer, ce dernier ayant fait un bond de près de 300 % entre 1900 et 2010 (Jones, Podolsky, & Greene, 2012). Entre 2005 et 2015, malgré une réduction d’en moyenne 10% du taux de décès standardisé pour l’âge, le nombre de décès attribuable à plusieurs types de cancer a augmenté de plus de 20%, notamment le cancer du poumon (20,1%), du colon ou du rectum (23,2%), du pancréas (30,8%) et de la prostate (31,9%)(Global Burden of Disease Study, 2016). Les besoins médicaux étant en évolution, les soins de santé et les thérapies se doivent de faire de même afin de permettre de combattre les maladies modernes dont les subtilités sont à la base de leur gestion.
Bien que documentés depuis l’Égypte Ancienne, les cas de cancers n’ont commencé à n’être véritablement étudiés qu’au 16ie siècle, où ils furent attribués à maintes causes hypothétiques allant des infections aux poisons. Les balbutiements quant aux causes du cancer ne furent émis en 1775 alors que le Dr Percivall Pott réalisait la première association entre le cancer du scrotum et le métier de ramoneurs. Après quoi, il apparut clair que l’excision des tumeurs des patients permettait une apparence de guérison lorsque les conditions d’hygiène et d’asepsies n’occasionnaient pas d’infections qui étaient souvent fatales à l’époque. Il fallut attendre le 19ie siècle pour que les premiers traitements émergent, notamment la découverte de la radioactivité par Marie et Pierre Curie. Le 20ie siècle fut par la suite marqué par le développement des premières approches pharmacologiques pour le traitement du cancer. Souvent combinés à la chirurgie, ces agents furent principalement générés avec l’objectif d’interrompre le cycle cellulaire sachant que les cellules cancéreuses proliféraient à un rythme plus rapide que la plupart des cellules normales (Dutcher et al., 2013). Bien qu’efficaces, ces médicaments ont tous démontré une forte toxicité due au manque de spécificité pour les cellules à traiter. À partir des années 1940, les connaissances médicales ont accru substantiellement et ont permis le développement de thérapies plus ciblées vers l’organe ou même la tumeur à traiter. Ces traitements furent baptisés thérapies ciblées (en anglais : targeted therapies).
Les premiers exemples de thérapies ciblées furent l’utilisation de la castration ou encore de l’iode radioactif (131I) pour les cancers de la prostate et de la thyroïde respectivement. Les découvertes biomédicales ont
ensuite pris un essor très rapide, notamment grâce à la l’élan qu’a permis la découverte de l’ADN en 1953 et de la biologie moléculaire, permettant de cerner des cibles thérapeutiques de plus en plus dirigées vers les cellules cancéreuses. Maintes thérapies firent leur apparition à partir de la fin des années 1990 (Figure 1) résultats de recherches scientifiques se traduisant en gains considérables dans le taux de survie des patients comme le montre l’évolution de la survie tous types de cancer confondus à la Figure 2 .
Figure 1 – Évènements marquants dans le développement de thérapies ciblées
Sélection de certains évènements marquants relevant des premières chimiothérapies et de la thérapie ciblée. Les années 1997 à 2016 sont élargies pour présenter le plus de découvertes, par manque d’espace, seuls quelques évènements sont montrés. [Figure originale sans droit d’auteur].
Les origines et bases du cancer
Il y a environ un milliard d’années, les premiers organismes dits métazoaires (pluricellulaires) ont fait leur apparition (Blair et al., 2009). À ce moment, les cellules eucaryotes ont « signé le pacte » leur permettant une collaboration bénéfique basée sur la spécialisation et la répartition des tâches pour subvenir au bien de l’organisme commun au fort coût de l’immortalité individuelle. Initialement sous la forme de regroupements cellulaires rudimentaires (protométazoaires), les cellules ont entamé leur coopération de façon simpliste, s’assemblant en réseaux cellulaires tout en échangeant des informations par l’entremise de médiateurs chimiques pour s’organiser. L’évolution sur plusieurs millions d’années entraîna ensuite la division plus claire des tâches via la différenciation des cellules en tissus et organes spécialisés dans l’exercice de fonctions très précises. Cette coordination fut rendue possible via l’instauration de mécanismes de régulation sophistiqués
au niveau génétique et épigénétique afin de réguler fermement les mécanismes initialement utilisés par les cellules eucaryotes protozoaires et protométazoaires pour, à l’origine, remplir l’objectif ultime guidé par la vie cellulaire : la division pour le maintien et la continuation de l’espèce.
Advenant une perte de ces mécanismes de protection instaurés par ces milliers d’années d’évolution, la survie de l’organisme entier serait mise en péril au bénéfice d’une seule cellule qui, d’une façon ou d’une autre, lèverait ces contraintes. Ainsi, le cancer tel que défini par l’Organisation mondiale de la Santé (World Health Organization, 2015) est ; « la croissance incontrôlée de cellules provenant de n’importe quelle partie du corps et dont la croissance peut envahir des tissus environnants et métastaser à des sites distants ». En déstabilisant les mécanismes de régulation génétiques, les cellules cancéreuses réactivent des proto-oncogènes en proto-oncogènes et/ou désactivent des suppresseurs de tumeurs. La résultante est qu’elles se dédifférencient du tissu duquel elles originent pour générer un nouvel assemblage cellulaire similaire à ceux primitivement générés et échappant à l’ensemble de mécanismes mis en place pour limiter la formation de telles tumeurs. Au moyen de mutations génétiques ou de bouleversements épigénétiques occasionnés par l’exposition à des carcinogènes menants à une instabilité génétique augmentée, les cellules acquièrent la capacité de former une tumeur. Pour y arriver, les changements induits doivent permettre aux cellules d’acquérir certaines caractéristiques propres aux cellules cancéreuses. Ces caractéristiques inclues 1) la perte de gènes suppresseurs de tumeurs, 2) la résistance aux mécanismes d’apoptose et d’anoïkose, 3) l’arrêt de la sénescence par la réparation des télomères, 4) l’évasion de la surveillance immunitaire par le retrait des récepteurs de surfaces, 5) l’acquisition de motricité cellulaire, 6) la sécrétion d’enzymes dégradant l’environnement cellulaire, permettant l’entrée des cellules dans la circulation sanguine ou lymphatique et possiblement l’apparition de métastases, 7) la survie en conditions hypoxiques/acides par le remplacement de la phosphorylation oxydative par la glycolyse (aussi connu sous le nom d’effet Warburg (Warburg, 1956)), 8) la production indépendante et autocrine de facteurs de croissance mitotiques et 9) l’accélération de l’instabilité génétique (Hanahan & Weinberg, 2011).
Alors que les cellules cancéreuses sont soumises à la sélection naturelle dès l’acquisition des premières mutations (plusieurs mutations étant souvent létales pour les cellules), une forte pression adaptative s’installe chez les cellules survivantes. Cette pression leur force d’acquérir rapidement plusieurs caractéristiques assurant une survie suffisamment longue pour soutenir la formation d’une tumeur à l’intérieur de laquelle chaque cellule évoluera rapidement de façon divergente. Celle-ci permet la résistance croisée aux différentes agressions (immunitaires, radiations, pharmacologiques), mais aussi de faire face aux « culs de sacs » génétiques (soit l’inactivation définitive de la progression du cycle cellulaire) que la génération hasardeuse de mutations est vouée à générer. Sur cette base, il n’est donc pas surprenant de voir que l’apparition de cancers
cliniquement significatifs soit plutôt rare; des mutations génétiques apparaissant par centaines chez des milliers de cellules de nos organismes quotidiennement, témoignant de l’efficacité des contraintes instaurées au fil de l’évolution et de l’efficacité des mécanismes de protection et de réparation.
Les cancers sont retrouvés chez l’ensemble des mammifères et mêmes chez plusieurs autres métazoaires ex. la drosophile, l’hydre, les chordés (comme le poisson-zèbre) et les amphibiens (Gateff, 1978; Gerhard et al., 2002; Martínez, 1998), bien qu’il soit difficile et rare d’y en observer. Des tumeurs ont même été documentées chez des mammifères préhistoriques (Rothschild et al., 2003).
Les bases théoriques du cancer classeraient cette maladie en tant que « maladie rare », néanmoins chez l’humain, les raisons de l’apparition d’autant de cas de cancers sont multiples et résultent de la combinaison de plusieurs facteurs. D’abord l’espèce humaine compte désormais un nombre très important de membres (> 7,5 milliards d’individus) qui ont maintenant une espérance de vie beaucoup plus longue que maints mammifères (Worldometers, 2017). Sur le plan statistique, ces deux facteurs expliquent déjà une bonne partie de l’observation si fréquente de cancer au sein de nos populations. Lorsque l’on y ajoute les nombreux facteurs environnementaux trouvés en cause pour accroitre les chances de cancer (ex. UV, polluants carcinogènes, infections virales et radiations (Société canadienne du cancer, 2017)), dont les durées d’exposition sont également plus longues vue l’espérance de vie grandissante, il devient plausible que la probabilité de développer cette maladie puisse être d’environ 50% au cours de l’espérance de vie (tout type de cancer et de sexe confondu au Canada (Société canadienne du cancer, 2017).
Les statistiques du cancer
Dans l’ensemble, depuis le début des années 1990, une tendance à l’augmentation de la survie des patients atteints de cancer est évidente avec une stabilisation suivie d’un déclin du nombre de décès annuel, et ce, malgré l’augmentation du nombre de nouveaux cas par année. En l’espace de moins de 30 ans, la survie moyenne sur 5 ans des patients atteints du cancer a augmenté de 20 % (passant de 49 à 69 % entre 1980 et 2007 respectivement). En parallèle, le nombre de publications scientifiques portant sur le cancer par année a plus que triplé sur cette période (passant de 32,199 en 1980 et à 98,473 en 2007). Ces statistiques en plus des nombreuses avancées significatives (Figure 2) témoignent de l’efficacité de l’essor des connaissances médicales de par la répercussion concrète sur la vie des patients atteints de la maladie. Malgré ces statistiques encourageantes, lorsque pris individuellement, plusieurs types de cancers sont actuellement confrontés à des problématiques particulières quant à leurs besoins spécifiques pour accroître davantage la gestion de ces maladies. À titre d’exemples, la survie relative des patients atteints de cancer du pancréas a,
encore aujourd’hui, un taux de survie sur 5 ans d’environ 7-8% comparativement au cancer de la thyroïde, où ce taux avoisine 95% (Société canadienne du cancer, 2017). Dans ce cas particulier, l’absence de traitements efficaces demeure l’obstacle majeur à franchir. Dans d’autres cas, les méthodes de dépistage et les méthodes diagnostiques sont la clé pour améliorer les chances de réponse aux traitements, comme c’est le cas pour le cancer du sein où l’implantation du programme canadien de dépistage a permis de réduire la mortalité des femmes admissibles d’environ 11% en 10 ans d’implantation (Vandal et al., 2008). La thérapie ciblée, par exemple l’Herceptin (anticorps monoclonal anti-HER2), a également permis d’accroitre la survie globale sur 10 ans des patientes de 9% pour les sous-types HER2-positifs (représentant 20% des cas). Somme toute, les avancés biomédicales tendent a corréler avec l’augmentation du taux de survie lié au cancer témoignant de l’efficacité de l’approche globale. La recherche de pointe se doit alors de définir les besoins permettant d’accroitre non seulement la survie, mais surtout la façon de prendre en charge les patients pour en assurer la qualité de vie, surtout face à une telle augmentation de l’espérance de vie. L’avènement de la thérapie ciblée impose toutefois des limitations importantes comparées aux chimiothérapies conventionnelles. En effet, les traitements étant dépendants de la présence ou de l’absence de leurs cibles au niveau des tumeurs, toute une gamme de connaissance et de technologies se doivent d’être mises en place pour catégoriser chaque tumeur pour en définir les traitements personnalisés.
Figure 2 – Statistiques relatives au cancer aux États-Unis depuis 1975
A – Superposition temporelle des courbes de nouveaux cas annuels, des décès dus au cancer et du taux de survie sur 5 ans (ordonnés de droite) pour tous les types de cancer confondus. Les données relatives aux nouveaux cas sont issues du SEER 9 (Howlader et al., 2016) tous facteurs confondus. B – Superposition temporelle du nombre de publications relative au cancer avec les taux de survie sur 5 ans (ordonnés de droite) Recherche Pubmed faite en date du 29 août 2017. [Figure originale sans droit d’auteur].
Le théranostic, soit la combinaison du diagnostic et de la thérapie, se veut le terme utilisé pour définir l’action de déterminer à l’aide d’une procédure le potentiel de réponse d’un patient à une thérapie donnée (DeNardo & DeNardo, 2012). Cette procédure peut être, par exemple, un essai sérique, une immunohistochimie sur biopsie ou encore un examen d’imagerie moléculaire. Le plus ancien essai théranostique est probablement l’usage d’iode radioactif afin de prédire la réponse au traitement d’un patient atteint d’hyperthyroïdie ou de cancer de la thyroïde. Le théranostic se veut ainsi la pierre angulaire de la médecine personnalisée au sens où il permet l’individualisation des traitements (le type de traitement, les doses, la possible toxicité, l’évaluation de la réponse), souvent avant même l’initiation du régime thérapeutique. Cet essor de la médecine personnalisée, bien que nécessaire pour améliorer encore plus la réponse aux traitements des patients, se voit contraint de combler une multitude de demandes telles que l’identification de cibles, la conception d’armes pharmacologiques et des méthodes diagnostiques associées à ces traitements spécifiques (théranostic).
Le cancer de la prostate
Statistiques du cancer de la prostate
Selon les statistiques récentes, le cancer de la prostate représente 20,7% des nouveaux cas de cancer chez les hommes, le plaçant au quatrième rang en termes d’incidence dans la population (hommes et femmes confondues) (Siegel et al., 2017; Société canadienne du cancer, 2017). Uniquement aux États-Unis, on estime à plus de 180,000 le nombre de nouveaux cas diagnostiqués par année et le nombre de décès reliés au cancer de la prostate à environ 26,000 (Siegel et al., 2016). Ainsi, selon ces statistiques, un Canadien sur huit développera un cancer de la prostate, mais seulement un sur 29 en décédera, ce qui représente tout de même 10% des décès reliés au cancer chez les hommes (Société canadienne du cancer, 2017).
Une particularité importante de ce type de cancer est la très forte prévalence des cas non diagnostiqués trouvés lors d’autopsies chez des patients décédés d’autres causes (Jahn et al., 2015). En effet, la prévalence observée est de 22 à 45% chez les hommes ayant entre 50 et 59 ans et il a même été établi qu’il existe une corrélation directe avec l’âge (Haas et al., 2008), toutefois ces cancers sont dans la vaste majorité des cas de bas grades souvent complètement asymptomatiques. Tout comme les lésions néoplasiques, les lésions précancéreuses sont également retrouvées en forte prévalence lors d’autopsies et celle-ci augmente également avec l’âge des sujets (Sánchez-Chapado , 2002). Ces statistiques sont alarmantes puisque cette forte incidence, malgré ce faible taux de mortalité relatif, entraîne une forte charge en termes d’intervention clinique auprès des patients diagnostiqués vue l’absence de modalités permettant de discriminer les cancers les plus agressifs nécessitant une intervention immédiate.
La prostate Anatomie
La prostate est une glande sexuelle accessoire la plus volumineuse chez les mâles. Elle est localisée dans le compartiment subpéritonéal, entre le plancher pelvien et la cavité péritonéale (Figure 3). Sa face postérieure repose sur la face antérieure du rectum alors que sa face antérieure repose sur le fascia du levator ani (le muscle pelvien ancestralement responsable de la motricité de la queue). Comme montré à la Figure 3, la prostate entoure complètement l’urètre et s’y connecte via le les ductes prostatiques au niveau du colliculus séminal (aussi appelé vérumontanum) qui recueille les spermatozoïdes matures avant l’éjaculation. En son centre se trouve l’utricule prostatique qui consiste en un résidu de la structure embryonnaire appelée canaux de Müller qui, chez la femme, forment l’utérus. Chez l’homme, cette structure reste indéveloppée sous l’influence de l’hormone anti-müllerienne testiculaire. Cette hormone induit également la différenciation du canal de Wolff vers les différentes structures urogénitales mâles, dont les épididymes, le vas déférents, les vésicules séminales et la zone centrale de la prostate (Figure 3). Chez la femme, en l’absence de l’hormone anti-müllerienne, qui est encodée sur le chromosome Y, cette structure entre en apoptose. Le reste de la prostate, lui, découle de la différenciation du sinus urogénital, tout comme la vessie, l’urètre et le pénis sous l’influence des androgènes. Sous l’influence des hormones sexuelles féminines, ces structures se différentieraient en une structure de transition nommée bulbe sinovaginal qui formerait ultimement le vagin. Avant la puberté, la prostate pèse environ deux grammes chez l’enfant, toutefois sous l’action de l’augmentation des niveaux d’androgènes, elle peut atteindre une moyenne de 20 g à l’âge adulte pour continuer d’augmenter avec l’âge jusqu’à des volumes de 40 à 80 g après 60 ans (Collins et al., 1993; Hayward & Cunha, 2000).
Figure 3 – Anatomie uro-génitale
Planche anatomique de l’appareil uro-génital mâle. Les zones de la prostate indiquées sont; le stroma fibromusculaire antérieur (SFA), la zone centrale (CZ), la zone de transition (TZ) et la zone périphérique (PZ) Adaptation française avec permission de (Timms & Hofkamp, 2011).
Structures et fonctions
Lors de l’éjaculation, les spermatozoïdes présents dans les canaux déférents sont propulsés au travers de l’urètre par la contraction des muscles puboccocygeus, ces contractions entraînent également la sortie des liquides présents dans les vésicules séminales et la prostate qui entraîne ainsi les spermatozoïdes. Les composantes fournies par les vésicules séminales, notamment de la séminogéline, de la fibronectine et de la lactoferrine (Lilja, 1985), vont entraîner la coagulation rapide de l’éjaculat après l’éjaculation. Au niveau fonctionnel, la prostate fournit, de par ses sécrétions, plusieurs facteurs permettant la liquéfaction ultérieure du sperme permettant la libération des spermatozoïdes. Sa fonction de glande dite accessoire est associée au fait qu’en absence de prostate, la fertilité n’est pas complètement nulle, mais diminuée. Les sécrétions prostatiques contiennent environ 1% de protéines et une quantité importante de zinc, qui sert d’antioxydant, et de citrate qui contribue à la motilité des spermatozoïdes et aux propriétés antimicrobiennes (Kelleher et al., 2011). Le zinc a notamment pour fonction de prévenir l’oxydation du citrate par l’aconitase via son effet inhibiteur sur cette enzyme (Costello et al., 2004). Il n’est donc pas surprenant de voir que l’épithélium prostatique ait pour fonction unique d’accumuler le zinc jusqu’à des concentrations jusqu’à 10 fois supérieures à l’ensemble des autres tissus via des transporteurs spécialisés. Parmi les protéines sécrétées, l’antigène prostate spécifique (aussi appelé kallikréine 3 ou APS) a pour fonction de liquéfier le coagulât par le clivage
des protéines, notamment la séminogéline (Lee et al., 1989). Ces protéines sont sécrétées par les cellules épithéliales contenues dans les acini prostatiques.
Au niveau organisationnel, la partie glandulaire de la prostate se divise en trois zones, soit la partie périphérique, de transition et centrale (Figure 3), ayant respectivement 70, 5 et 25 % des glandes sécrétrices. Au niveau histologique, ces glandes se présentent sous la forme d’acini tapissés de cellules épithéliales qui remplissent la fonction sécrétrice, la lumière des acini recueillant ainsi les sécrétions qui se rejoignent pour former des ductes de plus grandes dimensions se regroupant au niveau du colliculus séminal (vérumontanum) où les fluides prostatiques sont mélangés avec le reste de l’éjaculât. L’épithélium acinaire se compose de deux types de cellules importantes, les cellules épithéliales luminales exerçant la fonction sécrétrice et les cellules basales, accolées entre la membrane basale et les cellules luminales (Figure 4). Les cellules basales, elles, ont un rôle plutôt méconnu plutôt surtout associé au renouvellement épithélial de par la sous-population de cellules souches qui la compose (Bonkhoff, 1996; Bonkhoff et al., 1994).
Figure 4 – Histologie de la prostate
Histologie des structures prostatiques à différents grossissements mettant l’emphase sur les types de cellules présents au niveau des glandes prostatiques. Adaptation partielle avec permission de (Costello et al., 2011).
Origines et évolution du cancer de la prostate Étiologie du cancer de la prostate
Les cellules à l’origine des tumeurs de la prostate sont, dans plus de 90% des cas, les cellules de nature épithéliales, donnant ainsi naissance à des adénocarcinomes prostatiques. Les autres types de cancers de la prostate sont beaucoup plus rares ;
• Les adénocarcinomes ductaux, soient des cellules tumorales résidentes dans la lumière même des ductes prostatiques. Ces cancers représentent environ 5% des cas de cancer de la prostate, bien
qu’ils soient souvent retrouvés concomitamment dans les cas d’adénocarcinomes classiques (Martorana et al., 2016).
• Les cancers urothéliaux de la prostate, soient des tumeurs se formant dans le compartiment urétral au niveau de la prostate, ces cancers originent principalement de la vessie, mais se développent parfois dans la prostate (Liedberg et al., 2007). Ces cancers représentent environ 2,7% des cas, toutefois puisque ces tumeurs émanent souvent de la vessie, leurs taux d’incidence sont donc difficiles à calculer.
• Cancer de la prostate à cellules squameuses (représentant de 0,5 à 1% des cas) soient les tumeurs originant des cellules non glandulaires qui entourent la prostate (Mott, 1979).
• Les cancers à petites cellules (aussi appelés neuroendocriens, représentant entre 0,5 et 2% des cas), soient des tumeurs originant des cellules neuro-sécrétrices de la prostate (Beltran et al., 2011). Bien que rares, il est rapporté que des cellules cancéreuses vont initier une différenciation vers un phénotype neuroendocrinien dans virtuellement 100% des cas à la suite des traitements.
• Les sarcomes prostatiques soient les cancers originant des cellules musculaires de la prostate. Ce type de tumeur a une incidence faible (environ 1% de tous les types de cancers) qui est similaire pour l’ensemble des tissus musculaires.
Environ 95% des adénocarcinomes de la prostate vont se retrouver dans les zones périphériques et de transition de la prostate (Figure 3), le reste se retrouvant dans la zone centrale. Il est intéressant de remarquer que d’emblée la zone centrale a une source embryonnaire totalement différente de celle des deux autres zones, soient respectivement le sinus urogénital et le canal de Wolff, distinguant ainsi à la fois leurs programmes de différenciation et leurs susceptibilités relatives à la carcinogenèse (Lee et al., 2011).
Une des particularités du cancer de la prostate est la présence de lésions dites précancéreuses appelées néoplasies intraépithéliales prostatiques (mieux connues sous l’acronyme anglophone PIN; Figure 5). Bien que parfois controversée, la nature précancéreuse du PIN est bien acceptée. Les foyers de PIN se composent essentiellement de cellules épithéliales prostatiques qui ont commencé à accumuler des altérations génomiques sans toutefois être considérées comme étant cancéreuses (Qian et als., 1995). Au fil des modifications, dont les causes sont difficiles à identifier, mais semblent être une combinaison entre le vieillissement (l’âge), les facteurs génétiques héréditaires, l’exposition à des carcinogènes ou des agents infectieux et l’inflammation (De Marzo et al., 2007; Deutsch et al., 2004). Les cellules gagnent alors la capacité de proliférer plus librement. Les cellules des lésions de PIN restent toutefois confinées dans les ductes prostatiques, comme en témoigne la préservation des cellules basales dans ces glandes et l’augmentation du nombre de cellules épithéliales dans la lumière (Figure 5). Les lésions de PIN
s’accompagnent néanmoins de changements progressifs dans le stroma environnant où la densité vasculaire s’accroît, parfois accompagné d’accumulations de macrophages témoignant d’inflammation locale (Siegal et al., 1995). La présence de ces lésions précancéreuses est souvent multicentrique dans l’organe, témoignant ainsi de changements induits de façon progressive et spatialement délocalisée dans l’organe. Les lésions de PIN vont graduellement perdent des marqueurs de différenciation qui sont connus pour être également perdus dans les cas d’adénocarcinomes, témoignant de l’était précurseur de ces lésions. Au sein d’une même prostate, les foyers de PIN sont reconnus pour évoluer différemment, résultant souvent en de multiples lésions cancéreuses avec des phénotypes et des génotypes différents (Andreoiu & Cheng, 2010), dans certains cas, les cellules d’un même foyer peuvent aussi être hétérogènes.
Figure 5 – Histologie des adénocarcinomes prostate
Caractéristiques histologiques des lésions prétumorales et tumorales de la prostate selon la classification de Gleason. [Figure originale sans droit d’auteur].
Malgré cette hétérogénéité (Boyd et al., 2012), certaines altérations ont une forte prévalence au sein des adénocarcinomes de la prostate, notamment la perte du suppresseur de tumeur PTEN et la fusion entre les gènes TMPRSS2 et ERG (Bismar et al., 2011; Phin et al., 2013)(Figure 6). PTEN (phosphatase and tensin homolog gene) encode un régulateur négatif de la signalisation cellulaire promitotique passant par les voies PI3K-Akt, sa perte mono-allélique est retrouvée dans plus de 60% des adénocarcinomes de la prostate, ce qui en fait l’altération la plus commune pour le cancer de la prostate. La fusion génétique entre le gène TMPRSS2, qui encode une protéase a sérine, et celui du facteur de transcription ERG est retrouvée dans 40-80% des adénocarcinomes prostatiques (Boyd et al., 2012). Il est toutefois reconnu qu’elle n’est pas suffisante pour soutenir la prolifération cellulaire (Tomlins et al., 2008) (Figure 6).
Figure 6 – Étapes précoces de la carcinogenèse de l’épithélium prostatique
Au fil de la transformation oncogénique de l’épithélium, la perte de PTEN, soit via des mutations, la perte du locus ou encore via des modifications épigénétiques (Whang et al., 1998), mène à la perte de la régulation négative sur les voies de signalisation pro-prolifératives PI3K-Akt. Chez les lésions de PIN, la perte de PTEN est souvent hétérozygote, toutefois sa suppression complète est souvent retrouvée chez les tumeurs de hauts grades. Le gène TMPRSS2 a, quant à lui, pour particularité d’être régulé positivement par les niveaux d’androgènes via des androgen-response elements (ARE sur la figure), ainsi lorsque la fusion avec ERG, ce dernier se retrouve fortement exprimé. ERG est un facteur de transcription classé comme proto-oncogène strictement exprimé chez les métazoaires et ayant notamment pour fonctions la gestion de la sénescence et de l’entrée en différenciation (Sharrocks, 2001). Concomitamment à ces deux évènements, une multitude d’autres altérations sont connues pour arriver, mais avec des incidences plus faibles, par exemple l’amplification de l’oncogène Myc, ou des mutations du récepteur aux androgènes (AR)(Boyd et al., 2012). Figure tirée avec permission de (Squire, 2009).
Une fois les altérations génétiques suffisantes pour transformer les cellules, un des premiers signes de malignité est la perte des cellules basales au niveau des lésions (Figure 5). Les cellules cancéreuses, alors encore bien différenciées en épithélium prostatiques prolifèrent, générant ainsi une forte densité de glandes bien reconnaissables dans le tissu (Figure 5). Néanmoins, l’état de différenciation tend à se perdre au fil de l’évolution des tumeurs ainsi formées, les cellules dont la densité accroît significativement dans le tissu étant de moins en moins organisées sous la forme d’u épithélium glandulaire. Chez les tumeurs de hauts grades, l’état de différenciation est faible, voire perdu, et les cellules cancéreuses forment des amas sans structure histologique apparentée à l’épithélium prostatique (Figure 5).
Causes du cancer de la prostate
La multiclonalité du cancer de la prostate ainsi que son fort taux d’incidence dans la population sous-tend inéluctablement que ce type de cancer a des causes en mesure d’expliquer ce phénomène. Comme pour
plusieurs autres types de cancers (ex. du colon et du sein (Kinzler & Vogelstein, 1996)), les antécédents familiaux ont été explorés pour le cancer de la prostate. Certains loci ont été associés avec le cancer de la prostate héréditaire, notamment la région HPC1 (Hereditary prostate cancer 1) du gène RNASEL qui augmente la susceptibilité au cancer de la prostate, néanmoins la prévalence de cette mutation dans la population est très faible (Grönberg, 2003). Une méta-analyse faite chez 775 familles présentant un historique de cancer de la prostate héréditaire n’a démontré qu’un faible lien (Bailey-Wilson et al., 2012). D’autres loci ont été étudiés sans que de forte association est été trouvée, la plupart des mutations suggérées étant très rarement retrouvées dans la population en général (Deutsch et al., 2004). Une analyse de cohorte de paires de jumeaux suggère qu’au mieux, les facteurs héréditaires pourraient expliquer 29 à 50% de l’occurrence du cancer de la prostate (Lichtenstein et al., 2000).
Alors qu’aucune forte association génétique prédisposant au cancer de la prostate n’a été trouvée, la composante environnementale a, en contrepartie permis davantage d’associations. En effet, selon les régions du monde, le taux d’incidence du cancer de la prostate varie considérablement, aux États-Unis et au Canada les taux sont plutôt hauts (entre 64 à 137 cas par 100 000 habitants) comparativement à la Chine et le Japon (2 à 11 cas par 100 000 habitants)(Grönberg, 2003). Dans les études de migration, les Asiatiques ayant déménagé aux États-Unis voient leur taux d’incidence du cancer de la prostate augmenter bien qu’il reste tout de même inférieur à celui des Américains (Cook et al., 1999; Grönberg, 2003), ce qui suggèrent d’importantes composantes associées au style de vie et à l’environnement. En ce sens, plusieurs associations en relation avec le régime alimentaire ont été rapportées dans la littérature, notamment l’apport alimentaire en phyto-estrogènes, comme les flavones et lignanes, particulièrement retrouvés dans la diète orientale. Ces composés ont une activité inhibitrice envers la 5-réductase qui permet la conversion de la testostérone en dihydrotestostérone, ce qui pourrait expliquer leurs effets sur la progression tumorale des adénocarcinomes prostatiques (Goetzl et al., 2007). L’apport alimentaire en antioxydants comme le -tocophérol (vitamine E) ainsi certains oligoéléments comme le sélénium (Clark et al., 1998; Nomura et al., 2000) et les lycopènes provenant des diètes riches en tomates (Gann et al., 1999) ont également été montrés comme étant capables de réduire le risque de développer le cancer de la prostate. Les propriétés antioxydantes de ces molécules sont jusqu’à maintenant l’explication la plus plausible de cette protection qui préviendrait les dommages causés au génome en conditions de stress. L’inflammation s’avère être en fait au cœur des explications de la progression au fil des étapes précarcinogéniques (Nelson et al., 2004), ce qui est notamment appuyé par l’effet protecteur conféré par la prise d’anti-inflammatoire non-stéroïdien (Perron et al., 2003; Roberts et al., 2002), qui sont également en mesure de réduire la production de espèces réactives de l’oxygène (ROS) par les cellules inflammatoires. Il s’avère en fait que l’épithélium prostatique est particulièrement sensible aux ROS qui sont produits en grandes quantités lors par les processus inflammatoires (Reuter et al., 2010). Cette
susceptibilité vient de la fonction spécialisée de ces cellules qui produisent du citrate en forte quantité voué à la sécrétion. Cette production massive passe par une inhibition de l’aconitase par les hautes concentrations de zinc intracellulaire accumulées. Chez ces cellules, l’oxydation du citrate étant prévenue, le cycle de Krebs et une partie de la phosphorylation oxydative sont bloqués (Paschos et al., 2013). La résultante est une baisse considérable du métabolisme ainsi que des capacités de prolifération de ces cellules, et donc également du taux de production de ROS par la respiration oxydative, soit la principale source physiologique de ROS. Les cellules épithéliales se retrouvent donc à être exposées à des niveaux de ROS plutôt bas en conditions physiologiques. Ceci accentue donc leur susceptibilité face aux conditions de stress, et donc leur potentiel d’accumuler des dommages génomiques lors d’augmentations sporadiques ou chroniques, comme c’est le cas lors d’inflammation. Cohérent avec ce phénotype, dès les étapes précoces de la carcinogenèse, les lésions de PIN régulent déjà négativement le transporteur de zinc ZIP1 pour permettre la levée de l’inhibition sur le métabolisme cellulaire (Franklin et al., 2005).
Les infections bactériennes et virales ont également été suggérées comme étant causale d’une certaine proportion des cancers de la prostate. En effet, les patients ayant un historique de prostatites, de syphilis et de gonorrhée présentent un risque accru de développer un cancer de la prostate (Dennis et al., 2002). De plus, des analyses par PCR sur des tissus cancéreux de prostate ont montré la présence de virus Epstein-Barr (Grinstein et al., 2002) et d’herpèsvirus de type 2 (Baker et al., 1981). Ces infections pourraient, en partie, expliquer la forte prévalence de lésions d’atrophie prolifératives inflammatoires et d’infiltration de cellules immunitaires mononucléaires dans les zones périphériques aux foyers cancéreux chez les spécimens cliniques de cancer de la prostate (Platz & De Marzo, 2004). La contribution réelle des infections dans la causalité des adénocarcinomes prostatiques reste toutefois difficile à établir vus les biais de détection (possibles contaminations fécales, faible abondance) et les faibles associations dans la littérature (Wagenlehner et al., 2007).
Dans l’ensemble, bien que les causes exactes semblent hautement variables et mal définies, les causes environnementales et l’inflammation semblent être importantes pour le développement des lésions cancéreuses de la prostate (Platz & De Marzo, 2004). S’y ajoutent le style de vie, les pratiques sexuelles à risque, la diète, l’activité physique (Friedenreich & Thune, 2001) et le tabagisme (Honda et al., 1988) qui semblent également jouer un rôle, bien que les résultats soient souvent inconsistants avec des corrélations souvent faibles.
Gestion clinique du cancer de la prostate Dépistage
En clinique, les outils de dépistage pour le cancer de la prostate sont principalement limités au toucher rectal et au dosage sérique de l’APS. Le toucher rectal, lorsque pratiqué par les médecins, permet de détecter des anomalies par palpation au travers de la paroi du rectum telles que des nodules ou des conditions pathologiques comme la prostatite. Cette méthode est toutefois fortement basée sur l’interprétation faite par le médecin et les cancers détectés par touchers rectaux sont souvent déjà avancés (Epstein, 1994). L’APS est une enzyme de la famille des kallikréine exprimée et sécrétée par les cellules épithéliales prostatiques dont la fonction est de cliver les protéines responsables de la coagulation du sperme telles que la galectine-3 ou la semenogéline-1 (Robert et al., 1997; Saraswati et al., 2010). Bien qu’elle soit habituellement sécrétée vers la face apicale des cellules épithéliales, et donc vers la lumière des ductes prostatiques, une infime proportion de l’APS se retrouve dans le sang où elle est principalement liée à l’1-antichymotrypsine qui l’inactive enzymatiquement (Lilja et al., 1992). Les niveaux plasmatiques sont ainsi mesurables par méthode ELISA et, selon les tranches d’âges, des seuils limites ont été établis pour permettre le dépistage du cancer de la prostate (Balk et al., 2003). En effet, avec l’augmentation de la densité de cellules épithéliales lors de la formation de tumeurs ou lors d’hyperplasies, les taux d’APS circulants augmentent également, ce qui permet de dépister la présence probable de cancer chez les patients ayant des concentrations supérieures à 4 ng/mL. Néanmoins, le seuil d’APS à 4 ng/mL n’a, à lui seul, qu’une sensibilité d’environ 20% (Thompson et al., 2005) par contre, plus les niveaux d’APS sont élevés, plus le risque de cancer de la prostate est élevé (Schröder et al., 1998). Lorsqu’utilisée en combinaison avec le toucher rectal, la sensibilité de détection augmente avec la concentration de l’APS (Schröder et al., 1998). Par exemple, chez les patients avec de faibles niveaux d’APS (0 à 9,9 ng/mL) la capacité de détection de l’APS oscille entre 0 et 20% alors que celle du toucher rectal oscille entre 0 et 10,5%, mais elles ont respectivement des sensibilités de 52,9 et 30% chez les cas avec des niveaux supérieurs à 10 ng/mL. Une grande variété de conditions bénignes peuvent être la cause d’augmentations des niveaux d’APS circulant telles que la prostatite, l’hyperplasie bénigne, les infections urinaires, l’âge, l’activité sexuelle et même l’activité physique (Nadler et al., 1995), ce qui peut interférer considérablement avec les capacités de dépistage de ce biomarqueur, principalement dans les cas avec de faibles concentrations plasmatiques.
L’utilisation de l’APS pour le dépistage systématique est une pratique courante, notamment chez les patients de quarante ans et plus dans l’objectif de prévenir le développement de cancers avancés. Depuis le début des années 90, le dépistage systématique via l’APS sanguin a permis d’augmenter de façon importante le dépistage du cancer de la prostate. Cette pratique avait pour objectif de diminuer la mortalité associée à ce
cancer et d’accroître la qualité de vie des patients atteints via des traitements précoces. Plus de 20 ans plus tard, le constat des études est toutefois décevant et à mener à maintes critiques après la démonstration que le dépistage ne permettait pas de réduire la mortalité reliée au cancer de la prostate (Andriole et al., 2009; Schröder et al., 2014). Le dépistage systématique serait en fait la cause de morbidités accrues chez les patients lorsque des cancers indolents sont dépistés, diagnostiqués et traités malgré la faible mortalité qui leur serait possiblement associée (Schröder et al., 2009). En fait, le nombre de patients à dépister pour sauver une vie serait d’environ 1300 (number needed to screen; NNS, après 9 ans de suivi) et d’environ 503 (après 12 ans de suivi) alors que le nombre de patients à traiter (number needed to treat), lui, serait d’environ 43 et de 18 (après 9 et 12 ans respectivement) (Loeb et al., 2011; Schröder et al., 2009). Si on compare ces chiffres à la mammographie pour le dépistage du cancer du sein, où le nombre de femmes à inviter à un examen mammographique pour sauver une vie est de 1339 (NNS : 322 à 7455; chez les femmes âgées de 50 à 59 ans) et de 377 (NNS : 230 à 1050; chez les femmes agées de 60-69 ans) (Rembold, 1998; U.S. Preventive Services Task Force, n.d.), le même constat peut être établi (Welch et al. 2016). En soi, l’usage ou encore l’établissement de tout outil de dépistage systématique ou de modalités permettant de discerner fortuitement des lésions tumorales (ex. l’imagerie médicale) vient avec le risque de surdiagnostiquer des lésions inopinément (Welch & Black, 2010). Le cas du cancer de la prostate n’est pas isolé puisque des situations semblables s’appliquent aussi bien au cancer du sein, du rein, de la thyroïde et même aux mélanomes (Figure 7), où des augmentations importantes du taux de diagnostic ont été observées sans qu’il y ait pour autant de variations proportionnelles dans les nombres de décès associés à ces cancers, soit le critère de base du surdiagnostic. Dans ces cas, l’option pour remédier au surtraitement inhérent est de proposer une surveillance active plus serrée des cas/lésions suspicieuses, mais à risques apparents faibles et de proposer la prostatectomie aux patients ayant le plus de chance d’en bénéficier (Dall’Era et al., 2012). Bien sûr, la surveillance active vient avec son lot de désagréments pour le patient, dont le risque d’avoir sous-estimé la tumeur ainsi que le stress et l’anxiété de se savoir atteint du cancer et de devoir avoir des suivis médicaux serrés.
Figure 7 – Taux de diagnostic et de mortalité du SEER “Surveillance, Epidemiology, and End Results ” de 1975 to 2005 aux États-Unis
Courbes montrant les taux de nouveaux diagnostics de cancers pour le cancer de la thyroïde (A), le mélanome (B), le cancer du rein (C), le cancer de la prostate (D) et le cancer du sein (E) au cours des années 1975 à 2005. L’absence de parallélisme entre les courbes suggère la présence de surdiagnostic. [Figure tirée avec permission de (Welch & Black, 2010)]
Alors que l’utilisation de l’APS pour le dépistage reste débatable malgré son manque de spécificité et l’absence de capacité de discrimination pour les cancers non indolents, ce biomarqueur reste d’une utilité indéniable pour le suivi du traitement et des récidives (Wallace et al., 2014). En effet, normalement, après l’ablation chirurgicale de la prostate pour éradiquer une tumeur, les taux sériques d’APS passent sous la limite de détection technique (<0,01 ng/mL). Dans les cas où les tumeurs ont dépassé les marges chirurgicales et/ou ont déjà envahi des tissus avoisinants ou distants, les niveaux d’APS resteront détectables (échec biochimique) et permettront de rapidement intervenir par des traitements appropriés de radiothérapies ou de chimiothérapies. De plus, advenant la récidive de lésions primaires ou métastatiques restantes, mais encore