Thèse de doctorat de l’Université Sorbonne Paris Cité
Préparée à l’Université Paris Diderot
Ecole doctorale
Bio Sorbonne Paris Cité – ED 562
Laboratoire de Biologie Fonctionnelle et Adaptative
Equipe RéGLYS (Régulation centrale de la glycémie)
Le métabolisme des céramides hypothalamiques
induit une résistance à l’insuline centrale et une
dérégulation de l’homéostasie glucidique durant
l’installation de l’obésité
Par Mélanie CAMPANA
Thèse de doctorat de Biologie cellulaire et physiologie
Dirigée par le Pr. Hervé LE STUNFF
Présentée et soutenue publiquement à l’université Paris Diderot le 29 Septembre 2017
- Président du jury : ROUILLER-FABRE, Virginie / Pr / Université Paris Diderot - Rapporteurs : CORMONT, Mireille / DR / Inserm, Nice LEVADE, Thierry / PU-PH/ Inserm, Toulouse - Examinateurs : FIORAMONTI, Xavier / CR / INRA, Bordeaux RUTTER, Guy / Pr / Imperial college of London - Directeur de thèse : LE STUNFF, Hervé / Pr / Université Paris Diderot
Thèse de doctorat de l’Université Sorbonne Paris Cité
Préparée à l’Université Paris Diderot
Ecole doctorale
Bio Sorbonne Paris Cité – ED 562
Laboratoire de Biologie Fonctionnelle et Adaptative
Equipe RéGLYS (Régulation centrale de la glycémie)
Le métabolisme des céramides hypothalamiques
induit une résistance à l’insuline centrale et une
dérégulation de l’homéostasie glucidique durant
l’installation de l’obésité
Par Mélanie CAMPANA
Thèse de doctorat de Biologie cellulaire et physiologie
Dirigée par le Pr. Hervé LE STUNFF
Présentée et soutenue publiquement à l’université Paris Diderot le 29 Septembre 2017
- Président du jury : ROUILLER-FABRE, Virginie / Pr / Université Paris Diderot - Rapporteurs : CORMONT, Mireille / DR / Inserm, Nice LEVADE, Thierry / PU-PH/ Inserm, Toulouse - Examinateurs : FIORAMONTI, Xavier / CR / INRA, Bordeaux RUTTER, Guy / Pr / Imperial college of London - Directeur de thèse : LE STUNFF, Hervé / Pr / Université Paris DiderotRésumé :
Des études montrent que l’accumulation de lipides dans l’hypothalamus serait responsable de l’installation d’une lipotoxicité centrale : phénomène qui pourrait jouer un rôle dans l’apparition d’une insulino-résistance périphérique et du diabète de type II en dérégulant le contrôle nerveux de l’homéostasie glucidique. L'accumulation des céramides est connue pour être impliquée dans le développement d’une lipotoxicité des tissus périphériques. L’objectif de cette étude est de déterminer le rôle du métabolisme des céramides hypothalamiques dans l’installation d’une insulino-résistance centrale et d'étudier les mécanismes impliqués mais également de déterminer le rôle du métabolisme de ces céramides hypothalamiques dans la dérégulation de l’homéostasie glucidique induite par l’obésité.
L’installation d'une insulino-résistance centrale est étudiée à l'aide d'approches in
vitro, en utilisant des cellules hypothalamiques de souris GT1-7 traitées avec du palmitate
pendant 24h. Pour une approche in vivo, des rats Zucker obèses ont été perfusés avec la myriocine (inhibiteur de la synthèse de novo des céramides) en ICV pendant 21 jours, des tests de sensibilité à l'insuline et de tolérance au glucose sont réalisés. A la fin du traitement, les rats Zucker reçoivent une injection ICV d'insuline, puis la sensibilité à l’insuline ainsi que les taux de céramides sont quantifiés dans l’hypothalamus, les îlots de Langerhans sont isolés pour des tests de sécrétion d'insuline.
Dans les cellules GT1-7, le palmitate induit une insulino-résistance qui s’accompagne d’une accumulation de céramides. En présence de myriocine, les céramides ne sont plus accumulés et l’insulino-résistance induite par le palmitate est contre-carrée. En utilisant un inhibiteur de la PKC ζ et un adénovirus codant pour un dominant-négatif de la PKC ζ, nous avons montré que le palmitate n'est plus capable d'induire une insulino-résistance et ce malgré la présence d'une accumulation de céramides. Chez le rat Zucker obèse, l’accumulation de céramides hypothalamiques est contre-carrée par la myriocine. Ce résultat est associé avec une amélioration de la sensibilité à l’insuline dans l’hypothalamus. De façon, intéressante, ces animaux améliorent leur tolérance au glucose, amélioration liée à une augmentation du tonus parasympathique conduisant à une augmentation de la sécrétion d’insuline. Les îlots de Langerhans isolés à partir de ces rats présentent une capacité sécrétoire augmentée lors du traitement avec la myriocine.
Notre étude révèle que la lipotoxicité hypothalamique est associée à une accumulation de céramides dans cette structure, responsable de l’installation d’une insulino-résistance. Ces résultats mettent également en évidence le rôle clé du métabolisme des céramides au niveau de l’hypothalamus dans la dérégulation du contrôle nerveux de l’homéostasie glucidique induit par l’obésité.
Les acides gras polyinsaturés ω3, tel que l’acide eicosapentaénoïque (EPA) et l’acide docosahexaénoïque (DHA), à l’inverse des acides gras saturés, sont connus pour avoir des effets bénéfiques sur la sensibilité à l’insuline à la périphérie. Chez des souris sous régime enrichi en lipides, nous avons montré qu’une supplémentation en DHA permet de retarder l’installation du DT2. Dans les cellules GT1-7, le DHA, dont l’enzyme responsable de sa production est l’élongase 2 (Elovl2), et l’EPA sont capables de contre-carrer les effets délétères induits par le palmitate en modulant le métabolisme des céramides.
Mots clefs : Céramides, hypothalamus, insulino-résistance, obésité, lipotoxicité, cellules β-pancréatiques, nerf vague, sécrétion d’insuline, acides gras polyinsaturés ω3, élongase 2.
Abstract :
Studies show that hypothalamic lipid accumulation in the hypothalamus is responsible for the development of central lipotoxicity, a phenomenon that could play a role in the installation of peripheral insulin resistance and type II diabetes by deregulating the nervous control of glucose homeostasis. It is known that the accumulation of ceramides is involved in the development of lipotoxicity of peripheral tissues. The objective of this study is to determine the role of the hypothalamic ceramide metabolism on the installation of a central insulin resistance and to study the mechanisms involved on this phenomenon. We also determined the role of hypothalamic ceramide metabolism in the deregulation of obesity-induced glucose homeostasis.
The installation of a central insulin resistance is studied using in vitro approaches using hypothalamic GT1-7 mouse cells treated with palmitate for 24 hours. An in vivo approach use Obese Zucker rats were perfused with myriocin (an inhibitor of de novo synthesis of ceramides) in ICV for 21 days. Insulin sensitivity and glucose tolerance tests are performed. At the end of treatment, they receive an ICV injection of insulin, insulin sensitivity and ceramide levels are quantified in the hypothalamus. Islets of Langerhans are isolated for insulin secretion tests.
In GT1-7 cell line, palmitate induces insulin resistance which is accompanied by an accumulation of ceramides. In the presence of myriocin, ceramides are no longer accumulated and the insulin resistance induced by palmitate is counteract. Using an inhibitor of PKC ζ and an adenovirus encoding a dominant-negative of PKC ζ, we have shown that palmitate is no longer able to induce insulin resistance despite the presence of an accumulation of ceramides. In the obese Zucker rat, we have demonstrated an accumulation of hypothalamic ceramides which is counteract by myriocin. This is associated with an improvement in insulin sensitivity in the hypothalamus. Interestingly, these animals improve their glucose tolerance which is associated with an increase in parasympathetic tone leading to an increase in insulin secretion. Islets of Langerhans isolated from these rats have increased secretory capacity when treated with myriocin.
Our study reveals that hypothalamic lipotoxicity is associated with an accumulation of ceramides in this structure, responsible for the installation of insulin resistance. These results also highlight the key role of ceramide metabolism at the hypothalamus level in the deregulation of nervous control of obesity-induced carbohydrate homeostasis.
Ω3 polyunsaturated fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are known to have beneficial effects on peripheral insulin sensitivity to saturated fatty acids. In mice fed with high fat diet, we shown that DHA supplementation can delay the installation of DT2. In GT1-7 cell line, DHA, the enzyme responsible for its production, elongase 2 (Elovl2), and EPA are able to counteract deleterious effects induced by palmitate by modulating the ceramide metabolism.
Keywords : Ceramides, hypothalamus, insulin-resistance, obesity, lipotoxicity, pancreatic β-cell, vagus nerve, insulin secretion, ω3 polyunsaturated fatty acids, elongase 2.
Ce travail est dédié
à mes chers grands-parents, Angèle et Constantin
ainsi qu’à ma mère, Mathilde.
Remerciements
Je tiens tout d’abord à remercier tous les membres du jury qui ont accepté de juger ce travail. Je remercie le Professeur Virginie ROUILLIER-FABRE qui m’a fait honneur en acceptant de présider ce jury de thèse. Je remercie également le Dr Mireille CORMONT ainsi que le Professeur Thierry LEVADE pour l’intérêt qu’ils ont manifesté pour ces travaux en acceptant d’être rapporteurs et pour l’attention qu’ils porteront à la lecture de ce manuscrit. Je remercie enfin, le Docteur Xavier FIORAMONTI et le Professeur Guy
RUTTER de me faire bénéficier de leurs grandes compétences en acceptant d’examiner ce
travail scientifique.
Je tiens à remercier sincèrement le Professeur Christophe MAGNAN pour avoir eu la gentillesse de m’accueillir dans son laboratoire de recherche et pour son optimisme en toutes circonstances.
Je remercie et adresse ma profonde reconnaissance au Professeur Hervé LE STUNFF pour tout le savoir qu’il m’a transmis, l’expérience professionnelle et enrichissante qu’il m’a fait vivre durant ces trois ans de thèse. Merci pour toute la confiance que tu m’as accordée et pour avoir cru en moi. Grâce à toi j’ai pu acquérir un large panel de compétences essentielles à ma formation professionnelle. Mon esprit critique ainsi que ma démarche scientifique ne sont que l’aboutissement de tes conseils.
C’est avec une sincère gratitude que je remercie mon Poussinou, Claude, pour son grand investissement dans ce projet, pour sa patience, sa gentillesse, sa personnalité inimitable, sa bienveillance et sa bonne humeur en toutes circonstances. Et surtout Claude, merci pour ton écoute ! Tu as été comme une seconde maman pour moi au laboratoire. Rassure-toi, tu seras bientôt libérée de tes fonctions ! Notre complicité de sudistes exilées me manquera !
Ovviamente, questo lavoro non avrebbe mai funzionato senza il tuo aiuto, per cui tutti i miei profondi ringraziamenti sono per te, Lara. Ti ringrazio tanto per tutto quello che mi hai fatto imparare, per il tuo sostengo ogni giorno, per la tua presenza, per le tue battute e cazzate e anche per i tuoi capelli sparati sulla testa :) Grazie di avere resa questa tesi più bella e colorata di buon umore.
Un très grand merci au bureau 504A, Jess, Kelly et Axelle, mes compatriotes de pauses rédaction de thèse/manips et toujours présentes pour m’écouter rouspéter, pour partager une histoire ou un fou rire. Merci pour vos tous conseils de la vie de tous les jours.
Je tiens à remercier tous les membres de mon équipe ainsi que les membres de l’équipe C2OFFEE pour leurs conseils, leur gentillesse au quotidien et tous nos déjeuners partagés
sur la passerelle.
Un immense merci à mes parents, Alain et Mathilde, à ma sœur, Stéphanie, ainsi qu’à mes grands-parents, Angèle et Constantin qui, même s’ils n’ont pas toujours compris mon travail de recherche, ont toujours su me soutenir à leur manière. Papi et mamie, vous êtes un modèle de force et de courage pour moi. J’espère que ce travail vous rendra tous fiers de moi et que je serai à la hauteur de vos attentes.
A i mei amichi di à Corsica, Ghjulia, Sandrine, Sophie, Isabelle, Seb, Nicolas, Petru, Max
et Emilie. Per falla in corte parulle, vi ringrazziu di u vostru sustegnu e di tutti i nostri
mumenti di alegria inseme. Seti à mo famiglia d’aduzzione e vi scurderaghju mai. Demu ci l’avvene !
Merci à ma petite cousine, Elisa, qui m’a toujours soutenue et avec qui j’ai passé des moments mémorables et ce depuis notre plus tendre enfance.
Je remercie également ma chère marraine, Isabelle, qui malgré tous les problèmes que l’on peut rencontrer dans la vie, a toujours su être présente et disponible pour me prêter une oreille attentive et rassurante.
Merci également à tous mes amis parisiens et à ceux que je n’ai pas cité mais qui ont pris part à cette aventure de près comme de loin !
Table des matières
Remerciements _____________________________________________________ 9
Table des illustrations ______________________________________________ 17
I INTRODUCTION BIBLIOGRAPHIQUE _______________________________20
1
Diabètes, obésité et conséquences ________________________________ 22
1.1 Le diabète de type I _______________________________________________ 22 1.2 Le diabète de type II _______________________________________________ 22 1.3 Le syndrome métabolique __________________________________________ 23 1.4 L’obésité ________________________________________________________ 23 1.4.1 Epidémiologie : ________________________________________________________ 23 1.4.2 Définition : ____________________________________________________________ 24 1.4.3 Causes : ______________________________________________________________ 24 1.4.4 Conséquences : _______________________________________________________ 25 1.5 La Lipotoxicité ___________________________________________________ 28
2
L’insuline _____________________________________________________ 31
2.1 L’insuline : Généralités, structure et biosynthèse ______________________ 31
2.1.1 Généralités ____________________________________________________________ 31 2.1.2 Structure ______________________________________________________________ 31 2.1.3 Biosynthèse ___________________________________________________________ 31 2.1.4 Rôles physiologiques de l’insuline dans les tissus périphériques ______________ 32 2.1.5 Rôles physiologiques de l’insuline au niveau central _________________________ 32
2.2 Voie de signalisation de l’insuline ___________________________________ 33
2.2.1Le récepteur de l’insuline ________________________________________________ 35
2.2.2 Les substrats du récepteur de l’insuline (IRS) _______________________________ 35 2.2.3 La Phosphoiniositide-3-kinase ____________________________________________ 37 2.2.4 La Phosphoinositide Dependent Kinase 1 __________________________________ 37 2.2.5 La protéine kinase B (Akt) ________________________________________________ 38
Isoformes d’Akt ____________________________________________________ 38
2.2.1.1
Structure et mécanismes d’action d’Akt _______________________________ 38
2.2.1.2
Voies métaboliques régulées par Akt __________________________________ 40
2.2.1.3
2.2.6 Les protéines kinases C atypiques ________________________________________ 42 2.2.7 Régulations négatives de la voie de signalisation de l’insuline en conditions
physiologiques _____________________________________________________________ 44 2.2.7.1 L’internalisation du complexe hormone/récepteur _______________________ 44 2.2.7.2 Régulation négative du récepteur de l’insuline __________________________ 44 2.2.7.3 Régulation négative des IRS _________________________________________ 45 2.2.7.4 Régulation négative de la PI3K _______________________________________ 45 2.2.7.5 Régulation négative d’Akt ___________________________________________ 46
2.3 Résistance à l’insuline, obésité et diabète de type II ____________________ 46
2.3.1 Généralités ____________________________________________________________ 46 2.3.2 Modèles d’étude ________________________________________________________ 48 2.3.3 Régulations négatives de la voie de signalisation de l’insuline en conditions
pathologiques ______________________________________________________________ 48 2.3.3.1 Régulation négative de la signalisation insulinique par les acides gras _____ 48
2.3.3.2 Régulation négative de la signalisation insulinique par le stress du RE ___ 4949 2.3.3.3 Régulation négative de la signalisation insulinique par les céramides _______ 51 2.3.4 Effet des acides gras polyinsaturés ω3 sur la signalisation insulinique _________ 52
3
Les céramides _________________________________________________ 53
3.1 Structure et topologie des sphingolipides _____________________________ 53
3.2 Le métabolisme sphingolipidique ____________________________________ 54
3.3 Sphingolipides et signalisation cellulaire _____________________________ 57
3.4 Les enzymes impliquées dans la synthèse de novo des céramides ________ 57
3.4.1 La Sérine Palmitoyl Transférase (SPT) : ____________________________________ 57 3.4.2 La 3-cétosphinganine réductase : _________________________________________ 58 3.4.3 Les céramides synthases (CerS) : ________________________________________ 58 3.4.4 La dihydro-céramide désaturase __________________________________________ 61
3.5 Interaction entre céramides et protéines ______________________________ 62
3.6 Rôles physiologiques et physiopathologiques des céramides ____________ 63
3.6.1 Rôle des céramides dans le cancer _______________________________________ 63 3.6.2 Rôle des céramides dans l’inflammation ___________________________________ 65 3.6.3 Rôle des céramides dans l’obésité et le DT2 ________________________________ 66
3.6.4 Mécanismes impliqués dans l’inhibition de la voie PI3K/AKT par les céramides __ 67
Action des céramides au niveau des IRS _______________________________ 67 3.6.4.1
Action des céramides au niveau de la PI3K _____________________________ 68 3.6.4.2
Action des céramides au niveau d’Akt _______________________________ 6969
3.6.4.3
3.6.5 Divergence des mécanismes impliqués dans l’IR par les céramides dans les
différents organes ___________________________________________________________ 70 3.6.4.1 Au niveau du foie __________________________________________________ 70 3.6.4.2 Au niveau du tissu adipeux __________________________________________ 71 3.6.4.3 Au niveau du muscle squelettique ____________________________________ 72 3.6.6 Rôle des céramides dans le système nerveux central/cerveau _________________ 74
4
Régulation centrale de l’homéostasie énergétique ___________________ 76
4.1 Généralités sur l’homéostasie énergétique ____________________________ 76
4.2 L’hypothalamus __________________________________________________ 76
4.2.1 Anatomie et généralités _________________________________________________ 76 4.2.2 Populations neuronales impliquées dans la prise alimentaire __________________ 78
4.2.3 Rôle de l’insuline dans la régulation de la prise alimentaire __________________ 7979
4.2.4Intégration des signaux lipidiques par le SNC _______________________________ 81
4.2.4.1 Transport des acides gras libres dans le cerveau et dans les neurones _____ 81
Le « lipid sensing » _________________________________________________ 81
4.2.4.2
4.2.5Régulations nerveuses impliquées dans la sécrétion d’insuline ________________ 85
4.3 La lipotoxicité centrale _____________________________________________ 87
4.3.1Lipotoxicité et insulino-résistance hypothalamiques _________________________ 88
Lipotoxicité hypothalamique et inflammation ___________________________ 88
4.3.1.1
Lipotoxicité hypothalamique et stress du RE ___________________________ 89
4.3.1.2
Lipotoxicité hypothalamique et sphingolipides _________________________ 90
4.3.1.3
4.3.2Du métabolisme lipidique à l’homéostasie énergétique _______________________ 91
Lipotoxicité centrale et effets sur le foie _______________________________ 92
4.3.2.1
Lipotoxicité centrale et effets sur le tissu adipeux _______________________ 92
4.3.2.2
Lipotoxicité centrale et effets sur le pancréas ___________________________ 93
4.3.2.3
II OBJECTIFS DE LA THESE _______________________________________97
III ARTICLE SCIENTIFIQUE _______________________________________101
Résumé de l'article ________________________________________________135
IV RESULTATS EXPERIMENTAUX SUPPLEMENTAIRES_______________139
V DISCUSSION / CONCLUSIONS / PERSPECTIVES ___________________153
1
Lipotoxicité centrale et mécanismes impliqués dans l’insulino-résistance
hypothalamique __________________________________________________ 153
2
Lipotoxicité centrale et homéostasie énergétique ___________________ 168
3
Effets des acides gras polyinsaturés ω3 sur l’insulino-résistance _____ 175
4
Conclusion générale ___________________________________________ 176
VI MATERIELS ET METHODES ____________________________________179
VII REFERENCES BIBLIOGRAPHIQUES _____________________________193
VIII ANNEXES ___________________________________________________229
Liste des principales abréviations :
aCDase : CéramidasesAG : Acides gras AGL : Acides gras libres AGPI : Acide gras polyinsaturé AgRP : Agouti-related peptide ALA : α-linolénique
ARC : Noyau Arqué AS160 : Akt substrat 160
aSMase : Sphingomyélinase Acide BHE : Barrière Hémato-Encéphalique BMI : Body mass index
C1P : Céramide-1-phosphate
CART : Cocain and amphetamine regulated transcript CerS : Céramides synthases
CERT : Protéine de transport des céramides CRH : Corticotropin Releasing Hormone CRP : Protéine C réactive
CSF : Liquide cérébro-spinal DAG : Diacyl-glycérol
Degs 1 et 2 : Dihydro-céramides désaturase 1 et 2 DH-sph : Dihydro-sphingosine
DHA : Acide Docosahexaénoïque DMN : Noyau dorso-médian DT2 : Diabète de type II Elovl2 : Elongase 2
EPA : Acide eicosapentaénoïque FOX01 : Forkhead Box 1
FSV : Fraction stroma vasculaire GCS : Glucosylcéramide synthase GlcCer : Glucosylcéramides
GLP-1 : Glucagon like peptide-1 GLUT : Transporteur de glucose Grb : Growth factor receptor-bound
Grb2 : Growth Factor Receptor-Bound Protein 2 GSIS : Glucose-stimulated insulin secretion GSK3 : Glycogen synthase kinase
HFD : High Fat Diet
ICV : intra-cérébro-ventriculaire Il : Interleukine
IMC : Indice de masse corporelle IR : Insulino-résistance
IRE1 : Inositol-requiring enzyme 1 IRS-1 : Insulin Receptor Substrate 1
IRS-2 : Insulin Receptor Substrate 2 JNK : protéine Janus Kinase JNK KO : Knockout
LZR : Rat Zucker Mince
MAPK : Mitogen-activated protein kinases MCP-1 : Monocyte chemotactic protein-1 MLK-3 : mixed lineage kinase-3
mTOR : mammalian Target Of Rapamycin NASH : Stéatose hépatique non alcoolique NLR : Nod Like Receptor
NPY : Neuropeptide Y
OMS : Organisation mondiale de la santé OZR : Rat Zucker Obèse
PDK1 : Phosphoinositide-Dependent Kinase 1 PI : Phosphatidylinositol
PI3K : Phopshoinositide-3-kinase
PIP2 : Phosphatidylinositol-4,5-biphosphate PIP3 : Phosphatidylinositol-3,4,5-triphosphate PH : Pleckstrin homology domain
PKC : Protéine Kinase C
PKR : double-strand RNA-dependent protein kinase PHLPP : PH-domain leucine rich repeat protein phosphatase PP : Polypeptide pancréatique
POMC : pro-opiomélanocortine PP2A : Protéine Phosphatase 2A
PTEN : Phosphatase and Tensin homology PTP1B : Protéine tyrosine phosphatase-1B PVN : Noyau para-ventriculaire
RE : Réticulum Endoplasmique
RER : Réticulum Endoplasmique Rugueux RI : Récepteur de l’insuline
S1P : Sphingosine-1-phosphate S6K1 : S6Kinase 1
SAPs : Protéines Activatrices des Sphingolipides Ser-473 : Sérine 473
Shc : Src-homology and collagen homology SHIP-2 : SH2-containing 5’inositol Phosphatase SM : Sphingomyéline
SMase : Sphingomyélinase
SNA : Système Nerveux Autonome SNC : Système Nerveux Central
SNP : Système Nerveux Parasympathique SNS : Système nerveux sympathique SOCS : Suppressor Of Cytokines Signaling SphK1/2 : Sphingosines kinases 1 et 2 SPT : Sérine Palmitoyl Transférase
SPTLC 1/2/3 : Sérine Palmitoyl Transférase Longue Chaîne 1/2/3 TA : Tissu adipeux
TG : Triglycérides Thr-308 : Thréonine 308 TLR4 : Toll Like Receptor 4 TNF-α : Tumor Necrosis Factor α TLR : Toll Like Receptor
TRH : Thyrotropin Releasing Hormone UPR : Unfolded protein response VMN : Noyau ventro-médian WAT : Tissu adipeux blanc WT : Wild type
Table des illustrations
Figure 1 : Conséquences de l’hyperlipidémie ___________________________________ 26 Figure 2 : Effets des acides gras libres sur l’entrée de glucose dans les cellules musculaires.
_______________________________________________________________________ 29
Figure 3 : Voies de signalisation impliquées en réponse à l'insuline. __________________ 34 Figure 4 : Phosphorylation et structure de la protéine Akt. __________________________ 40 Figure 5 : Schéma récapitulatif des différents rôles de la protéine Akt. ________________ 42 Figure 6 : Structures des différents isoformes des PKC. ___________________________ 43 Figure 7 : Voies impliquées dans le métabolisme des sphingolipides. _________________ 56 Figure 8 : Schéma des différences d'affinité de substrats des CerS. __________________ 59 Figure 9 : Mécanisme d’inhibition de la voie de signalisation de l’insuline par les céramides dans différents types cellulaires. ______________________________________________ 73 Figure 10 : Représentation schématique des différents noyaux hypothalamiques. _______ 77 Figure 11 : Populations neuronales impliquées dans le contrôle de l’homéostasie
énergétique dans le noyau arqué. ____________________________________________ 79 Figure 12 : Implication des voies de signalisation de la leptine et de l’insuline sur la prise alimentaire dans l’hypothalamus. _____________________________________________ 81 Figure 13 : Innervation par le système nerveux autonome. _________________________ 86 Figure 14 : Voie de signalisation impliquée dans l’induction de la lipotoxicité hypothalamique par les acides gras saturés. _________________________________________________ 91 Figure 15 : Schéma récapitulatif des résultats obtenus in vivo chez les rats Zucker obèses traités ou non avec la myriocine. ____________________________________________ 135 Figure 16 Schéma récapitulatif des résultats obtenus dans les cellules GT1-7. ________ 136 Figure 17 : Effet de la supplémentation en DHA sur le poids corporel et la masse corporelle des souris. ______________________________________________________________ 140 Figure 18 : Effet de la supplémentation en DHA sur la sensibilité à l’insuline. __________ 141 Figure 19 : Effet de la supplémentation en DHA sur l'homéostasie glucidique. _________ 142 Figure 20 : Sécrétion d’insuline par les îlots de Langerhans des souris C57Bl/6J en fonction des différents régimes. ____________________________________________________ 143 Figure 21 : Quantité de céramides hypothalamiques induite par un régime HFD supplémenté avec 5% de DHA. ________________________________________________________ 144 Figure 22 : Effet de la supplémentation en DHA sur la sensibilité à l'insuline hypothalamique en réponse au régime HFD. ________________________________________________ 144 Figure 23 : Effet du DHA et de l’EPA sur la signalisation insulinique en réponse au palmitate dans les cellules GT1-7. ___________________________________________________ 145
Figure 24 : Effet du DHA et de l’EPA sur la quantité de céramides dans les cellules GT1-7 en réponse au palmitate. _____________________________________________________ 146 Figure 25 : Effet de l’inhibition de la β-oxydation par l’étomoxir sur les effets protecteurs du DHA dans les cellules GT1-7. _______________________________________________ 147 Figure 26 : Effet de la surexpression virale d’Elovl2 sur l’ARNm de la protéine Elovl2 dans les cellules GT1-7. __________________________________________________________ 148 Figure 27 : Effet de la surexpression virale d’Elovl2 sur la signalisation insulinique en
réponse au palmitate dans les cellules GT1-7. __________________________________ 148 Figure 28 : Effet du palmitate sur la signalisation insulinique dans les cellules GT1-7. ___ 154 Figure 29 : Schéma illustrant les deux entrées possibles d’incorporation du palmitate D4 dans la voie de synthèse de novo des céramides. _______________________________ 157 Figure 30 : Quantité de céramides-D4 à longues et à très longues chaînes dans les cellules GT1-7. _________________________________________________________________ 161 Figure 31 : Effet de l’acide okadaïque sur la signalisation insulinique en conditions de
lipotoxicité dans les cellules GT1-7. __________________________________________ 164 Figure 32 : Effet du FTY-720 sur la signalisation insulinique dans les cellules GT1-7. ___ 165 Figure 33 : Schéma récapitulatif du métabolisme du palmitate dans les cellules GT1-7 ainsi que ses différents modes d’action sur Akt. _____________________________________ 166 Figure 34 : Schéma des mécanismes potentiellement impliqués dans l’amélioration de la sécrétion d’insuline chez les rats Zucker Obèses traités avec la myriocine. ___________ 172
Tableau 3 : Liste des différents anticorps utilisés ainsi que leur dilution. ______________ 186 Tableau 4 : Liste des différents primers utilisés. _________________________________ 187
1 D
IABETES
,
OBESITE ET CONSEQUENCES
Le diabète est une pathologie chronique définie par une augmentation anormale de la glycémie basale. En 2040, la Fédération Internationale du Diabète (http://www.diabetesatlas.org) prévoit qu’un adulte sur dix sera atteint par cette maladie. Au cours des dernières décennies, sa prévalence a augmenté de manière constante faisant d’elle un problème de santé majeur. Il existe plusieurs types de diabète dont le diabète de type I et le diabète de type II (DT2).
1.1 L
E DIABETE DE TYPEI
Le diabète de type I, défini comme « diabète insulino-dépendant », est caractérisé par une hyperglycémie due à un déficit de sécrétion d’insuline par le pancréas. Ce déficit en insuline est causé par une destruction auto-immune des cellules β-pancréatiques (Pickup et al., 1998). Les causes du développement de cette maladie ne sont pas totalement connues. Le diabète de type I dépend de facteurs environnementaux qui interagissent avec des gènes de prédisposition qui va conduire à l’agression auto-immune des cellules β (De Beeck and Eizirik, 2016). Dans le monde, cette pathologie représente 10% des personnes atteintes de diabète.
1.2 L
E DIABETE DE TYPEII
Le diabète de type II (DT2), dit « diabète sucré », est caractérisé à la fois par une diminution de sécrétion d’insuline et également par une diminution de son efficacité d’action sur ses tissus cibles tels que le tissu adipeux, le foie et le muscle (Defronzo et al., 2009). De plus, une potentielle diminution de la masse cellulaire β-pancréatique se rajoute à ses facteurs. Ce type de diabète est fortement commun, en effet, il représente 90% des patients atteints de diabète et touche environ 5% des personnes âgées de plus de quarante ans à travers le monde. L’apparition de cette maladie est relativement lente.
L’étude de cette pathologie est complexe à cause des paramètres multifactoriels pouvant entrer en jeu. De nos jours, l’étiologie du DT2 reste toujours mal définie de par les
nombreux arguments controversés existants à son sujet. Malgré cette controverse, il est généralement admis que l’association de facteurs génétiques et environnementaux est impliquée dans l’installation de cette pathologie (Unger et al., 1995).
1.3 L
E SYNDROME METABOLIQUELe syndrome métabolique est défini comme un ensemble de symptômes résultants de troubles du métabolisme. Ces perturbations peuvent être morphologiques, physiologiques, biochimiques en lien avec l’obésité et prédisposant l’individu au DT2 et aux maladies cardiovasculaires. Il a été admis que pour parler du syndrome métabolique au moins trois des facteurs de risques suivants doivent être associés chez le même individu (Alberti et al., 2009) :
o Une hyperglycémie à jeun (≥ 100 mg/dL) o Une hypertension artérielle (140/90 mm Hg)
o Une obésité abdominale (dépendante de la population et du pays) o Une augmentation du taux de triglycérides (TG) (≥ 150 mg/dL)
o Une diminution du taux de cholestérol HDL (< 50 mg/dL chez la femme et < 40 mg/dL chez l’homme).
De nombreux facteurs de risques pourraient se rajouter à la liste décrite ci-dessus. Cependant le risque de développer un syndrome métabolique semble étroitement lié à l’obésité et à la résistance à l’insuline dite « insulino-résistance » (IR) (Kahn et al, 2006). En effet, il a été montré que l’obésité est associée à un risque accru de développer une insulino-résistance. De ce fait, l’obésité et l’insulino-résistance sont considérées comme étant les causes primaires et fondamentales du syndrome métabolique.
1.4 L’
OBESITE1.4.1 Epidémiologie :
L’obésité et le surpoids sont considérés comme l’épidémie du siècle par l’OMS. En effet, en 2014, deux milliards d’adultes étaient en surpoids, équivalant à environ 39% de la population adulte mondiale dont 13% d’obèses. D’ici 2030, le nombre de personnes atteintes
de surpoids devrait atteindre plus de trois milliards. Ainsi le surpoids et l’obésité représentent le cinquième facteur de risque de décès au niveau mondial.
En France, une enquête épidémiologique (ObEpi) réalisée tous les trois ans depuis 1997, a permis de montrer qu’en 2012, un tiers des Français adultes sont en surpoids dont 15% sont atteints d’obésité. La proportion de personnes atteintes d’obésité est passée de 3 356 000 d’obèses en 1997 à 6 922 000 d’obèses en 2012.
1.4.2 Définition :
L’obésité a été définie par l’OMS comme étant « une accumulation anormale ou excessive de graisse qui présente un risque pour la santé ». La mesure du surpoids et de l’obésité la plus communément utilisée est l’indice de masse corporelle (IMC) ou body mass
index (BMI). Celui-ci est égal au poids (en kilogramme) divisé par la taille (en mètre) au carré
de l’individu et est fréquemment utilisé pour estimer la surcharge pondérale. L’OMS a donc établit une classification concernant l’IMC :
o Dénutrition : IMC < 16,5 ; o Maigreur : 16,5 ≤ IMC ≤ 18,5 ; o Normalité : 18,5 ≤ IMC ≤ 25 ; o Surpoids : 25 ≤ IMC ≤ 30 ; o Obésité : IMC ≥ 30.
L’obésité étant elle-même divisée en trois classes : o Obésité modérée : 30 ≤ IMC ≤ 35
o Obésité sévère : 35 ≤ IMC ≤ 40
o Obésité massive ou morbide : IMC ≥ 40.
1.4.3 Causes :
Au-delà de ce simple constat d’excès de masse grasse, la cause fondamentale du surpoids et de l’obésité résulte dans un déséquilibre de la balance énergétique c’est-à-dire une inégalité entre les calories consommées et celles dépensées. Ce déséquilibre est notamment dû à une augmentation de la consommation d’aliments très caloriques riches en lipides ou en sucres mais également à une augmentation de la sédentarisation de l’individu (évolution des modes de transport, urbanisation croissante, manque d’activité physique).
A ces facteurs environnementaux viennent s’ajouter des facteurs génétiques, psychopathologiques (troubles du comportement alimentaire) faisant de l’obésité une pathologie complexe.
1.4.4 Conséquences :
La hausse de l’IMC augmente le risque de contracter certaines pathologies chroniques telles que les maladies cardiovasculaires (principalement les cardiopathies et les accidents vasculaires cérébraux), les troubles musculo-squelettiques (arthrose), des troubles du sommeil (apnées), certains cancers (de l’endomètre, du sein, des ovaires, du foie, de la prostate, de la vésicule biliaire, du rein et du colon) et de l’hypertension artérielle. Parmi les effets délétères de l’obésité, de nombreux déséquilibres énergétiques conduisent notamment, à un défaut d’efficacité de la sécrétion d’insuline mais également à une résistance à l’action de celle-ci aboutissant au DT2 (Cota D et al., 2007).
Chez un individu normal, la nourriture ingérée va être métabolisée et l’énergie provenant de la dégradation des acides gras (AG) va être stockée sous forme de triglycérides (TG) dans le tissu adipeux (TA). Le TA, sous l’action de l’insuline, participe à la clairance des lipides de la circulation sanguine en les stockant sous forme de TG et seront mobilisés en fonction des besoins nutritionnels de l’individu (Unger, 2002). Chez un patient obèse, la quantité d’énergie ingérée est supérieure à la quantité d’énergie dépensée. Cette consommation excessive de graisse va conduire à un surplus de lipides chronique et va induire une saturation du TA en TG. En effet, lorsqu’un excès d’AGL circulants apparaît, le TA compense en augmentant la taille de ses cellules (hypertrophie) ainsi que leur nombre (hyperplasie) (Jo et al., 2009). A long terme, si cet excès devient chronique, le TA deviendra alors incapable de stocker l’excédent d’AGL circulants et ces lipides seront stockés de manière ectopique dans différents tissus tels que le foie, le muscle ou bien le pancréas (Van Herpen et al., 2008). La séquence d’évènements conduisant à une accumulation ectopique d’AGL a été définie comme « l’hypothèse du surplus » (overflow hypothesis) (Danforth et al. 2000 ; Bugianesi et al. 2005). Selon cette hypothèse, l’insulino-résistance (IR) est la conséquence de l’incapacité du TA d’augmenter et de stocker l’excès de calories. La quantité d’AG stockés dans le muscle et dans le foie va interférer avec la signalisation de l’insuline, le transport du glucose et la synthèse de glycogène dans le muscle et va augmenter la néoglucogenèse hépatique (Bugianesi et al., 2005). L’accumulation de lipides dans le foie résulte d’une dérégulation entre l’apport, la synthèse, l’export et l’oxydation des AG. Dans le pancréas, ces AG sont souvent associés à un dysfonctionnement et à
Figure 1 : Conséquences de l'hyperlipidémie
Chez un patient sain, les lipides provenant de l’alimentation vont être stockés sous forme de triglycérides (TG) au sein du tissu adipeux. Chez un patient obèse, l’excès de lipides dû à une augmentation de l’apport calorique va conduire à une saturation du tissu adipeux, provoquant une inflammation de celui-ci ainsi qu’une augmentation de la quantité d’acides gras circulants. Cette hyperlipidémie va conduire à un stockage ectopique d’acides gras dans différents tissus tels que le muscle, le foie ou le pancréas dans lesquels ils auront des effets délétères tels que la stéatose hépatique, le dysfonctionnement ou l’apoptose des cellules β-pancréatiques. Ils induiront également une diminution de la synthèse de glycogène et du transport de glucose musculaire ainsi qu’une augmentation de la néoglucogenèse favorisant l’apparition d’une hyperglycémie. Ces acides gras provoqueront également une résistance à l’insuline musculaire, hépatique et dans le tissu adipeux. Dans un premier temps, pour palier à l’hyperglycémie, le pancréas va adapter sa réponse physiologique et en augmentant sa sécrétion d’insuline pour ramener la glycémie à un niveau correct. A long terme, le pancréas ne sera plus capable de réguler correctement la glycémie. Ainsi, l’hyperglycémie chronique et la résistance à l’insuline onduisent à l’apparition du diabète de type II.
Abréviations : AG = Acides gras, IR = insulino-résistance, TA = Tissu adipeux, TG = Triglycérides,
l’apoptose des cellules β-pancréatiques (Heni et al., 2010). Dans le TA, l’excès d’AG va notamment conduire à une IR et à une inflammation (Sears, 2015) (Figure 1).
Les raisons pour lesquelles le TA ne parvient plus à stocker ce surplus d’AGL restent pour l’instant en partie inconnues. Pendant longtemps, le TA a été considéré comme étant une masse inerte de stockage d’énergie. Récemment, il a été défini comme étant un régulateur majeur de l’homéostasie énergétique (Powell, 2007). En effet, le TA permet de gérer une grande partie de la réponse à l’insuline. Par exemple, l’absence du transporteur de glucose 4 (GLUT4) ainsi que l’ablation du gène GLUT4 à la surface des adipocytes de souris conduisent toutes deux à l’installation d’une IR musculaire et hépatique (Abel et al., 2001). A l’inverse, la surexpression de GLUT4 est corrélée à une meilleure sensibilité globale à l’insuline (Tozzo et al., 1997 ; Kim et al., 2000). Des interactions entre les adipocytes et les autres cellules de l’organisme semblent donc cruciales dans l’installation de l’IR.
En cas de stress dû à l’obésité, une infiltration du TA par des macrophages a été observée. Les adipocytes et ces macrophages peuvent sécréter une multitude d’adipokines capables de communiquer avec les hépatocytes/cellules musculaires et ainsi induire un état inflammatoire dans ces tissus.
Basé sur cette observation, des chercheurs ont supposé que l’obésité induit un état inflammatoire à bas grade qui mène à la désensibilisation de l’action de l’insuline. En effet, ces deux types cellulaires sont capables d’agir en synergie afin de sécréter des adipokines inflammatoires telles que le TNFα (Tumor Necrosis Factor α), les interleukines-6 (IL-6) et 8 (IL-8), la protéine chimio attractante de monocyte (MCP-1), la protéine C-réactive qui ont été définies comme étant des régulateurs clé de l’IR (Anghel and Wahli, 2007). Dans le plasma de patients obèses, une augmentation de TNFα, IL-6, MCP-1, protéine C-réactive a été observée (Dandona et al., 2004 ; Juhan-Vague et al., 2003 ; Kamei et al., 2006). Ces adipokines sont capables d’agir localement en modulant le métabolisme de l’adipocyte : ainsi MCP-1 et le TNFα augmentent la lipolyse adipocytaire et diminuent la synthèse de TG (Guilherme et al., 2008).
Une administration exogène de TNFα chez des patients humains provoque une diminution de la signalisation à l’insuline et du transport du glucose dans le muscle squelettique ; un effet similaire a pu être observé avec une administration d’AGL (Bouzakri and Zierath., 2007). Dans ces deux cas, la défaillance de la fonction adipeuse conduit à une augmentation de la quantité d’AG circulants et au stockage ectopique de lipides dans des tissus non-adipeux (Unger, 2002 ; Mittra et al., 2008 ; Van Herpen and Schrauwen-Hinderling, 2008).
Lorsque la quantité d’AG surpasse la capacité des cellules non-adipeuses à les estérifier en TG et/ou à les oxyder, une pléthore de lipides délétères s’accumule dans les cellules non-adipoctytaires (Kuminski et al., 2009 ; Mittra et al 2008 ; Van Herpen et al.,
2008). Dans ces tissus non adipocytaires, l’élimination de l’excès d’AG va emprunter une voie de type non oxydative et va conduire à la synthèse de composés tels que le diacyl-glycérol (DAG) et les céramides (Cowart, 2009 ; Chaurasia and Summers, 2015). Lorsqu’ils sont produits de façon dérégulée et présents dans ces cellules comme dans la circulation sanguine, ces deux composés lipidiques deviennent toxiques. Ce phénomène est appelé la « lipotoxicité » (Unger, 2005).
1.5 L
AL
IPOTOXICITED’un point de vue clinique, chez des patients diabétiques, le rôle de la lipotoxicité associée à l’obésité a pu être démontré dans des cas d’insulino-résistance du muscle squelettique et de stéatose hépatique (Slawik and Vidal-Puig, 2006). Ainsi, dans le muscle, l’action de l’insuline qui permet l’entrée du glucose dans la cellule est inhibée par les AGL car ceux-ci interfèrent avec la translocation de GLUT4 vers la membrane plasmique (Kusminski et al., 2009 ; McGarry, 1992 ; Shulman, 2000). Dans le foie, les AGL sont connus pour inhiber l’action de l’insuline qui, dans des conditions physiologiques, permet d’inhiber la glycogénolyse et la néoglucogenèse (Boden et al., 2002). Dans le TA, ces AGL vont inhiber l’action de l’insuline qui va normalement activer la lipogenèse de novo (Sears, 2015).
Dans les cellules musculaires et hépatocytaires, l’accumulation de DAG permet également l’activation de Protéines Kinase C (PKC) spécifiques, déclenchant ainsi la phosphorylation des sites de sérine/thréonine des substrats du récepteur de l’insuline (IRS-1 et IRS-2) ainsi que la déphosphorylation de la tyrosine, ce qui conduira à l’inhibition de la signalisation de l’insuline. (Van Herpen and Schrauwen-Hinderling, 2008 ; Meshkani and Adeli, 2009) (Figrue 2).
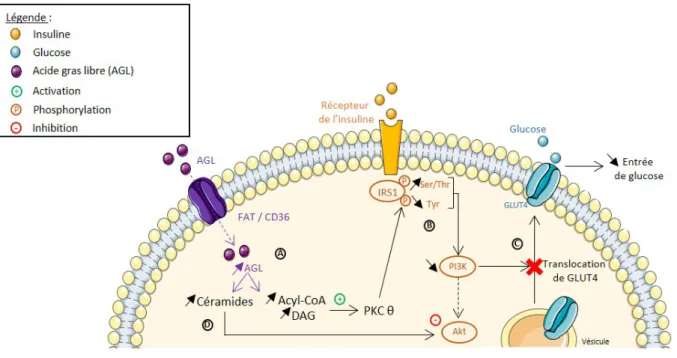
Figure 2 : Effets des acides gras libres sur l’entrée de glucose dans les cellules musculaires.
Chez un patient obèse, l’excès d’acides gras libres va pénétrer au sein des cellules musculaires par l’intermédiaire de FAT/CD36. Ces acides gras en excès seront ensuite métabolisés en acyl-CoA et en DAG. L’augmentation du DAG est responsable de l’activation de la PKC θ (A). La PKC θ va permettre d’inhiber la protéine IRS1 notamment en augmentant la phosphorylation des résidus sérines/thréonines et en diminuant celle des résidus tyrosines (B). IRS1 étant inhibée, la PI3K sera à son tour inhibée et empêchera la translocation de Glut4 à la membrane plasmique et diminuera ainsi l’entrée de glucose dans la cellule (C). L’augmentation d’acides gras libres peut également conduire à l’augmentation de la teneur en céramides qui inhibent la protéine Akt et donc la signalisation insulinique (D).
Abréviations : AGL = Acides gras libres, DAG = Diacylglycérol, FAT/CD36 = Récepteur des acides gras, GLUT4 = Transporteur de glucose 4, IRS1 = Insulin receptor substrate 1, PI3K = Phosphoinositide 3 kinase, PKC = Protéine Kinase C, Ser/Thr = Sérine/Thréonine, Tyr = Tyrosine.
Il a été également proposé que les céramides soient des médiateurs importants de la lipotoxicité (Poitout and Robertson, 2008). Une augmentation de la quantité de céramides a été observée aussi bien dans le plasma que dans les cellules musculaires de patients diabétiques, mais également dans le foie, le plasma et les muscles de souris obèses (Ussher et al., 2010 ; Wigger et al., 2017).
Durant l’obésité, les céramides qui s’accumulent dans le muscle sont les lipides les plus toxiques (Adams et al 2004 ; Coen et al., 2010). L’association entre l’accumulation de céramides et l’IR a bien été montrée à la fois dans les muscles de modèles de rongeurs et chez l’homme (Holland et al., 2008 ; Hage Hassan et al., 2014 ; Mahfouz et al., 2014). De même, la relation entre l’augmentation du contenu en céramides musculaires et la sensibilité à l’insuline a également été démontrée in vitro soit en exposant des cellules musculaires à un excès d’AG (tel que le palmitate) soit directement avec des analogues des céramides (Chavez and Summers 2003 ; Hajduch et al., 2001).
Des études ont suggéré que le foie est un organe clé dans la synthèse des céramides (Yamaguchi et al., 2004), mais leur contribution à l’installation de l’IR hépatique n’a toujours pas été clairement établie. Malgré le fait que certaines études n’aient pas trouvées de réelles corrélations entre l’accumulation de céramides hépatiques et la sensibilité à l’insuline (Monetti et al., 2007), d’autres ont démontré qu’un régime riche en lipides (High Fat Diet = HFD) induit une IR hépatique qui est contrecarrée par l’inhibition de la voie de synthèse des céramides ou par l’augmentation de leur dégradation dans les hépatocytes (Holland et al., 2011). De plus, une étude récente a montré que le foie, pour se protéger, est capable de sécréter les céramides (Watt et al., 2012).
Les céramides ont également un rôle important au niveau du pancréas endocrine en jouant sur la survie des cellules-β pancréatiques. En effet, des expériences ont montré que la voie de production des céramides, la voie de synthèse de novo, est impliquée dans l’apoptose des cellules-β pancréatiques (Poitout and Robertson, 2008 ; Véret et al., 2011). De manière intéressante, une diminution de la masse β-cellulaire et la perte de sécrétion d’insuline ont été corrélées à une augmentation de la teneur en céramides dans des îlots de Langerhans de rats Zucker diabétiques (ZDF) (Unger and Orci, 2011).
2 L’
INSULINE
2.1 L’
INSULINE:
G
ENERALITES,
STRUCTURE ET BIOSYNTHESE2.1.1 Généralités
L’insuline est une hormone peptidique anabolique sécrétée par les cellules β-pancréatiques des îlots de Langerhans. Son rôle majeur réside dans le maintien de l’homéostasie glucidique en régulant la balance entre la production de glucose hépatique et son entrée dans le muscle et le tissu adipeux. Elle favorise le métabolisme des lipides et des protéines ainsi que la division et la croissance cellulaire grâce à ses effets mitogéniques.
2.1.2 Structure
L’insuline est une hormone de 6 kDa. C’est un polypeptide contenant deux chaînes α et β reliées entre elles par deux ponts disulfures. La chaîne α se compose de 21 acides aminés contre 30 pour la chaîne β. Ce monomère d’insuline constitue la forme active de la protéine. L’insuline est cependant capable de se dimériser. L’association de trois dimères forme des héxamères qui constituent la forme de stockage majeure de l’insuline au sein des granules de sécrétion (Derewenda et al., 1989).
2.1.3 Biosynthèse
Classiquement, il est admis que l’expression du gène de l’insuline est exclusive aux cellules β-pancréatiques. Néanmoins, il existerait une production extra-pancréatique. En effet, des études ont montré dans différents modèles d’animaux diabétiques qu’elle pourrait être synthétisée par certaines cellules hépatiques, de la rate ou encore du tissu adipeux (Kojima et al., 2006). Des études plus récentes ont également mis en évidence qu’au niveau du cerveau en cours de développement, l’insuline pourrait y être synthétisée localement et serait utile au guidage axonal (Suckale and Solimena, 2010). Certains neurones de l’hypothalamus expriment également le gène de l’insuline et seraient capables de la produire (Gerozissis, 2003).
L’insuline est initialement synthétisée sous la forme d’un polypeptide immature composé d’une seule chaîne de 110 acides aminés : la pré-pro-insuline. Cette pré-pro-insuline sera transformée en pro-insuline suite à un clivage d’un domaine contenant 24 acides aminés au sein du réticulum endoplasmique rugueux (RER). Cette pro-insuline sera ensuite transportée du RER vers l’appareil de Golgi dans lequel la pro-insuline sera transformée en insuline. Cette transformation s’accompagnera d’une libération du peptide-C. Lors de la sécrétion d’insuline qui se fait par exocytose, le peptide-C et l’insuline seront co-sécrétés de façon équimolaire (Wilcox, 2005). Cette sécrétion d’insuline est déterminée par la quantité de glucose présente dans le sang. Suite à un jeûne, lorsque la glycémie est basse, le pancréas ne sécrètera pas ou peu d’insuline. A l’inverse, suite à un repas, lorsque la glycémie augmente, la stimulation de la sécrétion d’insuline sera maximale.
2.1.4 Rôles physiologiques de l’insuline dans les tissus périphériques
Dans le muscle squelettique, l’insuline favorise la capture du glucose ainsi que sa conversion en glycogène. Ce tissu est la première cible de l’insuline, car il constitue le site majeur de biodisponibilité du glucose in vivo (Kahn and Flier, 2000) et est connu pour être responsable de la capture de 70 à 80 % du glucose en réponse à l’insuline (Hegarty et al., 2003). Dans le foie, l’insuline stimule la synthèse de glycogène en inhibant la néoglucogenèse et la glycogénolyse, et empêche la sortie de glucose hépatique. Au sein du tissu adipeux, l’insuline stimule l’entrée de glucose et sa conversion en triglycérides (TG) et inhibe la lipolyse (Ahmadian et al., 2009). En plus de jouer un rôle important dans le métabolisme du glucose, l’insuline intervient également dans de nombreux processus cellulaires tels que le transport d’acides aminés, la synthèse de protéines ou bien encore la mitogénèse. La place centrale de l’insuline dans le contrôle du métabolisme du glucose explique la complexité des systèmes régulateurs de cette hormone. Elle a un rôle important car elle constitue le seul facteur hypoglycémiant de l’organisme. Elle pourrait être impliquée dans la régulation du « glucose-stimulated insulin secretion » (GSIS) en jouant sur la sensibilité au glucose ou sur la prolifération des cellules β-pancréatiques car ces dernières possèdent des récepteurs à l’insuline. (Kulkarni, 2004).
2.1.5 Rôles physiologiques de l’insuline au niveau central
Des récepteurs de l’insuline ont été découverts dans différentes structures cérébrales, comme par exemple, dans le bulbe olfactif, l’hypothalamus, le cortex cérébral et l’hippocampe (Stockhorst et al., 2004, Ghasemi et al., 2012).
L’insuline, au niveau central, agirait plutôt comme un neuropeptide impliqué dans les phénomènes de satiété, de régulation de la prise alimentaire, de l’olfaction, de la mémoire et de la cognition (Gerozissis, 2004 ; Kim and Feldman, 2015). Une étude a d’ailleurs montré qu’une délétion des récepteurs de l’insuline dans les neurones favorise l’installation de l’obésité chez des souris femelles (Brüning et al., 2000). D’autres travaux réalisés chez des rongeurs ont montré qu’une injection centrale d’insuline réduit la prise alimentaire et induit une perte de poids (Air et al., 2002). L’insuline peut être transportée dans le cerveau par l’afflux sanguin ou bien y être synthétisée localement. Elle semble agir sur l’appétit à travers la régulation d’autres neurotransmetteurs (tel que le neuropeptide Y) et peptides (tel que α-MSH (α-Melanocyte-stimulating hormone)). D’autres hormones sont capables d’agir au niveau de l’hypothalamus pour réguler la prise alimentaire comme notamment la leptine et la ghréline (Yu and Kim, 2012). La leptine et l’insuline, pour exercer leurs effets satiétogènes, semblent partager la même voie de signalisation dans l’hypothalamus (Devaraj et al., 2004).
D’autres expériences ont mis en évidence qu’une injection d’insuline dans le troisième ventricule diminue la glycémie en inhibant la production hépatique de glucose. Lorsque la signalisation de l’insuline dans l’hypothalamus est inhibée, l’injection d’insuline ne permet plus de diminuer la production de glucose. De ce fait, l’IR hypothalamique pourrait contribuer à l’hyperglycémie observée dans le DT2 (Obici et al., 2002).
Les récepteurs de l’insuline ont été montrés comme étant plus nombreux dans les neurones par rapport aux cellules gliales et ils sont principalement situés au niveau post-synaptique (Unger et al., 1991). Une administration intranasale d’insuline augmente les capacités d’apprentissage aussi bien dans des études réalisées chez l’Homme que chez les animaux (Benedict et al.,2011). Il a également été montré qu’une injection d’insuline directement dans l’hippocampe améliore la mémoire spatiale (McNay et al., 2010). Cependant, une perturbation de la voie de signalisation de l’insuline rend les neurones vulnérables au stress métabolique accélérant leur dysfonctionnement. Un défaut d’action de l’insuline cérébral est associé à une diminution des capacités cognitives et au développement de démences comme pour comme pour la maladie d’Alzheimer (De la Monte, 2009). De faibles performances cognitives dans des pathologies comme le diabète ou la maladie d’Alzheimer, sont associées à une diminution de l’expression des récepteurs à l’insuline mais également à une diminution de la quantité d’insuline présente dans le liquide cérébro-spinal (CSF) (Duarte et al., 2012 ; Moloney et al., 2010). Une étude récente a d’ailleurs mis en évidence une diminution de la phosphorylation de certaines protéines faisant partie de la voie de signalisation de l’insuline à la fois dans des cerveaux de patients diabétiques et de patients atteints de la maladie d’Alzheimer, et cette diminution était plus sévère chez des patients atteints simultanément par ces deux pathologies (Liu et al., 2011).
2.2 V
OIE DE SIGNALISATION DE L’
INSULINEL’insuline possède deux fonctions importantes ayant des voies de signalisation différentes tout en prenant naissance au niveau de son récepteur :
- Une fonction métabolique médiée par la voie de signalisation dont les substrats du récepteur de l’insuline (IRS) et la phopshoinositide-3-kinase (PI3K) font partie ;
- Une fonction mitogénique permettant la prolifération ainsi que la différenciation cellulaire par l’intermédiaire de la voie de signalisation des Mitogen-activated protein kinases (MAPK).
Il est à noter que la voie des MAPK peut être activée par l’intermédiaire des protéines IRS ou bien directement par la phosphorylation de la tyrosine 960 du récepteur de l’insuline (Gustafon et al., 1995 ; Langlais et al., 2004) (Figure 3).
Figure 3 : Voies de signalisation impliquées en réponse à l'insuline. L’insuline se fixe sur son récepteur et permet l’activation de deux voies de signalisation :
- Une voie dépendante de Shc, Grb2 et SOS qui vont permettre l’activation des MAPK et ainsi induire des fonctions mitogéniques comme la croissance cellulaire ou la différenciation. Cette voie de signalisation peut être activée par IRS ou bien directement par la phosphorylation de la tyrosine 960 du récepteur de l’insuline (A).
- Une autre voie dépendante d’IRS, de la PI3K, de PIP2 et PIP3, de PDK1 et d’AKT qui permet l’induction de fonctions métaboliques (B).
Abréviations : Akt = Protéine Kinase B, Grb2 = Growth factor receptor-bound protein 2, IRS = Insulin receptor substrate, MAPK = mitogenic-activated protein kinase, PI3K = Phosphoinositide 3 kinase, PIP2 = Phosphatidylinositol-4,5-biphosphate, PIP3 = Phosphatidylinositol-3,4,5-triphosphateShc = Src-homology-2-containing protein, SOS = Son-of-sevenless.
2.2.1 Le récepteur de l’insuline
La première étape d’activation de la voie de signalisation de l’insuline est la fixation de celle-ci avec son récepteur membranaire, le récepteur à l’insuline (RI). Le RI est un complexe hétérométrique composé de deux sous-unités α à la surface de la membrane cellulaire et deux sous-unités transmembranaires β. Ces sous-unités sont reliées entre elles par des ponts disulfures adoptant ainsi une forme cylindrique. Le RI appartient à la famille des récepteurs à activité tyrosine kinase. L’insuline, par l’intermédiaire des sous-unités α, va se fixer à son récepteur entraînant un changement de conformation rapide de celui-ci. Ce changement de conformation va, à son tour, stimuler l’activité tyrosine kinase intrinsèque des sous-unités β qui permettra une trans-autophosphorylation des résidus tyrosine 1158, 1162 et 1163 dans la région intracellulaire de ces mêmes sous-unités (Lee et al., 1997). Suite à cette autophosphorylation, le RI deviendra actif et conduira à la phosphorylation des résidus tyrosines de nombreuses protéines cellulaires cytoplasmiques dont les six isoformes d’IRS et les protéines Src-homology and collagen homology (Shc) (Taniguchi et al., 2006). Cette dernière est capable de se lier et d’activer la Growth Factor Receptor-Bound Protein 2 (Grb2) qui, à son tour, interagira avec une protéine appelée son-of-sevenless (SOS) ayant pour conséquence l’activation de la voie de signalisation des MAPK (Taniguchi et al., 2006). Cette cascade de signalisation, faisant intervenir ces molécules, est connue comme étant la voie de signalisation mitogénique de l’insuline.
La voie de signalisation métabolique régulant entre autre le métabolisme du glucose, des lipides et des protéines, quant à elle, fait appel à la voie des IRS et de la PI3K.
2.2.2 Les substrats du récepteur de l’insuline (IRS)
Ces protéines sont les premières cibles du récepteur lorsque celui-ci est activé. Ces IRS sont responsables des effets métaboliques de l’insuline en permettant la transduction du signal de celle-ci ainsi que le maintien de l’homéostasie glucidique (Biddinger and Kahn, 2006). Comme évoqué précédemment, il existe six isoformes de la protéine IRS. En revanche, seulement deux isoformes (IRS-1 et 2) ont été majoritairement étudiés et identifiés comme étant impliqués dans l’action de l’insuline (Withers et al., 1998 ; Tamemoto et al., 1994 ; McGettrick et al., 2005). Dans le muscle et dans le tissu adipeux, IRS-1 est l’isoforme médiant l’action de l’insuline tandis qu’IRS-2 joue un rôle très important au niveau hépatique, en contrôlant les fonctions métaboliques relatives à l’insuline, et au niveau du pancréas en régulant la prolifération cellulaire et la régénération (Kubota et al., 2000). Un knockout (KO) total pour IRS-1 a permis de mettre en évidence un défaut d’action de l’insuline dans le
muscle (Araki et al., 1994) et dans l’adipocyte (Tamemoto et al., 1994). En revanche, dans le foie, IRS-2 compense l’absence d’IRS-1 en augmentant son expression par rapport à des souris contrôles (Taniguchi et al., 2005). Des souris KO pour IRS-2 présentent une IR hépatique ainsi qu’une diminution de la masse β-cellulaire, aboutissant au DT2 (Kubota et al., 2000).
Dans le cerveau, les protéines IRS-1 et 2 sont exprimées (Lavin et al., 2016). Même si la balance entre ces deux protéines est actuellement toujours méconnue, il est généralement admis que ces deux protéines sont importantes pour induire les effets de l’insuline au niveau central. Par exemple, une étude réalisée chez des souris mises sous régime HFD montre une diminution de la phosphorylation de la protéine IRS-1 dans le cerveau (Liu et al., 2015). Une autre étude a montré qu’une délétion d’IRS-2 dans l’hippocampe de souris conduit à un défaut de signalisation de la voie PI3K (Costello et al., 2012). Des souris invalidées pour IRS-2 développent un phénotype diabétique (Lin et al., 2004) ainsi qu’une diminution de la taille de leur cerveau de 40% (Schubert et al. 2003).
Chez les rongeurs, l’isoforme IRS-3 est majoritairement présent tandis qu’il est absent chez l’homme (Sesti et al., 2001). La protéine IRS-4 est, quant à elle, exprimée dans de nombreux organes tels que le foie, le muscle, le thymus, le pancréas et notamment dans le cerveau (Sesti et al., 2001). Les deux derniers isoformes (IRS-5 et 6) ont été principalement identifiés dans le foie pour IRS-5 et dans le muscle squelettique pour IRS-6 (Cai et al., 2003).
Tous les isoformes d’IRS ont une structure similaire pouvant être divisée en deux domaines : 1) un domaine PTB (phospho tyrosine binding) qui est responsable de la reconnaissance des résidus tyrosine phosphorylés et donc de l’interaction avec la sous-unité β du récepteur de l’insuline (Eck et al., 1996 ) ; 2) un domaine PH (Pleckstrin homology domain) responsable de l’ancrage de la protéine à la membrane plasmique par l’intermédiaire de phosphoinositides (Jacobs et al., 2001) et capable de stabiliser l’interaction entre le RI et IRS (Yenush et al., 1996). Cette association permet au RI d’induire une phosphorylation sur potentiellement vingt résidus tyrosine différents sur chaque protéine IRS (White, 2002). Ces sites de phosphorylation sont responsables de la reconnaissance et du recrutement d’autres protéines contenant un domaine SH2 (Src Homology domain 2) telles que la PI3K ou Grb2 et permettant ainsi la transduction du signal insulinique (Whitehead et al., 2000).