FACULTE DES SCIENCES
THESE PRESENTEE
A L'ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
POUR
L'OBTENTION
DU
GRADE DE DOCTEUR ES SCIENCES
PAR MARCEL GUAY
BACHELIER - ES,-SCIENCES (CHIMIE)
DE
L'UNIVERSITE DE MONTREAL
SPECTRES .DE VIBRATION DE L-'HYDRAZINE ET DE QUELQUES-UNS DE SES SELS
A. L'ETAT-CRISTALLIN
remerciements
Je désire exprimer ma profonde gratitude au professeur Rodrigue Savoie± qui m'a aooueilli et guidé avec compétence dans ta réalisation de- ce travail de recherches.
Je tiens à remercier toute l 'équipe du laboratoire de spectroscopie pour leur aide précieuse et très amicale.
Mes remerciements s'adressent aussi â Monsieur R. Dubey et au professeur André Beauchamp de l’Université de Montréal pour leur contribution dans l'identification des axes cristal
lographiques de monocristaux; j'e remercie vivement le profes seur Aidée Cabana pour un stage qu'il m’a permis d'effectuer dans son laboratoire à Sherbrooke
Je suis reconnaissant à Madame Jocelyne Rêzo'let et c? Mademoiselle Andrée Marcoux qui ont collaboré à la dactylogra phie de cette thèse.
Je remercie le Conseil National de Recherches du Canada pour l'octroi d'une Bourse en Sciences (1967-1971).
Liste des figures vii
Liste des tableaux ... vi
INTRODUCTION ... „ 1
CHAPITRE I: INSTRUMENTATION... 7
1- SPECTROMETRES 1.1- Infrarouge moyen . 8
1.2- Infrarouge lointain ... 9
1.3- Spectromètre Raman, Cary 81... 9
1.4- Raman à source laser He - Ne... 10
1.4.1- Système d1 Illumination... 10
1.4.2- Mono chroma te ur... 12
1.4.3- Système de détection ... 13
1.4.4- Système d'amplification ... 14
1.5- Raman à source laser à l'argon ionisé ... 15
2- MATERIEL OPTIQUE 2.1- Pour 11 infrarouge ... 16 2.2 Pour le Raman 16 * 1 2 TALLIN 1- INTRODUCTION... 18 2- PARTIE EXPERIMENTALE ... 22
V
3- STRUCTURES CRISTALLINES ET SPECTRES DE VIBRATION DE % ... 4- RESULTATS ET DISCUSSION ...
4.1- Région des basses fréquences ... 4.2- Région des hautes fréquences .... 5- CONCLUSION i ...
CHAPITRE III: .SPECTRES DE VIBRATION DE L'OXALATE
ACIDE ' D 'HYDRAZTNTUM {N^E^HC^O^ ' SON ANALOGUE DEUTERIE HDgDC^O
ET DE
4* * 1- INTRODUCTION ...
2- EFFET RAMAN EN LUMIERE POLARISEE'
3- PARTIE EXPERIMENTALE . ... 4- RESULTATS...
5- DISCUSSION... ...
5.1- Structure cristalline et spectres de vibra tion de N^H^HC^O^... ... . .
5.1.1-
r
Région des basses fréquences (v < 500 cm "*") 5.1.2- Région des hautes fréquences (v> 500 cm5.1.2.1 Vibrations du pont 0...H.T.0 5.1.2.2 Vibrations des ions oxalate 5.1.2.3 Vibrations des ions hydrazinium 5.2- Spectres de vibration de N^D^DC^O^ a basse
température . .
CHAPITRE IV: SPECTRES DE VIBRATION DU SULFATE D’HYDRA- ZINIUM- (.N2HgS04) A L ’ ETAT . CRISTALLIN .
1- INTRODUCTION... 2- PARTIE EXPERIMENTALE ... 24 28 28 37 49 page 50 51 54 57 61
68
69 73 76 76 82 84 85 93 94 95 3- RESULTATS 954- DISCUSSION ... . 102
4.1-Structure cristalline et spectres de vibration page de N2H6S04... ... 102
4.2-Vibrations des ions SO ~~... 105
4.3- Vibrations des ions ^H^.44... 110
4.4- Vibrations de réseau ... 113
5- CONCLUSION ... 114
RESUME ... ... 116
Liste des figures
page
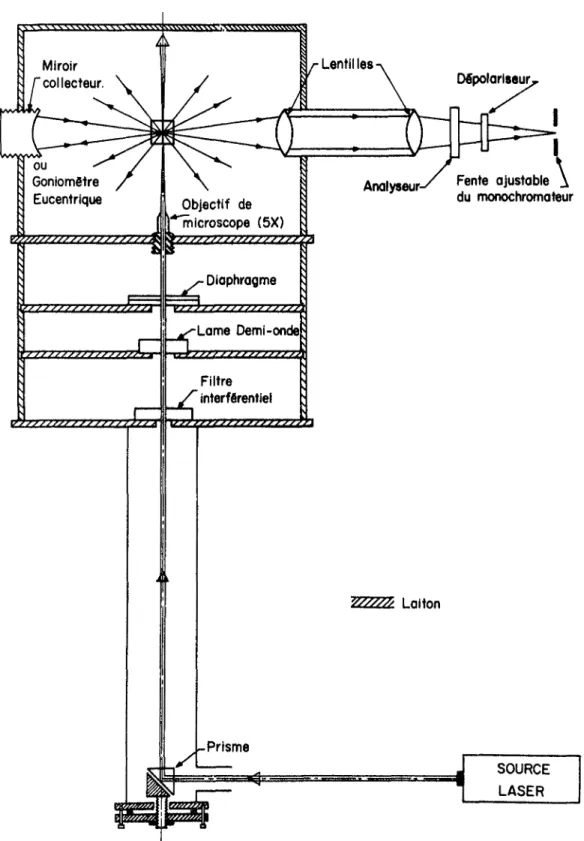
Fig. 1 Schéma du système d1 illumination du Raman
à source laser ... 11 Fig. 2 Isomères structuraux de 1'hydrazine . 20 Fig. 3 Montage pour la préparation de N2H4 anhydre 23 Fig. 4 Spectres Raman de (A) et de (B) cris
tallins à -195°C dans la région des basses
fréquences... ... 29 Fig. 5 Spectre infrarouge de cristallin dans
la région des basses fréquences .... 31 Fig. f6 Corrélation entre les fréquences des modes
de réseau observées en Raman et en infrarou
ge pour N2H4 et N2D^ cristallins .... 35 Fig. 7 Spectres Raman de N2H^ (A) et (B) cris
tallins à - 19 5°C dans la région des hautes
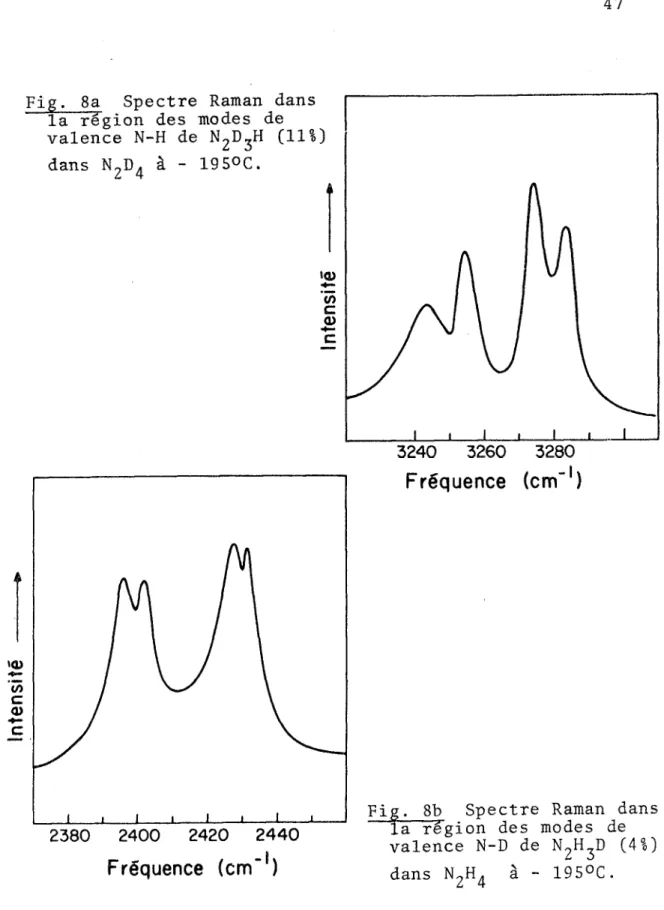
fréquences ... 38 Fig. -8 a Spectre Raman des modes de valence N-H de
N2D3H(11%) dans N2D4 à - 195°C .... 47 Fig. 8b Spectre Raman des modes de valence N-D de
N2H_D (4%) dans à - 195°C .... 47 Fig. 9 Notation de Porto pour l'effet Raman en
lumière polarisée ... . . 58 Fig. 10 Profil d'un monocristal de N9HgHC2C>4 et
orientation des axes cristallographiques. 60 Fig. 11 Spectres de vibration de N-H^HC-O.entre 40
et 700 cm . . . .... . . .
62
Fig. 12 Spectres de vibration de N-Hj-HC-O. entre
700 et 2000 cm-1 . . .... . . . 63 Fig. 13 Spectres de vibration de N-H^HC-O. entre
2000 et 3500 cm-1 . . .... 64
Fig. 14 Spectres de vibration de N-DcDCo0. entre 40 et 700 cm-1 % ^ 2 4
Spectres de vibration de NnDcDCo0, entre i-l
700 et 2000 cïrf 2 5 2 4
Spectres de vibration de N„D_DC„0. entre 2000 et 3500 cm"1. . . ... . . Spectres Raman de entre 50 et 350 cm et entre 2200 et 2600 cm-1, à 30, -35, -45 et - 75°C... Fig. 15 Fig. 16 Fig. 17 Fig. 18 Fig. 19a
Fig. 19b Environnement d'un ion sulfate dans ^HgSO^ cristallin, d'après Jflnsson et Hamilton ...
Fig. 20 Spectre infrarouge de N^R^SO, cristallin Spectres Raman de N2D,-DC20^,
350 cm ^ et entre 2300 et 2600 cm -190 ,«*-70 , -35 et 5°C . . . .
entre 50 et -1 _
Environnement d'un ion hydrazonium dans N2HgSO^ cristallin, d'après Jflnsson et Hamilton ...
à -180°C Fig. 21
Fig. 22
Spectre Raman de K^H^SO^ cristallin à - 190°C Spectre Raman en lumière polarisée d'un mono cristal de N2H_sq à 30°C ...
66
6786
87 96 96 98 99 page 100Liste des tableaux
page Tableau 1 Tableau de corrélation pour N2H4
cris-tallin, d'après la structure proposée
à partir de la diffraction des rayons X 25 Tableau 2 Tableau de corrélation pour
cris-tallin d'après la structure proposée a
partir de la diffraction des neutrons 27 Tableau 3 Fréquence en cm 1 des bandes infrarouges
et Raman de N^H^et cristallins (v <700 cm 1)... ...
30 Tableau 4 Tableau de corrélation pour cris
tallin d'après la structure que nous
proposons ... 33 Tableau 5a Fréquences en cm 1 des bandes infrarouges
et Raman de N2H4 > 700 cm--*-) . 39 Tableau 5b Fréquences en cm -1 des bandes infrarouges
et Raman de N2D4 > 700 cm""1) . 41 Tableau 6 Diagrammes des corrélations de symétrie pour
N2HgHC20^ à l'état cristallin 70 Tableau 7 Fréquences en cm 1 des bandes infrarouges
et Raman de N„HcHC„0, et de N-DuDC-O,
2 b 2 4
2 5 2 4
dans sa phase I (v < 500 cm i) 74 Tableau 8 Fréquences en cm 1 des vibrations fondamen
tales de l'ion hydrazinium N2Hg . 77 Tableau 9 Fréquences en cm des bandes infrarouges
et Raman de IS^Hj-HC^O^et de N^D^DC^O^dans sa phase I (v > 500 cm 1) ....
78 Tableau 10 Diagramme de corrélation pour N^D^DC^D^
cristallin dans sa phase II, d'après la
structure que nous proposons .... 90 Tableau 11 Fréquences en cm des bandes infrarouges
et Raman de N2DgDC20^à basse température 91 Diagrammes des correlations de symétrie
pour N2HgSO^ cristallin ... 103 Tableau 12
Tableau
Tableau
-1
13 Fréquences en cm des vibrations fonda mentales de N2H6++ ...
-1
14 Fréquences en cm des bandes infrarouges et Raman de à l'état cristallin
page 106
Les méthodes spectroscopiques, en particulier celles de la spectroscopie' infrarouge et Raman, comptent parmi les plus importantes pour 11 élucidation des structures moléculaires et cristallines. Dans les cas simples, 11 analyse des spec
tres infrarouges à très haute résolution permet, en principe, une détermination très précise des paramètres structuraux des molécules à l'état libre. Cependant, lorsque les moments
d'inertie sont élevés ou lorsque la résolution est trop faible, il devient impossible d'isoler les raies rotationnelles asso ciées aux transitions vibrationnelles et on n'observe plus que des envèloppes rotationnelles qui élargissent appréciablement les bandes de vibration. Cela comporte certains désavantages ; il devient notamment difficile de déterminer avec précision les fréquences vibrationnelles et de séparer les bandes rappro chées . On- a donc avantage dans beaucoup de cas à étudier les spectres d'échantillons en phases condensées plutôt qu'à l'état gazeux.
Des variations importantes se produisent dans les spectres de vibration lors de la condensation d'un gaz. Les rotations moléculaires deviennent fortement empêchées dans le liquide
3
et les transitions rotationnelles ~ disparaissent. Les modes
internes de vibration se caractérisent alors par des bandes plus ou moins larges suivant 1'importance des interactions entre
molécules dans ce milieu très désordonné. La spectroscopie infrarouge et Raman devient alors la technique d'excellence pour l'étude des interactions moléculaires, surtout lorsqu’il y a association par liens hydrogène, puisque les effets sont plus importants dans ces cas ^.
En passant de l'état liquide à l'état solide, les spec tres de vibration se modifient encore de façon appréciable. On note souvent des variations dans les fréquences et les intensités des bandes, ou même dans les activités de certains modes internes. Les vibrations internes se caractérisent en général par des bandes fines et on observe souvent un écla
tement de ces bandes en plusieurs composantes. Cela peut pro venir de 1'anisotropie locale (effet de site) ou encore du couplage entre vibrations identiques de molécules adjacentes dans le cristal (2,3,4) . Les translations et rotations
(librations) empêchées dans le solide peuvent provoquer 1'ap parition de bandes dans la région des basses fréquences. Ces modes de réseau sont importants lorsqu'il s'agit d'élucider
une structure cristalline et contribuent largement à la capacité calorifique des solides.
utilisées pour 1 * étude"des"structures cristallines, par exem ple la diffraction des rayons X et des neutrons, la spectres- copie infrarouge et Raman a pour base la dynamique des solides. Etant donné que les règles de sélection pour l'activité opti que dépendent des symétries moléculaires et cristallines, cet te méthode peut être d'un grand secours pour l'élucidation des structures cristallines. Quoique, en général, cette tech nique ne permet pas de déterminer le groupe spatial exact et les distances interatomiques dans un cristal donné, elle peut être utilisée avantageusement pour éliminer certaines structures, ou encore pour choisir entre plusieurs possibi lités . Ceci est particulièrement.important dans le cas des substances-hydrogénées parce que la faible section efficace des atomes.d'hydrogène ne permet pas de les localiser par la diffraction des rayons X.
Notre -intérêt pour la molécule, d'hydrazine (N^H^) a été soulevé au-départ, par les données contradictoires déjà
publiées sur la structure éristalline de ce composé. En ef fet, les résultats obtenus par diffraction des rayons X étaient en désaccord avec ceux déduits de la diffraction des neutrons. De plus, ce composé nous semblait intéressant du fait qu'il peut en principe, exister sous plusieurs conformations, sui vant l'orientation relative des deux groupements - NH2. Quoi qu'il était généralement admis que la forme la plus stable était la forme gauche, avec un angle azimutal de 90° environ ,
5
la possibilité qu'une petite fraction des molécules existât sous une autre configuration (par exemple, sous la forme trans/ ^ n * avait pas été définitivement écartée. On pouvait
aussi penser que la géométrie des molécules dans le cristal était différente, de celle en.phase gazeuse, par suite des
interactions moléculaires et du facteur empilement des molécules„
Nous nous sommes également intéressés à la structure de quelques sels de 1'hydrazine, en particulier, ceux répon dant aux formules stoéchiométriques H^SO^et H2C2°4"N2H4* Des études par diffraction avaient indiqué que ces complexes existent en fait sous forme de sels d'hydrazinium et d'hydra- zonium: N2H,_+ et N2H^ + SO J""" respectivement. La
spectroscopie infrarouge et Raman semblait être la méthode tout indiquée pour étudier les modifications apportées aux liaisons N-N et N-H par suite de 1'addition de un ou deux protons à la molécule d!hydrazine. L'oxalate acide d'hydrazinium offrait un attrait supplémentaire du fait qu'il est un des rares solides où l'on retrouve un pont hydrogène symétrique.
Un autre critère qui a motivé le choix de ces deux sels d'hydrazine.pour notre étude a été la facilité avec laquelle on a pu obtenir des monocristaux de taille raisonnable de ces com posés. De tels cristaux se prêtent merveilleusement bien à des études par spectroscopie Raman en lumière polarisée lorsque la raie excitatrice provient d'une source laser. En effet, le
faisceau laser se distingue par sa complète polarisation en plus de ses dimensions restreintes et de sa faible divergence. Dans de telles conditions, en variant 11 orientation relative des plans de polarisation de la lumière incidente et diffusée par rapport à 1'orientation d'un cristal unique, on peut déter miner la symétrie de toutes les vibrations actives en Raman de ce cristal. On espérait de cette façon vérifier 1'exactitude des structures cristallines proposées pour le sulfate d'hydrazo- nium et 1'oxalate acide d'hydrazinium.
Chapitre I
Nous donnerons dans ce chapitre, un aperçu général des techniques expérimentales employées dans la réalisation de cet te recherche. Les méthodes de préparation, de cristallisation et d1 analyse particulières à chacun des composés seront décri tes plus loin.
1- SPECTROMETRES
1.1- Infrarouge moyen
Les spectres dans le proche infrarouge (région 200-_3_
4000 cm ) ont été enregistrés sur un appareil de Perkin-Elmer, modèle 621. Cet appareil à double faisceau et à réseaux gravés nous permettait d'obtenir, dans les conditions normales d'opéra tion, une résolution de l'ordre de 1 cm . Le calibrage de cet appareil a été effectué à l'aide d'étalons gazeux et on estime que la précision sur les fréquences citées plus loin est de l'ordre de 1 cm dans le cas des bandes fines.
9
1.2- Infrarouge lointain
Dans 11 infrarouge lointain (région 20-400 cm ^), les speptres ont été obtenus à l'aide d'un interféromêtre de type Michelson, modèle FS-720, produit par Research and Industrial Instruments Co. Cet appareil était directement
relié à un petit ordinateur (Fourier Transform Computer, modèle FTC-100/7) et les spectres automatiquement reproduits sur un enregistreur X-Y. La résolution maximum (théorique) de cet
-1 -1
instrument est de 2.5 cm pour la région de 40-400 cm et de
-1 -1
1.25 cm pour la région de 20-200 cm . La précision sur les fréquences dans ces deux domaines est de l'ordre de 2 et 1 cm x respectivement.
1.3- Spectromëtre Raman, Cary 81
Les spectres Raman des échantillons polycristallins ont été enregistrés sur un spectromëtre Cary, modèle 81, muni d'une lampe hélicoïdale au mercure conventionnelle (Toronto arc source). La raie excitatrice (4358 R ) était isolée par solution filtre d'orthonitrotoluène (125 ml) et d'éthyl
violet (1.75 g) dans l'alcool isopropylique (3 litres). Les fentes du monochromateur étaient ajustées dans chaque région de façon à obtenir des bandes d'intensité raisonnable. La calibration de ce spectromëtre a été effectuée à l'aide de diverses lampes à décharge et les fréquences rapportées plus
-1
loin sont précises a ± 2 cm dans le cas des bandes fines.
1.4- Raman à source laser He-Ne
Les spectres Raman des monocristaux d'oxalate acide d'hydrazinium et de sulfate d1hydrazonium ont pu être obtenus en lumière polarisée grâce à un spectromêtre à source laser.
Notre appareil a été assemblé à partir de modules de provenances diverses et nous donnerons ici une description détaillée de
ses principales composantes.
1.4.1- Système d'illumination (Fig. 1)
Une source laser hélium-néon (Spectra Physics, modèle 125) émet la raie excitatrice (6328 £) à une puissance maximum d1 environ 70 milliwatts. Le rayon lumineux est réfléchi verti calement à l'aide d'un prisme et pénètre dans une chambre d'il lumination . Les raies secondaires émises par le laser sont éliminées à l'aide d'un filtre interférentiel (Spex 1430-5). Une lame demi-onde, déposée sur un rapporteur d'angle, permet de tourner de 90° le plan de polarisation du rayon lumineux. Un objectif de microsope(5 X) focalise ensuite la lumière sur 1'échan tillon qui peut ainsi être étudié en très faible quantité.
Le faisceau Raman diffusé par 1'échantillon est recueilli à 90° de l'axe d'éclairage par un miroir concave et un système de lentilles. L'une de ces dernières est montée sur une glissière horizontale et ajustée de façon à projeter l'image de la partie éclairée de 1'échantillon sur la fente d'entrée du monochrometeur.
11 Lentilles Miroir collecteur. Dépolarlseur ou / Goniomètre Eucentrlque Fente ajustable X du monochromateur Analyseur-Objectif de microscope (5X)
œwz&œ///y////zyA
Y/S/S/////SSS/S/SZ. Diaphragme Lame Demi-onde r/ Filtref
interfôrentielW///Z/
Laiton Prisme SOURCE LASERFig. 1: Schéma du système d'illumination du Raman â source laser.
Entre la deuxième lentille et la fente du monochromateur s1in tercalent un analyseur pour étudier la polarisation du fais ceau diffusé et un coin de quartz permettant de dépolariser ce même faisceau qui autrement serait réfléchi de façon différente par les réseaux, suivant son plan de polarisation.
Pour l'étude de monocristaux, le miroir collecteur est remplacé par un goniomètre eucentrique (Electronics and Alloys Inc., Mark II, modèle 63-6-MIE). Avec ce goniomètre, l'aligne ment d'un monocristal suivant ses axes peut atteindre une
précision de ± 0.2° .
Afin d'éliminer tout danger pour la vue de l'opérateur, le faisceau laser se propage à l'intérieur de tubes de laiton. La chambre d'illumination se compose de plaques de laiton gar nies de feutre noir à l'intérieur, pour éliminer les réflexions qui ont pour effet d'augmenter le bruit de fond dans les spectres.
1.4.2- Monochromateur
La décomposition du faisceau Raman est effectuée par un monochromateur double (Spex Industries Inc., modèle 1400), muni de réseaux Bausch et Lomb, gravés à 1200 lignes/mm et à
réflectivité maximum vers 5000 8 . Cet appareil muni de fentes ajustables de quelques microns à 3 mm d'ouverture, permet une résolution maximum de l'ordre de 0.1 8 , soit l'équivalent de 0.25 cm dans la région de 6000 8 . L'entraînement des
13
réseaux dans ce monochromateur étant linéaire en longueurs d'onde, il nous a fallu établir des tables de conversion, qui donnent la correspondance entre les longueurs d'onde en X, telles que lues sur 1 ' appareil, et les fréquences Raman (en cm "*") cor rigées pour le vide.
Ce monochromateur était périodiquement calibré à l'aide de diverses lampes à décharge gazeuse et les fréquences citées plus loin sont précises à i 1 cm 1.
1.4.3- Système de détection
A la sortie du monochromateur, se trouve une photomul- tiplicatrice (International Telephone and Telegraph Corporation, modèle FW-130) activée par une source de tension (Hamner
Electronics, modèle NV-19), variable de 0 à 3000 volts en cou rant continu, mais habituellement ajustée à 2000 volts. Cette photomultiplicatrice particulièrement sensible opère par cas
cades et libère à la sortie un très faible courant auquel s'a joute un courant parasite. Ce dernier peut être réduit d'un facteur 100 en refroidissant le tube à - 20°C. A cette fin, le phototube est entouré d'un manchon isolant dans lequel on fait circuler un courant d'azote gazeux refroidi. La tempé rature de 1'enceinte est ajustée automatiquement par un appa reil électronique qui régularise le débit d'azote froid.
1.4.4- Système d'amplification
Le signal à la sortie de la photomultiplicatrice peut être amplifié de deux façons, soit par un système de comptage de photons (Hamner Electronics), soit par un amplificateur à courant continu (Victoreen, modèle VTE-1). Ce dernier mode d'amplification s'est révélé supérieur au premier dans presque tous les cas et a été presqu'exclusivement utilisé.
Les spectres sont enregistrés à l'aide d'un appareil YEW (modèle LER 12 A) branché directement sur le système d'am plification . Cet enregistreur peu coûteux offre certains
avantages, notamment un choix de six vitesses de déroulement du papier (de 0.5 à 20 cm/min), une réponse rapide (0.8 sec pour une traversée complète) et finalement, il peut couvrir seize gammes de puissance (1 mV à 100 V). Il est habituellement à 10 mV pour une traversée complète de la plume.
1.5- Raman à source laser à l'argon ionisé
Notre spectromètre Raman avec source laser He-Ne offre certains avantages par rapport au Cary 81: il permet d'utiliser des échantillons plus petits, de poursuivre des études au voisi nage de la raie excitatrice et d'enregistrer facilement les spectres de monocristaux en lumière polarisée. On déplore cependant la faible énergie du faisceau laser. Dans les
15
conditions normales d'opération, 11 amplificateur à courant di rect doit être ajusté sur l'échelle de 10-10 ampere, le temps de réponse fixé à 3 secondes et les fentes ouvertes de 80 à 300 microns pour obtenir une intensité raisonnable des bandes
-1
Raman. On obtient alors une résolution de 1 â 3.5 cm et le spectre est marqué d'un bruit de fond appréciable. L'effet est d'autant plus important que l'on se situe à des fréquences plus élevées, par suite de la moins grande réflectivité des réseaux et la sensibilité plus faible de la photodétectrice.
A cause du peu de puissance de la source laser He-Ne et le faible pouvoir de diffusion des monocristaux que nous avons étudiés, nos spectres dans la région des hautes fré quences (au delà de 1500 cm-1) étaient masquées par un bruit de fond trop important. Les spectres en lumière polarisée étaient alors difficiles à obtenir et très imprécis. L'utili sation d'une source laser plus puissante s'avérait nécessaire. Nous nous sommes rendus à l'Université de Sherbrooke, dans les laboratoires du professeur Cabana, et nous avons repris les spectres des monocristaux avec un spectromètre Raman muni d'une source laser à Argon ionisé (Coherent Radiation, modèle 52). La raie â 4880 %, d'une puissance de 1 watt, a été uti lisée comme raie excitatrice. A l'exception de la source laser, les composantes du spectromètre de Sherbrooke sont analogues aux nôtres et le monochromateur (Jarrel-Ash) utilisé est de per formance sensiblement égale à notre Spex. Lors de
11 enregistrement des spectres, les fentes spectrales étaient ajustées entre 30 et 60 microns, ce qui donne une résolution de 1.0 a 2 cm 1. Les fréquences observées sur ce spectromêtre sont précises â ± 1 cm .
2- MATERIEL OPTIQUE
2.1- Pour 11 infrarouge
Les cryostats utilisés pour 1'enregistrement des spectres dans 1 ' infrarouge étaient de type conventionnel^. Dans 1 ' in
frarouge lointain, nous avons utilisé des fenêtres de quartz, qui peuvent transmettre jusqu'à 200 cm , ou de silicium,
uti--1
lisables jusqu'à 400 cm environ. Dans le proche infrarouge, nous avons utilisé des fenêtres d'iodure de césium (200-4000 cm "*") . Les mesures de température ont été effectuées à l'aide d'un thermocouple
cuivre-constantan,
branché à un thermomètre digital (Digitec, modèle 5611 , permettant une lecture directe en degrés Centigrade. Les températures citées plus loin étaient stables à ± 5°C.2.2- Pour le Raman
Les cellules Raman utilisées, étaient elles aussi de type conventionnel : cellules de Pyrex cylindriques (19 mm de
17
diamètre) pour les liquides et cellules coniques pour les
(7)
solides â température ambiante . Pour 11 enregistrement
des spectres à basse température, les cellules étaient placées à -1'intérieur d'un Dewar de Pyrex et refroidies par un
cou-(
8
) rant d1 azote gazeux tel que décrit dans la littérature v .SPECTRES RAMAN DE N^ ET N^ A L'ETAT CRISTALLIN
SPECTRES RAMAN DE N^ET N^ A L'ETAT CRISTALLIN
Dans ce chapitre, nous allons présenter et discuter les spectres Raman de 1'hydrazine (N^H^) et de son analogue deutérié.
1- INTRODUCTION
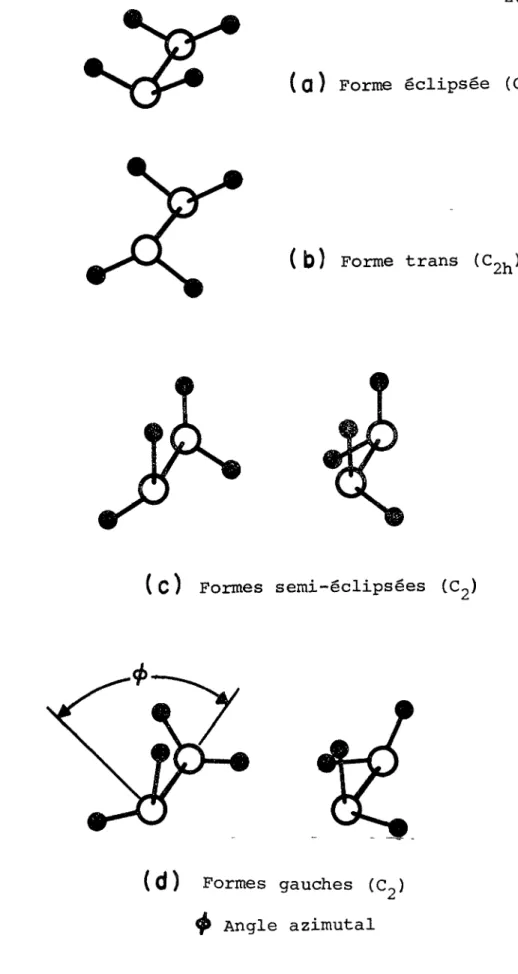
La molécule d'hydrazine (N^H^) est d'un intérêt tout par ticulier du fait que sa symétrie est déterminée par 1'orienta tion relative des groupements NH^ (Fig. 2). La structure
( 9 )
GAUCHE de symétrie , déduite par Penney et Sutherland n'a pu être confirmée par diffraction des électrons . Cepen dant, une analyse détaillée du spectre infrarouge de la vapeur
(
12
), centré à dans la région du mode de torsion interne
376.74 cm"\ a indiqué que l'angle azimutal dans cette molécule est voisin de 90°. Ce résultat a été vérifié par le spectre
(13,14)
micro-onde , qui a de- plus, permis d'évaluer les barrières de potentiel empêchant 1'inversion et la torsion interne au- tour de la liaison N-N à 990 et 1100 cm respectivement.
La structure GAUCHE est également en accord avec le moment^dOTti^ dipolaire de 1.75 Debye obtenu à partir du spectre micro-f<ype^
UVÏtS «ARES ^—X
( Q) Forme éclipsée (C2v)
(b)
Forme trans (C 2h)( d) Formes gauches (C2) $ Angle azimutal
21
Il n'est pas impossible, cependant, qu'une petite fraction des molécules existent sous une autre configuration, tant en
phase vapeur qu'en phase liquide. Les nombreux spectres infra rouges (12,17,24) e£ Raman(22,30) publiés ne permettent
pas d'éliminer cette possibilité.
Il existe une certaine confusion quant à la structure cristalline du solide. La mesure des chaleurs spécifiques et le calcul des fonctions thermodynamiques à partir des données spectroscopiques a conduit à une entropie résiduelle de 0.44
( 31 )
unités entropiques au zéro degré absolu . Cette valeur ne permet cependant pas de conclure à une structure désordonnée du solide puisqu'elle est moindre que la somme des impré cisions sur les valeurs calculées et obtenues expérimentale ment. Une étude du cristal par diffraction des rayons X,
(31)
par Collin et Lipscomb , suggère une maille élémentaire à 2
2
deux molécules définie par le groupe spatial C2^-P2^/m.
Dans cette structure, les molécules occupent des sites de symé trie Cg et doivent par conséquent, exister sous la forme CIS
(éclipsée) de symétrie C2 ou sous la forme TRANS de symétrie C2h* Cela n'implique pas nécessairement que l'une des configu rations soit la plus stable dans la molécule libre puisque, dans le cristal, d'autres facteurs (liaisons hydrogène,etc...)
(33) entrent en jeu. Une étude par diffraction des neutrons a conduit à une structure légèrement différente de celle obtenue
2
suggéré, avec deux molécules par maille élémentaire. Il est à noter que les seuls sites possibles dans ce groupe sont de symétrie . Baglin, Bush et Durig^^) Qnt récemment enre gistré les spectres de et NgD^ cristallins dans 1'infra rouge lointain en vue de choisir entre les deux structures cristallines proposées. En se basant sur le nombre de bandes observées, ils favorisent la structure obtenue à partir de
la diffraction des neutrons. Afin de vérifier cette déduction, nous avons étudié les spectres Raman de 1'hydrazine et de son analogue deutérié à diverses températures (- 70°, - 195°C).
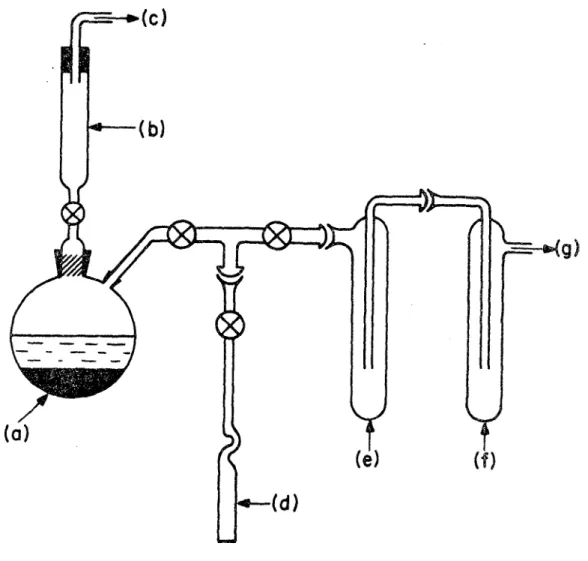
2- PARTIE EXPERIMENTALE
L'hydrazine (N^H^) utilisée a été obtenue à partir du produit commercial (Matheson, Coleman et Bell, à 97% anhydre) ,
avec séchage sur hydrure de calcium et distillation sous vide, suivant la méthode de
Lucien
(Fig. 3). Un dosage de l'é- chantillon par une solution aqueuse d'iodatev ' ' a révélé une pureté de 100% à 0.2% près. Le composé deutérié (concen tration en deutérium 97%, <1% D^O )a été obtenu de Merck, Sharp et Dohme et utilisé sans aucune purification.Des échantillons de ~16 ml de N2H^ et ~12 ml de N2D^ ont été cristallisés dans des cellules cylindriques en Pyrex de 19 mm de diamètre qu'on laissait descendre lentement
23
Fig. 3: Montage pour la préparation de N0H4 anhydre.
(a) Ballon à reaction contenant N0H_4 et CaH2 (b) Laine de verre et
(c) Dégagement de (d) Cellule Raman (e) Piège à - 20°C
(f) Piège à azote liquide (g) Pompe à vide mécanique
N.B. Tous les joints rodés ont été lubrifiés avec de la graisse à base de silicone
quelques degrés au-dessous du point de fusion (1.4°C). La cristallisation devait être amorcée avec de la glace sèche car 1'hydrazine montre une tendance marquée à la surfusion. De plus, nous avons placé un anneau chauffant autour des cel lules, de façon à éviter que la cristallisation ne se produise d'abord sur les parois. Lorsque les échantillons étaient com plètement cristallisés, on abaissait lentement la température du bain jusqu'à - 12°C. Les cellules étaient ensuite transfé rées dans le Dewar de Pyrex préalablement refroidi, pour étude
avec le Cary 81. Dans tous les cas, 1'abaissement de la tempé rature jusqu'à celles d'analyse 070°,- 195° C) était assez lent, afin d'éviter le bris des cristaux.
3- STRUCTURES CRISTALLINES ET SPECTRES DE VIBRATION DE N^H^ * 2
L'analyse des spectres vibrationnels de 1'hydrazine à l'état cristallin, à la lumière de la théorie des groupes^'4)
devrait permettre de distinguer entre la structure proposée
2
d'après la diffraction des rayons X (C^) et celle obtenue par
2
diffraction des neutrons (C2). Les deux études concluent à la présence de deux molécules par maille élémentaire. Dans le
2
premier cas (C,^), la symétrie locale dans le cristal (Cg) implique une symétrie moléculaire C2v (forme éclipsée, cis) ou
25
.♦j"
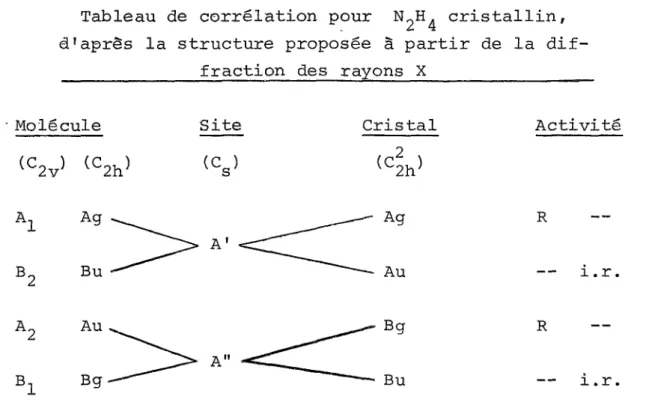
C2h. ^orme trans) , puisque le groupe de symétrie du site doit être un sous-groupe de celui de la molécule. Pour ces deux symé tries , les modes de vibration de la molécule se transforment en especes A' ou A" dans 1'approximation du site; celles-ci se scindent ensuite en deux composantes, l'une active en Raman et l'autre en infrarouge, par suite de l'effet de couplage dans le cristal (Tableau 1). D'après ce modèle, chacune des vibrations
Tableau 1
Tableau de corrélation pour N2H4 cristallin, d’après la structure proposée à partir de la dif-
fraction des rayons X
Molécule Site Cristal Activité
O
t o (C2h) (Cs} tC2h> A1 Ag A ' Ag R B2 Bu ' ~~~~-- Au i.r. a2 Au a" Bg R B1 Bg Bu i.r.Le groupe de symétrie du site est le groupe comprenant toutes les opérations engendrées par les éléments pie
symétrie ponctuelle passant par le centre de gravité d'une molécule dans le cristal.
internes de même que les trois modes de librations (Rx,Ry,R^,) devraient donner une composante en diffusion et une autre en absorption, les bandes ayant-des fréquences différentes dans les deux types de spectres puisqu'elles correspondent à des modes différents. Quant aux modes de translation empêchée, ils ne devraient être actifs qu'en Raman, les composantes infrarouges correspondant aux modes acoustiques, de fréquence zéro à la limite (k = 0). Cette structure implique donc neuf modes de réseau au total, trois actifs en infrarouge (libra tions) et six autres (3 librations et 3 translations) actifs en Raman. La présence de quelque neuf bandes observées par Baglin, Bush et Durig(^4) dans le spectre infrarouge entre 100 et 400 cm les a conduits à rejeter cette structure cristalline.
Si le cristal était du groupe C9 - P2^ , tel que déduit (
33
)de l'étude par diffraction des neutrons , les deux molécules dans une maille cristalline devraient se situer dans les posi tions générales, occupant ainsi des sites de symétrie C^. Les espèces vibrationnelles de la molécule isolée, de même que les rotations et les translations, se transformeraient alors en espèces A dans la symétrie du site, pour ensuite se dédoubler en composantes de symétrie A et B (actives à la fois en Raman
2
et en infrarouge) dans le groupe facteur C_ (Tableau 2).
Parmi les 12 modes de réseau prévus (6 translations et 6 libra tions) , trois (A + 2B) correspondent aux translations de
27
Tableau 2
Tableau de.correlation pour N2H4 cristallin, d'après la structure proposée à partir de la dif
fraction des neutrons
(modes acoustiques) et doivent être éliminés, Cette struc
ture qui se caractérise par un dédoublement de toutes les bandes, sauf celles correspondant aux modes de translation, et par la coïncidence des fréquences Raman et infrarouges, implique donc neuf modes de réseau (9R et 9 i.r.). Le spectre infrarouge lointain de 1'hydrazine cristallin^4) a clairement indiqué que cette structure cristalline était plus probable que celle déduite de la diffraction des rayons X.
4- RESULTATS' ET'DISCUSSION
4.1- Région des basses fréquences
On observe dans la région des basses fréquences du spec tre Raman de N2H4 cristallin une douzaine de bandes qui se retrouvent, décalées d'un facteur variant de 0.73 à 0.96, dans le spectre du composé deutérié (Fig. 4, Tableau 3). De plus, la plupart des bandes infrarouges n'ont pas d'équivalent en Raman. La variation des fréquences avec la température n'est pas responsable de cette disparité. Nous avons pensé que
celle-ci pouvait provenir d'une mauvaise cristallisation des
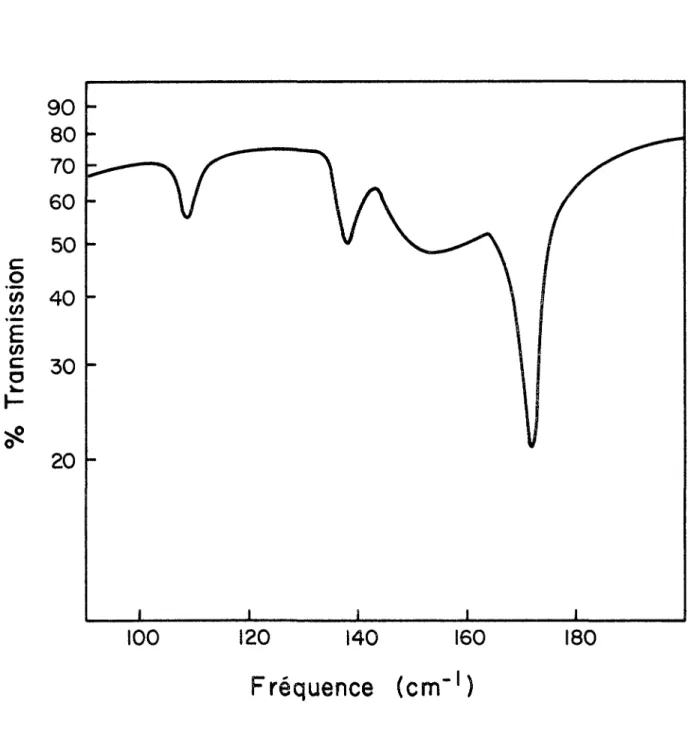
échantillons étudiés en infrarouge sur films de polyéthylène^^ . Afin d'éliminer cette possibilité, nous avons réexaminé le
spectre infrarouge entre 20 à 200 cm de N^H^, cristallisé lentement à partir du liquide entre deux fenêtres de quartz cristallin. Le spectre obtenu (Fig. 5) est sensiblement meil leur que celui déjà publié, mais on n'y retrouve aucune nouvelle bande. On note cependant, dans nos spectres que les deux pics aux fréquences les plus basses ont leur maxima à 108.5 et
138 cm ^ (comparativement à 106.7 et 136.3 cm , et que la bande à 172 cm est relativement intense par rapport à celle de 154 cm ^.
En se basant sur le grand nombre de bandes dans la région
-1
non-IN
T
E
N
S
IT
E
500 540 60 80 100 120 140 160 180 200 220 240 260 280 300 360 400 FREQUENCE(cm'1)
Fig. 4; Spectres Raman de N2H4 et de N2D4 (B) cristallins à - 195°C dans la région des basses fréquences.
Fente spectrale < 3 cm .
M va
Fréquences en cm de
-1
des bandes infrarouges et Raman (v < 700 cm--*-) et cristallins
*
Infrarouge Raman vdA+ Attribution
% % N2H4 N 2°4 <-130°C <-130°C -70'°C -195°C r
0
0U
-1 -19 5°C 94 97 87 90 0.928 Translation 106.7 101.5 0.951 Translation ~109 109 -101 101 0.927 Translation 112.5 10 7 108 0.960 Translation 122 116.5 0.955 Translation 126 132.5 117 123 0 .928 Libration 136.3 123 0 .902 Libration 154 136.9 0.889 Libration 154 142.3 0.924 Translation 172.5 156.5 0 .907 Libration 166 172 156.5 162.5 0.945 Translation 180.5 169.5 0.9 39 Translation 198 181 182 0 .919 Libration 226 197.5 0.874 Libration 212 226 209 217.5 0.962 Translation - 260 ~ 245 0.942 2 x 132.5 ? 274.9 243 0.884 Libration 278 289 254 264 0.913 Libration 405 277 0.684 Libration ou 405 298 0.736 Torsion 496 50 8 375 0.738 interne 524 533 39 4 0.739 Il II 6 50 492 0.757 Il II * Référence 34 .T
r
a
n
s
m
i
s
s
i
o
n
31
Fréquence (cm”1)
Fig. 5: Sgec.tre infrarouge de cristallin dans la région des basses fréquences.
coïncidence des fréquences Raman et infrarouges, il semble bien que nos résultats ne soient en accord avec aucune des deux
structures cristallines obtenues par diffraction. De plus, compte tenu des quelques douze bandes observées en Raman, en plus de celles à d'autres fréquences en infrarouge, nos ré sultats indiquent que la maille primitive du cristal contient plus de deux molécules, alors que le nombre maximum de modes de réseau serait de neuf. Il se peut également que la struc ture du cristal soit désordonnée par suite de la présence dans
/ 3Q\ le solide d'isomères de structure, comme dans le cas de N9F^v Cette possibilité ne peut être écartée, quoiqu'elle semble as sez improbable, vu la barrière de potentiel relativement éle vée à la position trans ( ~ 3 kcal) . De plus, la finesse des bandes dans le spectre Raman suggère une structure cristal
line bien ordonnée. Par conséquent, si la structure est bien MÔNOCLINIOUE tel qu'établi par la diffraction des rayons X et des neutrons, elle doit se situer dans un des groupes de
/ 39 )
la classe C2h ’ avec Quatre molécules dans des positions générales (sites de symétrie C^) de la maille primitive. Cette conclusion, basée sur la liste des positions générales et spé ciales des groupes du système monoclinique, n'est valable que si les molécules n'occupent qu'un seul ensemble de sites dans la maille cristalline. Il est à noter que pour un cristal moléculaire, comme c'est le cas ici, le contraire serait exceptionnel.
33
L' attribution des modes de réseau de N.H. et N„D. 2 4 2 4
cristallins s'explique assez bien dans 1'hypothèse d'une maille primitive de symétrie , contenant quatre molécules (symé trie locale ). La symétrie moléculaire est vraisemblable ment Cg dans ces conditions et il se pourrait que l'angle azimutal varie très peu en passant du gaz au solide. En se référant au tableau de corrélation (Tableau 4), ce modèle
im-Tableau 4
Tableau de corrélation pour N„H. cristallin, d'après la structure que nous proposons
Molécule Site Cristal Activité
(c1) (C2h)
/-Ag R —
?x' Ty, T,\ — Au i. r.
*x' %y' — Bg R
^ Bu i.r.
plique que chacun des degrés de liberté de translation et de rotation d'une molécule isolée devrait donner lieu à quatre modes de réseau dans le cristal, les espèces Ag et Bg n'étant
actives qu'en Raman et les deux autres (Au et Bu) en infrarouge seulement. Après avoir éliminé les trois modes acoustiques
(Au +2 Bu) , qui correspondent aux déplacements de 11 ensemble de la maille primitive suivant chacun des axes, on prévoit douze bandes actives en Raman (6 librations et 6 translations) et 9 en infrarouge (6 librations et 3 translations).
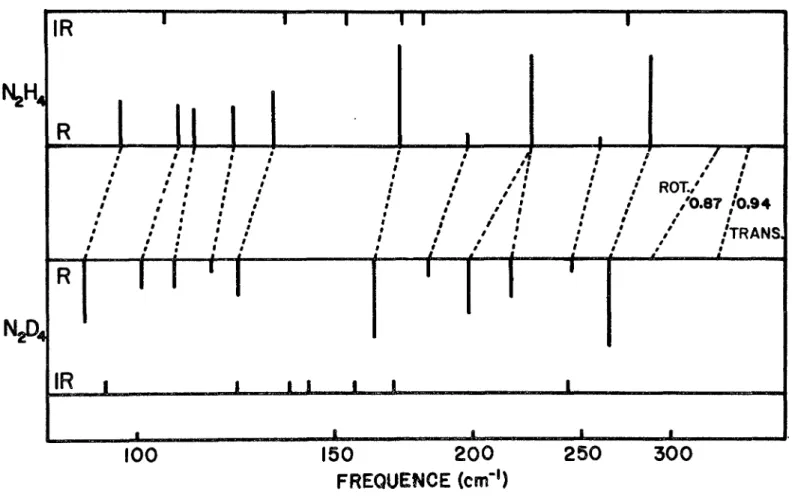
L1 attribution des modes de réseau de cristallin est facilitée par les résultats obtenus à partir du composé deutérié. La forme des bandes obtenues dans les deux cas et la mesure du décalage isotopique nous servent de guide. En première approximation, le décalage isotopique des fréquen ces devrait être proportionnel à la racine carrée du rapport des masses moléculaires (0.9 4) ou des moments d1 inertie, sui vant qu'il s'agit de translation ou de libration. D'après les paramètres de structure de la molécule libre, le rapport isotopique des fréquences devrait être de 0.72 pour les libra tions autour de l'axe mineur et de 0.87 pour celles autour des deux axes majeurs de la molécule. Les bandes de fréquences inférieures à 300 cm ^ qui montrent un décalage isotopique va riant entre 0.87 et 0.96, peuvent être attribuées soit à des modes de translation soit à des modes de libration autour des deux axes majeurs des molécules (Fig. 6). Le spectre infra
rouge lointain s'explique assez bien à partir du modèle que nous proposons et tous les modes prévus peuvent être attribués de
/TRANS,
>0 200
FREQUENCE (cm-1)
Fig. 6: Correlation entre les frequences des modes de réseau observés en Raman et en infrarouge pour N2H4 et N2D4 cristallins.
w U1
plus" compliquée du fait que six modes de translation sont pré vus (en comparaison de trois seulement en infrarouge), en plus de quatre librations autour des axes majeurs. On obser ve au-dessous de 300 cm , une dizaine de pics. Les diffé rences entre les rapports isotopiques calculés et observés sont importantes et indiquent un couplage appréciable entre ces
divers modes de réseau. C'est ainsi que la distinction entre modes de translation et libration qui apparaît dans le Tableau 3 et qui est basée sur le calcul des décalages isotopiques n'est que très approximative.
Par analogie avec le cas de H2°2 solide'41), on de vrait retrouver dans la région de 400 à 800 cm ^ des spectres de 1'hydrazine deux séries de bandes, correspondant aux modes de torsion interne et à ceux de libration autour de l'axe mineur de la molécule. Ces bandes devraient toutes se retrouver,
décalées par un facteur 0.72 environ, dans le spectre du com posé deutérié. Les deux doublets prévus en infrarouge se
-1 -i
situent probablement à 650 cm (492 cm dans ) et a
405 cm (277/298 cm ^ dans ^D^) . Les derniers ont été attri bués par Baglin, Bush et Durig au mode de torsion interne, à la suite d'une analyse vibrationnelle du solide. Ces auteurs ont également expliqué la bande forte à 650 cm ^ par une combinai-
son de celle à 405 cm avec un mode de réseau. Il semble plus probable que les deux bandes fortes observées en infrarouge soient toutes deux des fondamentales, à cause du décalage
37
isotopique important en passant au produit deutérié. Il est cependant difficile de déterminer laquelle provient de la tor sion interne. S'il s'agissait du pic de plus basse fréquence, on pourrait en conclure que 1'orientation relative des groupe ments . NH^ des molécules dans le cristal est différente de celle des molécules libres. En effet, si l'angle azimutal dans le solide est identique à celui de la molécule libre, l'effet de la barrière de potentiel s'opposant à la torsion interne devrait s'ajouter à celui des liaisons hydrogène et conduire à une fréquence de torsion plus élevée que celle de la libration correspondante. Dans le spectre Raman du solide, un des doublets prévus apparaît faiblement à 537/50 8 cm ^
(394/375 cm ^ dans N2D^), et l'autre n'a pas été localisé.
4.2- Région des hautes fréquences
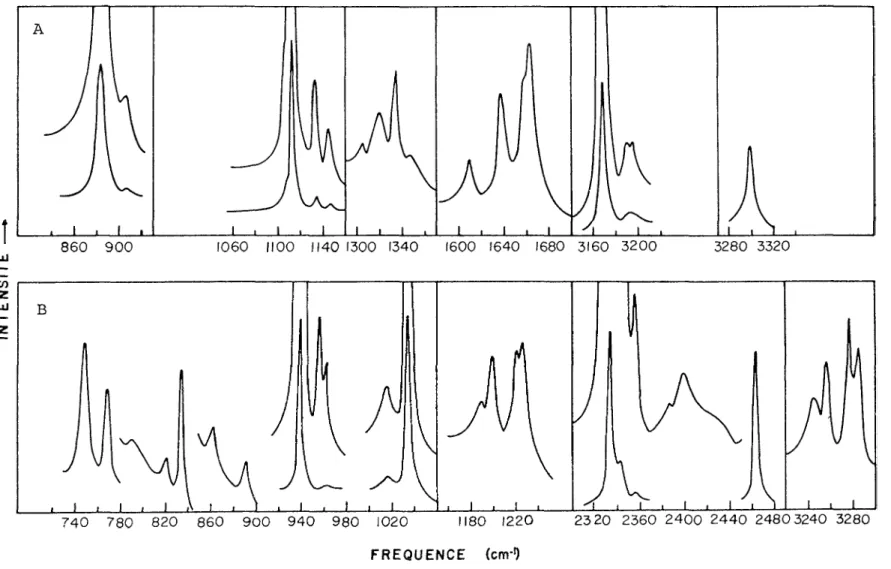
Le nombre des bandes observées dans la région de 700 à 3500 cm ^ des spectres Raman de N^H^ et N2D4 ^ 1'état cristallin (Fig. 7) est de beaucoup supérieur à celui obtenu en infrarouge. Notre attribution des bandes Raman (Tableaux 5a et 5b)rejoint celle des bandes infrarouges faite par Durig et ses collaborateurs(24). Nos résultats complètent les leurs et semblent être en accord avec la structure cristalline que nous avons déjà proposée. Selon ce modèle, chacune des fondamentales devrait donner des doublets tant en diffusion qu'en absorption,
IN T E N S 2320 2360
2400 2440
24803240 3280 180 1220 740 780 820 860 900 940 980 1020 FREQUENCE (cm'1)Fig. 7: Spectres Raman de N0H^ (A) et N2D4 ^ cristallins à - 195°C.
Fente spectrale < 3 cm \ U)
Tableau 5a
Fréquences en cm des bandes infrarouges et Raman de N2H4 (v 700 cm-1)
Vapeur* Liquide* _________ Solide
i. r. i. r. R i . r. * R(-70°C) R(-195°C) Intensité"*" Attributiont 780 871 872 884 883 883 82 Déformation N-N-H 901 905 3 (Au, Bg) 933 9 36 966 1042 988 1066 1098 1098 -1100 -1106 15 1100 1111 1113 237 Valence N-N (Ag) 1126 1130 1133 18 et déformation N-N-H (Au, Bg) 1133 1144 9 1275 1283 1304 1305 1304 1 1316 1319 3 Déformation N-N-H 1324 1329 1333 7 (Bu, Ag) 1350 1345 1347 1 suite page 40 U)
Vapeur* Liquide* Solide
i.r. i.r. R i.r.* R(-70°C) R(-19 5°C) Intensité' Attribution^
1587 1608 1614 1603 1610 1609 6 1628 16 39 1637 14 Déformation H-N-H 1655 -1658 11 (Bu, Ag) 1661 1663 21 30 85 3174 3168 796 3139 3188 58 3280 3189 3186 3200 3195 3194 64 Valence N-H
3314 3248 (Au, Bu, Ag, Bg)
3325 3264
3350 3332 3333 3310 3300 3299 428
* Référence 24 .
t Intensité approximative basée sur la hauteur relative des pics dans le spectre Raman à -19 5°C.
* Les symétries indiquées entre parenthèses sont celles des vibrations de la molécule libre de forme trans (0-%)•
Tableau 5b
Fréquences en cm des bandes infrarouges et Raman de ^4 (v 700 cm "*")
Vapeur* Liquide* _________ Solide
i.r. i.r. R i.r.*
0
0
0
r -1 R(-195°C) Intensité^* Attribution^ 688 727 733 744 748 749 11 Déformation N-N-D 722 766 769 7 785 791 1 Déformation N-N-D (n2d3h) ? 787 80 6 814 822 821 O 827 834 11 Déformation N-N-D 860 862 3 Déformation N-N-D(N2D-H) 890 891 3 15 14 ^ Valence ND2~ ND2 9 36 938 938 940 9 39 530 Valence N-N 956 956 9 962 962 5 Déformation N-N-D 985 987 1004 1015 1016 7 Valence N-N (N„D_H) 1032 1030 1041 1031 1035 91 Z j 1140 1184 1192 1190 2 Déformation D-N-D H 119 7 1194 119 8 1202 1200 8 Suite page 42Vapeur* Liquide* Solide Intensité"*" Attribution t i. r. i.r. R i.r.* R(-70°C) R(-195°C) 1213 1221 1221 6 Reformation D-N-D 1226 1225 7 2338 2333 540 2353 2348 2347 2348 2342 72 Valence N-D 2358 2355 19 2392 2386 5 2415 2411 2407 2409 2399 12 Valence N-D (N^D^H) 2431 2469 2430 2435 4 2493 2487 2479 2467 2463 460 Valence N-D 3254 3244 4 3263 3256 8 Valence N-H ( N„D-H) 3278 3275 10 1 5 3283 3284 7 * Référence 24,
- Intensité approximative basée sur la hauteur relative des pics dans le spectre Raman à - 195°C.
43
les composantes ayant des fréquences différentes dans les deux types de spectres. Dans la région des quatre vibrations de valence N-H(N-D) du spectre Raman des solides, on observe quatre pics alors qu'en infrarouge^4) ^eux seulement ont été
décelées. La non-coïncidence des fréquences ne permet pas de tirer de conclusion quant à la structure du cristal, puisque toutes les composantes n'ont pas été observées.
La situation est plus claire dans la région des deux modes de déformation H-N-H où quatre bandes sont clairement visibles dans le spectre Raman. Le fait que chacun de ces modes soit dédoublé est incompatible avec la structure déduite de
l'étude par diffraction des rayons X, pour laquelle une seule composante Raman est prédite pour chacune des fondamentales. La distinction entre la structure obtenue à partir de la dif fraction des neutrons et celle à quatre molécules par maille primitive proposée plus haut est Cependant, plus difficile. Dans le premier cas, et au contraire du second, il devrait y avoir coïncidence des fréquences des doublets prévus en infra rouge et observés en Raman. Une comparaison entre les fré quences des bandes infrarouges et celles des composantes Raman les plus proches montre dans plusieurs cas des diffé rences de l'ordre de 6 cm ^. Ces différences assez faibles, qui peuvent s'expliquer en partie pàr les imprécisions dans la mesure des fréquences, favorisent légèrement la structure à
quatre molécules par maille, de préférence à celle obtenue par la diffraction des neutrons.
La région de 700 à 1350 cm"^ du spectre Raman est assez complexe. L1 attribution des bandes provenant des quatre modes faisant intervenir les déformations N-N-H est rendue diffi cile par suite du couplage de certaines de ces vibrations avec celle de valence N-N. Cet effet, différent dans N^H^ et
, explique certains décalages isotopiques différents de ceux prévus. Les rapports isotopiques des fréquences de vraient être de l'ordre de 0.7 pour les vibrations de défor mation N-N-H et de 0.9 environ pour la vibration de valence N-N. Deux des modes de déformation N-N-H apparaissent sous la forme de quatre bandes vers 1325 cm ^ dans le spectre de
cristallin. Les bandes correspondantes dans N^D^ sont obscurcies par celles, très fortes de valence N-N. Un troisième mode de déformation N-N-H apparaît à une fréquence très voisine de celle de la vibration de valence N-N. Les pics sont cependant bien séparés dans le spectre du composé
deutérié, où le mode de déformation se présente sous forme de doublet à 821/834 cm ^. Il s'agit là sans doute d'un mode antisymétrique, vu le peu d'interaction avec le mode de valence. Il n'en va pas de même du dernier mode de déformation N-N-H
— 1 w 1
apparaissant à 883/905 cm et qui est déplace à 749/769 cm dans le spectre de N^D^. Le facteur isotopique observé (0.85) diffère nettement de celui prévu théoriquement (0.7) . De plus,
45
en passant au composé deutérié. On note un comportement in verse chez le mode de valence N-N. Il semble donc que le mode
-1
de déformation (mode symétrique) vers 890 cm dans le spectre de N^H^ soit fortement.couplé avec le mode de
valence N-N. Cela conduit à une redistribution des intensités au profit de la bande la moins intense. En passant au compo sé deutérié, les fréquences de ces vibrations sont plus diffé rentes au départ et le couplage moins grand, ce qui conduit à une distribution plus normale-des fréquences et des intensi
tés. On note également dans cette région une certaine disparité entre les fréquences infrarouges et Raman.
Le mode de valence N-N pose un problème puisqu’il semble donner un triplet en Raman. Cela peut s'expliquer dans le cas de NoH4 on attribue au mode de valence le doublet à 1113/1133 cm ^ qui est très peu influencé par la température alors que les fréquences des bandes faibles à 1106 et 1144 cm 1 qui pourraient provenir d’un mode de déformation N-N-H sont' abaissées de façon appréciable par une élévation de tempé rature. Dans le spectre de N^D^ cependant, en plus du dou blet à 939/956 cm ^ , logiquement attribuable au mode de valence N-N, on observe une autre bande très faible à 962 cm . Nous croyons que la ressemblance entre les spectres de N2H4 et N^D^ dans cette région n'est qu'accidentelle en ce qui re garde les pies faibles du côté des hautes fréquences. La bande
-1 -1
due à un mode de déformation N-N-D, lequel se retrouve vers 1325 cm dans . Les quelques' ;3%'"'d4Hydrogène dans r_l’échan tillon de ^2D4 devraient (statistiquement) donner environ
11% de , les autres impuretés isotopiques étant en con
centration négligeable. La seule bande dans le spectre Raman qui puisse être attribuée au mode de valence N-N de cette impureté est celle à 1035 cm ^ . Son intensité relative indi que une" teneur- un peu forte (17%) en N2D3H (5%H) , mais il se peut qu'elle soit superposée a une bande due à un mode de défor mation N-N-D.
Diverses autres bandes faibles sont apparues dans le spec tre Raman de N2D4 et sont explicables pour la plupart par
la présence de 1'impureté isotopique N^D^H. Les quatre
ban--1
des entre 3244 et 3284 cm , dues à des vibrations de valence N-H des molécules N^D^H , pour la plupart isolées dans la matrice solide de ^D^ sont d'un intérêt tout particulier (Fig. 8a). Le même phénomène se produit lorsqu'une petite quantité de N2H3D (4%) est présente dans (Fig. 8b) , alors que quatre bandes de valence N-D (à 2396, 2402, 2428 et 2431 cm 1) apparaissent dans le spectre Raman du solide. Ces pics ne sont donc pas dus à des harmoniques ou combinai sons, ou encore à un effet de couplage entre paires de molécu les de 1'impureté dans le cristal. Nous croyons plutôt qu'ils proviennent des vibrations de quatre groupements N-H (ou N-D) non-équivalents dans le cristal. Les travaux de R. Kopelman
In
te
n
s
it
é
47 Fréquence (cm" )Fig. 8b Spectre Raman dans la 'région des modes de valence N-D de (4%)
laissent supposer que des éclatements de bandes peuvent être causés par des orientations différentes d'une impureté iso topique isolée dans un cristal. En effet, si le champ cris tallin autour d'une molécule comme N2D^H dans N2D4 n'est pas isotrope, alors certaines orientations sont énergétiquement distinctes et chacune d'elles peut donner lieu à une composante vibrationnelle. Si les quatre liaisons N-H (ou N-D) ne sont pas équivalentes dans le cristal, les molécules doivent néces sairement occuper des sites de symétrie . Cette observa tion nous porte à refuter la structure obtenue par diffraction des rayons X (sites de symétrie C ), mais elle ne nous permet pas de distinguer entre celle obtenue par diffraction des neu trons et celle que nous avons proposée plus haut. Dans 1'hypo thèse d'une structure où deux types d'isomères structuraux
(présumément dans les configurations gauche et trans)seraient présents, on devrait attribuer deux des quatre bandes observées
â chacun des isomères. Etant donné que les quatre composantes Raman ont des intensités assez semblables, il faudrait que les deux isomères existent dans des concentrations à peu près égales. Cela est assez improbable du point de vue énergétique (barrière de 3 kcal/mole à la position trans) et devrait conduire à une entropie résiduelle appréciable au zéro degré absolu.
49
5- CONCLUSION
Même si les présents résultats s'expliquent de façon très satisfaisante en fonction d'une maille cristalline mono clinique de la classe C^^ , avec quatre molécules situées dans des positions générales, on ne saurait prétendre que nos conclusions sont d'une certitude absolue. En effet, ces déduc tions sont basées sur 1'hypothèse fondamentale que les bandes observées ont été interprétées de façon correcte. Cependant, 1'analyse des spectres Raman de N2H4 et N2D4 cristallins, compte tenu des résultats précédemment obtenus en infrarouge, semble incompatible avec les structures cristallines déduites des études par diffraction sur N2H4 solide. Il est probable que les positions des atomes d'azote dans la maille cristalline, telles que déterminées par diffraction, sont essentiellement correctes et que 1'incompatibilité avec les résultats spec troscopiques est reliée à la configuration et à 1'orientation des molécules dans le cristal. Une étude par diffraction des neutrons sur cristallin semble particulièrement indiquée.
SPECTRES DE VIBRATION DE L'OXALATE ACIDE D'HYDRAZINIUM (N^HC^)
SPECTRES DE VIBRATION DE L'OXALATE ACIDE D'HYDRAZINIUM (N^-HCgO.)
ET DE. .SON ANALOGUE. DEUTERIE.. .(N^DC^ )
1- INTRODUCTION
L'étude des spectres de vibration de 1'hydrazine à l'é tat cristallin nous avait amenés à faire un relevé complet des données spectroscopiques accumulées jusque-là pour cette molé cule. Une telle compilation nous a permis de classer les fré quences caractéristiques des modes fondamentaux de cette molé cule et nous a conduits à nous intéresser à la structure de quelques sels dérivés de 1'hydrazine.
Des études par diffraction^^ on^- indiqué que les complexes N^^-Acide peuvent exister sous forme de sels d'hydrazinium (N2H^+) et d'hydrazonium (N2Hg++ ). Les spec tres infrarouges et Raman de plusieurs sels de 1'hydrazine ^ ont permis de mettre en évidence les variations importantes des fréquences des vibrations N-N et N-H , suite à 1'addition de un ou deux protons à la molécule d'hydrazine. C'est dans le but de mieux caractériser cette influence que nous avons étudié les spectres Raman et repris les spectres infrarouges de 1'oxalate acide d'hydrazinium.
L'hydrazine peut former deux types de complexes avec l'acide oxalique: N>,H^.H2C20^ et 2N0H^.H2C20^ . La struc ture cristalline du premier composé a d'abord été étudiée par diffraction des rayons / et confirmée par diffrac tion des neutrons(50). ces travaux ont mis en évidence le caractère ionique du cristal (N2H5 + HC2°4 ^ ' ^ont la mail le élémentaire monoclinique à deux molécules est définie
2
par le groupe spatial P2 i/m - C^. L'étude par diffraction des neutrons(50) a permis de localiser les atomes d'hydro
gène dans le cristal où on distingue plusieurs types de
liaisons hydrogène. Les ions oxalate sont plans et enchaînés les uns aux autres en un réseau bidimensionnel par des ponts hydrogène symétriques et très courts (2.45 8). Les ions hydrazinium ( H ?N - sont reliés entre eux par des ponts hydrogène du type NH+...N (2.86 R). Les deux types d'ions sont reliés entre eux, d'une part, par des liaisons hydrogène triples du type NE ...O (2.99, 3.12 et 3.42 8), et,
d'autre part, par des liaisons hydrogène doubles du type NH+...0 (2.87 et 2.89 8).
Les ions hydrazinium ont une configuration décalée dans le solide. Les distances N-H du groupement -NH2 sont de
1.02 8 , alors que le groupement -NH^4- présente deux liaisons
N-H+ de 1.02 8 et une autre, dans un plan de symétrie, de 1.04 8. La symétrie de ces ions (ainsi que celle du site) est alors
0^. Les ions oxalate se caractérisent chacun par deux
liaisons C - O (1.236 8) et deux liaisons C-O...H (1.280 8), ce qui joint à leur planéité, leur confère une symétrie CL^.
53
Les sites qu'ils occupent sont de symétrie CL . L'atome d'hy drogêne du pont 0...H...0 occupe lui-même un site CL .
Lindgren, De Villepin et Novak ont récemment étu dié les spectres dans le proche infrarouge de NgH^HC^O^ et ^Dj-DC^O^ en suspension dans le nujol et le fluorolube, en vue de mettre en évidence les différents types de liaisons hydrogène dans ces solides. Leurs résultats confirment
1'existence d'un pont hydrogène 0...H...0 symétrique dans le composé hydrogéné. De plus, leur spectre à basse tempéra ture du composé deutérié semble indiquer une liaison 0-D...0 asymétrique. Dans le but de vérifier ces résultats et leur interprétation, nous avons repris les spectres infrarouges de N^Hç-HC^O^ et de N^D^DC^O^ et complété le travail en enregis
trant les spectres dans 1'infrarouge lointain et ceux en diffusion Raman. Nous avons également procédé à une étude détaillée du
spectre Raman en lumière polarisée d'un monocristal de N^H^HC^O^ ^ afin de vérifier 1'exactitude de la structure cristalline propo sée pour ce composé.
Au cours de notre travail, nous avons pris connaissance de la publication récente de Selvarajan sur les spectres Raman et infrarouge de 1'oxalate acide d'hydrazinium à 30°C. Nous avons choisi d'ignorer ces résultats de piètre qualité de même que 1'interprétation incohérente qu'on en donne.
2- EFFET RAMAN EN LUMIERE POLARISEE
D'après la théorie classique de la diffusion de la lu mière , lorsqu'il y a interaction entre une onde lumineuse et une molécule, cette dernière est assujettie à un champ électri que oscillant à la fréquence de la lumière, vQ . Sous 1'ef fet de ce champ électrique oscillant, il y a déplacement des électrons dans la molécule : un dipôle est ainsi induit. Ce dipôle peut irradier de la lumière dans toutes les directions sauf suivant sa ligne d'action. La valeur du moment dipolaire induit, M , dépend de 1'amplitude de l'onde lumineuse, E , et de la polarisabilité a de la molécule, qui est une mesure de la facilité avec laquelle les électrons peuvent être dépla cés à 1'intérieur de celle-ci:
M = a E fl]
Etant donné que le moment dipolaire induit, de même que le champ électrique inducteur, sont des vecteurs qui ne sont pas nécessairement parallèles, a doit avoir la forme d'un tenseur symétrique a a a
XX
xy XZ a a a xy yy yz a a a xz yz zzM = [t] ê
m
55
Le champ électrique oscille à la fréquence v et son amplitude maximum est E
o
M = [T] E
q cos(2TrvQt)
[4]
Si.le dipole oscillant réémet l'onde lumineuse absorbée sans variation de fréquence, on a.1'effet Rayleigh. Pour rendre compte de l'effet Raman, on doit admettre que la polarisabi lité de la molécule varie au rythme des déplacements nucléaires. En première approximation,
H-= Ho + Ç (i|~>0qi
m
L'indice zéro se rapporte à la configuration à 1'équilibre de la molécule, alors que q^ désigne l'une des coordonnées normales de déplacement qui décrivent les oscillations fon damentales des molécules. Ces vibrations normales sont au nom bre de 3N-6 (3N-5 dans le cas.des molécules linéaires), N étant le nombre d'atomes par molécule. On a donc,
q^ = Q° cos (2-iïv^ t) [ 6 ]
où v. désigne la fréquence de la vibration correspondant à la coordonnée normale q^
Finalement, en combinant les équations 4, 5 et 6, on obtient :
M = Eo [t] cos C2ttv t)' +
i E
ZQ?
4^-) /cos2ir(v
- v.)to L ■Jo O A 0.1 og. O X O 1
+ cos 2 it ( v - v . ) t
o 1 [7]
Le premier terme de 1'equation 7 décrit la diffusion Rayleigh, qui depend de la polarisabilité à l'équilibre jjrJQ . Le deuxiè me explique l'effet Raman dû aux oscillations fondamentales dont les -propriétés sont déterminées par le tenseur de polarisabi lité dérivé
tM Sg O *
Lorsque la molécule possède des éléments de symétrie, certains des coefficients a du tenseur de polarisabilité
[équation 2] peuvent s 1 annuler. Il en résulte que 11 intensité et le degré de polarisation des bandes Raman obéissent à des règles bien définies, dont découlent les règles de sélection pour 1'activité optique des vibrations normales. En effet, chaque mouvement de vibration (i) d'une molécule, lequel pos sède- une symétrie propre, peut être- décrit par une coordonnée de déplacement généralisée, q^. , qui est en fait une combinai son (vectorielle) des coordonnées de déplacements atomiques. La variation de g^ lors de la vibration entraîne [équation 5] une variation des seuls éléments a du tenseur de polarisabili té jVJ qui possèdent la même symétrie que q^. C'est en se basant sur cette relation que l'on peut déduire l'état de pola risation des vibrations qui donnent lieu aux raies Raman.
57
En utilisant un faisceau laser qui se caractérise par sa complète' polarisation, et en variant 1'orientation du plan de polarisation de la lumière incidente (à l'aide d'une lame demi- onde) et diffusée (à l'aide d'un analyseur) par rapport a
celle d'un cristal unique, on peut déterminer sans ambiguïté la symétrie de toutes les bandes Raman de ce cristal. Cela suppose évidemment que le cristal-est bien aligné suivant ses axes et que ses faces sont polies de façon à éviter toute réflexion parasite.
A titre d'exemple, si on illumine un cristal suivant son axe X , avec une lumière polarisée dans le plan XI , et
si on examine, suivant l'axe I , la lumière diffusée polari sée dans le plan IZ , seules les bandes résultant des vibra tions qui causent une variation de l'élément a (dcu *0) du
y z y z
tenseur de polarisabilité apparaîtront dans le spectre. Ces conditions peuvent se résumer par X(ÏZ)Y , en utilisant la notation de Porto (Fig. 9).
3- PARTIE EXPERIMENTALE
L'oxalate acide^d'hydrazinium-^H^H^O^) et son analogue deutérié ont été préparés selon la méthode décrite par
Turrentine, en mélangeant à chaud des solutions aqueuses contenant chacune des quantités équimoléculaires de N2H^.H20 et de H2C20^.2H20. Les produits de départ étaient de qualité commerciale ("Reagent Grade", The Fischer Scientific Co.).
X(IZ)X
X : direction de propagation de la lumière incidente. X(Y : plan de polarisation de la lumière incidente.
Z)Y : plan de polarisation de la lumière diffusée.
X : direction de propagation de la lumière diffusée. (IZ): seules les vibrations qui causent une variation de
11 élément a du tenseur de polarisabilité peuvent
donner lieu à une bande observable en Raman dans ces conditions.
Fig. 9; Notation de Porto polarisée.
(66)
pour l'effet Raman en lumière
Le produit hydrogéné final a été obtenu par évaporation lente d'une solution saturée dans l'eau chaude. Dans le cas du composé deutérié, les réactifs ont dû être préparés. Le mono hydrate NgD^.D^O a été obtenu en mélangeant à de l'eau lourde
(99% D^O) de 1'hydrazine deutérié acheté de Merck, Sharp
and Dohme (concentration en deutérium 97%). La solution d'acide oxalique deutérié a été préparée par la réaction d'oxalate de
59
calcium (CaC^O^) avec un mélange de DgSO^ dans D^O (1:1 en volume) et par filtration du CaSO^J à chaud. Le mélange D2S04//D2° a obtenu en faisant absorber très lentement par de l'eau lourde refroidie à 0°C des vapeurs d'anhydride sulfurique (SO^) provenant de la distillation d'un oléum com mercial. La pureté isotopique du N^D^DC^O^ n'a pas été dé terminée avec précision. Cependant, d'après 1'intensité des bandes infrarouges dues à l'espèce N^D^HDC^O^, on a estimé la teneur en deutérium de 1'échantillon à 95% environ.
Les spectres dans le proche infrarouge et dans 1'in frarouge lointain, à 30°C et - 180°C, ont été obtenus â par tir de suspensions dans le nujol et dans le fluorolube. Les spectres Raman d'échantillons polycristallins ont été enre gistrés à 30°C et - 195°C, à partir d'une fine poudre entas sée dans une cellule conique.
Les monocristaux de NgH^HCgO^ qui ont servi â 1'enre gistrement des spectres Raman en lumière polarisée, ont été ob tenus par évaporation lente à température ambiante, d'une solu tion aqueuse saturée. Ces cristaux avaient la forme de parallé lépipèdes réguliers de dimensions approximatives 1.5 x 1 x 5 mm
(Fig. 10).
L'identification des axes cristallographiques a, b, a été effectuée par diffraction des rayons X, grâce à la
Fig. 10 Profil d'un' monocristal de
et orientation des axes cristallographiques.
collaboration' du professeur André Beauchamp de 11 Université de Montréal. Plusieurs cristaux non-retouches ont été étudiés
sous deux orientations différentes-, Y(- -)X et Y(- -) Z Les variations des intensités relatives de certaines bandes dans: ces deux orientations ont: permis une plus grande certitude dans: la détermination des symétries: de vibration. Cependant, le fait qu'aucune face des cristaux n'était perpendiculaire â- 1:' axe - d ' observation dans ces conditions n'a pas permis d'obte nir: l'extietion complète de toutes: les bandes là où la symétrie le prévoyait. En effet, des. réflexions aux interfaces pro