FACULTÉ DES SCIENCES
THÈSE PRÉSENTÉE
A L'ECOLE DES GRADUÉS DE L'UNIVERSITÉ LAVAL
POUR OBTENIR
LE GRADE DE DOCTEUR ÈS SCIENCES
PAR
JACQUELINE MICHEL Licenciée es sciences
de la faculté des sciences d'Orsay (France)
CONTRIBUTION A L'ÉTUDE DE L'INDUCTION ASYMÉTRIQUE 1-3 SUR DES Ji-HYDROXYCÉTONES
REMERCIEMENTS
Je tiens â exprimer ma sincère reconnaissance et ma vive gratitude à Madame le Professeur P. Canonne pour l'aide et les encouragements constants qu'elle m'a apportés tout au long de mes recherches. Je désire la remercier tant pour les nom breux conseils judicieux et les suggestions qu'elle n'a cessé de me prodiguer pendant la réalisation de ce travail, que pour 11 assistance humaine dont elle a toujours su faire preuve à mon égard.
Mes remerciements vont également au Conseil des Arts du Canada, qui, par l'octroi d'une bourse, a rendu possible 1'ac complissement de ce travail.
Je suis reconnaissante envers tous mes camarades de chi mie organique et plus particulièrement ceux du laboratoire pour leur coopération et leur intérêt pour mon travail.
Je voudrais également exprimer tous mes remerciements à Madame C. Comeau pour la réalisation typographique de cette thèse qu'elle a menée avec compétence et dévouement.
Je ne saurais oublier Mademoiselle D. Thibault, dont j'ai pu apprécier la gentillesse et la disponibilité pour son assis
tance technique très utile.
C'est par l'octroi de subventions du Conseil National de Recherches du Canada que ce travail a pu s'accomplir, ce dont je l'en remercie.
V
TABLE DES MATIERES
Page
INTRODUCTION... 1
CHAPITRE I CONSTITUTION DES ORGANOMAGNESIENS ET ACTION SUR LES COMPOSES CARBONYLES . . 4
CHAPITRE II ETUDE DES 3-HYDROXYCETONES ... 10
II. 1 Introduction... 10
II. 2 Préparation... 16
II. 3 Mécanisme... 18
II. 4 Etude conformationnelle... 23
A Etude par Infrarouge ... 23
B Etude par R.M.N... 29
C Hypothèses conformationnelles . . 35
CHAPITRE III ETUDE DES 3-DIOLS BITERTIAIRES. ... 43
III. 1 Introduction... 43
III. 2 Préparation... 46
III. 3 Mécanisme... 51
111.4 Purification des 3-diols et séparation des diastéréoisomères... 54
111.5 Etude configurationnelle et conforma tionnelle...57
A Etude par Infrarouge ... 58
B Etude par R.M.N... 73
C Hypothèses conformationnelles . . 86
CHAPITRE IV INDUCTION ASYMETRIQUE 1-3 98
IV. 2 IV. 3
Induction asymétrique 1-3.
Induction asymétrique 1-3 sur des méthylcétones 3-hydroxylées .
A Dosage des mélanges de 3-diols diastéréoisomêres... B Proportion des mélanges de
3-diols diastéréoisomêres . C Proposition de modèles d'état
de transition ... Page . 106 . 117 . 118 . 119 . 125 CHAPITRE V V.l V. 2
SYNTHESES DE L'HYDROXY-3 TRIPHENYL-1,3,3 PROFANONS (15) ET DES 3-DIOLS
BITERTIAI-RES (48-53)... 131
Préparation et étude de 11 hydroxy-3, triphényl-1,3,3 propanone (l j...131
Préparation et étude des 3-diols biter- tiaires (48-53)... 134
CONCLUSION... 138
PARTIE EXPERIMENTALE ... 140
REMARQUES GENERALES... 140
CHAPITRE I PREPARATION DES SOLUTIONS D'ORGANOMA-GNESIENS MIXTES ... 143
CHAPITRE II PREPARATION DES 3-HYDROXYCETONES . . . 146
CHAPITRE III PREPARATION DES 3-DIOLS BITERTIAIRES . . 153
INTRODUCTION
Ce travail s'inscrit dans 1'ensemble des recherches en treprises actuellement sur les conformations de 3-diols (1-4) ou de g-aminoalcools (5) et se veut être une contribution à l'étude stérêochimique de réduction de 3-hydroxycétones pré sentant un centre asymétrique en 3 de la fonction carbonyle par des organomagnésiens (6). Par ce type de réactions, il se situe dans une suite logique des travaux commencés au labora^ toire.
Une étude réalisée par Bilodeau (7) portait sur 1'action des organomagnésiens sur des 3-dicétones symétriques et asymé triques et mettait en évidence la présence de B-hydroxycé tone s et de 3-diols bitertiaires.
Canonne et Leitch (8)se sont intéressés également à cette réaction dans le but d'obtenir une 3-hydroxycétone pure à subs tituant arylaliphatique afin de synthétiser, par deshydrocycli sation, des naphtalênes polyméthylés.
Les travaux de Castelli (9) visent à déterminer le méca nisme de réaction des organomagnésiens sur des 3-hydroxyesters substitués en a ou cycliques.
C'est dans ce cadre de 11 action des organomagnësiens sur des composés 3-dicarbonylés que les problèmes de diastéréoiso- mérie des B-diols nous ont semblé présenter un certain inté rêt, et à cette fin, nous avons réalisé la synthèse d'un grand nombre de B-diols, composés nouveaux, de formule:
R - C(OH)CHg - CHg - C(OH)R'. Les substituants R et R' étant des groupements phényles substitués, benzyliques également subs titués, cyclaniques, aliphatiques et allyliques, ils ont été introduits sur la pentanedione-2,4 puis sur les B-hydroxycéto ne s en fonction de 11 encombrement stérique et des effets élec troniques qu'ils laissent prévoir.
O ?
VRMgX
ch3-c-ch2-c -ch3 ____________ » 2)hydrolyse
OH
0
i
il
R-C-CH,-C-CH iL
ch3OH
OH
yRMgX
R •—C-CHy-C— R'<—---i z i
CHg
Ch^
2Jhydrolyse
Auparavant, nous nous étions intéressés à un nouveau mode de synthèse de 1'hydroxy-3 triphényl-1,3,3 propanone à partir du benzoylacétate d'éthyle. Cette hydroxycétone solide, donc facilement purifiable,nous a permis d'étudier certaines des conditions opératoires de la réduction des 3-hydroxycétones par des halogénures d1arylmagnésium.
3
avancées sur la constitution des organomagnésiens et sur les mécanismes de leur réaction sur les cétones, nous aborderons au chapitre II leur addition aux (3-dicétones menant aux 0-hydroxycétones. Apres que différents modes de synthèse auront été envisagés, l'étude spectrographique permettra de discuter des différentes hypothè ses conformationnelles.
Le troisième chapitre traitera des g-diols bitertiaires: leurs préparations, leurs études spectroscopiques, leurs con figurations - détermination des diastéréoisomères érythro et thréo - leurs conformations seront successivement décrites.
Un quatrième chapitre sur 11 induction asymétrique-1,3 tentera de discuter des mécanismes d1 attaque, de 1'influence relative des réactifs de Grignard et des 3-hydroxycétones.
Ensuite, au chapitre V, nous verrons la synthèse de l'hy- droxy-3 triphényl-1,3,3 propanone et des 3-diols en dérivant.
CONSTITUTION DES ORGANOMAGNESIENS ET ACTION SUR LES COMPOSES CARBONYLES
A deux stades de notre synthèse - réduction de la pentane dione-2,4, puis celle des B-hydroxycétones obtenues - nous fai sons intervenir des organomagnêsiens solvatés dans l'éther anhydre. Nous allons brièvement rappeler les dernières hypo thèses énoncées sur la constitution des organomagnêsiens qui, depuis Grignard, se sont révélés comme d'importants agents de synthèse.
Ces "Grignards" ont soulevé beaucoup de controverses de puis que Grignard (10) eût rapporté en 1900 que les halogénu-
res d'alcoyle réagissaient avec le magnésium dans l'éther pour produire des composés très actifs.
Grignard (10) tout simplement leur attribua la formule RMgX et Jolibois (11) en 1912 suggéra que la formule dimère
5
Ashby (12) en 1967 reprit le problème en faisant la syn thèse des précédentes hypothèses complétées par ses nouveaux travaux. Deux articles de Fréon (13) relatent le rôle des halogénures métalliques et des solvants sur la réactivité des organomagnésiens. Nous dégageons, en les rapportant, les points essentiels de ces études.
1) L'organomagnésien mixte RMgX est identique au point de vue structure et réactivité à RgMg + MgX^: équilibre de Schlenk (14) établi par R.M.N. (15) et échange statistique en tre le magnésium de R^Mg et de MgX^.
2) Des mesures de conductimétrie (16) ont mis en éviden ce la présence d'espèces ioniques, mais celles-ci n'existent qu'a de faibles concentrations.
La R.M.N. a permis de déterminer que peu de carbanions se formaient dans l'éther à partir des organomagnésiens (17).
3) La détermination des masses moléculaires fait appa raître des degrés d'association divers selon la nature de l'ha logène, les éthers oxydes et la concentration des solutions. Les chlorures sont plus associés que les bromures et les iodu- res, et de même les arylmagnésiens plus que les alcoylmagné- siens. Dans des solvants fortement donneurs et à faible con centration les organomagnésiens sont monomères et dans des
R^MgMgX
2
représentait mieux leur structure. Depuis lors, les discussions commencèrent sur l'exactitude de la représentation de ces "Grignards" par l'une ou l'autre de ces deux formules.solvants peu donneurs et a concentration élevée sont polymè res. On en conclut que 1'organomagnésien existe sous la for me de plusieurs espèces en équilibre (12,18) rapportées dans le schéma 1.
f^2 Mg Mg X 2 s
11
( R Mg X >2
Rg Mg
2
+ Mg Xg
il
RMgXRgMg MgXg pouvant s'écrire :
/*\ ^R-Mg Mg-R ou
Mg' Mg ou
R - Mg XMg -X
X R^ x" 'R MgX solvaté dans l ' éther ;
S
R-Mg-X
tS
R
R
R
i i iMg “X... Mg —X... Mg —X
t t tS
S
S
forme monomère et forme trimère proposées
par Ashby (12) et confirmées par Rayons X (19 )
Schéma 1
Les divers mécanismes d1 addition des organomagnésiens sur des composés carbonylés ont pu être vérifiés par des études cinétiques. Deux hypothèses en relation avec les deux struc tures dimère et monomère ont été proposées :
Hamelin (20) et Mosher (21) supportaient un mécanisme d1 or dre 2 avec un transfert à six centres faisant intervenir une
7
molécule de cetone et une molécule dimère d'organomagnésien.
Anteunis (22) appuyait le mécanisme de Swain et Boyles (23) avec un mécanisme de réaction d'ordre 3 et deux molécu les d'organomagnésiens.
Nous retenons le mécanisme d'Ashby qui a 11 avantage de tenir compte de 11"équilibre de Schlenk" et du complexe inter médiaire R -->C — 0 MgR' déterminé par spectroscopie (12) .
I4/* étape
\
r
C**0... Ma 4-S •(- S
RZ
X
R
S
K
C =* 0
■+■ R - Mg-X
...
R/
S
Schéma 2
Il y a d'abord départ d'une molécule de solvant et
établissement d'une liaison entre le carbonyle et le magnésium. Cette première phase constitue l'étape lente de la réaction,
(schéma 2).
Ensuite, il y a attaque du groupement carbonyle par une seconde molécule d'organomagnésien.(schéma 3)
\
?
C=>0-*-- Mg +- S
4-R
x
s
&R Mg X
RR-C-OMg R' 4- Mg Xo
i ^R' .
R'0"s
Mg
sz sx
?R— C—O-Mg-R'
R' 1
1
Mg-X
X PR-Ç-OMg X -f RMgX
R'
Schéma 3
La constante de vitesse de la réaction globale est don née par la formule (i)
%obs
(k F
)(K F )[G]2 i i 1 1 + (K F )[G] (I) i[G] étant la concentration en réactif de Grignard, Fi représentant la fraction de 11 espèce de Grignard existant dans l'état d'équilibre,
9
et Fu étant la fraction de 11 espèce de Grignard existant dans la seconde étape.
Si K[G] >>1 la réaction sera du second ordre.
Si K[G] «1 la réaction devient du troisième ordre.
Souvent l'ordre de grandeur de K est de 103, ce qui donne K[G] #1 lorsque la concentration du Grignard est de 10 3M, il convient de parler plutôt de molécularité que d'ordre de réac tion.
Les solutions organomagnésiennes utilisées dans la prépa ration des 3-hydroxycétones et des (3-diols sont obtenues à partir des bromures d'aryles et des chlorures d'alcoyles. Les chlorures sont préparés par chlorométhylation et chloration au moyen du chlorure de sulfuryle SC>2Cl2 ( 8) . Ces "Grignards" sont obtenus à basse température avec un léger excès de magné sium 10% et une dilution d'une mole d'halogênure pour sept moles d'éther afin d'éviter la réaction de duplication (de Wurtz):
RMgX + RX -> R-R + Mg%2
Ils sont ensuite dosés par la méthode acide base de Gilman (24) et les rendements atteignent 90-98%.
ETUDE DES B-HYDROXYCETONES
II. 1 Introduction.
Les 3-cétols et les g-aldols s'obtiennent couramment par duplication des aldéhydes et des cétones en milieu faiblement alcalin (KCN, CO^Na^, NaOH dilué, Ba(OH), mais dans ces con ditions, la réaction de crotonisation est particulièrement im portante.
Des modes de préparation des B-cétols,nous connaissons ceux de Colonge (25) qui étudia 11 autocondensation des méthyl- cétones au moyen d'acides halohydriques, puis d1aminomagnésiens mixtes, et chercha à condenser des cétones a-bromées, dont il préparait le magnésien, sur des cétones et aldéhydes. Dubois (26) reprit la méthode par cétolisation à l'aide de composés organomagnésiens mixtes a radical ramifié ralentissant la réaction principale d'addition: le chlorure d'isopropyl- magnésium fut retenu préférentiellement et ses travaux furent
11
étendus à des acetophenones substituées par Maroni (27) (sché ma 4). Cette méthode s'adapte mieux aux méthylcétones aliphati ques qu1 aromatiques : la condensation duplicatrice de l'acéto- phénone conduit à un rendement limité en g-cétols. Uniquement dans le cas où la méthylcétone de départ était la méthyléthy1- cétone,deux (3-hydroxycétones isomères pouvaient être obtenues, le produit majoritaire ayant généralement pour structure:
rch2c(oh)ch3-ch2-coch2r. C6H5-CO-CH3 (CtHc —N ICHj) M*Br) ou (t- C,W, MgCl j cè
H5
-C(OMgX)—CH2C6H5
CH3
I j_______ % Cj-L- C — CHL— C — Ce Hr
———
6 5,
2
i, 65
h
2U
OH
0
Schéma 4
Par ces méthodes de cétolisation, on n1 obtient que des CH3-CH2~COR. Notre but était de déterminer 11 influence des substituants portés par le carbone
dans 11 induction asymétrique 1-3; or, un groupement R en
a
du carbonyle autre qu'un méthyle aurait introduit un effet stérique notable lors de 1'approche de 1'organomagnésien.La littérature nous donne alors le choix d'une autre 3-cetols de formule
R-
o (OH)méthode: celle de 1'action d1organomagnésiens sur des $-cêto-nes symétriques. Cette voie a été ouverte par Zélinsky (28) qui prépara la diacétone alcool par action de l'iodure de mé thylmagnésium sur 1 ' acétylacétone et, par la suite , fut reprise, élargie et mise au point par Canonne et Leitch qui synthétisè rent des 8-hydroxycétones à substituants aromatiques à partir de 11acétylacétone afin d'obtenir par déshydrocyclisation des composés aromatiques à noyaux polycondensés.
En prévision de l'étude de réaction ayant trait à 1'induc tion asymétrique 1-3,nous avons préparé une série de B-hydroxy- cétones de formule RC(OH)CH^-CE^-COCH^.
Notre étude principale est axée sur les substituants R a caractère benzénique (groupements phênyles
1,
2,, 3_, 4_, J5 et benzyles substitués 6_,1_, B, 9_, 10_,
JLl);
désirant faire va rier la nature des substituants nous avons introduit sur 1'acé tylacétone des groupements cyclohexyle JL2,, isopropyle 1_3 et allyle 14.Pour une approche plus aisée, les 6-hydroxycétones concer nées dans 1'ensemble de l'étude réactionnelle ont été classées d'après leurs analogies et propriétés respectives selon 3 types
Dans le tableau I, nous donnons la nomenclature et les for mules développées des produits préparés.
13
TABLEAU I
Les g-hydroxycetones étudiées
No Type Formules Nomenclature
1 I OH 0 C - CH,- C - CH. i / J CH
3
hydroxy-4 phényl-4 pentanone-2 2 I OH 0 I HC - CH.-C-CHo hydroxy-4 g-tolyl-4 I *
*
pentanone-2 ch3 3 I Cl OH l C -i CH, 0 II CH2-C-CH3 hydroxy-4 £-chloro- phényl-4 pentanone-2 4 I Cl OH 0 I II C -CH2-C- CH3 CH3
hydroxy-4 m-chloro- phényl-4 pentanone-2 CH30 OH 0 I II C -ch2-c-cH3 hydroxy-4 g-méthoxy phényl-4 pentanone-2 CH 3TABLEAU I (suite)
No Type Formules Nomenclature
- "
o>
OH 0 ru r ru r ru hydroxy-4 méthy1-4 - CH2-Ç-CH2-C-CH3 p^nyl-5 pentanone-2 CH3 /CH3 OH OH
H
H
. CHfC-CH2-LcH3 ™^s.2 CH3CB
ïk
OH 0 1 IIa
11
O"- CH2~C - CH2~C — CHg hydroxy-4 methyl-4I m-tolyl-5 pentanone-2 ch3 i - CH,-©. OH 0 -CH.-C -CH^C-CH, hydroxy-4 méthyl-4 4 | 4
j
p-tolyl-5 pentanone-2 CH3CH3K
OH 0 1 II ^ 11 0H3^Q>—CH--C - CH0-C-CHg hydroxy-4 méthyl- 4 ^ | ^ (xylyl-3,4)-5 penta-CH3 none-2 CH3X11
11
O
OH 0 1 II—CH0-C -CH0-C-CHo hydroxy-4 méthyl-4 4
j
4 J (xylyl-3,5)-515
TABLEAU I (suite)
No . Type Formules Nomenclature
OH 0 II C-CH, 12 III
O
?"CH2" hydroxy-4 cyclo- hexyl-4pentano-%
ne-2 OH CH , 0 II C-CH2 13 III 'CH-C -CH9- / 11
CH3 CH, hydroxy-4 dime thyl- 4,5 hexano- ne-2 OH 0III CH2= CH-CH2-C - CH2- C - CH3 hydroxy-4 methyl-4 heptêne-6-one-2 ch3
II-2 Préparation.
Certaines de ces S-hydroxycetones ont été synthétisées par la méthode déjà décrite (8). Nous faisons réagir 3 moles de réactif de Grignard pour une mole de pentanedione-2,4, nous hydrolysons le mélange réactionnel par une solution saturée de chlorure d1ammonium et extrayons à l'éther. Se pose ensuite le problème de 11 isolement de la 0-hydroxycétone du mélange de composés obtenus.
En effet, une dizaine de produits outre la 8-hydroxycéto ne existent dans le mélange réactionnel: les hydrocarbures for més par libération de 1'hydrogène de l'énol (le benzène, le
toluène et les xylènes sont éliminés par évaporation sous vide) et ceux formés par duplication (réaction de Wurtz); les méthyl- cétones de scission et de déshydratation; les alcools prove nant de la réduction de ces cétones et de 1'hydrolyse de l'ha- logënure qui n'a pas réagi et les g-diols diastéréoisomëres.
Auparavant, excepté la diacétone alcool obtenue par
Zélinsky en 1902, par action des organomagnésiens sur les 0-di- cétones,aucune 0-hydroxycétone n'avait été isolée. Kohler en 1931 (29) et Freeman en 1958 (30) n'avaient recueilli comme produits de réaction que des cétones de clivage ou de déshydra tation, ceci é-liant dû à la fragilité des composés. Bilodeau en
1967 s'était heurté a des difficultés pour obtenir les hydroxy- cétones pures par distillation fractionnée ou par chromatogra phie en phase vapeur.
17
(CH^)NCH2-C0NHNH2 pour isoler les composés cétoniques des autres produits organiques, le complexe formé [(ay
3NCH2-CONHN=CRR'
]+Cl" étant soluble dans l'eau. Pour regénérer la cétone il évita 11 emploi d'acide chlorhydrique qui clive les hydroxycétones par rétroaldolisation. Il suivit la méthode de Teitelbaum (31)qui déplaçait les hydrazones formées par 1'addition de formal déhyde à 36%. OH 0 H„ OH +0H R-C-CHo-C-R" --- :--- ► R -C - CH->- C - R" t i ^ R' R' ---„ R COR' + CH3 COR" 4 H*
Donc, on traite le mélange brut avec le réactif de Gi rard T dans le méthanol absolu en présence d'une résine échan geur acide comme catalyseur. Les cétones sont regénérées de la solution aqueuse par addition de formaldéhyde et extraites en continu par un mélange éther-éther de pétrole dont la propor tion varie suivant les composés.
La séparation des 3-hydroxycétones des mé thy1cé tone s et des cétones g ,3-insaturée s s'effectue soit par distillation, soit par chromatographie sur colonne de gel de silice; parfois les deux méthodes sont combinées (types I et III) pour celles dont la présence de cétones et,3-insaturées est importante.
Nous avons cerné les limites de cette méthode de prépara tion des 3-hydroxycétones en faisant réagir des organomagnésiens
plus encombrants: les chlorures de naphtyl-1 méthylmagnésium, de phënanthry1-9 méthyl magnésium (32) et de ter-butylmagné sium, les bromures d'o-tolylmagnésium et de naphtyl-1 magné sium. Tous ont donné des rendements peu élevés sauf en ce qui
concerne le premier composé cité.
ÏI-3 Mécanisme.
L'étude de réaction des organomagnésiens sur les g-dicé- tones avait été entreprise par Bilodeau (7): des cétones de scission, des g-cétols et des g-glycols avaient été mis en évidence.
Il avait remarqué une différence de comportement entre les quatre g-dicétones étudiées:
Avec 1'acétylacétone, le benzoylacétone et la phénylben- zoylacëtone, il obtenait des g-hydroxycétones et des g-diols.
Avec le dibenzoylméthane seulement des g-hydroxycétones et aucun diol. Riendeau (33) dans une étude récente isole 1% de g-diols, mais ce résultat reste cependant conforme à la conclusion tirée du mémoire de Bilodeau formulant l'hypothèse que les diols sont obtenus à partir de la forme dicétonique et les hydroxycétones à partir de la forme cétoénolique; en outre, il suppose que les réactifs de Grignard réagissent suf fisamment vite sur les deux formes avant qu'il y ait déplace ment de l'équilibre cétoénolique.
19
examinons le mécanisme de réaction conduisant aux 3-hydroxycé- tones et aux g-diols (schéma 5).
L'acétylacétone existe sous deux formes tautomêres dans l'éther. Les mesures tirées de la R.M.N. par Rogers (34), don nent 90-95% de forme énolisée. Ce résultat implique une for mation prépondérante de (3-hydroxycétones. Les facteurs influant sur l'équilibre dans le sens de formation de i'ënol sont l'ac-- croissement de température, la dilution et la polarité du sol vant.
Il est rare d'obtenir ce haut pourcentage, car se forment au cours de la réaction des cétones de scission (10%) et de déshydratation (10%).
Kohler (29) explique la formation de cétones de scission lors de l'acidification du complexe magnésien, par le passage par
un intermédiaire monomagnésien qui subit facilement le cli vage : OMgX OMgX R - C = CH - C- R ---» i R
0
OMgX
H i R — C —Cl-L-C-R 4 i R OMgX r-c=ch2 -t- r2c-oNous pensons que le clivage se fait pendant la réaction, avant l'hydrolyse, par une réaction rétroaldol au niveau du complexe magnésien I sous l'influence basique des alcoolates. Le diacé-
O II /CX CH3
if
CH. CH, O' h /CX ch3 H X CH XcH3j
RMgX X„ Mg
O'' ^0 Il I C C — R ch3 ch2 ch3 RMgX X XMg
Mg
b O R-XAR
CH3 CHZ CH3 RH X O'" OA À
ch3 ch ch3 X Mcj R O=x
X
Mg 0 1c
CH CH] h2o, NH4C1 OH O R — C— CH*—^ — CH i / CH3ACTION des ORGANOMAGNÉSIENS
sur la PENTANEDlONE-2,4 Schéma 5 OH OH i i R — C - CH]— C - R X CH
21
2%) peut s'expliquer par le mécanisme inverse â partir de l'ion ënolate de 11 acétone. Les composés que nous obtenons à partir de la pentanedione-2,4 sont 1'acétone et une méthyl cétone.
Au sujet des cetones de déshydratation, Freeman en obte nait beaucoup car il travaillait avec six équivalents par mole de B-dicétones et a reflux de benzène. Cette réaction est sem blable à celle de la crotonisation et nous pouvons prévoir le même mécanisme d'élimination qui est le suivant:
MgX
0
Freeman, introduisant un grand excès de réactifs, suggéra que l'organomagnésien attaquait la cétone a,3-insaturêe,
0 R' - C-R' R II R — C — CH
2
OH R' R -C — CH=Zy
Rconditions opératoires nous n1 avons pas observés.
Nous avons remarqué que la proportion de cétones
a
, (3-éthylé- niques varie suivant 11 introduction du groupement. Elle augmente dans le cas d'un groupement aryle, cyclohexyle ou isopro- pyle. Nous pensons que pour les cétones correspondantes de ty pe I, cette formation est favorisée par résonance.
Ce He — CCH--C—0
° 3 i i
CHg
CH,
En ce qui concerne les autres groupements alcoyles, leur encombrement rend probablement possible la constitution d'une molécule plane.
En conclusion, nous soulignons les trois facteurs influen çant le rendement en 6-hydroxycétones préparées dans l'éther anhydre.
la nature du réactif de Grignard;
la durée du temps d'addition dont 1'augmentation fait baisser le rendement en hydroxycétone : produits de
clivage formés sous 1'influence de 1'organomagnésien en excès ;
la température réactionnelle lors de 1'addition de la 3-dicétone. En 1'abaissant, on évite 1'élimination de-OMgX, donc la formation de produits de déshydrata tion; par contre, 1'équilibre cétoénolique est légè rement déplacé vers la forme dicétonique (9).
23
II-4 Etude conformationnelle.
Nous avons examine les spectres d1 absorption dans 1'infra rouge et les spectres de résonance magnétique nucléaire des 14 |3-hydroxycétones étudiées. L1 exploitation de ces données spectrales nous permettra de proposer quelques hypothèses con formationnelles concernant ces méthylcétones g-hydroxylées.
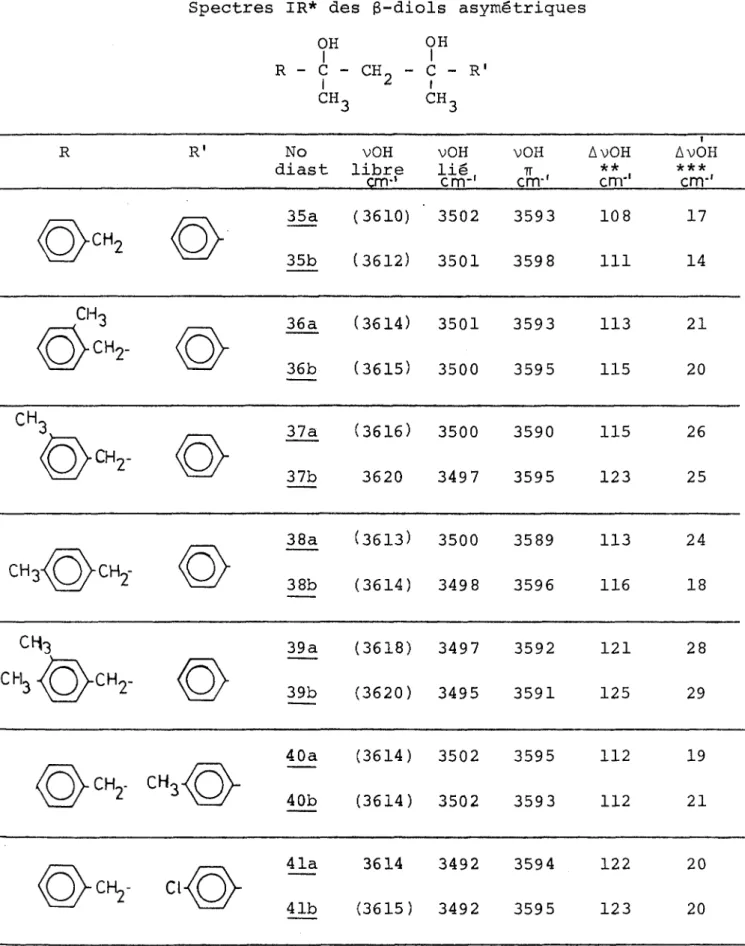
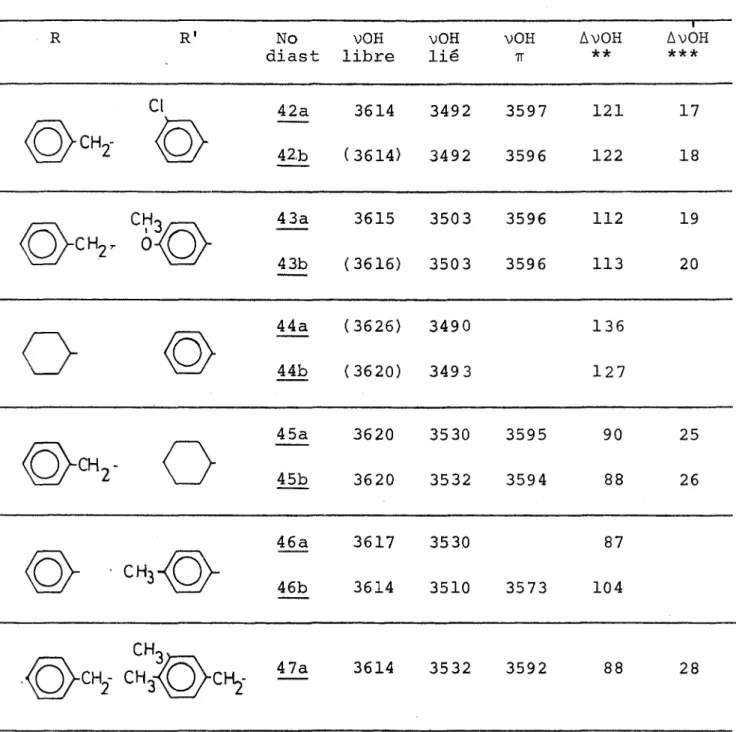
A. Etude par infrarouge.
Les fréquences vC - 0 dont nous rapportons les valeurs dans le tableau II varient dans de faibles proportions. Un lé ger effet hypsochrome est observé pour les g-hydroxycétones de type II, sans doute dû à 1'augmentation de longueur de la chaî ne. Les substituants électrodonneurs ou attracteurs (-OCH -CHg,-Cl) sont trop éloignés pour avoir une influence sur la bande du carbonyle comme on le constate pour des acétophénones substi tuées (35).
Pour ces composés une autre région nous a intéressés, cel le comprise entre 3300 et 3700 cm-1: zone de vibrations de valence des groupements hydroxyles. On remarque en général trois types de bandes OH:
VOH libre 3610 - 3640 cm 1
VOH lié intra ou dimérique 3500 -.3600 cm-1
vOH polymère 3300 - 3400 cm”1.
unique bande large variant entre 3440 et 3505 cm-1 (tableau II ) en solution diluée 10-2 - 10-3M les associations intermolécu laires disparaissent et on remarque deux ou trois bandes fines selon les hydroxycêtones :
vOH libre 3610 - 3625 cm-1
vOH lié intramoléculaire (hydrogène lié au noyau aro matique) 3570 - 3600 cm-1
vOH lié intramoléculaire (hydrogène lié a 11 oxygène) 3500 - 3550 cm-1.
On pourra donc être en mesure de calculer:
les écarts AvOH représentant la distance en cm-1 entre les bandes vOH libre et vOH lié intra, et Av'OH la distance en cm-1 entre les bandes vOH libre et vOH lié tt
.
Une relation em pirique a été établie par Kuhn (36), d’après une étude systéma tique sur des diols, entre AvOH et rO...H (distance entre l'oxy gêne et 1’hydrogène). Cette relation a été modifiée plus ré cemment par Brutcher (36) et donne42.5
AvOH = --- - 3.5 (II) Av en cm-1 rO...H - 1.4 rO...H en Â
Bien qu'approximative cette relation a pu être utilisée pour déterminer la conformation de 3-diols. Elle montre que AvOH est inversement proportionnel à la distance entre 1'hy drogène et 1'oxygène donc une valeur de AvOH élevée indiquera 1'existence d'une liaison forte, ce qui est le cas de
25
11 ensemble de nos composés (80 cm-1 <AvOH< 110 cm-1 ).
l'aire intégrée de chaque bande représentant l'absorp tion du vibrateur correspondant, d'après l'équation suivante:
A ou A'
A = coefficient d'absorption de la bande OH libre
A' = coefficient d'absorption de la bande OH lié
vi, v2 domaine de fréquence étudié
G concentration en mole/Z
t
trajet optique (épaisseur de la cuve) en cmIo énergie incidente de fréquence v
Iv énergie transmise de fréquence v
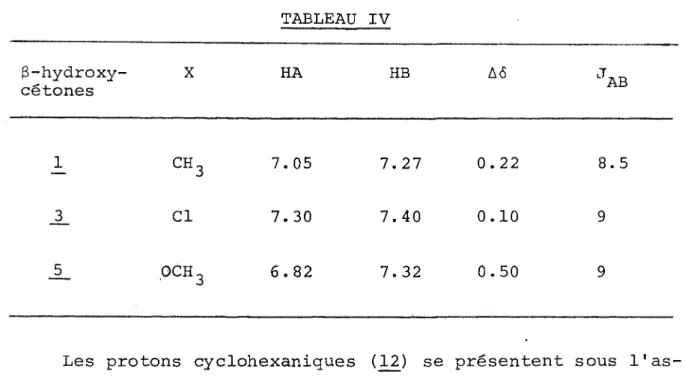
Dans tous les exemples étudiés (1-14) nous vérifions l'existence d'une liaison hydrogène intramoléculaire entre 1'hydroxyle et le carbonyle (tableau II). De plus, nous remar quons pour les g-hydroxycétones de type II une interaction in tramoléculaire entre 1'hydroxyle et le noyau aromatique. Pour 1 'hydroxycétone 1_4 nous prévoyions également l'existence d'une liaison ïï, mais seulement un très faible épaulement est visi ble (3592 cm-1).
Les hydroxycétones 1_2 et L3 présentent des spectres IR analogues par leur allure a ceux des 0-cétols de type I, excep té une force un peu moins grande de la liaison hydrogène
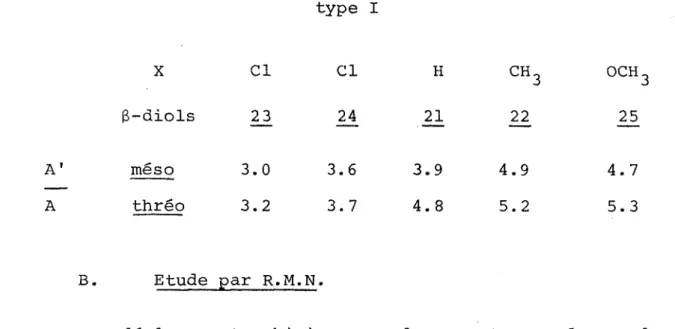
(AvOH = 95 cm i au lieu de AvOH - 105 cm~1) •
L ' absence de liaison tt pour les composés de type I permet de tenir pour acquis qu'il né peut y avoir de liaison intramo- léculaire entre 11 hydroxyle et le noyau aromatique lorsque ces deux substituants sont géminés.
A 1
Le rapport — des coefficients d'absorption intégrée des bandes OH lié (A") et OH libre (A) a été calculé pour trois hy dro xycé to ne s pouvant être considérées représentatives de chaque type (Tableau III).
Méthode de calcul.
En général, pour déterminer les coefficients d1 absorption intégrée, la densité optique D CD^ = log1Q (^) v = log1Q (^-)
T: transmission en %]est portée sur un graphique en fonction du nombre d'onde v, (38, 39). Ces bandes peuvent être assimi lées à des courbes de Lorentz.
La fonction lorentzienne étant considérée comme une
repré-Log(|°)^ (IV)
1 + a (v-vm) 2
[a n&t une constante. eX vm la fréquence, au maximum d'ab-ioaptlon]
sentation mathématique acceptable de la courbe de variation de 1'absorption en fonction de la fréquence.
L'aire délimitée par la courbe [log^Q (5^-) = f (v) ] peut être mesurée soit par la méthode des carreaux restants, soit par pe sée de la bande découpée, soit en utilisant l'aire du triangle
27
TABLEAU II
Spectres IR* des 3-hydroxycétones R-C (OH)CH^CH^-COCH
No R VC=0 cm-1 VOH libre crrr' vOH lié intra cm' vOH lié
TT
cm-' * * AvOH cm-1 *** Av ' OH cm-' 1#
1705 3618 3511 107 2 1704 3618 3508 110 2C‘-^
1705 3614 3504 110 4_ 1708 3610 3507 103 _5 ch3-° xgy
1704 3617 3508 109 2 1706 3618 3531 3594 87 24 7<g>CH?
1719 3619 3534 3597 85 22 8 1712 3624 3542 3592 82 32 2 CH3^§>CH2- 1711 3622 3535 3590 87 32 10 chH33-@>
ch 2-1708 3623 3532 3590 91 33 11 1710 3624 3536 3592 88 32 12o-
1705 3624 3530 94 13(cn^CH-
1708 3625 3529 96 14CH2- CH-CH2-
1705 3620 3531 (3592) 89 28en solution diluée dans le tétrachlorure de carbone (10-3M) Av OH - \)OH libre - vOH lié intra.
qui sous-tend la courbe (39), ce qui revient â calculer la densité optique correspondant aux maxima d'absorption et la largeur en cm-1 de la bande d1 absorption à mi-hauteur (AvOH)i. C'est la méthode que nous avons adoptée en utilisant la for mule (III) transformée en (V).
A ou A' = (AvOH)i log10 i (V) Tableau III Type No (AvOH)| cm- 1 A 104 (AvÔH)i — 1 cm A ' io4 A'" A I JL 18 0.09 96 0.78 8 II 6 16 0.02 80 0.51 32 III 13 24 0.04 104 0.69 17
Ces chiffres indiquent une forte proportion de formes ché-A1
datées pour nos composés. On constate un rapport plus fai ble dans le cas de 11 hydroxycétone à substituant phényle ]., or
c'est pour ce type de 8-cétol que l'on observe des valeurs de AvOH élevées. De ce résultat on peut déduire que les liaisons hydrogènes qui s'établissent dans cette hydroxycétone sont plus fortes mais en nombre plus restreint. Donc peut-être pour des raisons stériques la molécule n'existera pas que sous une forme cyclique comme on peut le prévoir pour les hydroxycétones de type II ou III.
29
Dans chaque série de g-hydroxycétones (types I, II et III) on ne note pas de différences sensibles dans la forme des spec tres qui auraient pu être apportées par les substituants élec-
tro attracteurs ou donneurs fixés sur le groupement phényle. Uniquement une diminution d'intensité de la bande OH libre est observée dans les composés de type I lorsque le phényle est substitué.
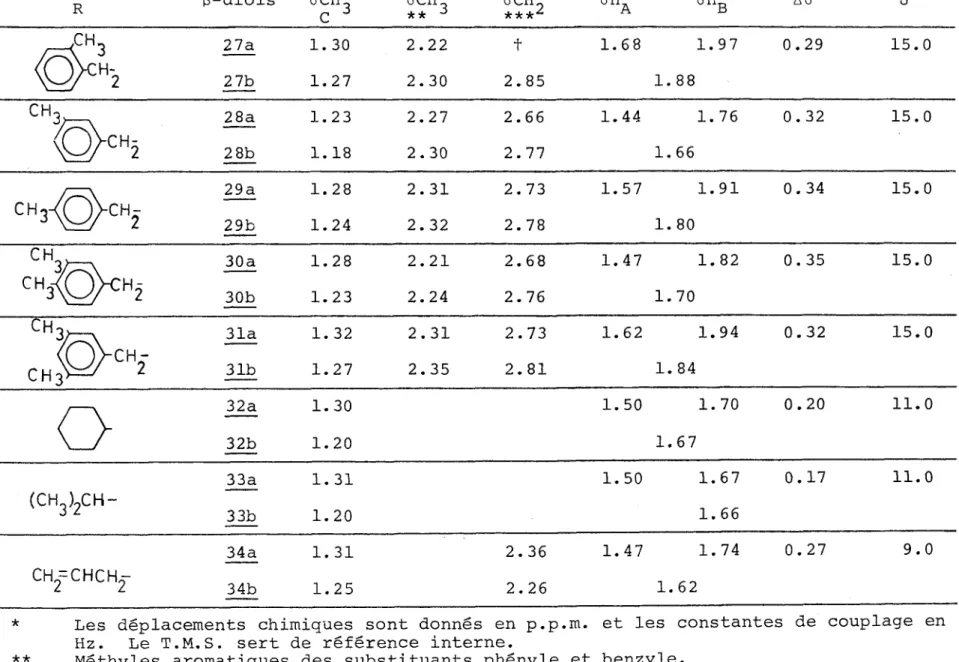
B. Etude par R.M.N.
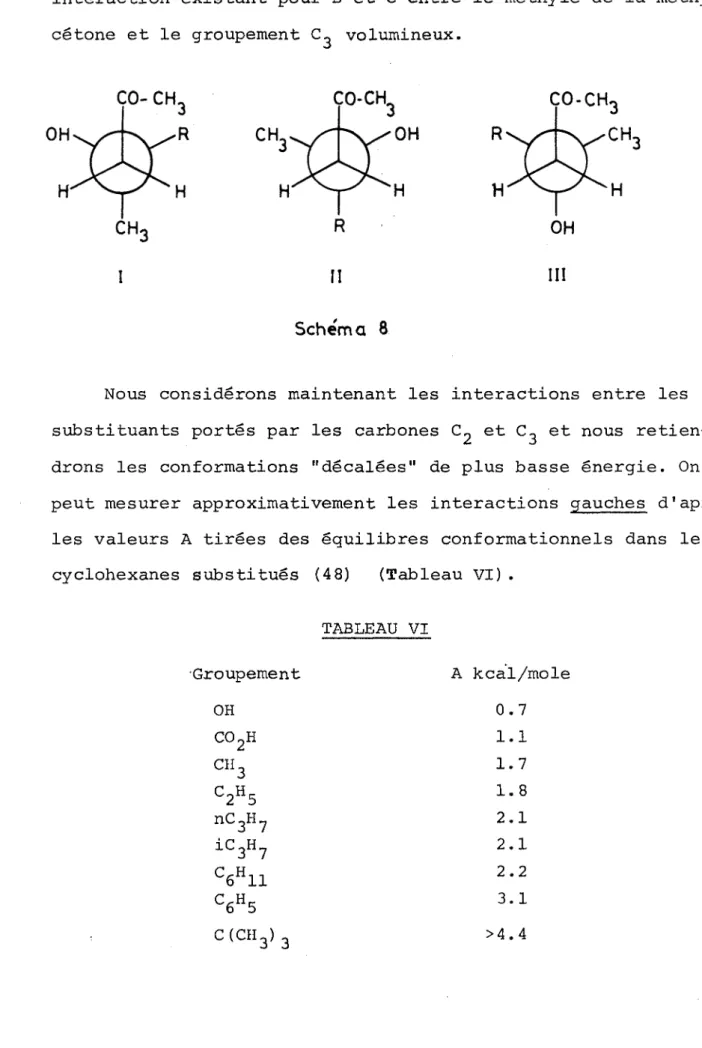
Les résultats donnés par la R.M.N. sont rassemblés dans le tableau V, les signaux des hydroxyles et des systèmes aro matique , cyclohexanique, isopropylique et allylique n'y sont pas rapportés*. Les hydroxyles se manifestent sous la forme d'un singulet vers 3.5 p.p.m. pour les hydroxycétones de types II et III et vers 4.5 p.p.m. pour celles de type I. Les si gnaux des protons aromatiques ont des déplacements chimiques de l'ordre de 7.0-7.5 p.p.m. Ce sont des singulets pour ceux appartenant au groupe benzyle a l'exception de celui substitué en méta par un méthyle, et des multiplets lorsque les phényles sont attachés a des carbones quaternaires. Dans les hydroxycé- tones
2_, 3,
et !5 les groupes benzéniques para-disubstitués met tent en évidence les signaux d'un système AgB- pour lequel on sait que les constantes de couplage ont généralement les ordres de grandeur suivants JAB = 6 à 9 Hz; = JBB - 1 à 3 Hz et J'aB = 0 à 1 Hz (41). Si on considère que notre système estCes valeurs figurent dans la partie expérimentale et sont données pour chaque composé.
sensiblement identique au système du p-chloroiodobenzène étudié par Richards et Schaefer (42) pour lequel = JBB et j'ab ~ 0 on peut déterminer approximativement la constante de couplage et les déplacements chimiques et <5b d'après les positions des 4 raies principales dont la disposition est qua siment analogue à celle d'un système AB légèrement perturbé par le couplage AA et BB. ( et Jgg sont petits par rapport a . Dans le tableau IV sont indiqués les déplacements chi-miques <5A et ôB en p .p.m. et la constante
TABLEAU IV de couplage J^g en Hz B-hydroxy-cétones X HA HB Aô JAB 1 ch3 7.05 7.27 0.22 8.5 _3_ Cl 7.30 7.40 0.10 9 _5_ OCH3 6.82 7.32 0.50 9
Les protons cyclohexaniques (12) se présentent sous l'as-pect de deux massifs situés entre 1.0 et 2 .0 p.p.m.
Dans le système isopropylique (13) on remarque un multi-plet correspondant à 1 ' hydrogène tertiaire et pour les méthyles on note une non équivalence de 2. 0 Hz (0.3 p.p.m.). La cons-tante de couplage entre les protons méthyliques et l'hydrogène tertiaire est évaluée à 7.0 Hz.
31
(4.92 p.p.m.) Hc ,Ha (5.85 p.p.m.)
(5.18 p.p.m.) Hg
C
CH2_(2.34 p.p.m.)
Les protons allyliques (14) apparaissent dans la zone de 4.7-6.2 p.p.m. selon un motif bien caractéristique que nous retrouverons pour le diol _34. Le massif le plus déblindé 5.4- 6.2 p.p.m. est attribué au proton tertiaire couplé avec les deux méthylènes voisins; les deux autres protons éthyléniques ne sont pas équivalents : le proton résonne dans un champ plus fort (4.92 p.p.m.) que le proton Hg (5.18 p.p.m.). Le méthylène couplé avec se manifeste sous la forme d'un dou blet *(J = 6.0 Hz) .
Dans notre discussion nous nous référons aux hydroxycéto nes 1_2 et 1^3 car nous supposons que les substituants cyclohexy- le et isopropyle influencent moins les autres protons des grou pements étudiés que ne peuvent le faire le phényle et le benzy- le dans les autres hydroxycétones. Il apparaît que sous 11 in fluence du champ extérieur HQ, la circulation diamagnétique des électrons tt des cycles benzéniques provoque un champ induit mo difiant la constante d'écran
a
des protons voisins.C'est ce que l'on constate principalement dans les hydro- xycétones de type I où l'effet de champ du phényle perturbe
* Ce doublet large ne permet pas de calculer la constante de couplage allylique avec le proton Hc dont la valeur est comprise entre o.5 et 2.5 p.p.m.
Spectres de R.M.N.* des (3-hydroxycé tones. OH Hj 0 I
lA II
R-C-C-C - CH., I I 3 CH3 HgC
D
No R 6CH] <5 CH 3 5CH0 ** ^ 6 CH ***J sha shb AôAB J C D 1.42 1.89 2.66 3.04 0.38 16..02
1.39 1.89 2.23 2.63 2.97 0.34 16.,52
c'^ 1.45 1.99 2.78 3.02 0.24 16..0 4 1.48 2.07 2.84 3.12 0.28 17.,5 5_ ch3°^> 1.42 1.98 2.71 2.97 0.26 16., 56
‘©’CH2
- 1.12 2.01 2.75 2. 42 1 1.16 2.06 2.77 2.33 2.,51 8. CH3^ <oy^2- 1.07 2.01 2.74 2.27 2,,43 U> tvTABLEAU V (suite) No R 5CH3 C ôCH3 D
&CE0
** ^ <5 CH., A**-3 6H 6H AôAB J9
CH3-^5>CH2-
1.18 2.06 2.76 2.28 2.46 10 c^§}ch2-1.18 2.00 2.73 2.15 2.42 11 1.10 2.00 2.76 2.15 2.42 12a
1.11 2.16 2.54 13(
ch3)2
ch-
1.12 2.18 2.60 14CH2=CH-CH2-
1.18 2.13 2.52Les déplacements chimiques sont donnés en p.p.m. et les constantes de couplage en Hz pour des solutions à 25% dans CCl^. Le T.M.S. sert de référence interne. Méthylène du groupe benzyle.
1'ensemble des protons dans la molécule. On observe la non- équivalence magnétique des protons et Hg du méthylène, cel le-ci se traduit par les 4 raies caractéristiques d'un systè me AB de constante de couplage de 16.0 Hz. Tandis que les pro tons méthyléniques des hydroxycétones des types II et III appa raissent sous la forme d'un singulet.
On note le blindage: 0.28 p.p.m. et 0.18 p.p.m. des si gnaux des méthyles _D des g-cétols 1_ et 6_, représentatifs des types I et II, relativement au signal de 1J3. En conséquence, on suppose le méthyle D situé dans le cône de blindage des phé- nyles. Dans le type I, le blindage est moins tangible pour _3, 4y 5_: hydroxycé tones ayant sur des phényles un substituant chlore ou méthoxvle, mais on peut remarquer que les glissements chimiques de tous les protons de ces hydroxycétones sont dé placés vers les champs faibles, cet effet est imputable au chlo re et au groupe méthoxyle.
On observe pour les cétols du type II et ceux du type III des déplacements chimiques d'ordre de grandeur comparable pour le méthyle (3, en conséquence, il est peu probable que le méthy le subisse l'effet de champ du noyau aromatique. Par contre les protons méthyliques des cétols de type I sont fortement déplacés vers les champs faibles (-0.30 p.p.m.), ce qui impli que pour le groupe méthyle sa situation à 1'extérieur du cône d'anisotropie paramagnétique du phényle. Il aurait été inté ressant d'observer la variation du déplacement chimique du si gnal correspondant au méthyle
C_
en fonction des groupements élect ro attracteurs ou donneurs portés par le phényle35
para-disubstitué (hydroxycetones de type I). Une légère dif férence existe mais elle est très minime de l'ordre de 0.03 p.p.m., ce qui correspond en fait à la marge d1 imprécision de
11 appareil A-60. L'effet donneur du méthyle et du méthoxyle fixés sur le noyau aromatique aura tendance a déplacer le si gnal du groupe méthyle C_vers les champs forts en renforçant la charge positive du carbone porteur de trois protons ; l'ef fet attracteur du chlore produira le phénomène inverse.
Pour deux hydroxycétones: 1_ et £ nous avons appliqué la méthode de double résonance magnétique nucléaire en irradiant les méthyles C et D, elle n'a provoqué aucune modification du spectre relative aux protons méthyléniques pour les deux exem
ples choisis.
C. Hypothèses conformationnelles.
Des études conformationnelles basées sur la R.M.N. ont été réalisées sur des aldéhydes de formule RCH^CHO et R2CHCHO par Karabatsos (43) et Lehn (44). Auparavant des études IR, Raman, et de diffraction électronique ont établi qu'une liai son simple éclipsait une double liaison. En s'appuyant sur la variation de la constante de couplage J avec la température et la polarité du solvant Karabatsos et Lehn déterminent que la conformation la plus stable dans les aldéhydes monosubstituées est non pas celle où le carbonyle éclipse une liaison c-H mais une liaison C-R, sauf si la conformation ainsi obtenue est ren due moins stable par suite d'un encombrement stérique lorsque
R est un groupement ter-butyle (schéma 6 ) H' F H H H R Ra.CH3,-C2^,^H2CH2CH3, -CH(CH ) ,CLH
J 2 6 5
R=-c6h5,-c(ch3)3 Schéma 6Dans le cas des aldéhydes disubstituées, uniquement lors que R est un méthyle, la liaison C-R éclipse le carbonyle, dans les autres cas les interactions stériques ne favoriseront pas cette conformation.
Karabatsos insiste sur le fait que des peti tes variations de l'angle dièdre <j> (fig. ci-0*
contre) peuvent être tolérées et n1 altèrent pas 11 interprétation des résultats.
H
Mais ces conclusions doivent être appliquées pour des cé- tones avec réserves car Fétizon a montré (45,46) en séries di- et triterpéniques que les méthylcétones n'avaient pas la même conformation que les aldéhydes correspondantes. De même,
les résultats de Jacques (47) ont prouvé que si des aldéhydes et des cétones conduisent au même alcool prédominant, ceci ne
37
tient pas à la différence de nature des nucleophiles (hydru- res ou magnésiens), mais sans doute à une conformation diffé rente des aldéhydes et des cétones: le méthyle des méthylcé- tones gênant plus la rotation de la molécule qu'un hydrogène aldéhydique.
Dans les conformations que nous proposons (schémas 7, 8, 9) nous conservons l'hypothèse du carbonyle s'éclipsant de pré férence avec une liaison carbone-carbone plutôt que carbone-hydrogène.
Afin de discuter des conformations des 6-hydroxycétones nous adoptons la projection de Newman suivant C]_C2* (schéma 7) puis (schéma 8) et une projection en dérivant suivant
C1C3 (schéma 9 ).
A
BSchéma 7
C
Des trois conformations A, B, C représentées (schéma 7) A est la plus favorisée car elle ne présente pas la forte
* Système cetone:
utilisé pour numéroter 0 C H - C - CH2
1
2
les OH 3 carbones R d'unehydroxy-interaction existant pour B et C entre le méthyle de la méthyl- cétone et le groupement volumineux.
II
III
Schéma 8
Nous considérons maintenant les interactions entre les substituants portés par les carbones et C3 et nous retien drons les conformations "décalées" de plus basse énergie. On peut mesurer approximativement les interactions gauches d'après les valeurs A tirées des équilibres conformationnels dans les cyclohexanes substitués (48) (Tableau VI) .
TABLEAU VI oupement A kcal/mole OH 0.7 COgH 1.1 ch3 1.7 % 1.8 nCf-7 2.1 iC3H7 2.1 C6H11 2.2 Vs 3.1 C (CH3) 3 >4.4
39
Les groupements benzyle (CH^C^Hg) et méthylcarbonyle (COCHg) ne figurent pas dans ces données mais leur encombre ment peut être supérieur pour le benzyle à celui du n-propy- le et pour le méthylcarbonyle à celui du groupement carboxyli- que.
Dans le tableau VII nous résumons les interactions existant dans les trois conformations en :ne faisant pas paraître les in-teractions communes a toutes les conformations •
TABLEAU VII
I II III
H «-- » CH 3 R H — OH
OH '-- ► COCH3 CH3 " CO CH 3 R COCH3
R *—► COCH3 OH 4—* COCH3 CH COCH3
Les conformations les plus stables en tenant compte des tableaux VI et VII devraient être dans l’ordre décroissant II, I, III.
En combinant les conformations proposées précédemment sui vant les axes C^C^ et nous allons examiner dans le sché ma 9 les positions que prend le groupe carbonyle par rapport aux
substituants du carbone asymétrique C3 en considérant la projec tion selon C^Cg.
Nous avons remarqué dans l'étude infrarouge que les liai sons hydrogènes étaient fortes, donc pour cette raison nous éli minons les conformations IIIA, IIB, IC dans lesquelles 1'oxygè ne et 1'hydroxyle sont en opposition. Les conformations IB,
OH
CH,
II B
I c ne HIC
CONFORMATIONS des J
3
-HYDROXYCÉTONES41
IIIB, IIC, IIIC ont le désavantage de présenter de très fortes interactions méthyle-R ou méthyle-méthyle du même ordre que les interactions diaxiales 1-3 dans le cyclohexane disubstitué (48)
(CHg *—» CH2 3.7 cal/mole). Deux conformations chelatées res tent possibles IA et IIA. Le schéma 10 donne leur représenta tion spatiale.
IA IIA
Equilibre conformationnel des /Shydroxycétones
Schéma 10
La conformation IA permet d1 expliquer le léger blindage du méthyle en a du C = O constaté pour les hydroxycétones du type I (schéma 11) ; la liaison C = O fait alors un angle de 30° avec la liaison
C2C3'
ceci restant en accord avec les con clusions de Karabatsos (43).Nous supposons un équilibre entre les deux conformations IA et IIA, déplacé vers IA pour les hydroxycétones de type I et vers II A pour celles des types II et III. Le groupement volumineux se trouve en position "quasi équatoriale" dans un
cycle"pseudo-chaise"(IIA) et comme on le constate dans les schémas 9 et 10 le groupement R est loin du groupement carbo- nyle. Lorsque R est un groupement phényle, assez volumineux si on se réfère au tableau VI, on peut expliquer sa position "quasi axiale" par la polarité du groupement phényle comme ce la a été établi par Karabatsos (43) pour des cetones avec un substituant phényle en a du C = O et par Bellamy (49) pour des a-chlorocétones.
Schéma 11
Dans les chapitres suivants nous allons montrer que la différence de comportement vis-à-vis des réactifs de Girard T entre les hydroxycétones du type I et celles du type II, peut être expliquée par la conformation IA préférentielle pour les cétols du type I.
CHAPITRE HI
ETUDE DES g-DIOLS BITERTIAIRES
III. 1 Introduction
Afin d1 interpréter le déroulement stérique de 1'action des organomagriésiens sur les 3-hydroxycêtones, nous les avons fait réagir sur les 14 g-cétols décrits au chapitre II possé dant un centre asymétrique inducteur en 3. Nous avons obtenu des 3-diols bitertiaires diastéréoisomères (schéma 12) et leur
OH <^H RIim-C - CH0- C"iR' OH 0 t H Ri"-C -CH2-C -CH
3
-(- R'MgXcV
d OH CH-, ▼ ▼ 3 RII-C - CH2- C-"R' CH OH CH3
CH3
hro Schéma 12préparation/ leur configuration et leur conformation font l'ob jet de ce chapitre.
Dans ce présent travail, nous convenons de désigner par le terme érythro le diastéréoisomêre dont la configuration est telle que, dans une des conformations possibles, les groupe ments méthyles d'une part, et les groupements hydroxyles d'au
tre part, sont simultanément éclipsés et par le terme thréo l'autre diastéréoisomêre (schéma 13).
Dans la suite de l'exposé nous ne représenterons qu'un seul des énantiomères. Le terme méso sera utilisé a la place d'érythro lorsque la molécule possédera un plan de symétrie,
R = R' .
R ? CH^C^OH 13 i ch2 CH3kÇ-*OH R"érythro
thréo
Schéma >3 R OH^C ^CH3 ch2 I OH ►Ç«CH3 R' CH' R ► C-* OH i CH-'2 i OH ► C **CH. R' OH R -C. I CH. i ‘CH,
CH3»Ç«OH R'Bilodeau (7) sur les g-hydroxycêtones 1 et 6 avait fait réagir respectivement le bromure de phénylmagnésium et le chlo rure de benzylmagnésium et n'avait isolé qu'un seul diastéréo isomêre. Doutant qu'il y ait stéréospécificité totale de la réaction nous avons repris ces expériences et avons constaté la
45
présence de diastêrêoisomêres méso et thréo (21, 26).
Outre les 3-diols bitertiaires symétriques (20-34) de for mule générale: OH OH i l R - C - CH„ - C - R i ^ | CH 3 CH _
nous avons été amenés à préparer des 3-diols bitertiaires asy métriques (35-47) de formule générale:
OH OH
R - C - CH0 - C - R' I 2 |
CH 3 CH 3
après avoir déterminé que 11 action du bromure de phénylmagné- sium sur 1'hydroxy-4 phënyl-4 pentanone-2 (1) menait au dias- téréoisomêre majoritaire thréo et que 11 action du chlorure de benzylmagnésium sur 11 hydroxy-4 mëthy 1-4 phényl-5 (6_) condui sait au dias té réois ornere prépondérant méso (3-diols obtenus par Bilodeau). Le but, en préparant ces diols, était de véri
fier si la stéréosélectivité de la réaction était due à 11 in troduction d'organomagnésiens plus ou moins encombrants ou po laires ou dépendait de la différence de conformation des états de transition qui est fonction du groupement R substitué sur le carbone asymétrique inducteur.
Le tableau VIII rassemble les différents diols préparés dont il sera fait mention dans la suite du mémoire.
g-diols bitertiaires a déjà été entreprise par Wolf (4). Sur des g-hydroxycétones tertiaires de formule générale
R - C(OH)CH^ - CH2 COR préparées par duplication de méthylcé- tones par Maroni (27) est introduit 11iodure de méthylmagné sium; des 3-diols de formule générale:
OH OH
R - C - CH0 - C - R
1 / 1
CH. CH.
sont obtenus. A partir des 3-hydroxycétones 1, 6y L3 nous avons préparé les couples de diastéréoisomêres des 3-diols 21, 26, 33 (R = CçHç., CH2CgHg, CH (CH^) ayant des constantes physiques identiques à celles rapportées par Wolf, mais la proportion des couples de diastéréoisomêres obtenus est dif férente.
III.2 Préparation
Des 3-diols bitertiaires ont déjà été préparés par 1'action de RMgX sur des esters maloniques (50) diméthylmaloniques (51), éthyl-2 acétyl acétique (52) sur les 3-dicétones (7,28,29) avec des rendements médiocres ; 5 à 10%. On obtient des 3-diols avec un rendement appréciable en faisant agir des magnésiens d'al- coyle (4,53) et d'aryle (4,7,54) sur des g-hydroxycétones ter tiaires .
Les g-diols symétriques 20-34 ont été synthétisés par deux voies.
47 TABLEAU VIII g-diols symétriques No R R' g-diols de type I 21
22
23 24 25 g-diols de type II 26 27 28 29 30 31g-diols de type III
^ ^"ll 33 CH2-CH=CH2
X
Y
Z
OH OH ^3 ^3 H P-Œ3 p-Cl m-Cl 2-OCH2 OH OH ch3 ch3 H o-CH3 m-CH, m-CH., E.-CH3 m-CH, OH OH 1 1 R - G - CI L - G - R 1 2 / ai, en. H H H H m-CH, m-CH, 34TABLEAU VH I (suite) g-diols asymétriques No R R' g-diols de formule
35
36
37
38
39
40
41
42
43
g-diols de formule44
C6H11
C6H5
45
CH^C^
C6H11
46
C6H5
£"CH3C6H4
47
Œ2C6H5
CH2(CH3)2-3,4 C6U
X Y Z OH OH C CH -i 2 9#
CH3 œ3 H H H H o-Œ^ H H m-CHj H H p-ffl3 H m-CHj E-tH3 H H HE-œ3
H H p-Cl H H m-Cl H H 2~och, OH OH |R
- C - CH-1 z - C - R'\
™3
CH349
directement à partir de la pentanedione-2,4, avec un ren dement de 5 à 10%, ils sont alors des sous-produits de la réac tion de préparation des hydroxycétones Çl-14);
à partir des hydroxycétones tertiaires de formule
R - C(OH)CHg - CHg - CO - CHg avec un rendement de 20 à 65% selon les cas, (schéma 14).
Cette dernière méthode de préparation fut celle utilisée pour la synthèse de tous les g-diols bitertiaires (20-47).
Les B-diols sont préparés dans des conditions similaires à celles des B-hydroxycétones: 3 moles de réactif de Grignard pour une mole de composé carbonylé, à une modification près; elle concerne la température du mélange réactionnel qui est maintenu à environ -20° pour la synthèse des hydroxycétones afin d'éviter la formation de produits secondaires (voir II), tandis qu'il est conservé lors de la préparation des diols en tre 15 et 20°, car à une température plus basse, nous avons constaté une baisse appréciable du rendement.
Comme produits secondaires de la réaction, on observe les hydrocarbures de types R'-H et R'-R', les méthylcétones de scis sion et de déshydratation les alcools de réduction de cétone et d'halogénure non réagi et parfois 1'hydroxycétone de départ.
CHg-C —CHg- C —CHg
R <**• C — CH n~ C RA 2 A
CHj CH3
OMgX O i h RMgX R — C — C H2~C — CHg+
CH. R H"C - CHL- C -ilRA 2 A
CH3 CH3 H2 O, NH4Cl+
méso
OMgX CH-, ▼ f J Rv-C - CHo-C 'ii RA
CH-
OMgXA
OH CH-, ▼ f 3 R H- C - CH?-C -iRA
À
CH, OHthréo
ACTION des ORGANOMAGNÉSIENS sur la PENT ANEDlONE-2/ et sur les p-HYDROXYCÉTONES
SYNTHESE des p-DlOLS B1 TERTIAIRES
Schéma H
Ul O
51
III.3 Mécanisme
Le mécanisme de réaction est fort simple ainsi qu'en té moigne le schéma 14. L'action du Grignard RMgX sur 1'acétyl- acétone ou sur la 3-hydroxycétone substituée par un groupe ment R conduit à une même proportion du 3-diol diastéréoiso me re prépondérant. On en déduit que les 3-diols bitertiaires résultent de 1'addition sur la forme non énolisée de l'acétyl- acétone avant que celle-ci puisse se tautomériser et que 1 ' in termédiaire, en 1' occurrence 1 ' alcoolate de la 3-hydroxycétone, est le même. Sur ce complexe métallique le mécanisme d'atta que par une seconde molécule de réactif de Grignard conduit à un état de transition dont la conformation sera étudiée dans le chapitre IV portant sur 1'induction asymétrique 1-3.
La formation des composés de scission s'explique facile ment par un mécanisme de transfert circulaire (schéma 15) favo risé dans un milieu basique. Pour une même réaction le rempla cement d'un organomagnésien par un composé lithien plus basi que augmente la proportion de produits de clivage (6). Esa- fov (55) a démontré que les 3-diols se clivaient plus aisément en milieu acide qu'en milieu basique; ceci explique pourquoi on évite d'effectuer 1'hydrolyse en milieu acide.
Les produits de clivage isolés du mélange réactionnel sont la méthylcétone de scission et 1'acétone qui se condense par aldolisation pour donner la diacétone alcool (schéma 15).
R-Cs ch
3
H c-
ch3
0
9
“CH3C-CH2 CH-C-CH3
0 OH
CH^-C-CHg-C -
ch3
Formation des composes de scission
Schém a 15
* I u IR-
cx^
c-
ch3
CH3
è
-OMgX
C
/ \R1
R2
£OMgX 0
R-C - CH - C - CH
I 3 ch3
0 IICH-C
ch3
Formation des composes de déshydratation
53
groupe carbonyle part facilement sous 1'influence de l'al- coolate jouant le rôle de base; ainsi se forme la cétone de déshydratation (schéma 16).
Nous avons indiqué dans la préparation des B-diols (III.2) que que nous retrouvions 1'hydroxycétone de départ. A ce propos,
Cram suggère que 1'hémicétalisation de 11alcoolate de magné sien donne un composé insoluble dans le milieu réactionnel qui ne peut réagir puisque la fonction carbonyle est de ce fait blo quée (schéma 17) .
III .4 Purification des g-dioTs et séparation des diastéréo- isomêres.
Les 3-diols bitertiaires à substituants isopropyliques (13) et allyliques (14) sont liquides, nous les avons purifiés par distillation sous un vide de 0.1 mm de Hg, la fraction corres pondante aux 3-diols contient le mélange des deux diastéréoiso- mêres.*
Les 3-diols bitertiaires à substituants aromatiques (31, 35-46) ou cyclohexaniques (3.2/ £4, 45) sont pour la plupart solides. Ils sont fortement solubilisés dans le milieu réac tionnel ce qui implique une vitesse de cristallisation faible. Le diastéréoisomêre existant en quantité prépondérante cristal lise le premier. On constate une cristallisation plus rapide pour le thréo que pour 11érythro.
Avant de procéder à la séparation des diastéréoisomêres
,
nous avons purifié les diols des produits secondaires obtenus lors de leur synthèse. La méthode quantitative, qui nous a semblé la plus adaptée a ce problème, est la chromatographie sur colonne de gel de silice.Sont élués dans l'ordre: les hydrocarbures, les cétones
La détermination de la configuration des diols est expo sée dans l'étude par R.M.N. (III.5) , mais par commodité nous désignerons dès maintenant les diastéréoisomêres par les termes méso ou érythro et thréo qui leur ont été attribués.
55
de scission et de déshydratation souillées parfois des alcools de réduction, les g-diols (une première séparation des diasté- réoisomêres est alors amorcée), 1'hydroxycétone de départ.
Pour obtenir une bonne séparation du mélange des diasté- réoisomêres on introduit celui-ci sur une colonne de gel de si lice a raison de 1 g de produit pour 200 g de gel de silice. La pureté des diastéréoisorneres est contrôlée par chromatogra phie sur couches minces et par R.M.N.
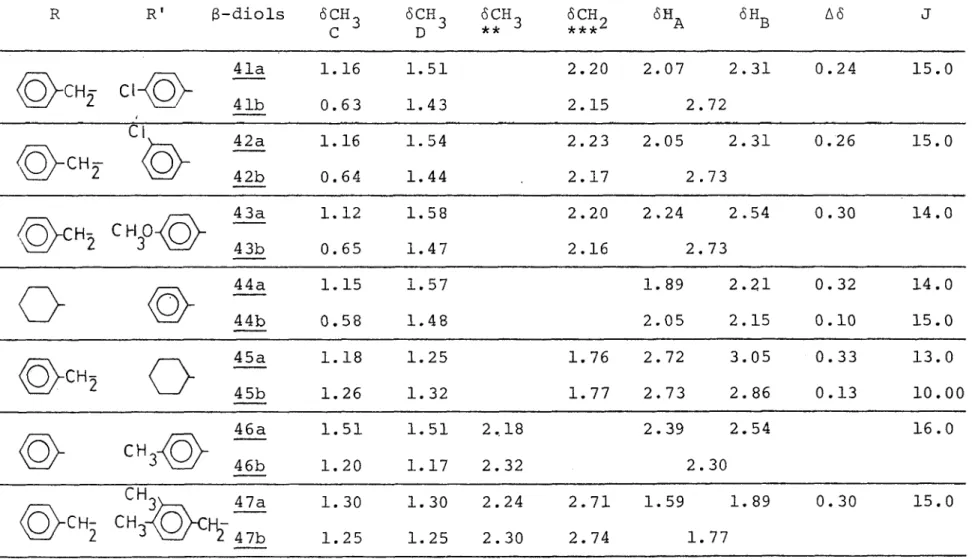
Les diols du type II (26-31) , les diols 33 et 34. du type III et le diol asymétrique 4_7 ont des temps de rétention trop voisins pour être séparés. Nous avons cherché à isoler les diastéréoisomères des diols 33. et 34. liquides par chromatogra phie en phase vapeur sans résultat. Des essais de séparation par distillation sur colonne à bande tournante d'une centaine de plateaux théoriques (Nester-Faust) et par chromatographie en phase vapeur ont également été tentés pour le diol 3_3 par Wolf (4) et se sont avérés inefficaces. Pour les diols du ty pe I (26-31) , les diols
32.
et 46., le rapport des facteurs de rétention Rf thréo/Rf méso est d ' environ 1.3 à 1.5.En considérant l'ordre d'élution d'alcools diastéréoisomè- res RCH( CHg )CHOHR' par chromatographie en phase vapeur Felkin (56) a essayé d'élaborer une règle afin de déterminer leur configu ration. Par extension, les g-diols suivent la même règle que les alcools du premier groupe (molécules chélatées) à savoir que généralement le composé thréo est élué le premier. La liai
diastéréoisomère, on est tenté de penser qu'il est moins suscep tible de former des liaisons intermoléculaires avec les groupes polaires de la phase stationnaire. Les facteurs de rétention relativement voisins pour les couples de diastéréoisomêres étu diés à substituants benzyles nous permettent de supposer que les hydroxyles du méso et du thréo sont engagés également dans des associations intramoléculàires.
En ce qui concerne les 8-diols asymétriques, il semble difficile d'élaborer une règle définie d'après leur élution sur colonne de gel de silice . Chacune des deux préparations de ces B-diols conduit à un diastéréoisomère majoritaire différent et généralement nous avons constaté que l'isomère le plus abondant est le plus retenu. Jacques (5) avait déjà formulé cette remarque dans le cas d'aminoalcools isomères. Leur élution par chromato graphie sur couche mince permet de calculer le rapport des fac teurs de rétention Rf thréo/Rf érythro qui se trouve être pour ces composés inférieur ou égal à 1.
La chromatographie sur colonne est une bonne voie de puri fication puisqu'elle nous a permis d'obtenir des diols spectro- scopiquement et analytiquement purs. Les isomères thréo des diols symétriques de type II sont les seuls à n'avoir pu être purifiés.
Des essais de séparation d'isomères ont déjà été réalisés par l'intermédiaire de leurs dérivés notamment pour des 8-diols bisecondaires (2,38,5 7,5 8). Les diacétates, les borates, les sulfites, les dioxanes-1,3 de ces diols s'obtiennent facilement.
57
Les g-diols bitertiaires comme tous les alcools tertiaires per mettent moins facilement de telles réactions,cependant Wolf (4) a préparé quelques diacétates et dioxanes-1,3 mais n'a pas da vantage séparé les isomères des dérivés des diols 2_6 et
23_
que nous ne l'avons fait pour les diastëréoisomères de ces diols.De 1'ensemble des expériences réalisées, il résulte que dans la série des g-diols symétriques le rendement des
réactions dépasse 50% pour les g-diols du type I (sauf pour 25 ). Et on constate que les g-diols asymétriques sont obtenus avec un rendement de 60% à partir des hydroxycétones du type II en introduisant un magnésien phénylique et avec un rendement in férieur à 50% à partir des hydroxycétones des types I et III réagissant avec un magnésien benzylique ou aliphatique. De ces résultats nous pouvons conclure à 1'influence de la nature de 1'entité nucléophile sur le rendement de la réaction.
III.5 Etude configurationnelle et conformationnelle
Parmi les g-diols diastéréoisomères que nous avons ob tenus par réduction de méthylcétones g-hydroxylées, seuls les diols ,21 et
26_
antérieurement préparés par une autre méthode (4) ont fait l'objet d'une détermination de configuration et de con formation.La configuration thréo ou ërythro de g-diols en série aromatique de formule
R2COH-CH(CH3)-CHOH-C6H5 R=C2H5,CgHg
au moyen d'une série de réactions stéréospécifiques. Notre étude par contre a été réalisée en utilisant les propriétés spectroscopiques de chaque diol diastérëoisomêre.
Dans certains cas (2,5,38,60), l'étude par spectroscopie infrarouge des liaisons hydrogènes intramoléculaires peut se révéler suffisante pour déterminer à la fois la configuration et la conformation des composés. Ce qui n'a pas été possible pour les B-diols étudiés dont les comportements different se lon les séries. La R.M.N. a pu établir de façon univoque 1'at tribution des configurations en se basant sur les signaux des méthyles et des méthylènes en a des hydroxyles.
A. Etude par infrarouge
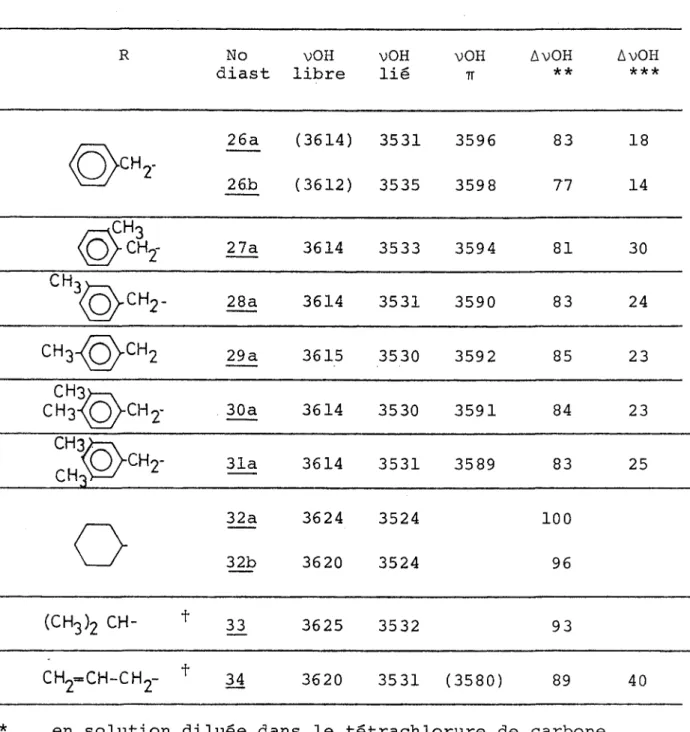
Les spectres complets de nos divers diols à l'état liqui de ou solide demeurent assez analogues pour des couples de diastéréoisomêres* et il est difficile d'en faire 1'interpréta tion, donc nous nous sommes limités à l'examen de la région comprise entre 3300 et 3700 cm""1, zone des vibrations de va lence des groupements hydroxyles.
Les différents types de vibration vOH ont été signalés précédemment (chapitre II) au sujet de l'étude par infrarouge des 8-hydroxycétones qui sera menée de façon parallèle en ce qui concerne les B-diols.
En solution peu diluée 10“dans le tétrachlorure de * Les valeurs des vibrations de valence vC-0 et de déforma
59
carbone on distingue trois types de bandes correspondant à des groupements OH, soit libres, soit en association intra- moléculaire, soit en associations dimérique ou polymérique. La fréquence vOH libre dépend des effets inductifs des substi tuants et de la classe de la fonction alcool.
En solution plus diluée 10""2 à 10“3M les associations intermoléculaires disparaissent et on mesure;
l'écart AvOH qui traduit la force de la liaison hydrogène
AvOH = vOH libre - vOH lié intra
l'aire intégrée de chaque bande qui représente l'absorp tion du vibrateur correspondant. De cette dernière mesure, plusieurs auteurs (2,38) ont tiré une évaluation semi-quan titative des concentrations relatives des espèces liées et non liées. L'étude la plus fondamentale a été réalisée par Bue sur une série de diols-1,3 primaires-secondaires et bisecondaires. Ce dernier détermine les coefficients d'absorption intégrée des bandes caractéristiques des hydroxyles libres (A) et liés (A1) calculées selon l'équation III (chapitre II).
L'auteur fait 1'hypothèse,dans le cas de g-diols symétri ques, que tout oscillateur OH (^=1 ou 2) non engagé dans un pont hydrogène a le même coefficient d'absorption intégrée A. que le monoalcool obtenu en saturant l'autre fonction alcool, ce qui permet d'écrire :
représentant le nombre d'OH libres d'espèce -i par mole de diol. Le nombre de ponts hydrogènes par mole est alors:
= 2 - Z^ (VII)
A une température donnée Bue observe que Z^n'^ demeure propor tionnel au coefficient d1 absorption intégrée A' de la bande OH lié:
A' = X'Zn^ (VIII)
X1 représente le coefficient d1 absorption intégrée d'un hydroxy- le totalement lié, Bue l'a déterminé égal à 1.17 ± 0.03 unité internationale. Cazaux (2) a appliqué cette méthode à une sé rie de 3-diols bisecondaires, ce qui lui a permis de montrer que X' n'est pas indépendant de la structure du diol. Il aug mente légèrement avec l'effet inductif mais demeure constant pour un même couple de diastéréoisomères.
Par ailleurs, il suppose dans le cas de 3-diols portant deux fonctions alcool qui ne sont pas de même ordre, que 1'hy drogène engagé dans la liaison intramoléculaire est celui qui appartient à la fonction alcool primaire.
Les valeurs spectroscopiques des fréquences v et des écarts Av sont consignées dans les tableaux VIII (3-diols symétriques) et IX (3-diols asymétriques). Les fréquences moyennes des ban des d'élongation des hydroxyles libres et liés sont récapitulées dans le tableau X. Le classement présent des 3-diols tient
61 TABLEAU VIII OH OH j Ç - CH, -- C • CH
R No vOH vOH vOH A vOH AvOH
diast libre lié TT ** ***
cm*' cm-1 crrr' crrr' cm"1 /PSv ~ 3616 3533 83 Gr 21b 3614 3509 (3574) 105 40 CH3' 22a 3615 (3529) 84 22b . 3612 3508 3572 104 40 Cl 23a 3615 3528 87 2 3b 3606 3507 (3570) 99 38 Cl CH3° 24a 3618 3524 94 24b 3609 3511 98 25a 3616 3526 80 25b 3614 3510 3574 104 40
TABLEAU VIII (suite)
R
NovOH
vOH
vOH
AvOH
AvOH
diast libre lié
ir
**
***
<0>CHr 26a 26 b (3614) (3612) 3531 3535 3596 3598 83 77 18 14 27a 3614 3533 3594 81 30 CH3)—v 0.ch2. 28a 3614 3531 3590 83 24 CH3^0>CH2 29a 3615 3530 3592 85 23 30a 3614 3530 3591 84 23
SËK
31a 3614 3531 3589 83 25 /"A 32a 3624 3524 100v>
32b 3620 3524 96 (CH3)2 ch- t 33 3625 3532 93 ch2=ch-ch2- t 34 3620 3531 (3580) 89 40 en solution diluée dans le tétrachlorure de carbone(2.510'3M)
AvOH = vOH
libre -vOH
lié intra.*** AvOH = vOH
libre -vOH
lié ttt spectre du mélange de diastéréoisomêre a = érythro
b = thréo