DISTRIBUTION CHEZ LA SOURIS GESTANTE DU
TRANSGÈNE GFP INTRODUIT AVEC UN VIRUS
ADÉNO-ASSOCIÉ DE SÉROTYPE 9
Mémoire
Ken Bisabu Kelu
Maîtrise en Biologie cellulaire et moléculaire
Maître ès sciences (M.Sc.)
Québec, Canada
iii
Résumé
La thérapie génique utilisant les vecteurs viraux est une des approches thérapeutiques possible pour le développement d’une thérapie pour les maladies génétiques mono-géniques dont l’ataxie de Friedreich (FRDA). Dans le but de vérifier l’efficacité des virus adéno-associés (AAV) à transférer la copie normale du transgène dans les tissus qu’ils infectent, nous avons insérer le gène codant pour la forme améliorée de la protéine fluorescente verte (eGFP) dans les AAV sérotype 9 (AAV9). Ces AAV recombinants (AAVr) ont été ensuite administrés aux souris femelles gestantes par voie intraveineuse et intra-péritonéale. Les résultats obtenus montrent que le transgène a été transféré à la fois dans les tissus des souris femelles ainsi que dans ceux des souriceaux.
v
Abstract
Gene therapy using viral vectors is one of the therapeutic approaches possible for the development of genetic therapies for monogenic diseases including Friedriech's Ataxia (FRDA). In order to verify the effectiveness of adeno-associated virus (AAV) to transfer a transgene in the tissues they infect, we inserted the gene encoding the enhanced form of the green fluorescent protein (eGFP) in AAV Serotype 9 (AAV9). These recombinant AAV (rAAV) were then administered by intravenous and intraperitoneal to pregnant female mice. The results obtained show that the transgene was transferred in both in the female’ mouse tissues and in those of young mice.
vii
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des tableaux ... ix
Liste des figures ... xi
Liste des abréviations... xiii
Dédicace ... xvii
Remerciements ... xix
INTRODUCTION ... 1
Chapitre 1 : Transfert des gènes et développement d’une thérapie possible pour l’ataxie de Friedrich ... 3
1.1. La thérapie par transfert des gènes ... 3
1.1.1. Définition ... 3
1.1.2. Historique ... 3
1.1.3. Systèmes de distribution des gènes ... 4
1.2. Développement de la thérapie par transfert des gènes pour l’ataxie de Friedreich ... 18
1.2.1. Aspects fondamentaux de l’ataxie de Friedreich ... 18
1.2.2. Étude préliminaire chez des souris modèles de l’ataxie de Friedreich. ... 25
1.2.3. Avantages de l’administration des vecteurs viraux adéno-associés chez des femelles en gestation. ... 25
Chapitre 2 : Distribution d’un transgène introduit par le virus adéno-associé sérotype 9 ... 27
2.1. Hypothèse et Objectifs ... 27 2.1.1. Hypothèse ... 27 2.1.2. Objectifs... 27 2.2. Matériels et Méthodes ... 27 2.2.1. Matériels ... 27 2.2.2. Méthodes ... 28 2.3. Résultats ... 30 2.3.1. PCR du gène eGFP ... 30
2.3.2. Immunobuvardage de la protéine eGFP ... 33
2.4. Discussion ... 36
Conclusion ... 41
ix
Liste des tableaux
Tableau 1: Caractéristiques des principaux vecteurs viraux utilisés en thérapie génique (Howarth, Lee, & Uney, 2010). ... 5 Tableau 2: Tropisme tissulaire préférentiel des AAV selon les sérotypes associés à leurs recepteurs et
corecepteurs correspondants (Buning, Perabo, Coutelle, Quadt-Humme, & Hallek, 2008), (Nonnenmacher
& Weber, 2012). ... 10 Tableau 3: Paramètres de la PCR et séquences des amorces pour amplifier l’eGFP ... 29
xi
Liste des figures
Figure 1: Organisation du génome de l'AAV sauvage (Drouin & Agbandje-McKenna, 2013). ... 9 Figure 2: Principales étapes du trafic intracellulaire des particules AAV sauvages (Schultz &
Chamberlain, 2008). ... 12 Figure 3: Illustration de l'organisation génomique d'AAVr. Forme standard (a). Forme complexe de la
cassette d'expression sous le contrôle d'un promoteur bidirectionnel (b) et dans un AAVr
bicistronique (c) (Le Bec & Douar, 2006). ... 13
Figure 4: Analogie des génomes AAVr et scAAV dans le noyau de la cellule hôte (Le Bec & Douar, 2006). ... 16 Figure 5: Taille de répétitions des triplets GAA dans l’intron 1 du gène de la frataxine corrélée aux
conséquences phénotypiques (Santos et al., 2010). ... 20
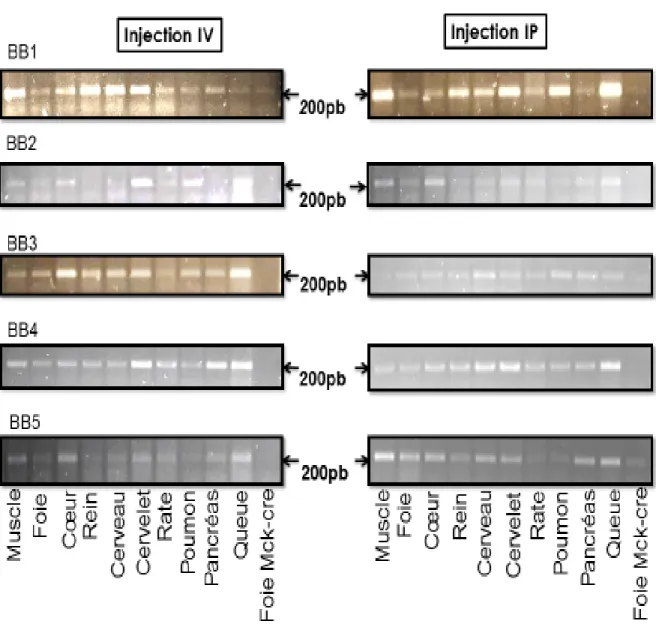
Figure 6: Physiopathologie de l’ataxie de Friedreich et différentes cibles thérapeutiques possibles (Santos et al., 2010). ... 22 Figure 7: Électrophorèse sur gel d'agarose 2% des produits PCR du gène eGFP dans les tissus
maternels. Foie MCK-Cre: Foie de la souris modèle MCK-Cre pris comme contrôle négatif. ... 31
Figure 8: Électrophorèse sur gel d'agarose 2% des produits PCR du gène eGFP dans les tissus des
bébés (BB1, BB2, BB3, BB4 et BB5). Foie MCK-Cre: Foie de la souris modèle MCK-Cre pris comme contrôle négatif. ... 32
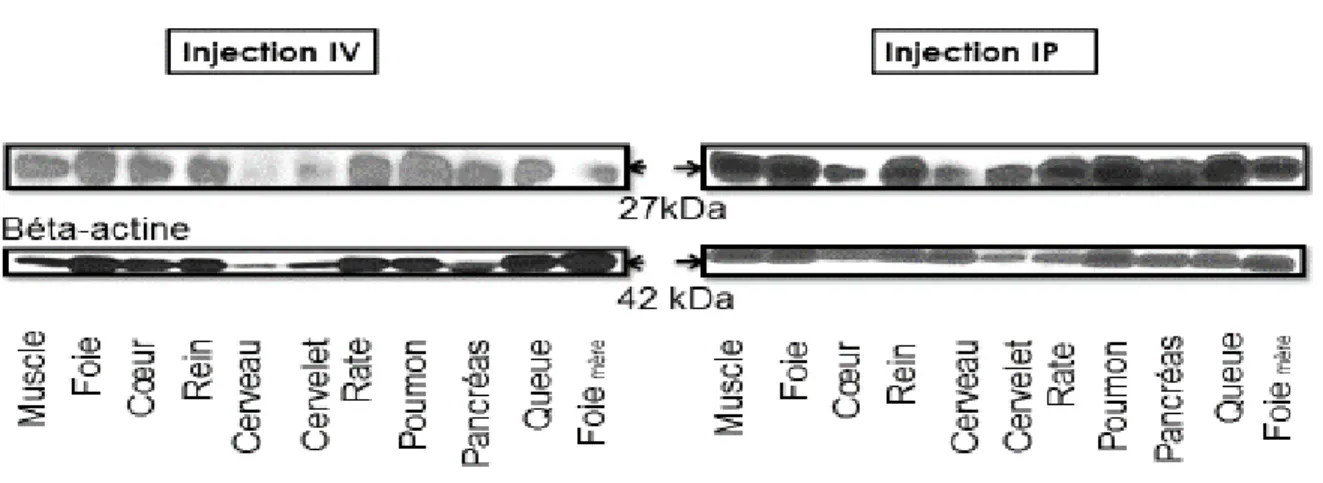
Figure 9: Immunobuvardages de la protéine eGFP faits dans les tissus maternels. Foie mère: Foie
d'une des femelles C57BL10J pris comme contrôle positif. Béta-actine: protéine cytoplasmique de référence prise comme contrôle interne. ... 33
Figure 10: Immunobuvardages de la protéine eGFP faits dans les tissus des bébés (BB1, BB2, BB3,
BB4 et BB5). Foie mère: Foie d'une des femelles C57BL10J pris comme contrôle positif. Béta-actine: protéine cytoplasmique de référence prise comme contrôle interne... 35
Figure 11: Immunobuvardages de la protéine eGFP pour le contrôle négatif. Foie mère: Foie d'une des
femelles C57BL10J pris comme contrôle positif, Foie MCK-Cre: Foie de la souris modèle MCK-Cre pris comme contrôle négatif, Foie: Foie d'une des souris C57BL10J. ... 36
xiii
Liste des abréviations
AAP : Assembly-Activating Protein
AAV : Adeno-Associated Virus (Virus Adéno-Associé)
AAVr : Recombinant Adeno-associated virus (Virus Adéno-Associé Recombinant) AAV9 : Adeno-associated virus Serotype 9 (Virus Adéno-Associé Sérotype 9) Ad : Adénovirus
ADA : Adénosine déaminase ADN : Acide désoxyribonucléique
ADNdb : Acide désoxyribonucléique double brin ADNsb : Acide désoxyribonucléique simple brin
ALV : Avian Leukemia Virus (Virus de la leucémie aviaire) ARN : Acide ribonucléique
ARNm : Acide ribonucléique messager BB : Bébé
BPF : Bonnes Pratiques de Fabrication CMV : Cytomégalovirus
CTS : Central Terminaison Sequence (Séquence de terminaison centrale) DAB : Diaminobenzidine tetrahydrochloride
eGFP : Enhanced Green Fluorescent Protein (Protéine fluorescente verte améliorée) EDTA : Ethylenediamine Tetra acetic acid (Acide Éthylène Diamine Tétra acétique)
ELISA : Enzyme-Linked Immunosorbent Assay (Dosage d’immunoadsorption par enzyme lié) emCBA : Enhanced mini Chicken Beta-Actine
FIX : Facteur anti-hémophilique IX FIV : Feline Immunodeficiency Virus
xiv
FRDA : Friedreich’s ataxia (ataxie de Friedreich) FXN : Frataxine
FIX : Facteur anti-hémophilique IX GAA : Guanine Adénine Adénine GMP : Good Manufacturing Practices
HAd : Adenovirus human type (Adénovirus de type humain)
HAd2 : Adenovirus Serotype 2 Human type (Adénovirus de sérotype 2 de type humain) HAd5 : Adenovirus Serotype 5 Human type (Adénovirus de sérotype 2 de type humain) HDAC : Histone déacétylase
HEK : Human Embryonic Kidney
hFXN : Human Frataxin (Frataxine humaine)
HIV-1 : Human immunodeficiency virus-1 (Virus de l’immunodéficience humaine de type 1) HFV : Human foamy virus
HRP : Horse-radish peroxydase HSV : Herpès simplex virus
HSV-1 : Herpès simplex virus de type 1 HSV-2 : Herpès simplex virus de type 2
HTLV : Human T-Lymphotropic Virus (Virus de lymphocyte T humain) 5-HTP : 5-Hydroxytryptophane
IRE : Iron Response Element (Élement de réponse à l’atome de fer) IRP : Iron Regulatory Protein (Protéine de régulation de l’atome de fer) ISCU : Iron Sulfate Cluster Assembly
ISD11 : Desulfurase Interacting Protein 11
xv
KDa : Kilo Dalton KIKI: Knock-in Knock-in KIKO : Knock-in Knock-out
LTR : Long Terminal Repeat (Longue répétition terminale) MCK-Cre : Muscular Creatinine Kinase-Cycling recombinase Nfs1 : Nitrogen Fixation 1
NLS : Nuclear Localization Signal (Signal de localisation nucléaire) NSE-Cre : Neurone Specific Enolase-Cycling recombinase ORI : Origine de réplication
OriS : Origine de réplication de la région unique courte p53: Protéine 53
PBS : Primer Binding Site (Site de liaison d’amorces) PBS : Phosphate Buffer Salin
PCR : Polymerase Chain Reaction (Réaction de polymérisation en chaine) pH : Potentiel à l’Hydrogène
PPAR : Peroxysome Proliferator Activated Receptor (Récepteur activé par les proliférations des peroxysomes) PPT : Poly Purine Tract
Rb : Protéine Rétinoblastome RBS : Rep Binding Protein Rev : Regulator of virion protein RRE : Rev Response Element
RSV : Red Sarcoma Virus (Virus de sarcome de Roux) Rb : Protéine Rétinoblastome
xvi
SDS : Sodium Dodecyl Sulfate
SDS-PAGE : Sodium Dodecyl Sulfate Polyacrylamide (Électrophorèse en gel de polyacrylamide contenant du
dodécyl sulfate de sodium)
SIV : Simian Immunodeficiency Virus SMN : Survival Motor Neuron SNC : Système nerveux central SRI : Séquence répétée inversée TBE : Tris Boric Acid EDTA TRS : Terminal Resolution Site UL : Région unique longue
US : Région unique courte
VHH : Virus herpétique humain
X1-SCID: Type 1 of X-linked Severe Combined Immunodeficiency (Déficit Immunitaire Sévère de type 1 lié à l’X)
xvii
Dédicace
À mes parents,
À ma famille biologique,
À tous ceux et celles qui me sont chers, Je dédie ce mémoire.
xix
Remerciements
<<Celui qui reçoit la connaissance la recueille comme un don inestimable et jamais comme une contrainte pénible>> disait Albert Einstein. L’accomplissement de cette maitrise à l’université Laval dans le programme de biologie cellulaire et moléculaire vient d’apporter une touche particulière à mon parcours professionnel de médecin-clinicien.
Au premier regard, je voudrais sincèrement remercier mon directeur de recherche, le Dr jacques P. Tremblay, pour non seulement m’avoir accepté dans son laboratoire mais aussi et surtout pour sa preuve de patience et de compréhension à mon égard en dépit de mes imperfections techniques. Grâce à lui et à son équipe, j’ai pu bénéficier d’un nouveau bagage scientifique qui complète mes connaissances sur la compréhension et la prise en charge des maladies. A travers ces quelques lignes, veuillez trouver l’expression de ma profonde gratitude. Je voudrai aussi remercier mon organisme boursier, le Programme Canadien de Bourses de la Francophonie / le Bureau Canadien de l’Éducation Internationale (PCBF/BCEI) et son Agence d’exécution, l’Association des Universités et Collèges du Canada (AUCC), tous mandatés par le Ministère des Affaires étrangères, Commerce et Développement Canada (MAECD) pour le soutien financier dont j’ai pu bénéficier pour ma formation.
Je voudrais particulièrement témoigner ma reconnaissance à Madame Jeanne Gallagher, ancienne gestionnaire principale du PCBF, pour sa rigueur et son soutien moral durant toute la période de ma formation. Ma gratitude est aussi dirigée vers toutes/ tous celles/ceux qui ont contribué d’une manière ou d’une autre à l’aboutissement de cette formation.
1
INTRODUCTION
L’introduction d’un transgène dans la cellule à des fins thérapeutiques est une des stratégies de la biologie moléculaire exploitée en thérapie génique d’addition qui consiste à l’introduire la copie normale d’un gène muté dans les cellules cibles dans le but de restaurer les fonctions cellulaires déficientes. Cette stratégie de complémentation génique est à priori adaptée aux maladies génétiques mono-géniques à mode de transmission récessive mais son champ d’application est également étendu à certaines maladies acquises dont le traitement curatif peut nécessiter l’apport d’une protéine fonctionnelle exogène (Bunnell & Morgan, 1998).
Les systèmes de distribution qui accompagne cette approche de transfert des gènes restent à ces jours une des difficultés majeures qui limite la livraison du matériel génétique thérapeutique aux cellules d’intérêt (Bouard, Alazard-Dany, & Cosset, 2009). Plusieurs systèmes d’expression des gènes sont utilisés à cet effet, entre autres ceux utilisant les vecteurs viraux et non viraux dont les avantages et inconvénients de chacun ont été évalués dans plusieurs expériences de thérapie génique aussi bien in vivo qu’ex-vivo (Razi Soofiyani, Baradaran, Lotfipour, Kazemi, & Mohammadnejad, 2013), (Wang & Gao, 2014).
Dans le présent projet, les vecteurs viraux ont particulièrement retenu notre attention, singulièrement le sérotype 9 des AAVr eu égard de son efficacité prouvé dans de nombreux essais précliniques et cliniques réussis (Daya & Berns, 2008). Il constitue à l’heure actuelle un vecteur de convenance capable d’infecter multiples organes y compris ceux du système nerveux central (SNC) (Rahim et al., 2011) , le cœur (Pacak et al., 2006) et le pancréas (Griffin et al., 2014) qui sont les tissus cibles affectés par plusieurs maladies neurodégénératives d’origine génétique dont l’ataxie de Friedreich qui nous intéresse particulièrement comme maladie modèle sur laquelle peut s’appliquer cette actuelle approche thérapeutique de transfert des gènes. Les travaux réalisés dans le présent mémoire ont poursuivis deux grands desseins, celui de tester l’efficacité de ce système utilisant l’AAV9 comme vecteur de choix, à infecter les tissus et d’évaluer le degré de distribution du virus dans les tissus selon les voies et le moment d’administration de celui-ci. Pour ce faire, nous nous sommes servis d’un AAV9 codant pour le gène eGFP sous le contrôle du promoteur emCBA, que nous avons administré à des femelles en gestation dans le but de rendre disponible précocement le transgène dans les tissus des souriceaux.
3
Chapitre 1 : Transfert des gènes et développement
d’une thérapie possible pour l’ataxie de Friedrich
1.1. La thérapie par transfert des gènes
1.1.1. Définition
La thérapie génique, abordée depuis plus d’une quarantaine d’années, est une stratégie thérapeutique basée sur les connaissances de la biologie moléculaire et de la génétique humaine, qui repose sur l’introduction et l’expression spécifique d’un ou de plusieurs gènes thérapeutiques dans les cellules, dans le but soit de suppléer le dysfonctionnement d’un gène anormal ou absent, soit d’apporter une nouvelle fonction aux fins d’une thérapie curative ou préventive.
1.1.2. Historique
La découverte de la molécule d’acide désoxyribonucléique (ADN) le 25 avril 1953 par James Watson et Francis Crick (Watson & Crick, 1953) et le décryptage de l’information génétique véhiculée par cette dernière, sont au centre de la thérapie génique. Cette macromolécule contient des informations utiles à l’accomplissement des diverses fonctions cellulaires et elle peut être introduite dans la cellule à l’aide des vecteurs d’expression des gènes appropriés.
Il a fallu alors attendre environs vingt ans après la caractérisation des enzymes de restriction que les premières expériences de biologie moléculaire exploitant le potentiel des virus dans le transfert des gènes voient le jour (Friedmann, 1992). L’évolution à grande amplitude de cette nouvelle technologie de la biologie moléculaire a nécessité en 1974, la tenue de la conférence d’Asilomar aux États-Unis qui a réuni les chercheurs de la biologie moléculaire et les médecins dans l’objectif d’encadrer et de sécuriser la faisabilité de cette nouvelle approche dans le respect des lois éthiques comme précédemment établies par le code de Nuremberg (1947) et la déclaration d’Helsinki (1964) (Berg, 2008), (Jin, Yang, & Li, 2008). Dans ce nouveau terrain des lois et principes contrôlant la recherche médicale impliquant les êtres humains, le Dr Martin Cline de l’université de Californie à Los Angeles a été le premier à se mouiller en tentant d’introduire pour la toute première fois le gène fonctionnel de l’hémoglobine à deux patients souffrant de la thalassémie (Mercola & Cline, 1980).
A l’heure actuelle, avec le développement de la biologie moléculaire et de la génétique humaine, la matérialisation de cette nouvelle approche passe par la compréhension et l’optimisation d’un système de distribution capable de livrer le matériel génétique thérapeutique avec sécurité et efficacité. En effet,
4
l’utilisation de la macromolécule d’ADN à nu comme matériel génétique thérapeutique est limitée par son incapacité à traverser les membranes cellulaires et son instabilité dans un milieu biologique. Ces contraintes biologiques conditionnent son incorporation dans un vecteur qui permettra non seulement de la compacter et recouvrir sa charge négative mais aussi et surtout de servir de système de distribution pour l’acheminer dans la cellule d’intérêt.
1.1.3. Systèmes de distribution des gènes
Plusieurs systèmes de vectorisation pour le transfert des gènes ont été développés. Les vecteurs viraux particulièrement ceux dérivés des rétrovirus, des adénovirus (Ad), des virus de l’herpès simplex et des AAV sont les plus utilisés dans les expérimentations de transfert des gènes aussi bien in vivo qu’ex vivo (Wang & Gao, 2014), (Evans, Ghivizzani, & Robbins, 2012). Les vecteurs non viraux ou synthétiques (les lipides cationiques, les nanoparticules et les exosomes) et les méthodes physiques (l’electroporation, le gene gun et les injections hydrodynamiques) font parties aussi de système de transfert des gènes mais avec une efficacité moindre par rapport à ceux d’origine virale (Wang & Gao, 2014) .
Le choix sur le système de distribution des gènes est déterminé pour chaque maladie en fonction de la nature de l’anomalie génétique en cause, les cellules et les organes à cibler (Lundstrom & Boulikas, 2003). À ce jour, les vecteurs viraux restent les principaux vecteurs utilisés en thérapie génique pour les maladies génétiques mono-géniques (Bouard et al., 2009). En effet, les premiers succès de la thérapie génique l’ont été avec l’utilisation de vecteurs viraux. C’est en 1990 et 2000 respectivement, que les équipes de French Anderson, Michael Blaese et collaborateurs (Anderson, Blaese, & Culver, 1990) et de Alain Fisher (Fischer, 2000), ont tenté de façon séparée, le tout premier essai clinique réussi, utilisant un rétrovirus codant pour le gène de l’adénosine déaminase dans les lymphocytes T et B des fillettes souffrant d’un déficit immunitaire en adénosine déaminase (ADA), une des formes des déficits immunitaires combinés sévères d’origine génétique. Ensuite, la guérison des nourrissons atteints du déficit immunitaire combiné sévère de type 1 lié au chromosome X (X1-SCID) avec l’équipe du Professeur Alain Fisher, Marianne Cavazzana-Calvo et collaborateurs (Cavazzana-Calvo et al., 2000) qui introduisit dans les cellules précurseurs hématopoïétiques d’origine médullaire (les CD34+), le gène normal codant pour la sous-unité ɣc commune aux récepteurs de certaines cytokines, à l’aide d’un vecteur rétroviral. Beaucoup d’autres essais cliniques utilisant les vecteurs viraux ont montré l’efficacité de transduction du transgène avec ces derniers dans plusieurs maladies génétiques mono-géniques comme l’Amaurose congénitale de Leber (Maguire et al., 2009), l’hémophile B (Nathwani et al., 2011), l’adrénoleucodystrophie (Cartier et al., 2010), la mucoviscidose (Flotte et al., 2003) et les myopathies de Duchenne (Bowles et al., 2012).
5 1.1.3.1. Vecteurs viraux recombinants
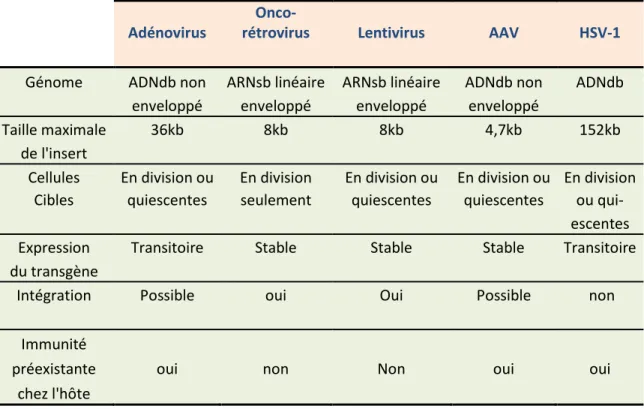
Cette section portera essentiellement sur les principaux types des vecteurs viraux recombinants utilisés en thérapie génique et dont les caractéristiques (Tableau 1), avantages et inconvénients de chacun ont été évalués par des nombreuses expériences de transfert des gènes conduites aussi bien in vitro qu’in vivo. Tableau 1: Caractéristiques des principaux vecteurs viraux utilisés en thérapie génique (Howarth, Lee, & Uney, 2010).
Adénovirus
Onco-rétrovirus Lentivirus AAV HSV-1
Génome ADNdb non ARNsb linéaire ARNsb linéaire ADNdb non ADNdb
enveloppé enveloppé enveloppé enveloppé
Taille maximale 36kb 8kb 8kb 4,7kb 152kb
de l'insert
Cellules En division ou En division En division ou En division ou En division Cibles quiescentes seulement quiescentes quiescentes ou qui-
escentes
Expression Transitoire Stable Stable Stable Transitoire
du transgène
Intégration Possible oui Oui Possible non
Immunité
préexistante oui non Non oui oui
chez l'hôte
1.1.3.1.1. Vecteurs rétroviraux
Les rétrovirus sont une famille des virus à ARN diploïde simple brin et à transcription inverse, appartenant à la classe de rétroviridae qui regroupe en son sein plus d’une centaine de genres, entre autre les onco-rétrovirus, les lentivirus, les alpha rétrovirus et les spumavirus, exploités en thérapie génique et particulièrement les onco-rétrovirus et les lentivirus desquels dérivent les vecteurs les plus fréquemment utilisés dans le système de distribution des gènes (McKay et al., 2011).
1.1.3.1.1.1. Onco-rétrovirus
1.1.3.1.1.1.1. Onco-rétrovirus sauvages
Le virus de la Leucémie Murine de Moloney (MLV) représente le genre de gamma rétrovirus. Il est pathogène pour la souris et possède un génome viral simple, de nature ribonucléique (ARN) qui comprend trois cadres de
6
lecture ouverts; les gènes GAG, POL et ENV. Ils partagent en commun avec les lentivirus d’autres éléments structuraux du génome agissant en cis, notamment la séquence ψ se rapportant au signal d’encapsidation de l’ARN viral, la séquence PBS (Primer Binding Site), la séquence PPT (Poly purine tract), la séquence CTS (central terminaison sequence) intervenant dans le processus de rétro transcription et les séquences LTR (Long Terminal Repeat) qui encadrent en 5’ et 3’ le génome viral et qui contiennent un site de polyadénylation. Les LTR sont impliquées dans la rétro transcription, l’intégration du génome viral et servent de région promotrice de la transcription (Burmeister, 2001), (Escors & Breckpot, 2010). Ces virus avec leurs vecteurs correspondants sont réputés pour avoir un pouvoir oncogénique lié à la fois à l’induction par le virus de l’activation d’oncogènes cellulaires à partir de la région promotrice des LTR et à l’activation à distance par les séquences activatrices dites «enhancers» des LTR, d’un promoteur oncogénique de la cellule hôte (Burmeister, 2001), (Deichmann et al., 2011).
1.1.3.1.1.1.2. Onco-rétrovirus recombinants
Les vecteurs onco-rétroviraux dérivent particulièrement du MLV de souris (Blaese et al., 1995). Du génome viral sauvage, ne sont conservées que les séquences LTR qui contrôlent l’expression du transgène et les séquences ψ, PBS et PPT. Ces vecteurs n’infectent que les cellules en mitose car ils profitent de la dissolution de la membrane nucléaire lors de la prophase mitotique pour accéder au noyau et interagir avec l’ADN de l’hôte, et sont donc incapables de transduire les cellules en interphase et celles qui ne se divisent pas (Verma & Somia, 1997), ce qui empêcherait leur application dans le transfert des gènes pour les cellules du SNC différenciées (Blomer et al., 1997).
1.1.3.1.1.2. Lentivirus
1.1.3.1.1.2.1. Lentivirus sauvages
Les HIV-1 (Human Immunodeficiency Virus-1), SIV (Simian Immunodeficiency Virus) et FIV (Feline Immunodeficiency Virus), respectivement pathogènes pour l’homme, les primates non humains et les félins, représentent le genre lentivirus (Razi Soofiyani et al., 2013). Il possède un génome complexe constitué des trois cadres de lecture de base auxquels s’ajoutent les séquences de régulation (tat et rev) et les séquences accessoires (nef, vif, vpu et vpr) (Frankel & Young, 1998). Les lentivirus par contre ne sont pas associés à la genèse des tumeurs chez l’hôte, néanmoins ils sont réputés pour être à la base d’un plus grand nombre de transformations cellulaires rencontrées surtout avec le virus de l’immunodéficience simien (VIS) (Maggiorella et al., 1998).
1.1.3.1.1.2.2. Lentivirus recombinants
Les vecteurs lentiviraux dérivent particulièrement des HIV-1, SIV ou FIV. Leurs génomes diffèrent de celui du vecteur onco-rétroviral par la présence de la séquence RRE sur laquelle se fixe la protéine régulatrice Rev qui
7 sert à l’exportation vers le cytoplasme des ARNs constitutifs du génome viral et des ARNm viraux codant pour les protéines de structure et les enzymes virales (Lee, Culver, Carpenter, & Dobbs, 2008). Ces vecteurs infectent à la fois les cellules en état de quiescence (phase G0) et celles qui se divisent (Lewis, Hensel, &
Emerman, 1992) car ils possèdent un signal de localisation nucléaire (NLS) qui leur permet d’accéder au noyau de la cellule hôte et d’interagir avec l’ADN (Zennou et al., 2000), (Neshati, Liu, Zhou, Schalij, & de Vries, 2014). Le vecteur a l’avantage d’avoir une capacité d’encapsidation élevée (7 à 8 kb) mais le pouvoir pathogène associé à la forme sauvage dont il dérive, limite son utilisation comme vecteur d’expression (Uren, Kool, Berns, & van Lohuizen, 2005).
1.1.3.1.2. Vecteurs Adénoviraux 1.1.3.1.2.1. Adénovirus sauvages
L’Adénovirus appartient à la famille d’adénoviridae regroupant plusieurs genres dont le mastadenoviridae qui comprend l’adénovirus de type humain (HAd), pathogène pour l’homme et reparti en 51 sérotypes dont le 5 et le 2 sont les plus fréquemment utilisés en thérapie génique et en protocole de vaccination clinique (Yu et al., 2012). Il possède un génome à ADN double brin de 36 kb composé de trois régions ( précoce, intermédiaire et tardive) codantes flanquées des séquences ITR, dont l’expression est synchronisée selon les phases du cycle infectieux (Robinson, Seto, Jones, Dyer, & Chodosh, 2011). Ils sont hautement immunogènes (Tatsis & Ertl, 2004) et réputés des propriétés de transformation cellulaire et tumorigéniques chez le modèle animal à la suite de l’inhibition des protéines suppresseurs de tumeurs (p53 et Rb) par les régions E1a et E1b (Talbot & Crawford, 2004). Ces propriétés de transformation cellulaire peuvent évoluer vers le cancer lorsqu’en plus de l’inhibition des protéines suppresseures de tumeurs, s’ajoutent d’autres facteurs susceptibles d’activer les oncogènes cellulaires.
1.1.3.12.2. Adénovirus recombinants
Les vecteurs Adénoviraux dérivent essentiellement du sérotype 2 et 5 du virus sauvage de type humain (HAd2 et HAd5) car ils ne sont pas responsables des pathologies humaines graves. Trois générations de vecteurs existent et sont caractérisées par des régions délétées spécifiques à partir du génome sauvage. Le vecteur de troisième génération, dénué de tous les gènes viraux exceptées les séquences ITR, sont moins immunogènes et moins toxiques (Tatsis & Ertl, 2004) mais les essais précliniques sont encore très nécessaires pour s’assurer de la sécurité de ces virus comme vecteurs de thérapie génique (St George, 2003),(Brunetti-Pierri & Ng, 2008).
1.1.3.1.3. Vecteurs herpétiques (Virus de l’herpès simplex)
Le virus de l’herpès simplex (HSV) est un Virus Herpétique Humain (VHH) appartenant à la famille des Herpesviridae, à la sous famille des Alphaherpesvirinae et au genre de Simplex Virus qui comprend le virus de l’Herpès simplex de type 1 (HSV-1) et 2 (HSV-2), le cytomégalovirus et le Varicellovirus (vacella-zoster-virus)
8
(de Silva & Bowers, 2009), (de Oliveira & Fraefel, 2010). Le HSV-1 est parmi les virus qui présentent un intérêt particulier pour le transfert des gènes du fait entre autre de son tropisme naturel pour les organes du système nerveux central (SNC) (de Silva & Bowers, 2009), (Ho et al., 1995).
1.1.3.1.3.1. Herpès simplex type 1 sauvage
Les HSV-1 sont des virus à ADN double brin linéaire, de 152kb de taille dont le génome est logé dans la capside virale entourée par le tégument et une enveloppe protéo-lipidique externe. Il est composé des séquences UL (unique long), Us (unique court) et de trois origines de réplication, OriL en une seule copie dans
la région UL et OriS en deux copies dans la région US, encadrées par des séquences répétées inversées (SRI)
aux extrémités 5’ et 3’ (de Oliveira & Fraefel, 2010).
1.1.3.1.3.2. Virus de l’herpès simplex recombinant de type 1
Au regard de la taille élevée du génome viral sauvage, trois types différents des vecteurs viraux dérivés de l’HSV-1 sont produits, les vecteurs HSV-1 recombinants (atténués et non réplicatifs) et les vecteurs HSV-1 amplicons (Jacobs, Breakefield, & Fraefel, 1999). Le vecteur a l’avantage d’infecter naturellement des types cellulaires bien spécifiques sans nécessité de modification de son enveloppe, notamment les cellules neuronales et épithéliales (Nicoll, Proenca, & Efstathiou, 2012) . En outre son utilisation n’est pas limitée par la taille du transgène mais l’immunogénicité et la cytotoxicité qui lui sont associées le rendent moins pratique en thérapie génique qu’en protocole de vaccination et stratégie anti-tumorale (Jacobs et al., 1999).
1.1.3.1.4. Vecteurs viraux Adéno-associés 1.1.3.1.4.1. Virus adéno-associé sauvage
Les AAV sont de petits virus non pathogènes pour l’homme qui appartiennent au genre de dependoviridae et à la famille de parvoviridae, découverts par Robert W. Atchison en 1965 comme contaminants de préparations adénovirales (Atchison, Casto, & Hammon, 1965). Ils possèdent une capside à symétrie icosaédrique d’environs 20 nm de diamètre recouvrant un génome à ADN simple brin d’environ 5 kilo bases (Kb) de taille, constitué de deux séquences terminales répétées inversées (ITR) délimitant deux régions codantes Rep et Cap qui codent respectivement pour les protéines non structurales (Rep 78, 68,52 et 40) sous le contrôle du promoteur p5 et p19 et les protéines structurales (VP1, VP2, VP3 et AAP) sous le contrôle du promoteur p40. Les ITR contiennent les sites RBS (Site de liaison de la protéine Rep) et TRS (Site de résolution terminale), indispensables pour l’encapsidation, l’intégration et la réplication du virus (Sonntag, Schmidt, & Kleinschmidt, 2010).
9 Figure 1: Organisation du génome de l'AAV sauvage (Drouin & Agbandje-McKenna, 2013).
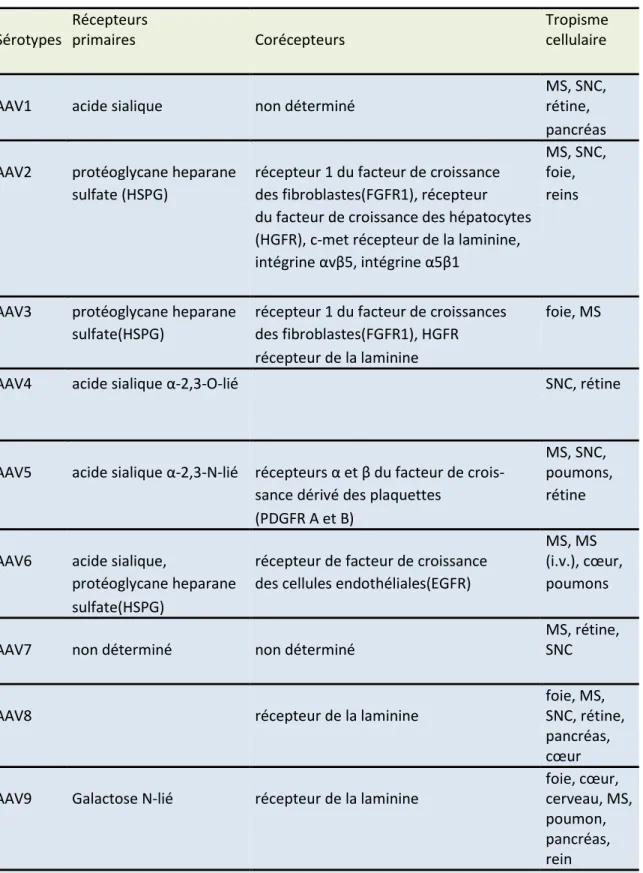
1.1.3.1.4.1.1. Répartition en Sérotypes et Tropisme tissulaire préférentiel
L’identification des sérotypes et variants des AAV ont été faites à partir de la mise en évidence de ces derniers dans un grand nombre de tissus d’espèces animales et humaines corroborant avec la haute prévalence anti- AAV dans ces espèces (Calcedo, Vandenberghe, Gao, Lin, & Wilson, 2009). Les propriétés physico-chimiques et différences biophysico-chimiques des acides aminés constitutifs des protéines de la capside virale sont à la base de cette diversité et permettent aux virus de reconnaitre de façon spécifique les récepteurs et corécepteurs membranaires de surface cellulaire avant leur internalisation dans la cellule hôte par endocytose (Agbandje-McKenna & Kleinschmidt, 2011). Plus d’une centaine de sérotypes et variants d’AAV ont été isolés des différentes espèces animales (Chiorini, Afione, & Kotin, 1999), (Wu, Asokan, & Samulski, 2006). Parmi les sérotypes identifiés, cinq ont été isolés du stock des préparations adénovirales alors que tous les autres sérotypes ont été découverts dans des tissus humains et non humains (Atchison et al., 1965), (Gao et al., 2004). Le sérotype 2, le premier à avoir été étudié, et les 9 autres premiers sérotypes sont, les plus utilisés en pratique pour la production des vecteurs recombinants. Chacun d’eux possèdent un tropisme préférentiel d’infection quoi que non exclusif, pour un organe ou un type cellulaire donné (Tableau 2). La découverte récente du sérotype 9 capable de reconnaitre des récepteurs cellulaires des tissus nerveux, cardiaques et d’autres tissus ouvre une nouvelle perspective pour son utilisation dans les pathologies héréditaires mono-géniques.
10
Tableau 2: Tropisme tissulaire préférentiel des AAV selon les sérotypes associés à leurs recepteurs et corecepteurs correspondants(Buning, Perabo, Coutelle, Quadt-Humme, & Hallek, 2008), (Nonnenmacher & Weber, 2012). Sérotypes Récepteurs primaires Corécepteurs Tropisme cellulaire
AAV1 acide sialique non déterminé
MS, SNC, rétine,
pancréas
AAV2 protéoglycane heparane récepteur 1 du facteur de croissance
MS, SNC, foie,
sulfate (HSPG) des fibroblastes(FGFR1), récepteur reins
du facteur de croissance des hépatocytes
(HGFR), c-met récepteur de la laminine,
intégrine αvβ5, intégrine α5β1
AAV3 protéoglycane heparane récepteur 1 du facteur de croissances foie, MS
sulfate(HSPG) des fibroblastes(FGFR1), HGFR
récepteur de la laminine
AAV4 acide sialique α-2,3-O-lié SNC, rétine
AAV5 acide sialique α-2,3-N-lié récepteurs α et β du facteur de crois-
MS, SNC, poumons,
sance dérivé des plaquettes rétine
(PDGFR A et B)
AAV6 acide sialique, récepteur de facteur de croissance
MS, MS (i.v.), cœur, protéoglycane heparane des cellules endothéliales(EGFR) poumons
sulfate(HSPG)
AAV7 non déterminé non déterminé
MS, rétine, SNC
AAV8 récepteur de la laminine
foie, MS, SNC, rétine,
pancréas, cœur AAV9 Galactose N-lié récepteur de la laminine
foie, cœur, cerveau, MS, poumon, pancréas, rein AAV
11
10, 11,12 non déterminé non déterminé
1.1.3.1.4.1.2. Cycle viral
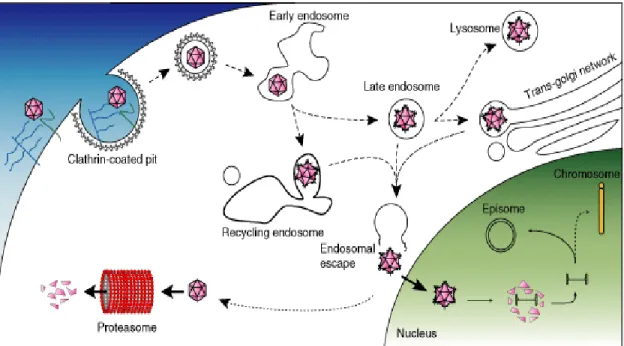
Le manque d’autonomie de réplication des AAV les soumet à un cycle infectieux reparti en deux phases selon l’assistance ou non d’un virus auxiliaire, le plus souvent un Ad qui est le premier à être décrit comme virus auxiliaire des AAV et à l’origine même de leur dénomination des «virus adéno-associés ». Par ailleurs d’autres virus en l’occurrence le HSV-1, le cytomégalovirus (CMV) et le virus du papillome humain (HPV) sont aussi capables de jouer la fonction d’assistance dans la réalisation du cycle réplicatif des AAV (Daya & Berns, 2008). En effet, l’entrée d’un AAV dans la cellule commence par la reconnaissance des récepteurs et corécepteurs de surface cellulaire par les protéines de structure de la capside virale. Cette interaction s’accompagne de la formation des vésicules à clathrine puis de l’internalisation du virus par endocytose. Le trafic intracellulaire du virus vers le noyau se fait par le transport endosomal allant de l’endosome précoce à celui tardif et actionné par les protéines motrices de la matrice intracellulaire le long du réseau des microtubules. L’environnement acide de l’endosome tardif favorise la libération des particules virales puis leur échappement dans une région cytoplasmique à proximité du noyau cellulaire hôte. L’entrée du virus dans le noyau n’est pas clairement connu mais les hypothèses évoqués par certains auteurs suggèrent une probable interaction avec les protéines des pores nucléaires (Hansen, Qing, & Srivastava, 2001), (Nonnenmacher & Weber, 2012). Parallèlement ce trafic peut être dévié vers le système Ubiquitine-Protéasome (UPS) initié par la phosphorylation des acides aminés polaires non chargés et à pH neutre (Tyrosine et Sérine), exposés à la surface des capsides virales (Aslanidi et al., 2013). Dans le noyau, il a lieu la décapsidation des particules virales suivie de la mise à nu de leur génome. En absence du virus auxiliaire, l’AAV entre dans la phase de latence où son génome simple brin est converti en ADN double brin soit sous l’action de la machinerie réplicative de la cellule hôte, soit à la suite de l’appariement de deux génomes simple brin de polarité opposée. Après cette conversion, le génome viral persiste en état de latence soit sous forme épisomal circulaire avec la formation des structures concatémères d’ADN de plus haut poids moléculaire ou il s’intègre dans le génome hôte sur un site spécifique, le locus AAVS1 localisé chez l’homme sur le chromosome 19q13.42 (bras long du chromosome 19, région 13, bande 42) sous la dépendance des protéines Rep et séquences ITR (Figure 2). Ces phénomènes intégratifs sont particulièrement prouvés pour le sérotype 2 d’AAV (AAV2) (Huser et al., 2010). Par ailleurs d’autres loci ont été récemment caractérisés, les loci AAVS2 sur le chromosome 5p13.3 et AAVS3 sur le chromosome 3p24.3 mais ils restent non spécifiques et aucune intégration n’y a été associée à ce jour. En présence du virus auxiliaire, l’AAV entre dans la phase réplicative caractérisée par la réplication de l’ADN simple du génome viral à polarité positive ou négative et à proportion égale, la synthèse des protéines Rep et Cap et l’assemblage de particules virales combinée avec la réplication
12
autonome du virus auxiliaire avec comme issue un éclatement de la cellule hôte suivi de la libération de particules virales.
Figure 2: Principales étapes du trafic intracellulaire des particules AAV sauvages (Schultz & Chamberlain, 2008).
1.1.3.1.4.2. Virus Adéno-associés recombinants
La structure des AAVr est faite des séquences ITR, nécessaires à la conversion double brin de l’ADN simple du virus et à son encapsidation. Les gènes Rep et Cap du génome sauvage sont retirés et remplacés par la cassette d’expression comportant le gène thérapeutique d’intérêt avec son promoteur en amont et une séquence de polyadénylation en aval, le tout dans la limite de 5 kb d’encapsidation du virus (Figure 3).
13 Figure 3: Illustration de l'organisation génomique d'AAVr. Forme standard (a). Forme complexe de la
cassette d'expression sous le contrôle d'un promoteur bidirectionnel (b) et dans un AAVr bicistronique (c) (Le Bec & Douar, 2006).
1.1.3.1.4.2.1. Conception, purification et optimisation des vecteurs viraux adéno-associés 1.1.3.1.4.2.1.1. Conception et purification des AAVr
Le protocole classique de production des AAVr utilise la méthode de production transitoire basée sur la tri-transfection dans des cellules des mammifères, généralement les 293 dérivées des HEK (Human Embryonic Kidney) de trois plasmides vecteurs apportant respectivement le transgène et les ITR, les gènes Rep et Cap et les séquences du virus assistant pour les fonctions auxiliaires (Shin, Yue, & Duan, 2012), (Mueller, Ratner, Zhong, Esteves-Sena, & Gao, 2012). Les autres protocoles utilisent un système d’expression reproductible dans les lignées cellulaires d’insectes, en particulier le baculovirus (Kohlbrenner et al., 2005). Cependant, pour
14
l’une ou l’autre des méthodes de production, le vecteur est obtenu sans le gène Rep du virus sauvage qui n’est utilisé que pour la phase de production et n’est pas délivré dans les cellules infectées. Ceci réduit fortement la probabilité de l’intégration du vecteur dans le génome hôte. Ce faible risque intégratif a été signalé dans un certain nombre d’études démontrant les phénomènes intégratifs localisés de manière élective sur le chromosome 12, à proximité des régions géniques qui codent pour des micro-ARN régulateurs (Donsante et al., 2007), des ilots CpG et des régions riches en Guanine et Cytosine (Li et al., 2011) et associés à un risque faible de formation des carcinomes hépatocellulaires. Par ailleurs beaucoup d’autres études réalisées sur un large éventail de modèles murins utilisant les AAVr n’ont pas fait état de ces phénomènes (P. Bell et al., 2005). On pense donc que d’autres facteurs intrinsèques à des modèles animaux et à des pathologies spécifiques seraient à la base de ces différences de constatations car Peter Bell et collaborateurs révèlent dans une étude menée chez les souris B6C3F1 que même les souris contrôles n’ayant pas été traitées avec l’AAVr développent des tumeurs au même titre que les souris traitées (P. Bell et al., 2006). Aux fins de l’utilisation clinique, chacune des méthodes de production devra être couplée à des meilleurs procédés de purification répondant aux exigences des bonnes pratiques de fabrication (BPF/GMP) (Allay et al., 2011).
1.1.3.1.4.2.1.2. Optimisation
1.1.3.1.4.2.1.2.1. Modifications des protéines de la capside virale
Dans le but d’obtenir la spécificité de reconnaissance des cellules d’intérêt, différentes stratégies sont utilisées pour créer des mutations au niveau des protéines de la capside virale, générer des capsides hybrides composées des protéines structurales dérivées de deux sérotypes différents ou encore des capsides chimériques ou mosaïques (Choi, McCarty, & Samulski, 2005). D’autres protocoles introduisent des mutations qui affectent les acides aminés de la capside virale substrats des kinases dont la phosphorylation dévie le trafic intracellulaire du vecteur vers la voie de dégradation protéasomale. Cette dernière stratégie améliore le trafic intracellulaire du vecteur et par voie de conséquence l’efficacité de transduction (Gabriel et al., 2013). 1.1.3.1.4.2.1.2.2. Amélioration de la capacité d’encapsidation
La capacité d’encapsidation des AAVr limitée à environ 5 kb constitue une limite pour l’empaquetage de transgène de taille élevée. Une des stratégies d’amélioration constitue à cliver le transgène en deux moitié et introduire chacun d’eux dans deux AAVr différents dont l’un doit contenir en plus du promoteur et de la moitié du transgène, un site donneur d’épissage et l’autre un site accepteur, l’autre moitié du transgène et la séquence de polyadénylation. Après infection, la taille du transgène est reconstituée en totalité à la suite de l’épissage des sites donneur et accepteur des deux vecteurs et de la recombinaison intermoléculaire de leurs séquences ITR. Un autre procédé utilise la recombinaison homologue pour la transduction de transgène de très haute taille après assemblage de deux vecteurs (Pryadkina et al., 2015).
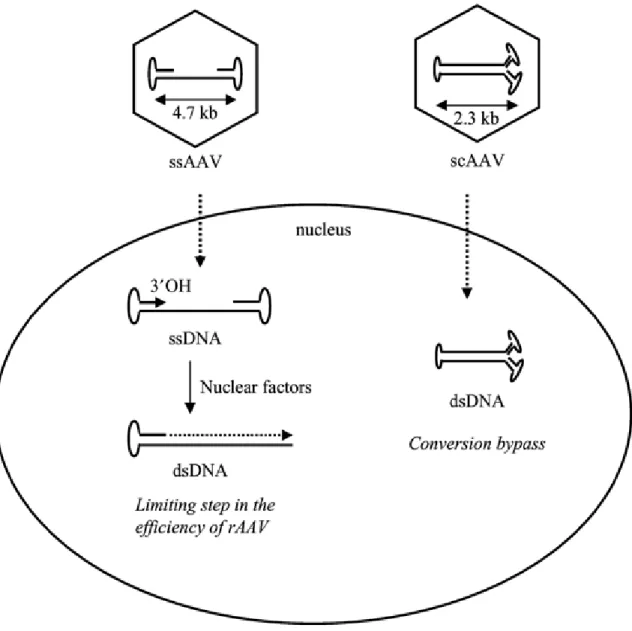
15 1.1.3.1.4.2.1.2.3. Construction de la forme auto-complémentaire des vecteurs viraux adéno-associées (scAAV)
La construction de la forme auto-complémentaire d’AAVr (scAAV) est une autre stratégie d’optimisation de l’efficacité de transduction par le vecteur. En effet, la conversion du génome simple brin d’AAVr conventionnel en génome double brin, forme transcriptionnellement active, est une des étapes limitantes de la transduction (Ferrari, Samulski, Shenk, & Samulski, 1996). D’où l’idée de construire une deuxième génération d’AAVr pour contourner l’obstacle. La structure du vecteur est faite de deux moitiés du génome simple brin d’AAVr, délimitées chacune à leurs extrémités libres par les séquences ITR et reliées à leurs parties internes par une ITR commune. Une fois dans le noyau, les deux génomes simple brin dimérisent sous forme d’une molécule d’ADN double brin active transcriptionnellement et capable de transduire efficacement le transgène (McCarty, Monahan, & Samulski, 2001), (McCarty, 2008), (Buie et al., 2010). Un essai clinique réalisé chez dix patients atteints d’hémophilie B, fait preuve de l’efficacité de cette forme auto-complémentaire d’AAVr dont la seule injection du sérotype 8 pseudotypé codant pour le facteur anti-hémophilique IX (FIX), a permis l’expression à long terme du transgène associée à une amélioration clinique (Nathwani et al., 2014). D’autres études rapportent les résultats similaires chez les modèles murins souffrant de l’hémophilie B et de l’amyotrophie spinale, traités respectivement avec le scAAV codant pour le FIX (Wu et al., 2008) et le gène SMN1 normal (Meyer et al., 2015).
16
Figure 4: Analogie des génomes AAVr et scAAV dans le noyau de la cellule hôte (Le Bec & Douar, 2006). 1.1.3.1.4.2.2. Avantages des vecteurs viraux adéno-associés
Depuis la découverte des AAV sauvages et la production des premiers AAVr, les études précliniques et cliniques utilisant ces vecteurs respectivement dans les modèles animaux et humains n’ont cessé de se multiplier, témoignant du bénéfice thérapeutique que procure ces derniers lorsqu’ils sont utilisés en thérapie génique. En effet, les AAVr figurent parmi les premiers succès cliniques de transfert des gènes avec notamment l’utilisation de l’AAV2 dans le traitement de la fibrose kystique (Tebbutt, 1999),(Daya & Berns, 2008). La première thérapie génique à être acceptée dans le monde l’a été avec un AAVr codant pour la lipoprotéine lipase (AAV-LPLS447X), l’enzyme déficitaire dans l’hyperlipoprotéinémie héréditaire de type 1, une maladie métabolique caractérisée par une élévation des triglycérides sériques (Kastelein, Ross, & Hayden, 2013), (Bryant et al., 2013), (Ferreira et al., 2014). Par la suite, plusieurs essais impliquant les AAVr
17 ont été menés avec succès dans les maladies génétiques mono-géniques et acquises comme la maladie d’Alzheimer, la maladie de Parkinson, le déficit en alpha-1 antitrypsine, le cancer de la prostate, l’insuffisance cardiaque et bien d’autres (Fish & Ishikawa, 2015), (Trepel et al., 2015). Toutes ces preuves d’efficacité restent fondées sur de nombreux avantages que possèdent les AAVr et qui les classent parmi les vecteurs de convenance en thérapie génique. Voici quelques-uns:
i. Leur large tropisme, particulièrement associé à la diversité structurale de leurs capsides naturelles, modifiables par les techniques de criblage (Benskey et al., 2015).
ii. Leur efficacité hautement élevée de transduction et à long terme des cellules en division et à l’état de quiescence (Mingozzi & High, 2011), (Peel & Klein, 2000), (Pan et al., 2013).
iii. Leur excellent profil de sécurité (Karda et al., 2014) car les vecteurs persistent dans le noyau essentiellement sous forme épisomal et n’ont aucune spécificité d’intégration. Les risques intégratifs rapportés par certains auteurs restent relativement rares et aléatoires.
iv. Leur faible pouvoir immunogène comparé à d’autres vecteurs (Sun, Anand-Jawa, Chatterjee, & Wong, 2003), (Buie et al., 2010) car ces réponses immunitaires, souvent dirigées contre les vecteurs et rarement contre le transgène, sont conditionnées par une infection préalable du sujet par le virus. Cette infection préalable est un évènement rare mais qui sensibilise le système immunitaire hôte pour une infection ultérieure. Aussi la séroprévalence anti-AAVr développée contre les capsides virales et associée à une activité neutralisante du vecteur, est proportionnelle à l’âge des sujets, d’où l’intérêt d’utiliser les vecteurs adéno-associés beaucoup plus tôt dans la vie des patients.
En dépit de ces éléments procurant les bénéfices à l’utilisation des AAVr, quelques inconvénients peuvent limiter leur usage en thérapie génique, notamment la faible capacité du cargo et les barrières rencontrées dans leur cycle infectieux. Néanmoins ces limites restent surmontables par l’utilisation des formes optimisées des AAVr.
1.1.3.1.4.2.3. Spécificités associées au Sérotype 9 des vecteurs viraux adéno-associés Le Sérotype 9 des AAVr récemment isolé avec son récepteur cellulaire correspondant, le galactose présente un tropisme particulier pour les tissus neurologiques et extra-neurologiques (C. L. Bell, Gurda, Van Vliet, Agbandje-McKenna, & Wilson, 2012). Voici quelques-unes de ses spécificités, raison de son utilisation préférentielle dans le présent travail :
18
ii. Sa spécificité à infecter naturellement le cerveau (Rahim et al., 2011), (Mattar et al., 2013) et le cœur (Pacak et al., 2006), (Pacak et al., 2006), (Bish et al., 2008), (Qi et al., 2010), (Okada et al., 2013), qui sont les tissus cibles visés dans ce travail.
iii. Sa capacité de traverser la barrière hémato-encéphalique, facilitant ainsi son accès au système nerveux central (Foust et al., 2009), (Gray, Matagne, et al., 2011).
iv. Son avantage d’infecter les cellules aussi bien dans leur état de quiescence et qu’à l’état post- mitotique, ce qui est un avantage pour les neurones et cardiomyocytes qui ne se divisent pas (Karda et al., 2014).
1.2. Développement de la thérapie par transfert des gènes pour
l’ataxie de Friedreich
1.2.1. Aspects fondamentaux de l’ataxie de Friedreich
1.2.1.1. Historique.
L’ataxie de Friedreich, du grec ataxia, signifiant trouble de l’équilibre, est la plus fréquente des ataxies héréditaires humaines d’origine génétique (mono-génique) qui doit son nom à Nicolas Friedreich, médecin et neuropathologiste allemand qui l’a décrite pour la première fois en 1863 (Koeppen, 2011) comme un trouble de la coordination des mouvements, caractéristique de l’atteinte du vermis cérébelleux. La prévalence de la maladie dans l’ensemble des ataxies cérébelleuses autosomiques récessives est estimée à environ 2 à 4 sur 100000 naissances dans la population de la race blanche (Palau & Espinos, 2006) avec une expérience de vie moyenne comprise entre 35 ans et 50 ans. Plus d’un siècle après la découverte et la description clinique de la maladie, soit en 1988 et en 1996, se succéderont respectivement la localisation du gène FRDA responsable de la maladie sur le chromosome 9q13.1 (bras long du chromosome 9, région 13, bande 1) par l’équipe de S. Chamberlain au Royaume Uni (Chamberlain et al., 1988) et son identification avec la protéine pour laquelle il code, la frataxine, par l’équipe conjointe franco-canadienne de Michel Koenig (France ) et Massimo Pandolfo (Canada) (Campuzano et al., 1996). Ces deux équipes ont à l’occasion démontré pour la toute première fois la relation de cause à effet entre l’anomalie génétique, l’expansion des trinucléotides GAA dans un gène et les conséquences phénotypiques qui en résultent et vont à cette même occasion mettre au point le test génétique de dépistage de la maladie.
19 1.2.1.2. Génétique de l’ataxie de Friedrich
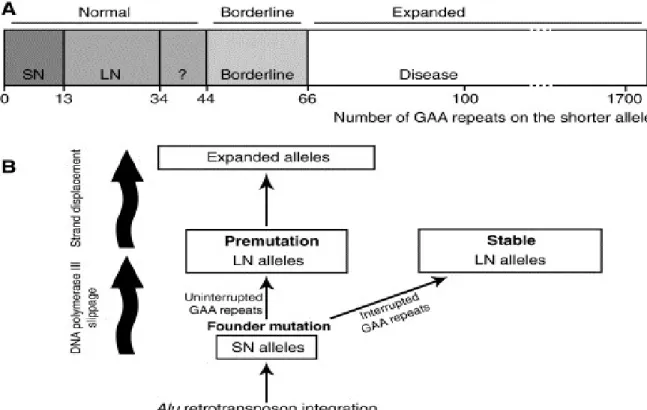
L’ataxie de Friedreich est une maladie héréditaire, mono-génique, à transmission autosomique récessive et dont le gène responsable est localisé sur le chromosome 9q13.1 et code pour un ARNm de 1,3 kb traduisable en une protéine de 216 acides aminés, la frataxine. Le gène FRDA est un petit gène de 40 kb possédant 5 exons. Il peut être le siège de plusieurs mutations, la plus fréquente (98% de mutations) étant une expansion des trinucléotides GAA (GAA)n dans l’intron 1 du gène (Delatycki, Williamson, & Forrest, 2000) (Figure 6). De cette expansion découle trois situations cliniques en fonction de nombres de répétitions GAA qui peuvent aller de 13 à 34, de 44 à 100 et de plus de 100, correspondant respectivement à un état normal, une permutation et un état pathologique. Environ 96% des patients sont homozygotes pour les répétitions trinucléotidiques GAA alors que les 4% restant sont hétérozygotes composites (Pastore & Puccio, 2013). L’expansion des triplets GAA entraine une déformation de la structure de l’ADN qui adopte une conformation en triple hélice affectant la vitesse de l’élongation de la transcription du gène FRDA et une hétérochromatinisation de la région promotrice du gène diminuant l’accessibilité de cette dernière par les facteurs de transcription et l’ARN polymérase II (Herman et al., 2006). Ces deux altérations conjuguées sont responsables de la diminution de la quantité de l’ARNm et par conséquence à la diminution de la protéine frataxine dont le déficit est à la base de toutes les manifestations cliniques associées à la maladie.
20
Figure 5: Taille de répétitions des triplets GAA dans l’intron 1 du gène de la frataxine corrélée aux
conséquences phénotypiques (Santos et al., 2010).
1.2.1.3. Frataxine, protéine mitochondriale
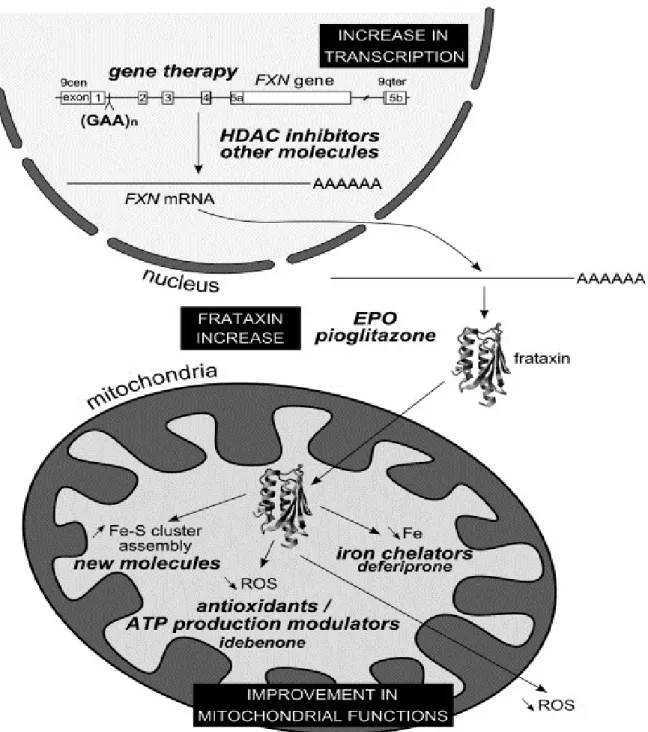
Le métabolisme du fer intra-mitochondrial est intimement lié à la concentration et à l’activité de la frataxine, une protéine mitochondriale hautement conservée dans les espèces et dont la structure est partiellement connue à la suite des études de similitude réalisées chez la levure et chez les mammifères. Elle est synthétisée dans le cytoplasme sous forme d’un précurseur de 210 acides aminés qui, après son importation dans la mitochondrie, subit un double clivage protéolytique sous l’action des peptidases mitochondriales engendrant la forme intermédiaire de la protéine (19 Kda) et la forme mature fonctionnelle (14 Kda) de structure globulaire ayant 2 hélices α et 7 brins β au cœur de laquelle sont concentrés les acides aminés hydrophobiques exposant à sa surface des acides aminés négativement chargés α1β1 qui reconnaissent et lient l’atome de fer (Schmucker, Argentini, Carelle-Calmels, Martelli, & Puccio, 2008), (Condo et al., 2007). Son rôle n’est pas clairement élucidé mais quelques hypothèses faisant état de dysfonctionnement mitochondrial consécutif au manque de la frataxine ont été avancées (Foury & Cazzalini, 1997), (Colin et al., 2013). En effet, le fer extracellulaire (fer ferrique) lié à la transferrine est incorporé par endocytose dans le
21 cytoplasme où il transit après réduction dans un pool de fer ferreux (fer+2) libre mis en réserve sous forme liée
à la ferritine. Selon le besoin de la cellule, une partie de fer+2 est utilisée pour la synthèse des métalloprotéines
à fer héminique. Une autre partie est recyclée vers le milieu plasmatique. Une troisième partie subit un transport facilité vers la mitochondrie sous l’action de la frataxine. Ce fer dans les mitochondries est utilisé pour la biogénèse des noyaux Fe-S. La frataxine s’associerait à la machinerie protéique qui intervient dans la mobilisation de l’atome de soufre et à l’assemblage des noyaux Fe-S, respectivement comme cofacteur et donneur du fer. Cette machinerie est faite des complexes protéiques Nfs1, ISD11, ISCU et de la frataxine, munie d’une activité cystéine Desulfurase catalysant la transformation de la L-cystéine en L-alanine et en soufre (Gerber, Muhlenhoff, & Lill, 2003), (Parent et al., 2015). Cette même machinerie intervient à la synthèse des métalloprotéines à fer non héminique, notamment les protéines à centre Fe-S intervenant dans les réactions de transfert d’électrons, essentielles au processus de la respiration (phosphorylation oxydative) et dont la fonction serait basée sur la capacité des atomes de fer des noyaux Fe-S à passer d’un état réduit (fer+2) à un état oxydé (fer+3) et inversement. Ainsi les centres Fe-S peuvent facilement accepter les électrons
ou les donner à d’autres entités. L’excès de fer intra-mitochondrial, provenant soit de l’échappement du fer de la chaine de transfert d’électrons, soit de la dégradation des complexes protéiques à centre Fe-S, est évacué hors de la mitochondrie pour éviter qu’il soit oxydé et qu’il puisse générer les radicaux libres. On pense que la frataxine jouerait un rôle dans l’exportation du fer à ce niveau (Figure 6). D’autres régulateurs sont également utiles dans la régulation de la concentration intra-mitochondriale du fer, ce sont des protéines régulatrices de fer (IRP) qui fonctionnent comme des senseurs du fer et du stress oxydant et existent sous deux formes selon la concentration du fer dans le milieu. En présence d’une forte concentration de fer, elles le captent et forment les centres Fe-S de type 4Fe-4S avec une fonction aconitase qui intervient dans le cycle de Krebs. En carence de fer, elles forment des centres Fe-S de type 3Fe-4S ou perdent totalement leur cofacteur, le fer, auquel cas elles modifient leur structure pour devenir capable de lier une région spécifique de l’ARNm appelée IRE (Iron Response Element) réprimant la traduction des protéines impliquées dans l’utilisation, le stockage et l’exportation du fer (Martelli & Puccio, 2014), (Rotig et al., 1997). Ces hypothèses confirment les constatations sur le dysfonctionnement mitochondrial associé au déficit de la frataxine identifiées pour la première fois chez les patients Friedreich et le modèle de la maladie chez la levure, se caractérisant par l’accumulation du fer intra-mitochondrial et la diminution de la synthèse des complexes protéiques de la chaine de transfert d’électrons, ce qui débalance le fonctionnent cellulaire en anaérobiose et crée un cercle vicieux (Rotig et al., 1997).
22
Figure 6: Physiopathologie de l’ataxie de Friedreich et différentes cibles thérapeutiques possibles (Santos et al., 2010).
23 1.2.1.4. Symptomatologie
Le tableau clinique de l’ataxie de Friedreich associe les signes d’atteintes neurologiques et extra neurologiques (Santos et al., 2010). Les manifestations cliniques sont liées à la souffrance cellulaire des tissus dont le fonctionnement dépend particulièrement de l’activité mitochondriale (Camara, Lesnefsky, & Stowe, 2010).
1.2.1.4.1. Signes d’atteintes neurologiques
i. Un syndrome cérébelleux tributaire de l’atteinte du vermis cérébelleux et qui se caractérise par une incoordination des mouvements et une démarche ébrieuse, d’où le terme ataxie.
ii. Un syndrome pyramidal consécutif à l’atteinte (lésion) du faisceau pyramidal et qui se caractérise par une fatigabilité des membres, une hémiparésie allant jusqu’à l’hémiplégie unilatérale ou bilatérale, une réapparition des certains reflexes archaïques (signe de Babinski) et l’abolition d’autres reflexes. iii. Un syndrome sensitif profond associé à l’atteinte des fibres du cordon postérieur de la moelle
épinière et caractérisé par des troubles de la sensibilité profonde et de l’équilibre.
1.2.1.4.2. Signes extra neurologiques
i. Une cardiomyopathie hypertrophique pouvant évoluer vers une insuffisance cardiaque congestive et un diabète sucré mixte (Harding & Hewer, 1983), (Koeppen, 2011).
ii. Des déformations osseuses (pieds creux, scoliose, etc…) et anomalies oculaires (Santos et al., 2010).
1.2.1.5. Modèles cellulaires et animaux
Les modèles cellulaires et animaux de l’ataxie de Friedreich ont été développés pour étudier l’expression de la frataxine, les désordres induits par le manque de cette dernière et l’identification des biomarqueurs (mesures des parois cardiaques, dosage de la frataxine) nécessaires pour mesurer la progression de la maladie et l’efficacité du traitement.
1.2.1.5.1. Modèles cellulaires
Ce sont des lignées cellulaires stables exprimant la forme mutée de la frataxine humaine (Calmels et al., 2009). Les autres types cellulaires sont porteuses du gène de la frataxine endogène réprimé par la machinerie
24
des micro-ARN et de l’expansion des triplets GAA endogène issue de la reprogrammation des cellules différenciées des patients Friedreich (Martelli, Napierala, & Puccio, 2012).
1.2.1.5.2. Modèles animaux
Les modèles animaux sont des souris transgéniques chez qui la frataxine endogène a été supprimée à l’aide du système LOX P/Cre-recombinase (modèles MCK-Cre et NSE-Cre). Ces modèles établissement l’implication de la frataxine de façon primaire dans la biogénèse des noyaux Fer-Soufre (Puccio et al., 2001). Les autres modèles animaux sont basés sur l’introduction de l’expansion des triplets GAA dans le gène de la frataxine à l’aide du chromosome artificiel dérivé de la levure (modèles KIKI, KIKO, YG8sR, YG22sR) (Martelli et al., 2012). Ces modèles sont utiles pour la compréhension de la maladie et l’évaluation des différentes stratégies thérapeutiques (Al-Mahdawi et al., 2006).
1.2.1.6. Avancées thérapeutiques
Des efforts sont fournis au niveau de la recherche fondamentale et de la collaboration entre scientifiques dans le but de mettre au point un traitement définitif (curatif) contre l’ataxie de Friedreich. Malheureusement il n’y a encore de traitement disponible. Néanmoins quelques avancées thérapeutiques visant à corriger la symptomatologie, les désordres métaboliques et l’étiologie de la maladie, sont actuellement exploitées par différentes équipes de recherche dans le but d’améliorer la qualité de vie des patients, les unes encore en phase préclinique et les autres, en phase d’essai clinique.
1.2.1.6.1. Traitement symptomatique
C’est un traitement multidisciplinaire, entrepris en général lorsque les bilans neurologiques, cardiaques et orthopédiques sont établis. Il consiste en la kinésithérapie basée sur la rééducation des troubles moteurs, orthopédiques et respiratoires, combinée à l’usage de certaines molécules dont la 5-hydroxytryptiphane (5-HTP), la Benzerazide et les vitamines, efficaces contre les troubles ataxiques.
1.2.1.6.2. Traitement des désordres métaboliques
On recourt à ce jour à l’utilisation de l’Idébénone, un analogue de la coenzyme Q10 (ubiquinone), utilisé pour ses doubles propriétés anti-oxydantes à la fois soluble et membranaire, neutralisant ainsi l’effet des radicaux libres dans les cellules. Cette molécule a fait preuve de son efficacité sur l’amélioration des fonctions cardiaques chez les patients Friedreich (Rustin et al., 1999), (Hausse et al., 2002). Cette amélioration n’est obtenue que lorsqu’elle est utilisée avant l’installation des lésions cardiaques et neurologiques irréversibles (Pineda et al., 2008). L’utilisation de la Déferiprone, un chélateur de fer et dont le bénéfice thérapeutique passe par la diminution de la concentration intra-mitochondriale du fer, se révèle être efficace surtout dans la réduction de l’atteinte cardiaque. L’usage de ce médicament reste entaché de quelques déceptions liées à
25 l’aggravation des troubles ataxiques et des manifestations idiosyncrasiques (Pandolfo et al., 2014). La Pioglitazone, un antidiabétique oral, est utilisé pour son effet activateur du récepteur nucléaire PPARγ (récepteur γ activé par les proliférations des peroxysomes) qui induit via l’activation des facteurs de transcription, l’expression de la super oxyde dismutase et de la frataxine mitochondriales dans les cellules pancréatiques et dans toutes les cellules en culture (Wilson, 2012).
1.2.1.6.3. Traitement étiologique (génétique et épi génétique)
Certaines équipes de recherche travaillent sur la production in vitro de la protéine frataxine recombinante et la thérapie génique à base des facteurs de transcription capables de stimuler la production endogène de la frataxine (Chapdelaine, Coulombe, Chikh, Gerard, & Tremblay, 2013). D’autres équipes recherchent des classes spécifiques d’inhibiteurs des histones déacétylases (HDAC) capables de cibler le gène de la frataxine et d’inverser la répression génique (Herman et al., 2006), (Rai et al., 2008). Par ailleurs, la piste la plus prometteuse à l’heure actuelle reste la thérapie génique consistant à transférer la copie normale du gène déficient de la maladie à l’aide d’un AAVr. Le récent essai préclinique sur l’ataxie de Friedreich utilise l’AAVrh.10 ciblant les cardiomyocytes et codant pour la frataxine normale chez des souris transgéniques reproduisant les symptômes cardiaques de la maladie (modèle MCK-Cre). Un rétablissement complet et rapide du cœur a été observé avec un effet positif sur l’arrêt de développement des atteintes cardiaques chez des animaux asymptomatiques (Perdomini et al., 2014).
1.2.2. Étude préliminaire chez des souris modèles de l’ataxie de Friedreich.
Une autre étude a consisté en la prise en charge thérapeutique des modèles sévères des souris pour l’ataxie de Friedreich essentiellement les souris NSE-Cre et MCK-Cre (Gerard et al., 2014). L’AAV9 codant pour la frataxine humaine a été conçu (AAV9-hFXN) et administré aux souris par voie intra-péritonéale. Dans le but de déterminer la dose efficace pour le traitement, une série des doses décroissantes ont été testées dans la première partie de l’étude. Une augmentation de la durée de vie a été observée chez ces modèles des souris après avoir reçu une dose de 6 x 1011 du virus accompagnée de la détection par la technique
immuno-enzymatique (ELISA) de la protéine frataxine dans le cœur, cerveau, muscle, rein et foie. L’échographie cardiaque réalisée chez le modèle MCK-Cre a montré une amélioration des fonctions cardiaques. Cependant les symptômes neurologiques ont persisté chez le modèle NSE-Cre. Ces résultats de la littérature suggèrent qu’un traitement précoce (in utero) pourrait être plus efficace.
1.2.3. Avantages de l’administration des vecteurs viraux adéno-associés chez des
femelles en gestation.
L’injection des AAVr chez des femelles en gestation comparée à l’administration postnatale, présente quelques bénéfices qui améliorent l’efficacité de transduction du transgène. Cette approche offre une
26
opportunité d’intervention précoce et limite le développement de la pathogénie chez le fœtus. Elle n’est pas encore étudiée chez les humains mais quelques essais précliniques réalisés chez les primates non humains présentant les maladies humaines comme celle de Criggler-Najjar de type I et l’Amorause congénitale de Leber, font preuve de l’efficacité de cette approche comparée à la thérapie postnatale (Karda et al., 2014). Voici quelques-uns des bénéfices associés à l’utilisation de cette approche:
i. La possibilité d’infecter avec des AAVr, une proportion élevée des cellules d’intérêt. ii. L’administration du virus se fait avant l’installation de la tolérance immunitaire des bébés.
iii. La chance de développer les réponses immunitaires est réduite car la sensibilisation du système immunitaire de l’hôte par une infection préalable est une condition sine qua none au développement des réactions immunitaires lors d’une administration ultérieure des AAVr. Cependant la présence des anticorps maternels neutralisants anti-AAVr préexistants pourraient avoir un impact négatif sur la quantité de virus à traverser chez le fœtus surtout lorsque ce dernier est administré par voie IV. iv. La diminution de risque de progression de la pathogénie avant le développement irréversible des