Moyens de prévention et contrôle d'une maladie
émergente infectieuse: cas de la maladie d'Aujeszky
Mémoire
Lisa Auclert
Maîtrise sur mesure
Maître ès sciences (M. Sc.)
Moyens de prévention et contrôle d'une maladie
émergente infectieuse :
cas de la maladie d'Aujeszky
Mémoire
Lisa Zoé Auclert
Sous la direction de :
Sylvain Bourgoin
Résumé
Ce mémoire de maîtrise porte sur l'étude de la maladie d'Aujeszky, de son évolution et des moyens de lutte contre cette maladie. La maladie d'Aujeszky est causée par le virus de la pseudo-rage (PRV). Ce virus touche principalement les porcs et sangliers et peut être transmis également à de nombreuses autres espèces, incluant l'homme.
Le virus de la pseudo-rage peut être divisé en deux souches principales avec de nombreuses recombinaisons inter-clade et intra-clade. Les souches variantes sont actuellement les plus répandues dans le monde. Elles sont impliquées dans des événements de transmission inter-espèces. Cette étude permet de déterminer si la présence de souche variante est en augmentation depuis 2011. Ainsi que de savoir si le PRV circule toujours dans les élevages de porcs et présente toujours un risque par rapport aux espèces sauvages, comme le sanglier, chez lesquelles le virus est fréquemment détecté.
Le travail effectué a permis de présenter l'évolution de ce virus grâce aux comparaisons et analyses génétiques. Cela permet de mieux appréhender les dynamiques de la transmission et l’évolution inter-espèces du virus chez un même hôte et entre hôtes différents. Ceci souligne l'importance de la mise en place de réseaux de surveillance, dans les élevages de porcs domestiques autant que dans les espèces sauvages, ainsi que la nécessité de mettre en place de mesures prophylactiques strictes et adaptées telles que la vaccination et les sacrifices d'animaux contaminés pour éviter la propagation de la maladie dans les élevages.
Abstract
This master's thesis focuses on the study of Aujeszky's disease, its evolution and ways to control it. Aujeszky's disease is caused by the pseudo-rabies virus (PRV). This virus mainly affects pigs and wild boars and can also be transmitted to many other species, including humans.
The pseudo-rabies virus can be divided into two main strains with many inter-clade and intra-clade recombinations. Variant strains are currently the most common in the world, involved in interspecies transmission events. The study of this virus has shown that populations carrying the variant strain have increased since 2011. Thus, PRV can still circulate in pig farms and still poses a risk compared to wild life species, such as wild boar, where the virus is frequently detected.
The work carried out has allowed us to present the evolution of this virus through genetic comparisons and analyses. This provides a better understanding of the dynamics of transmission and the interspecies evolution of the virus in the same host and between different hosts. This underlines the importance of the establishment of surveillance networks in domestic pig farms as well as in wildlife, as well as the need for strict and appropriate prophylactic measures such as and sacrifices of contaminated animals to prevent the spread of the disease on farms.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des tableaux ... v
Liste des figures ... vi
Liste des abréviations, sigles, acronymes ... vii
Remerciements ... x
Avant-propos ... xi
Introduction ... 1
0.1 Les maladies infectieuses émergentes ... 1
Définitions ... 1
Conditions favorisant l'émergence de maladies infectieuses ... 3
0.2 La maladie d'Aujeszky ... 5
Historique ... 5
Prévalence - Hôtes affectés ... 6
Transmission ... 8
Symptômes ... 9
Structure du virus de la pseudo-rage ... 9
Cycle de réplication et état de latence ... 11
Étiologie ... 13
Méthodes de lutte et de surveillance ... 14
Chapitre 1 : Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus ... 17
1.1 Résumé ... 18
1.2 Abstract ... 18
1.3 Introduction ... 19
Material and methods ... 20
Results ... 21
Discussion ... 31
References ... 33
Conclusion ... 37
Liste des tableaux
Tableau 0.1 - Détection d'anticorps anti PRV chez les sangliers dans de nombreux pays du monde... 6 Tableau 1.1 - Positive Selection and Adaptive Sites of PRV on gB, gD, gE and gC protein... 29
Liste des figures
Figure 1 Représentation des relations entre facteurs de risques pouvant mener à l'émergence de maladies
infectieuses ... 4
Figure 2 Frise Chronologique des infections présumées ou vérifiées par le PRV chez l’homme ... 8
Figure 3 Structure détaillée du virion du PRV, et gènes codant les protéines constituant le virus ... 11
Figure 4 Schématisation du cycle de réplication du PRV ... 12
Figure 5 Schématisation des évolutions possibles d'une épidémie avec et sans mesures préventives ... 40
Figure 1. 1 History, geographical distribution, and diversity of full-length PRV infections ... 23
Figure 1. 2 Recombination analysis of 29 pseudorabies virus (PRV) full-length genomes ... 25
Figure 1. 3 Maximum likelihood (ML) tree of the gB, gD, gE, and gC genes ... 26
Figure 1. 4 Maximum clade credibility tree of the gC segment ... 27
Figure 1. 5 Skyline plot of each pseudorabies virus clade ... 29
Figure 1. 6 Structure of gB and gC protein ... 30
Liste des abréviations
ADN Acide désoxyribonucléique ARN Acide ribonucléique
ELISA dosage d'immunoabsorption par enzyme liée
MERS coronavirus du syndrome respiratoire du Moyen-Orient MI maladie infectieuse
MIE maladie infectieuse émergente OMS Organisation Mondiale de la Santé PCR réactions en chaîne par polymérase
PRV Virus de la pseudo-rage, virus de la maladie d'Aujeszky, herpèsvirus porcin de type 1 SRAS Virus du Syndrome respiratoire aigu sévère qui a causé l'épidémie de 2003
" "
- Ed -
Remerciements
Merci aux chercheurs Dr Sylvain Bourgoin et Dr Marc Pouliot, qui m'ont permis d'écrire ce rapport de maîtrise. Merci à la Dr Josée Bastien pour son soutien pour ma poursuite d'études, merci à eux pour le temps qu'ils ont pris pour m'aider.
Merci à Dr Caroline Gilbert pour son aide. Merci aux travailleurs que j'ai rencontrés au cours de ma formation et qui m'ont soutenue et donné de bons conseils, en particulier à Matthew Wade, Hugues Fausther Bovendo, Jannie Pedersen, Hiva Azizi pour leur aide au laboratoire. Merci à Alice Berger, Matthew et Hugues, pour leur bienveillance. Merci à Myriam Aubin et Laurence Belley et bravo à vous deux pour vos diplômes. Merci à Alexandre Brunet pour m'avoir aidée en m'indiquant le meilleur chemin et m'avoir encouragée dans mes travaux, pour avoir été franc avec moi et aussi pour ses cours de cytométrie en flux.
Avant-propos
Depuis mon arrivée à Québec, j'ai eu l'occasion de travailler sur différents projets et de m'impliquer dans des expériences variées. J'ai eu la satisfaction d'avoir les formations en manipulation de souris, en analyse de résultats de cytométrie en flux, expériences in vitro et in vivo, détection, culture et purification de virus et j'ai découvert de nouvelles méthodes d'organisation du travail et d’éthique. J'ai travaillé à la publication de l'article qui fait l'objet du chapitre 1 et dont je suis co-premier auteur car j’ai réalisé des figures et travaillé à l’analyse des données obtenues ainsi qu’à la rédaction de l’article. Ce journal autorise l’utilisation des articles publiés par les auteurs dans le cadre de rapports de maîtrise. Outre ce projet, j'ai également participé à d’autres gros projets, notamment la mise en place d'un réseau de surveillance de virus aviaires chez les oiseaux migrateurs du Nord du Canada, uniquement grâce au soutien des collaborateurs qui ont bien voulu m’aider, dans le but de prévenir de futures épidémies (collecte et analyse de plus de 2000 échantillons provenant de régions reculées de Québec) et j’ai pu travailler sur le vecteur du virus de la stomatite vésiculaire en tant que plateforme de vaccination ainsi qu'au le séquençage de virus détectés chez des oiseaux morts que j'ai disséqués.
L'article intitulé " Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus" présenté au chapitre 1 a été publié dans le journal "The Journal of infectious diseases" le 28 December 2018 (DOI:10.1093/infdis/jiy731). La liste des auteurs de l'article est la suivante : Wanting He1, Lisa Zoé Auclert2, Xiaofeng Zhai1, Gary Wong2,5,7, Cheng Zhang1, Henan Zhu6, Gang Xing3, Shilei Wang1, Wei He 1, Kemang Li1, Liang Wang7, Guan-Zhu Han4, Michael Veit8, Jiyong Zhou3, Shuo Su1*.
1. MOE International Joint Collaborative Research Laboratory for Animal Health & Food Safety, Jiangsu Engineering Laboratory of Animal Immunology, Institute of Immunology, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China.
2. Département de microbiologie-infectiologie et d’immunologie, Université Laval, Québec, QC, Canada. 3. Key laboratory of Animal Virology of Ministry of Agriculture, Zhejiang University, Hangzhou, China. 4. College of Life Sciences, Nanjing Normal University, Nanjing, Jiangsu 210023, China.
5. Institut Pasteur of Shanghai, Shanghai, China.
6. MRC-University of Glasgow Centre for Virus Research, Glasgow, Scotland, United Kingdom.
7. CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China.
8. Institute for Virology, Center for Infection Medicine, Veterinary Faculty, Free University Berlin, Robert-von-Ostertag-Straβe 7-13, Berlin, Germany.
Introduction
0.1 Les maladies infectieuses émergentes
Définitions
Une maladie infectieuse (MI) est une maladie causée par la transmission d'un agent infectieux entre individus, pouvant être d'origine virale, bactérienne, parasitaire, fongique ou bien causée par un prion.
L'Organisation Mondiale de la Santé (OMS) donne la définition suivante d'une MI émergente (MIE) : il s'agit d'une "maladie infectieuse qui est apparue dans une population pour la première fois ou qui pourrait avoir déjà existé, mais dont l’incidence ou la portée géographique connaît une rapide expansion". Cette définition souligne l'importance de l'augmentation du nombre de cas détectés dans un espace et un temps donné. En effet l'incidence d'une maladie représente le nombre de cas apparus pendant une année au sein d'une population. L'augmentation de l'incidence peut être liée à une augmentation du nombre de personnes infectées, à une modification de l'expression d'une maladie (augmentant par exemple la gravité des symptômes et la visibilité du pathogène) ou à une augmentation des détections (non pas une augmentation du nombre de cas réel) due à l'amélioration des tests de diagnostic. L’émergence dépend aussi de la capacité à détecter la maladie. De plus un pathogène endémique déjà présent dans une zone peut être considéré comme émergent suite à son implantation dans une nouvelle région ou à son adaptation à une nouvelle espèce.
Des modifications, tant dans l'environnement que chez l'agent pathogène et ses hôtes, vont influencer les symptômes d'une maladie, le nombre et le type d'hôtes atteints. Certains facteurs sont reconnus pour favoriser l'émergence de maladies infectieuses émergentes et peuvent permettre le déclenchement d'une épidémie ou d'une pandémie chez l'homme. Parmi les caractéristiques retrouvées dans les déclenchements d'épidémies de maladies émergentes on retrouve une relation continue proche entre les hommes, une espèce intermédiaire (d'élevage ou destinée à la consommation) et une espèce sauvage constituant un réservoir pour le virus. L'homme est à proximité de l'espèce domestique d'élevage ou destinée à la consommation, et le contact entre l'espèce domestique et l'espèce réservoir (avec laquelle le virus a co-évolué) permet l'introduction de l'agent pathogène, lorsque le virus peut réaliser le saut d'espèce [1].
Pour les virus influenza par exemple, le virus se trouve chez les oiseaux sauvages ansériformes qui constituent le réservoir naturel du virus en constante évolution. Ces derniers peuvent transmettre le virus à des espèces animales domestiques rassemblées pour des élevages, comme les élevages de poulets et de porcs. Ces derniers représentent des hôtes intermédiaires dont les populations sont denses en élevage (favorisant l'amplification et la production de virus) et qui ont une proximité avec les hommes auxquels ils peuvent alors transmettre la maladie.
L'actuelle pandémie de coronavirus SARS-CoV-2 en cours a causé à cet instant plus de 183 000 morts mondialement en 5 mois. Selon les hypothèses prédominantes sur les circonstances ayant mené au déclenchement de la pandémie, elle a suivi le même schéma de transmission, en effet le virus était présent dans une espèce réservoir sauvage, la chauve-souris, dont le guano a permis la transmission à un hôte intermédiaire fouissant dans la terre pour se nourrir : le pangolin. Le pangolin, classé comme espèce "en danger critique d'extinction" (et l'espèce la plus braconnée au monde selon l'ONG WildAid), aurait constitué l'espèce intermédiaire en permettant au virus de s'adapter pour pouvoir infecter l'homme lors de la consommation de pangolin.
Le virus de la pseudo-rage (PRV) sur lequel ce rapport est centré, est un virus qui co-évolue avec les sangliers sauvages qui constituent l'hôte réservoir du virus, ce virus peut être transmis à des cochons d'élevage. Les cochons d’élevages peuvent ensuite transmettre le virus à l’homme. La première contamination rapportée date de 1914 et en tout 17 cas d’infections ont été rapportés. Chez l’homme également le virus peut infecter le système nerveux central (SNC) suite à un contact avec des animaux domestiques portant le virus [2], récemment, un cas d'infection du système nerveux central a été confirmé en 2017 chez un éleveur de porcs ayant manipulé des conteneurs pour animaux, étant en contact avec un élevage de porcs et présentant des maux de tête, des problèmes visuels et de la fièvre, et dernièrement en 2019 un nouveau cas d'infection du SNC a été confirmée [3].
A ces trois exemples de virus, s'ajoutent de nombreuses autres maladies passées ayant pu causer de graves épidémies (Rinderpestvirus, MERS, SRAS, influenza, Ebola notamment). Il est estimé que plus d'un quart des décès dans le monde entier est directement lié à des maladies infectieuses. Parmi les agents infectieux causant des maladies chez l'homme, plus de 61% sont des zoonoses (ce qui signifie qu'ils sont transmis naturellement entre les animaux et les humains) [4], [5].
Conditions favorisant l'émergence de maladies infectieuses
On peut classer en trois groupes les types de facteurs qui peuvent expliquer l'émergence d'une maladie infectieuse chez les animaux d'élevage ou chez l'homme : ceux correspondant à une
- évolution de l'agent infectieux
(en particulier les mutations pouvant affecter la virulence, la transmissibilité, conférant la capacité de s'adapter et d'infecter un nouvel hôte),
- modification de l'environnement et des contacts entre l'agent infectieux et différents hôtes (par exemple la destruction d'écosystèmes habitats de la faune sauvage favorisant le contact entre les espèces hôtes de pathogènes et l'homme ou les animaux domestiques, changements climatiques permettant l'extension géographique des vecteurs de maladies comme les moustiques, conditions sanitaires en particulier d'élevage et de mise sur le marché, pollution environnementale également)
- variation dans la vulnérabilité des hôtes à l'infection
(particulièrement l'existence de facteurs de co-morbidité, malnutrition, tabagisme, accès aux soins, capacités d'isoler les individus malades et mobilité des hôtes, surpopulation, affaiblissement immunitaire par des conditions d'élevages favorisant par exemple l'utilisation d'antibiotiques, individus immunodéprimés ou immunosupprimés pour des greffes)[4].
Ces trois éléments, en interagissant directement ou indirectement, permettent la formation de nouvelles niches écologiques pour les agents pathogènes (Fig.1).
Figure 1 Représentation des relations entre facteurs de risques pouvant mener à l'émergence de maladies infectieuses, adapté à partir de l'article [6].
Nous allons étudier plus précisément au chapitre suivant une maladie émergente dans les élevages de porcs, dont le réservoir sauvage permet l'évolution, et qui peut également s'adapter pour être transmise et infecter l'homme : la maladie d'Aujeszky.
0.2 La maladie d'Aujeszky
Historique
La maladie d'Aujeszky est une maladie découverte dans les années 1800 chez les bovins où cette maladie était observée et déclenchait des démangeaisons extrêmes chez les animaux [7]. Elle doit son nom au vétérinaire et chercheur hongrois Aladár Aujeszky qui a isolé pour la première fois le virus à l'origine de la maladie en 1902 et en a fourni des descriptions détaillées [8]. Il s'agit du virus de la pseudo-rage (PRV), aussi appelé le virus de la maladie d'Aujeszky, un virus de la famille des Herpesviridae et sous-famille des alphaherpesvirus. Le PRV est capable d'infecter les bovins, mais aussi un éventail d'hôtes varié incluant les porcs et les sangliers, qui sont les hôtes réservoirs naturels, des ruminants, carnivores, rongeurs et animaux domestiques tels que les chiens et chats [9]. Cette maladie est caractérisée par une atteinte du système nerveux central ainsi que des problèmes respiratoires, et peut provoquer d'importantes pertes économiques concernant les élevages porcins. Chez les porcs, le PRV peut persister sous une forme latente et favoriser la dissémination du virus. En effet dans les élevages les porcs adultes ne présentent pas toujours de signes cliniques, ils peuvent contaminer les animaux sensibles : les truies, ce qui déclenche des avortements, et les porcelets, chez lesquels on peut constater une mortalité élevée pouvant atteindre les 100% [10]. Ainsi la maladie d'Aujeszky a un impact économique considérable qui mène à d'importantes pertes pour la filière porcine, diminuant la productivité et entraînant des restrictions commerciales pour limiter la propagation du virus.
Ainsi la vaccination au sein des élevages a été mise en place rapidement et a démontré une très bonne efficacité dans de nombreux pays, notamment aux Etats-Unis, au Canada, en Allemagne, en Nouvelle-Zélande [11]–[13]. Cependant la vaccination ne prévient pas l'évolution du virus chez les sangliers sauvages qui restent de potentiels réservoirs, ni la réinsertion dans les élevages de nouveaux variants du PRV. De plus, la transmission entre espèces et l'infection d'animaux hôtes secondaires cause chez eux des maladies beaucoup plus graves, fréquemment mortelles. Ceci souligne l'importance de la mise en place de réseaux de surveillance des pathogènes chez les animaux sauvages [13].
Prévalence - Hôtes affectés
L'hôte naturel principal du PRV est le sanglier ainsi que le porc. Les dépistages de présence de PRV chez les sangliers (hôte naturel sauvage) sont peu souvent réalisés. Les vaccins permettent de canaliser et dans certains cas éradiquer le virus dans les élevages de porcs, cependant ce virus est très répandu chez le porc sauvage dans le monde entier [14]–[18] (Tableau 0.1).
Tableau 0.1 - Détection d'anticorps anti PRV chez les sangliers dans de nombreux pays du monde, extrait modifié de [13]. Pays Région sangli ers testés pourcen tage positifs présenta nt des anticorp s anti-PRV Source de l’étude Etats-Unis Californie 130 3,08
Clark RK, Jessup DA, Hird DW, Ruppanner R, Meyer ME (1983) Serologic Survey of California Wild Hogs for Antibodies Against Selected Zoonotic Disease Agents. J Am Vet Med Assoc 183:1248–1251
Hawaï 116 46 Nettles VF, Erickson GA (1984) Pseudorabies in wild swine. Proc Annu Meet U S Anim Health Assoc 88:505–506
Texas 124 37,1
Corn JL, Swiderek PK, Blackburn BO, Erickson GA, Thiermann AB, Nettles VF (1986) Survey of Selected Diseases in Wild Swine in Texas. J Am Vet Med Assoc 189:1029–1032
Ile Ossabaw 661 30,26
Pirtle EC, Sacks JM, Nettles VF, Rollor EA (1989) Prevalence and transmission of pseudorabies virus in an isolated population of feral swine. J Wildl Dis 25:605–607
Floride 1662 34,84
van der Leek ML, Becker HN, Pirtle EC, Humphrey P, Adams CL, All BP, Erickson GA, Belden RC, Frankenberger WB, Gibbs EP (1993) Prevalence of pseudorabies (Aujeszky’s disease) virus antibodies in feral swine in Florida. J Wildl Dis 29:403–409
Caroline du Sud 20 50
Corn JL, Cumbee JC, Barfoot R, Erickson GA (2009) Pathogen Exposure in Feral Swine Populations Geographically Associated with High Densities of Transitional Swine Premises and Commercial Swine Production. J Wildl Dis 45:713–721
Belgiqu
e Hainaut 47 15
Czaplicki G, Dufey J, Saegerman C (2006) Is the Walloon European Wild boar a potential reservoir of Pseudorabies virus for porcine live stock? Epidemiol et Sante Anim 49:89–101
Namur 403 22
Luxembourg 1422 17
Liège 983 15
Croatie Moslavacka gora 44 54,55
Zupancic Z, Jukic B, Lojkic M, Cac Z, Jemersic L, Staresina V (2002) Prevalence of antibodies to classical swine fever, Aujeszky’s disease, porcine reproductive and respiratory syndrome, and bovine viral diarrhoea viruses in wild boars in Croatia. J Vet Med B Infect Dis Vet Public Health 49:253–256
Républi que
Tchèqu Czech Republic 338 29,88
Sedlak K, Bartova E, Machova J (2008) Antibodies to selected viral disease agents in wild boars from the Czech republic. J Wildl Dis 44:777–780
France Corsica 492 32,32
Albina E, Mesplede A, Chenut G, Le Potier MF, Bourbao G, Le Gal S, Leforban Y (2000) A serological survey on classical swine fever (CSF), Aujeszky’s disease (AD) and porcine reproductive and respiratory syndrome (PRRS) virus infections in French wild boars from 1991 to 1998. Vet Microbiol 77:43–57
Allema gne
Baden-Wurttemberg 141 36,88
Heinritzi K, Aigner K, Erber M, Kersjes C, Von Wangenheim B (1999) Brucellosis and Aujeszky’s disease (pseudorabies) in wild boars within an enclosure (case report). Tierarztl Prax Ausg G Grosstiere Nutztiere 27:50–55
Mecklenburg-Western Pommerania
99 13,13 Kaden V, Lange E, Hanel A et al (2009) Retrospective serological survey on selected viral pathogens in wild boar populations in Germany. Eur J Wildl Res 55:153–159
Brandenburg 166 15,66
Italie Sardaigne 4027 27,81 Firinu A, Scarano C (1988) African swine fever and classical swine fever (hog cholgera) among wild boar in Sardinia. Rev Sci Tech Off Int Epi 7:901–908 Grosseto district 152 26,97 Lari A, Lorenzi D, Nigrelli D, Brocchi E, Faccini S, Poli A (2006) Pseudorabies
virus in European wild boar from Central Italy. J Wildl Dis 42:319–324
Campania 342 30,7
Montagnaro S, Sasso S, De Martino L et al (2010) Prevalence of Antibodies to Selected Viral and Bacterial Pathogens in Wild Boar (Sus scrofa) in Campania Region, Italy. J Wildl Dis 46:316–319
Maremma
(Toscane) 265 47
Guberti V, Ferrari G, Fenati M, Maroc MA, Pasquali T (2002) Pseudorabies in wild boar. In: Proc 4th Meet Eur Assoc Zoo- and Wildl Veterinarians (EAZWV)
Pologn
e Olstyn 34 11,8
Lipowski A, Mokrzycka A, Pejsak Z (2002) Seroprevalence of Aujeszky’s disease in Poland in the years 1998–2000. Med Weter 58:35–39
Rouma
nie Roumanie 357 55,18
Vuta V, Barboi G, Olvedi I et al (2009) The presence of antibodies tot Aujeszkys Disease, bovine viral diarrhoea and porcine reproductive and respiratory syndrome in wild boars, preliminary data. Rev Rom Med Vet 19:75–81 Russie Wladimirska, Moskowskaja Twerskaja, Belgorodskaja, Chabarowski Kraj, Charkowskaja
48 39,58 Shcherbakov AV, Kukushkin SA, Timina AM et al (2007) Monitoring of infectious diseases among wild boars. Vopr Virusol 52:29–33
Belgorodskaya, Vladimirskaya,Mosk ovskaya, Tverskaya, Smolenskaya, Kirovskaya Oblasts and Chabarovsky Krai 83 32,53
Kukushkin S, Baborenko E, Baybikov T, Mikhalishin V, Domskiy I (2009) Seroprevalence of antibodies to main porcine infectious pathogens in wild boar in some regions of Russia. Acta Silv Lignaria Hung 5:147–152
Slovéni
e Slovénie 672 17,11
Ministry of Agriculture, Forestry and Food Slovenia (2010) Information on the Aujeszky’s Disease Situation, Slovenia. Presentation at the SCfCAH meeting, Brussels, 1-2 June 2010
http://eceuropaeu/food/committees/regulatory/scfcah/animal_health/presentati ons/201006102_ad_sloveniapdf
Espagn
e Espagne Centrale 16 56,25
Gortazar C, Vicente J, Fierro Y, Leon L, Cubero MJ, Gonzalez M (2002) Natural Aujeszky’s disease in a Spanish wild boar population. Ann N Y Acad Sci 969:210–212
Asturia; Ebro
Bassin, Jaen 693 44,44
Vicente J, Ruiz-Fons F, Vidal D et al (2005) Serosurvey of Aujeszky’s disease virus infection in European wild boar in Spain. Vet Rec 156:408–412
Ainsi le sanglier sauvage en liberté peut agir comme un réservoir permettant l'évolution du PRV [19]. Les transmissions du sanglier aux carnivores consommant des carcasses de sangliers, aux porcs domestiques qui peuvent servir d'intermédiaire à la contamination occasionnelle d'hommes, peuvent ensuite se produire comme
Le réservoir sauvage naturel constitué par les sangliers permet l'évolution du virus puis sa ré-insertion dans les élevages malgré les campagnes de vaccination efficaces. Dans certaines régions du monde où les campagnes de vaccination obligatoires n'ont pas été correctement menées et les mesures de biosécurité sont insuffisantes, le virus reste endémique : c'est le cas de certaines régions en Chine [21].
Les porcs sont anatomiquement et physiologiquement très semblables à l'homme, ainsi ils peuvent servir de réservoirs pour les agents pathogènes zoonotiques, intermédiaires permettant la transmission d'un réservoir animal sauvage à l'homme, ce qui est observé occasionnellement avec le PRV (Fig.2) et plus fréquemment pour de nombreux autres virus (le virus de la grippe notamment et arbovirus, circovirus, flavivirus, herpèsvirus, nidovirus, orthomyxovirus, paramyxovirus et picornavirus) [22][23][24].
Figure 2 Frise Chronologique des infections présumées ou vérifiées par le PRV chez l’homme, extrait de [25]. Transmission
Le virus est transmis entre animaux via leurs sécrétions, excrétions et la propulsion d'aérosols, via les fomites et cette transmission peut commencer dès le 1er jour d'infection, sans que l'animal ne présente de signes cliniques de la maladie. La transmission oronasale est la transmission principale chez les porcs domestiques et une transmission conjonctivale est également possible. Le virus peut être transmis par voie vénérienne, qui est la principale voie de transmission chez les sangliers. Le PRV est propagé lors de la consommation de carcasses d'animaux infectés par des espèces carnivores (sauvages ou domestiques, il s'agit notamment d'une voie de contamination des chiens et chats). En effet les carcasses d'animaux infectés peuvent présenter d'importantes charges virales. Lorsqu'elles sont consommées non cuites par des animaux sensibles à cette infection, les carcasses permettent au virus d'infecter l'oropharynx et les voies respiratoires de ces animaux. Le virus pouvant en effet survivre jusqu'à plusieurs semaines dans la viande des carcasses d'animaux infectés [26].
1940 1914 2 lab technicians infected after exposure to infected material 1986 2 tourists infected after exosure to cats and domestic
animals 1910 1920 1960 1980 1990 2010 1940 2 lab workers infected after contact with an infected dog 1 watchman and 1 vet infected after contact with an infected dog at a pig breeding station 1963 1983 2017 1 swineherd infected with variant strain of PRV after contact with pigs 1 tourist infected after rescuing an infected cat 1983 1992 6 cattle workers infected after contact with infected cows
Le virus peut être présent chez son hôte à l'état latent, des modifications hormonales ou un stress peuvent réactiver son excrétion et ainsi mener à l'infection de nouveaux animaux [27]. Suite à l'infection, la réplication a lieu au niveau du système respiratoire puis les nerfs sensoriels sont infectés et le virus peut se propager aux neurones. Le virus peut infecter l'épithélium, l'endothélium vasculaire, les lymphocytes et macrophages, et persister dans les neurones à l'état latent [27]. Une fois ré-activé le virus peut être à nouveau transmis à de nouveaux individus.
Symptômes
Chez les porcelets, les décès sont très fréquents, précédés par des tremblements, convulsions, une paralysie. Chez les porcs adultes des troubles respiratoires sont apparents et une susceptibilité accrue aux infections bactériennes est observée, des co-infections pouvant mener à des pneumonies sévères. L'infection au PRV peut causer des avortements chez les truies. Chez les sangliers les symptômes nerveux et respiratoires sont moins observés, les souches présentes chez les sangliers sont moins virulentes que celles trouvées chez les porcs d'élevage [27].
Le PRV peut causer la mort chez les chats, chiens, rats, moutons, et affecter des espèces sauvages telles que les ratons laveurs, ours bruns, ours noirs, coyotes, cerfs, panthères et renard roux[27], [28].
Chez l'homme les rares infections sont en général peu sévères, causant une fièvre, faiblesse, infection de nerfs crâniens et des démangeaisons, ces cas sont liés à des contacts rapprochés avec des animaux, aucun cas de propagation communautaire n'a été décrit à ce jour chez l'homme [2].
Structure du virus de la pseudo-rage
De la famille des Herpesviridae, les virions (d'environ 225nm) sont des virus à ADN linéaire double brin protégés par une nucléocapside icosaédrique. La capside est entourée d'une protéine de matrice constituant le tégument ainsi que d'une enveloppe lipidique présentant des glycoprotéines en surface (Fig.3).
Le génome du PRV permet de coder 70 gènes différents dont 40 sont conservés parmi tous les herpesvirus et proviendraient d'un ancêtre commun. Ces gènes permettent au virus de se répliquer, ils sont trouvés dans la même région du génome viral chez les différents herpesvirus [29]. Le séquençage de souches de PRV en circulation ainsi que les analyses phylogénétiques ont permis de distinguer deux génotypes majeurs de PRV. Les souches de PRV dites classiques touchent de nombreuses espèces et ont été isolées dans les années 90. Elles se distinguent des souches de PRV variantes qui sont apparues en 2011 en Chine. Tandis que les PRV
classiques affectent un très large éventail d'hôtes, les souches variantes ont été détectées essentiellement chez les porcs [30].
La principale protéine constituant la capside est codée par UL19 qui permet la formation d'une capside de 125nm très conservée chez ces herpesvirus. Les protéines du tégument (plus de 14 protéines virales) ainsi que des filaments d'actine provenant des cellules infectées remplissent l’espace entre la capside et l'enveloppe, constituant le tégument qui permet l'entrée du virus dans les cellules et le maintien de sa morphologie. Enfin, l'enveloppe lipidique du PRV provenant des cellules infectées comprend les 16 protéines membranaires du PRV (gB, gC, gD, gE, gG, gH, gI, gK, gL, gM, gN, UL20, UL43, US9, UL24, UL34). Ces glycoprotéines permettent l'interaction entre les cellules hôtes et le virus ainsi que la modulation du système immunitaire [31]. Les séquences de ces glycoprotéines variant selon les sous-types de virus, elles permettent de comprendre son évolution par comparaison de séquences et analyses phylogénétiques, en particulier des gènes gB, gC, gD, gE très utilisés pour comprendre l'évolution du virus. En effet ces glycoprotéines sont impliquées dans la liaison aux récepteurs cellulaires (et à l'entrée du virus dans la cellule), à la pathogénicité ainsi qu'au déclenchement de la réponse humorale [25], [32].
Figure 3 Structure détaillée du virion du PRV, et gènes codant les protéines constituant le virus (source [29])
Cycle de réplication et état de latence
Le virus va dans un premier temps s'attacher à la membrane de la cellule à infecter. Ce sont les protéines gC exprimées en surface de l'enveloppe du virion qui, en se liant aux sulfates d'héparine des cellules cibles, permettent l'attachement du virion. Suite à cet accrochage, la glycoprotéine gD permet l'entrée du virus dans la cellule en interagissant avec le médiateur d'entrée d'herpèsvirus cellulaire. Les glycoprotéines gB, gH et gL facilitent l'entrée du virion à l'intérieur de la cellule.
Une fois entré dans la cellule, la capside et l'enveloppe du tégument sont libérés dans la cellule et vont interagir avec des protéines des voies de transport cellulaires. La protéine VP16 du tégument permet la translocation jusqu'au noyau et l'activation de l'ARN polymérase très rapidement suite à l'infection. En effet ce sont les microtubules qui permettent le transport jusqu'au noyau cellulaire. Dans un premier temps, les protéines UL5, UL8, UL9, UL29, UL30, UL42, UL52 permettent alors la réplication de l'ARN du virus via un mécanisme de cercles roulants, puis les protéines EP0, UL54 et US1 activent la transcription. Par le mécanisme de cercles roulants, le génome adopte une forme circulaire et, lors de la réplication, génère des génomes viraux concatémères qui nécessitent ensuite un clivage. L'ADN est ensuite synthétisé et les protéines UL19, UL18, UL25, UL38, et UL35 sont transportées au noyau où elles vont permettre de former une capside et d'encapsider le nouvel ADN synthétisé via la protéine UL6. Les protéines UL32, UL33, UL15, UL17 permettent ensuite le clivage des concatémères, l'ADN clivé formant ainsi les génomes du virus, et ces protéines sont impliqués également dans l'encapsidation de l'ADN. La nucléocapside formée sort ensuite du noyau puis les protéines formant le tégument ainsi que son enveloppe vont s'associer à la capside ainsi qu'à la membrane lipidique de l'appareil de golgi. Le virus complet est ensuite transporté à la membrane cellulaire en utilisant les voies cellulaires de transport de vésicules et est ensuite relâché hors des cellules [29].
Figure 4 Schématisation du cycle de réplication du PRV, extrait de [33].
En plus de se répliquer, le PRV peut aussi entrer en état de latence, qui correspond à un état ayant suivi une infection aigue à la suite de laquelle l'hôte infecté a survécu et contenu la transcription et réplication du virus.
La latence est caractérisée par la présence du génome viral en tant qu'épisome dans la cellule infectée neuronale, sans production de particules virales infectieuses. Les transcriptions détectables pendant la latence sont les transcriptions associées à l'état de latence. Le génome du virus reste présent dans les cellules infectées sous forme d'épisome. Ainsi le virus reste présent chez l'hôte qui ne présente pas de maladie. Le génome du PRV reste présent dans les noyaux des cellules infectées. Ce maintien sous forme latente a lieu essentiellement dans les neurones des ganglions trigéminaux pour le PRV. Le génome du virus reste présent mais n'est pas activé et la production de virions n'a pas lieu, permettant ainsi au virus de persister chez son hôte en cessant d'activer sa réponse immunitaire aiguë. Le virus peut cependant ensuite être réactivé, menant alors à la dispersion de nouveaux virus. Ceci permet la contamination de nombreux animaux à partir d'un hôte réactivant la production du PRV [34], [35].
Étiologie
Le virus de la pseudo-rage cause la maladie d'Aujeszky. Ce virus commence par infecter les poumons, où il cible dans un premier temps l'épithélium des voies respiratoires supérieures. La réplication du virus dans les voies respiratoires cause une inflammation qui se traduit par des lésions pulmonaires et des troubles respiratoires allant jusqu'à de graves pneumonies [36]. L'inflammation cause aussi la fièvre observée chez les animaux infectés et l'excès d'inflammation et les cytokines inflammatoires peuvent déclencher une réponse immunitaire excessive alors délétère pour l'individu infecté.
Le virus répliqué dans les voies respiratoires va pouvoir ensuite infecter les neurones sensoriels et être transporté par les nerfs sensoriels à travers le ganglion trigéminal jusqu'au cerveau par transport axonal rétrograde [37]. Au niveau du cerveau le virus cause une inflammation qui altère le développement du système nerveux chez les porcelets et des lésions importantes chez les porcs adultes développant des méningo-encéphalites et ganglionévrites, les neurones dégénèrent et se nécrosent [36]. Ces atteintes du système nerveux mènent à des symptômes tels que les tremblements et convulsions, pouvant aller jusqu'à des paralysies.
Le virus peut également à ce niveau rester un temps inaccessible au système immunitaire dans le tissu nerveux, et être conservé chez le porc sous forme latente. La réactivation du virus présent sous forme latente, causant une encéphalite. L'épithélium respiratoire peut ainsi être ré-infecté chroniquement par le virus qui est présent au niveau du système nerveux constituant le tissu réservoir pour ce virus [36]. Lors de la phase de latence, l’expression des gènes viraux impliqués dans la réplication virale est fortement inhibée et l'activité infectieuse diminue, grâce à la transcription de gènes associés à la latence (gènes LAT) activés uniquement au niveau neuronal, dus à un promoteur associé à la latence, LAP, activé au sein des neurones. Le virus n'est
cependant pas complètement "endormi", il inhibe notamment l’induction d'apoptose des cellules pour maintenir sa présence jusqu'à sa réactivation [38]. Ce mécanisme de latence est une caractéristique essentielle retrouvée également chez les autres herpesvirus. La réaction peut être observée suite à un stress, à une fatigue ou immunosuppression qui induisent une réactivation de l'expression des gènes viraux et de la production de virus.
Il a également été observé que le virus peut en faibles quantités se propager par la circulation sanguine et ainsi infecter de nombreux autres organes et y causer des lésions. Il provoque des dégradations au niveau du placenta des femelles en période de gestation, qui causent des décompositions des tissus des fœtus et leur mort in utero, des avortements, ainsi qu'un taux de mortalité élevé chez les porcelets infectés [36]. Méthodes de lutte et de surveillance
Afin d'éviter les pertes importantes dans les élevages, il est possible d'augmenter la biosécurité afin de réduire la contamination par l'espèce réservoir (mise en place de barrières physiques pour empêcher le contact entre sangliers et élevages de porcs). Actuellement il n'existe aucun traitement, par conséquent la prévention et la vaccination sont essentielles pour éviter les pertes dans les élevages en procurant aux animaux une immunité au PRV [39]. Finalement, il est possible de détecter rapidement la présence du virus en contrôlant l'état de santé des animaux d'élevage et en utilisant des tests de détection du PRV.
En ce qui concerne la biosécurité, pour le PRV plusieurs mesures peuvent être mises en place. L'introduction de nouveaux animaux dans un élevage (porcs reproducteurs par exemple) présente des risques d'introduire la maladie. Ces risques peuvent être réduits en sélectionnant les fournisseurs et en établissant une quarantaine pour les nouveaux animaux qui devrait être couplée idéalement à un test de détection des anticorps afin de détecter un infection latente (bien que les tests ne soient pas systématiquement effecués en pratique). Les individus visiteurs (ainsi que les animaux circulant librement) ou l'acquisition de nouveau matériel ayant été en contact avec l'environnement extérieur peut également introduire les virus dans l'élevage. Ainsi la réduction des visites, associées à des mesures d'hygiène comme des douches à l'entrée des élevages permettent de limiter ces risques [40].
Le vaccin viral atténué Bartha-K61, un vaccin utilisé depuis 1970, est un vaccin vivant atténué. Ce virus a une délétion de séquence qui est reliée à l'absence de virulence [41]. C'est le vaccin le plus utilisé pour l'éradication du PRV dans des élevages du monde entier, qui a été très efficace dans de nombreux pays contre les souches de PRV classiques. Cependant il n'a pas permis de prévenir totalement les infections par le PRV [42]–[44]. En 2011, des nouvelles souches de PRV apparaissent dans des élevages en Chine ayant pourtant reçu le vaccin Bartha-K61, on appelle ces souches les PRV variants (notamment les souches ZJ01,
ne semblait pas fournir une bonne protection. Un panel de vaccins est mis en place pour lutter contre ces nouvelles souches : le vaccin ZJO1 inactivé chez lequel gE et gI ont été déplétés, qui protège efficacement contre les souches de PRV classiques et les variants [46]. Deux vaccins atténués à partir de la souche TJ sont préparés, chez le premier le gène gE est déplété, chez le second gE, gI et TK sont déplétés [47, p.], [48]. D'autres vaccins basés sur la souches HN1201 (déplété pour gE, et déplété pour gE, gI et TK), sur la souche JS212 (déplétion des gènes gE et gI), sont mis en place et sont en cours d'évaluation [49]–[52]. Cependant malgré toutes ces préparations de vaccins qui ont été créés et ne sont pas encore actuellement utilisés dans les élevages car elles nécessitent des validations additionnelles, une étude récente souligne que les vaccins Bartha-K61 protègeraient efficacement contre les PRV variants et que l'émergence de nouveaux cas de PRV dans les élevages vaccinés pourraient aussi s'expliquer par des déviations des protocoles d'immunisation recommandés. Ainsi le vaccin Bartha-K61 pourrait être utilisé pour protéger des souches classiques et variantes efficacement, par une seconde immunisation dans les élevages [45]. De nombreux tests permettent de détecter le PRV en mesure préventive ou suite à l'apparition de lésions et à l'augmentation de la mortalité. Le test le plus répandu consiste à détecter la présence d'anticorps ciblant le PRV par dosage d'immunoabsorption par enzyme liée (ELISA), permet de détecter les anticorps qui ciblent la protéine gE du PRV. Ce test permet de détecter gE, qui est déplété dans la majorité des vaccins utilisés, cette technique permet donc ainsi de distinguer les porcs infectés des porcs vaccinés lors du test (les tests permettant cette distinction sont appelés DIVA : tests qui Différencient les individus Infectés des individus Vaccinés) [53]., Des réactions en chaîne par polymérase (PCR) peuvent également être utilisées pour détecter la présence du virus en amplifiant par PCR les gènes gB ou gD du PRV chez les animaux [54]. Ces tests sont également utilisables chez les sangliers afin de déterminer les zones où sont présents des sangliers porteurs du PRV et susceptibles d'entrer en contact avec les porcs et de leur transmettre le PRV. Cette détection environnementale est importante notamment pour les élevages en plein air, par opposition aux élevages confinés, dans lesquelles les risques de contamination entre sangliers et porcs d'élevages sont plus élevés [55]. Un test PCR ciblant le gène gE du PRV permet également de distinguer les animaux vaccinés des animaux infectés (test DIVA) [56]. Très récemment un nouveau test a été mis en place, qui permettrait la détection in situ très rapide (en environ 15 minutes) du PRV, ce test est une immunochromatographie à flux latéral permettant de détecter les virus par blocage de fluorescence, par absorption sur des bandelettes et permettrait de diagnostiquer très rapidement les infections à PRV [57].
L'éradication des infections au PRV par le vaccin Bartha-K61 a été efficace dans de nombreux pays qui ont appliqué un ensemble de méthodes de contrôle strictes telles que : l'abattage des élevages infectés, les programmes de vaccination, la restriction de l'importation de porcs et la séparation des porcs domestiques des sangliers (notamment par doubles clôtures électriques pour les élevages en plein air) [13]. Une fois le
virus éradiqué, une surveillance couplée à de strictes mesures d'abattage lors de détection d'animaux infectés dans les élevages a permis de contenir le PRV même dans les zones dans lesquelles des sangliers sauvages sont porteurs du PRV. Ainsi il semble important de mettre en place tant des mesures de protection (biosécurité) que des mesures de détection (tests de détection des virus PRV) et de prévention (vaccins). Problématique : Les mutations chez des virus émergents peuvent permettre à un agent infectieux ayant co-évolué avec une espèce (l’hôte naturel du virus), de cibler les cellules d’une autre espèce ayant une proximité physique avec l’hôte naturel du virus. Lorsque le virus acquiert la capacité d’infecter un nouvel hôte d’une espèce différente, on parle alors de saut d’espèce. Dans le cas du PRV, les souches variantes présentent des mutations qui permettent au virus d’être transmis à l’homme. Le PRV évolue tant dans un environnement sauvage (au sein des sangliers) qu’au sein d’élevages en proximité avec l’homme. Ainsi il est essentiel de suivre l’évolution des souches pouvant causer des zoonoses (souches variantes), suite aux campagnes de vaccination, afin de savoir si ce virus présente un risque considérable pour l’homme, si ce risque est stable ou en augmentation (selon la proportion de souches variantes en circulation proche de l’homme).
On pose l’hypothèse qu’au sein des populations animales, la présence de souches variantes plus susceptible de causer des zoonoses est en augmentation depuis les années 2011.
Les objectifs sont la comparaison des séquences de souches de PRV afin d’en comprendre l’évolution chez différentes espèces animales et d’évaluer les risques de zoonoses en déterminant si les souches variantes sont en augmentation ou diminution dans les populations sensibles à proximité l’homme.
Le prochain chapitre sera consacré à l'étude de la variabilité de PRV, soulignant son évolution entre différentes espèces animales et les variabilités génétiques y étant associées. L'étude de la dynamique d'évolution du PRV permet aussi de mettre en évidence l'importance évolutive des sauts d’espèces pour l'évolution des virus.
Chapitre 1 : Interspecies transmission, genetic
diversity, and evolutionary dynamics of
pseudorabies virus
Wanting He1, Lisa Zoé Auclert2, Xiaofeng Zhai1, Gary Wong2,5,7, Cheng Zhang1, Henan Zhu6, Gang Xing3, Shilei Wang1, Wei He 1, Kemang Li1, Liang Wang7,
Guan-Zhu Han4, Michael Veit8, Jiyong Zhou3, Shuo Su1*.
1. MOE International Joint Collaborative Research Laboratory for Animal Health & Food Safety, Jiangsu Engineering Laboratory of Animal Immunology, Institute of Immunology, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China.
2. Département de microbiologie-infectiologie et d’immunologie, Université Laval, Québec, QC, Canada.
3. Key laboratory of Animal Virology of Ministry of Agriculture, Zhejiang University, Hangzhou, China.
4. College of Life Sciences, Nanjing Normal University, Nanjing, Jiangsu 210023, China.
5. Institut Pasteur of Shanghai, Shanghai, China.
6. MRC-University of Glasgow Centre for Virus Research, Glasgow, Scotland, United Kingdom.
7. CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China.
8. Institute for Virology, Center for Infection Medicine, Veterinary Faculty, Free University Berlin, Robert-von-Ostertag-Straβe 7-13, Berlin, Germany.
1.1 Résumé
Contexte : Le virus pseudorabies (VRP) cause la maladie d’Aujeszky chez les porcs et peut être transmis à d’autres mammifères, y compris les humains. Ici, nous avons systématiquement étudié la transmission interspécifique et l’histoire évolutive du PRV.
Méthodes : Nous avons effectué l’analyse complète sur la phylodynamique, la sélection, et la biologie structurale pour résumer l’évolution phylogénétique et adaptative de PRV basée sur toutes les séquences pleine-longueur et principales disponibles de glycoprotéines.
Résultats : le PRV peut être divisé en deux clades principaux avec la recombinaison inter-clade et intra-clade fréquente. Clade 2.2 (variante PRV) est actuellement le génotype le plus répandu dans le monde, et le plus souvent impliqué dans des événements de transmission inter-espèces (y compris les humains). Nous avons également constaté que la taille de la population du clade 2.2 a augmenté depuis 2011 et que le nombre effectif de reproduction a été supérieur à 1 de 2011 à 2016, ce qui indique que le PRV peut encore circuler dans les élevages de porcs et qu’il présente toujours un risque de transmission entre espèces, notamment en Chine. Il convient de noter que nous avons identifié des sites d’acides aminés dans certaines glycoprotéines importantes gB, gC, gD et gE qui peuvent être associés à l’adaptation PRV à de nouveaux hôtes et l’évasion immunitaire aux vaccins.
Conclusions : Notre étude fournit un aperçu génétique important de la transmission et de l’évolution inter-espèces du VPR à l’intérieur et entre les différents hôtes, ce qui justifie une surveillance supplémentaire.
1.2 Abstract
Background: Pseudorabies virus (PRV) causes Aujeszky’s disease in pigs and can be transmitted to other mammals, including humans. Here, we systematically studied the interspecies transmission and evolutionary history of PRV.
Methods: We performed comprehensive analysis on the phylodynamics, selection, and structural biology to summarize the phylogenetic and adaptive evolution of PRV based on all available full-length and major glycoprotein sequences.
Results: PRV can be divided into two main clades with frequent inter-clade and intra-clade recombination. Clade 2.2 (variant PRV) is currently the most prevalent genotype worldwide, and most commonly involved in cross-species transmission events (including humans). We also found that the population size of clade 2.2 has increased since 2011 and the effective reproduction number was greater than 1 from 2011 to 2016, indicating
China. Of note, we identified amino acid sites in some important glycoproteins gB, gC, gD and gE that may be associated with PRV adaptation to new hosts and immune escape to vaccines.
Conclusions: Our study provides important genetic insight into the inter-species transmission and evolution of PRV within and between different hosts that warrant additional surveillance.
1.3 Introduction
Pseudorabies virus (PRV) is an alphaherpesvirus that causes Aujeszky’s disease in pigs, its host reservoir species [1]. Infections are a concern for the swine industry as Aujeszky’s disease leads to decreased reproduction in sows, respiratory disease in adult pigs, disorders of the nervous system, and high mortality in piglets. The virus also infects a wide range of other animals, such as ruminants, carnivores, and rodents [2]. Transmission occurs through the exchange of saliva, nasal discharge between animals, and airborne particles [2].
Discovered in the early 1900s, PRV has been distributed globally since the 1980s and is now prevalent in at least 44 countries [1]. PRV has a large genome (over 140,000 nucleotides) coding for over 100 proteins. The viral envelope proteins, including the well-characterized glycoprotein B (gB), gC, and gD play a central role in the induction of immunity and are involved in virus entry and virulence. In particular, gE is a virulence factor and determines tropism for the central nervous system [3]. The number of reported full-length PRV genome sequences has increased recently. However, phylogenetic analysis of partial sequences of the gC gene is a suitable approach for PRV genotyping as previously discussed [4, 5]. Moreover, given that the gB, gC, gD, and gE genes are involved in receptor binding, pathogenicity and the induction of antibodies, they are appropriate to study phylogenetic and epidemiologic features. Previous phylogenetic analysis based on the gC gene identified two main PRV genotypes [4]. Strains isolated in Eastern and Southern Europe, Latin America, Africa, and Asia belong to clade 1. Strains from clade 2 (also known as variant PRV) are found almost exclusively in China [4]. The emergence of variant PRV was first reported in China in 2011 [6] [7] [8] in pigs that had previously been vaccinated with a live-attenuated PRV vaccine (Bartha-K61) [9].
PRV infections of humans had been sporadically reported in the past. Typical symptoms include weakness, fever, sweating, dysphagia, and neurologic disorders [10] [11]. The most recent case of suspected PRV infection in humans occurred in China
in 2017, in which the genome of the virus isolate clustered with variant PRV Chinese variants (clade 2) [12] . The patient, a swineherd worker, presented fever, headaches, and visual impairment after exposure to sewage on a hog farm. While most symptoms resolved spontaneously, her vision was still compromised 6 months post
The diagnosis was supported by results from next generation sequencing (NGS), PCR, and serological tests [12]. This case highlights the importance of developing a vaccine providing protection against variant PRV and to increase infection control awareness among farm workers.
PRV is an important threat to animals and possibly humans. Indeed, many countries have also reported PRV interspecies transmission [13]. The interspecies transmission and evolution of PRV is different among different regions and until now, no studies have investigated the diversification patterns and evolutionary dynamics of PRV in depth. Here, we integrated phylogenetic, molecular selection analysis, and structural biology to address the following questions: (a) which animal species can PRV infect and whether adaptive evolution occurred during virus transmission from swine to new hosts and thus new environment; (b) what is the role of different PRV clades in interspecies transmission?; (c) is variant PRV a newly emergent pathogen in China or is it much older?; (d) what is the evolutionary dynamics of classical and variant PRV?; and (e) what is the role of recombination in PRV evolution?
Material and methods Sequence dataset
The gB, gC, gD and gE gene sequences and 29 PRV full-length genomes were collected from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) up to April 20th, 2018 (Table S1). Sequences were aligned using MAFFT7 and manually adjusted using MEGA7.0 [14, 15] to calculate the frequency of occurrence of cross-host transmission events in each country. To determine differences in diversity ratios of homologous protein-coding regions between Qihe547 (the emergent and latest genome in China in NCBI) with each clade, 8 PRV strains, i.e. SC, LA, Ea, Fa (clade 2.1), HNX, HNB (clade 2.2), Bartha and ADV32751 (clade 1) (GenBank accession numbers: KT809429, KU552118, KU315430, KM189913, KM189912, KT824771, JF797217, KU198433, respectively) were compared to the Qihe547 strain (accession number: KU056477). Recombination analysis and protein coding variations
The RDP4 program was used to analyze the occurrence of recombination [16]. A total of seven algorithms, including GENECONV [17], RDP [18], Chimaera [19], SiScan [20], 3Seq [21], MaxChi [22], and LARD [23], were employed. Recombination events were confirmed with a p-value cut–off of 0.05 and by at least four out of seven methods [24]. To clarify intra-clade and inter-clade recombination within PRV full-length genomes, SplitsTree (v4.14.6) was used to reconstruct networks [25]. In addition, bootscanning and similarity plot were inferred by Simplot (v3.5.1), with a sliding window of 1,500 nucleotides and moving in 750 nucleotide steps [26]
The best-fit nucleotide substitution model was chosen using jModelTest software (version 2.1.7) with the highest Bayesian information criterion [27]. RAxML software (version 2.4.8) was used to construct maximum likelihood (ML) phylogenies. The general time-reversible with gamma distribution (GTR+G) substitution model was applied. Support for phylogenies was assessed via 1000 nonparametric bootstrap replicates [28]. In addition, given that the gC gene has been previously used for phylogenetic and phylodynamic analysis of PRV owing to its high variability compared with other genes [5, 29] and has the most available sequences, Markov Chain Monte Carlo (MCMC) methods were used to analyze its evolutionary dynamics. BEAST software (version 1.8.4) was used to estimate the time to the most recent common ancestor (tMRCA) and evolu-tionary rates [30]. The best-fit model, GTR+G, a relaxed clock (log-normal), and a coalescent: Bayesian skyline model was set to estimate the efficient population size. The length of the chain was 1 × 109 generations, and samples were taken every 1 × 104generations. Two independent runs were applied and combined using LogCombiner software with a 10% burn-in. The final tree was summarized using TreeAnnotator software and displayed using FigTree software (version 1.4.3). Effective reproduction number estimation
The effective reproduction number (Re) represents the average number of secondary infections resulting from a typical infected person at a given time within a population in which not all hosts are necessarily susceptible. We estimated the Re of clade 2.2 based on the gC gene sequences using the following formula:
Re= λ ÷ (µ + ρ)
where Re is the effective reproduction number, λ is birth rate of PRV, µ is the death rate of PRV, and ρ is the sampling probability. BEASTv2.7 was used to estimate the Re of PRV with the HKY+G model and a relaxed lognormal clock. Tree prior was set birth death skyline contemporary model with one sampling proportion. The chain length was 1×108 generations and samples were taken every 1×104 generations. LogCombiner was used to combine the two runs.
Selection and adaptive evolution analysis
The methods used to address selection and adaptation evolution of the gB, gC, gD, and gE genes are summarized in Supplemental Text.
Results
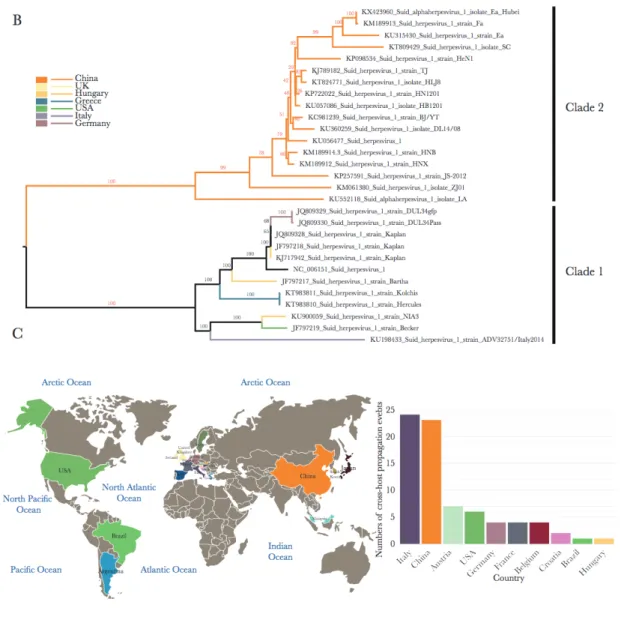
PRV has been suspected to infect humans since its discovery (Fig 1A) and has been reported in numerous countries (Fig 1B). We constructed a ML phylogenetic tree with all known full-length PRV sequences (Fig 1C). The ML tree shows that PRV is divided into two clades, 1 and 2. Clade 1 includes European and American sequences. All strains in clade 2 are from China. When the data is summarized by country, frequent occurrence of PRV interspecies transmission was observed (Fig 1C). The most frequent interhost transmission events were observed in Italy and China, with 24 and 23 events, respectively. In addition, the average amino acid difference between latest China emerging PRV Qihe547 strain and 8 other PRV strains was 1.93%. However, with two clade 1 strains this value becomes 4.94%, with four clade 2.1 strains 1.16%, and with two clade 2.2 strains 0.46%. Among the 69 protein coding regions, the regulatory protein ICP22 (US1) showed the highest differences, and the capsid triplex subunit 2 (UL18) encoding for the minor capsid protein maintained minimal differences (Fig S2).
Detection of recombination
Recombination signals were detected using RDP4 with a cut-off p-value of 0.05, with at least four out of seven methods showing that almost all PRVs have undergone recombination based on full genomes (data not shown). The phylogenetic network suggests that intra-clade recombination is less than that of inter-clade recombination (Fig 2A), but inter-clade recombination did occur. Analysis using Simplot confirmed that the clade 1 strains recombined with other clade 1 strains (Fig 2B). Moreover, earlier clade 2 strains, including SC (accession number: KT809429) and LA (accession number: KU552118), may have originated from recombination events between clade 1 strains (such as the Bartha vaccine) (Fig 2 and Fig S3) and early clade 2 strains. In addition, variant isolates, like HeN1 (accession number: KP098534) and Qihe547 (accession number: KU056477) may have originated from recombination between clade 1 isolates and clade 2 vaccine isolates, such as Ea and Fa (Fig 2 and Fig S3).
Figure 1. 1 History, geographical distribution, and diversity of full-length PRV infections. A). Timeline of suspected PRV infections of humans. B). Global distribution of PRVs and number of PRV interspecies transmission events occurring in each country. C). ML tree of PRV full-length genomes. Different countries are denoted by different markers.
Figure 1. 2 Recombination analysis of 29 pseudorabies virus (PRV) full-length genomes. A, Network tree was constructed using 29 PRV full-length genomes by SplitsTree software (version 4.14.6) with kimura 2-parameter model. Isolates in green regions correspond to clade 1, and isolates in pink regions to clade 2. B–D, Bootscanning analysis of PRV full-length genomes with a sliding window of 1500–base pair (bp) nucleotides moving in 750-nucleotide steps. Each query and compared sequence is labeled on the top of the picture.
Subtyping and interspecies transmission
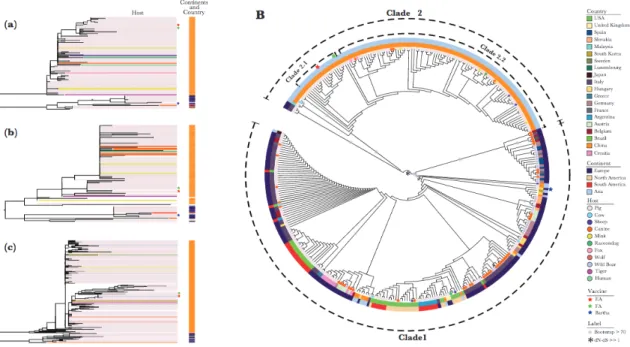
Since the gB, gD, and gE glycoproteins are involved in triggering the host humoral immune response against PRV, we used these genes to perform phylogenetic analysis (Fig 3A-C, respectively). We found that ML trees for the three genes shared a similar topology and PRV can also be divided into clades 1 and 2. Clade 1 is mainly composed of European, American, and early Chinese strains. In addition to domestic pigs, a PRV sequence from infected dogs in Italy also clustered with clade 1. Clade 2 was mostly composed of Chinese sequences, as well as 1 strain from Malaysia and 3 strains from Italy. In addition, interspecies transmission in clade 2 was found to be more frequent than in clade 1, with infections of sheep, mink, canine, fox, raccoons, tigers, wild boars, and humans. PRVs infecting other animals are scattered in the ML trees of gB, gD, and gE. Surprisingly, some sequences from infected pigs shared 100% identity at the genomic level with sequences from other infected animals (Fig 3). Based on the gB ML tree, the PRV sequence from the most recent human case in China [12] clustered with PRV sequences of Chinese pigs (Fig 3), suggesting that the human PRV isolate had a swine origin.
Figure 1. 3 Maximum likelihood (ML) tree of the gB, gD, gE, and gC genes. The ML tree was constructed using RAxML (v8.4.10) with the general time-reversible with gamma distribution substitution model and 1000 bootstraps. A, ML tree based on gB (a), gD (b), and gE (c) coding sequences. Colored lines represent the host, and vertical bars on the right represent countries or continents where strains are isolated. The star on the right of tree represents the vaccine strain. B, ML tree constructed based on the gC gene. Colored circles
represent the host; outer rectangular boxes, separate countries and continents; and stars, vaccine strains name. Ea (gG−/TK− vaccine) and Fa (gE−/gI−/TK− vac-cine) currently used in China. Pseudorabies virus could be divided into 2 main clades, clades 1 and 2; clade 2 is subdivided into clades 2.1 and 2.2.
Among the glycoprotein-encoding genes, gC has the highest mutation rate and the largest number of reported sequences [4]. We found no evidence for recombination in the gC gene, and better root-to-tips divergence against sampling date compared to the gB, gD, and gE genes (data not shown). Therefore, we further analyzed PRV genotypes using the gC gene [5, 29]. ML and MCC trees exhibit similar structures (Fig 3D and Fig 4), confirming that PRV can be divided into clades 1 and 2. Clade 1 is mainly composed of sequences from some European and American countries including Italy, Brazil, the United States, Germany, and parts of China.
Figure 1. 4 Maximum clade credibility tree of the gC segment, tree constructed using BEAST software (version 1.8.4). The general time-reversible with gamma distribution optimal nucleotide substitution model and the coalescent: Bayesian skyline model have a total chain length of 1 × 109 and are sampled every 1 × 104 times. Colored lines represent the host; rectangular boxes, isolated countries or continents; and circles, vaccine strains name. Ea (gG−/TK− vaccine) and Fa (gE−/gI−/TK− vaccine) currently used in China.
These sequences were collected from domestic pigs, wild boars, cows, dogs, raccoons, sheep, and the Bartha vaccine strain from China (accession number: JF797217). Interestingly, canine PRVs in clade 1 were shown to
cluster with isolates from pigs and wild boars. This suggests that canine PRV isolates may have originated from pigs [31]. Clade 2 can be further subdivided into clades 2.1 and 2.2. Sequences contained in clade 2.1 were mainly from Chinese strains prior to and during 2016. They are composed of pig-infected PRV, and the vaccine strains Ea (accession number: KU315430, for gG-/TK- vaccine HB-98) and Fa (accession number: KM189913, for gE-/gI-/TK- vaccine SA215) that have been used in China [32-34]. Clade 2.2 mainly includes one strain isolated in 2007 in China (accession number: KY398757) and other strains isolated after 2011 in China. Some cases of interspecies transmission were observed with isolates from this clade. Although gC has the highest mutation rate within the PRV glycoproteins, some gene sequences from infected pigs shared 100% identity with sequences from other infected animals.
Evolutionary dynamics
The MCC tree indicated that two clades have been independently circulating for a long time. The estimation of the tMRCA and evolutionary rate of PRVs by the MCMC method revealed that PRVs may have originated in 1419.13 (95% HPD: 367-1860.16), and the main trunk differentiated into clades 1 and 2 in 1659.22 (95% HPD: 1176.29-1871.18) and 1871.56 (95% HPD: 1598.63-1949.77), respectively. Additionally, the tMRCA of clade 2.1 is 1959.38 (95% HPD: 1945.71-1964.00), whereas for variant isolates (clade 2.2) it is 2002.96 (95% HPD: 1997.15-2007.00). This suggests that the variant PRV was present in China at least since 2002, but it did not cause a large-scale epidemic until 2011. The estimated overall PRV evolutionary rate was low (mean: 5.32×10-5 substitutions/site/year; 95% HPD: 2.94×10-5-7.83×10-5). In addition, we estimated the evolutionary rate of clade 1 to be 2.09×10-5 (95% HPD: 2.14×10-12-6.69×10-5 substitutions/site/year) and clade 2 to be 5.45×10-5 (95% HPD: 1.01×10-5-1.01×10-4 substitutions/site/year) (Table S2). Net is also a measure of the effective population size of the virus. To understand the population size of clade 1 and clade 2, the Bayesian skyline coalescent reconstructed revealed that the relative genetic diversity of clade 1 was more or less constant (Fig 5A). However, from 2004 to 2010, the decreasing efficient population size suggested that the diversity of clade 2 PRV declined, while a dramatic increase in diversity was observed from 2010 to mid-2012 and remained at a high level until 2016 (Fig 5B). This phylodynamic pattern is present in clade 2.2 isolates, indicating that the variant PRV population in China was affected. This is also consistent with the widespread prevalence of clade 2.2 strains in China from 2012. Estimation of the Re value in clade 2.2 from 2011 to 2015 indicated that the annual Re was the lowest in 2011 with a mean value of 1.38 (95% HPD: 0.16-3.8) and the highest in 2014 with a mean value of 4.17 (95% HPD: 0.88-10.06) (Figure S1). Selection and adaptive evolution analysis
in the gC protein were under positive selection according to FEL, MEME, and FUBAR. Residue 348 in gE was confirmed by SLAC and FUBAR to have undergone positive selection, promoted by factors such as vaccine selection, etc. However, no positively selected sites were observed in gD. Analyses of positively selected branches of the gB, gC, and gD genes suggested that the virus may be undergoing host adaptation (Fig 3). The analysis of selection on branches of the gB and gC genes indicated that positive selection was also observed for many European and American PRV strains. This may indicate cross host transmission and gradual adaptation to the new host.
Figure 1. 5 Skyline plot of each pseudorabies virus clade. A, Clade 1. B, Clade 2. The skyline plots were constructed using BEAST software (version 1.8.4). The general time-reversible with gamma distribution optimal nucleotide substitution model and the coalescent: Bayesian skyline model have a total chain
![Figure 1 Représentation des relations entre facteurs de risques pouvant mener à l'émergence de maladies infectieuses, adapté à partir de l'article [6]](https://thumb-eu.123doks.com/thumbv2/123doknet/2886125.73550/17.918.135.701.100.773/figure-représentation-relations-facteurs-risques-émergence-maladies-infectieuses.webp)
![Tableau 0.1 - Détection d'anticorps anti PRV chez les sangliers dans de nombreux pays du monde, extrait modifié de [13]](https://thumb-eu.123doks.com/thumbv2/123doknet/2886125.73550/19.918.130.795.404.1088/tableau-détection-anticorps-sangliers-nombreux-monde-extrait-modifié.webp)
![Figure 2 Frise Chronologique des infections présumées ou vérifiées par le PRV chez l’homme, extrait de [25]](https://thumb-eu.123doks.com/thumbv2/123doknet/2886125.73550/21.918.137.796.419.656/figure-frise-chronologique-infections-présumées-vérifiées-homme-extrait.webp)
![Figure 3 Structure détaillée du virion du PRV, et gènes codant les protéines constituant le virus (source [29])](https://thumb-eu.123doks.com/thumbv2/123doknet/2886125.73550/24.918.132.790.110.661/figure-structure-détaillée-virion-gènes-codant-protéines-constituant.webp)
![Figure 4 Schématisation du cycle de réplication du PRV, extrait de [33].](https://thumb-eu.123doks.com/thumbv2/123doknet/2886125.73550/25.918.131.669.521.881/figure-schématisation-cycle-réplication-prv-extrait.webp)