HAL Id: hal-01611606
https://hal.archives-ouvertes.fr/hal-01611606
Submitted on 20 Oct 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Carbonated hydroxyapatite starting from calcite and
different orthophosphates under moderate hydrothermal
conditions: Synthesis and surface reactivity in simulated
body fluid
Doan Pham Minh, Ange Nzihou, Patrick Sharrock
To cite this version:
Doan Pham Minh, Ange Nzihou, Patrick Sharrock. Carbonated hydroxyapatite starting from
cal-cite and different orthophosphates under moderate hydrothermal conditions: Synthesis and surface
reactivity in simulated body fluid. Materials Research Bulletin, Elsevier, 2014, 60, p. 292-299.
�10.1016/j.materresbull.2014.08.052�. �hal-01611606�
Carbonated
hydroxyapatite
starting
from
calcite
and
different
orthophosphates
under
moderate
hydrothermal
conditions:
Synthesis
and
surface
reactivity
in
simulated
body
fluid
Doan
Pham
Minh
*
,
Ange
Nzihou,
Patrick
Sharrock
UniversitédeToulouse,MinesAlbi,CNRSUMR5302,CentreRAPSODEE,CampusJarlard,F-81013Albicedex09,France
Keywords: Structuralmaterials Chemicalsynthesis Infraredspectroscopy Surfaceproperties ABSTRACT
Theone-stepsynthesisofcarbonatedhydroxyapatite(CAP)usingcalciteanddifferentorthophosphates was investigatedina closedbatchreactor. Onlyorthophosphoric acidcould leadto thecomplete decompositionofcalciteparticles,whenthereactiontemperaturewassetat80!C.Ontheotherhand,the
reactiontimeandthedilutionof theinitialcalcitesuspensionhadnosignificantinfluenceonthe formationofthesolidproducts.CAPwasformedasthemaincrystallinecalciumphosphatewiththe carbonatecontentintherangeof4.2–4.6wt.%.ThethermaldecarbonationofthesynthesizedCAPstarted at750!Cbutitwasonlysignificantat1000!Cunderairatmosphere.Thisthermaldecarbonationwas
totalat1200!Corabove.AllCAPsamples andproductsfollowingthermaltreatmentswerefound
bioactiveinthetestusingsimulatedbodyfluid(SBF)solution.
1. Introduction
Theinorganicmineralphaseofhumanteethandboneismainly composed of calcium hydroxyapatite (Ca-HA, Ca10(PO4)6(OH)2),
withdifferentsubstitutionssuchassodium,magnesium,chloride, fluoride, carbonate,etc. [1–3].This explainsthe interestin the developmentofCa-HAandsubstitutedCa-HAinthebiomaterials field,duetotheirbiocompatibilityandbioactivity[4,5]. Substitut-ed Ca-HA is formedwhen calcium,phosphate,and/or hydroxyl groups of Ca-HA structure are replaced by other metals and functionalgroups.Thus,bonemineralhasusuallythemolarratio ofcalciumtophosphatevaryingintherangeof1.37–1.87compared withthatof1.67forstoichiometricCa-HA[3].Ingeneral,carbonate isfoundtobethemostabundantsubstitutionanditscontentin bonemineralvariesintherangeof3–8wt.%[6].Carbonategroups caninsertintheapatiticstructurebyreplacingphosphateand/or hydroxyl groups to form carbonated hydroxyapatite (CAP). Conventionally,CAPisA-typeCAPwhencarbonategroupsreplace phosphate groups;B-type CAP when carbonate groups replace hydroxyl groups; and A-B-type CAP when carbonate groups replacesimultaneouslyphosphateandhydroxylgroups.
ThesynthesisofCAPcallsusuallyfortheclassicalwetanddry methods.Inthewetprocess,solublecalciumandphosphatesare
precipitatedinanaqueoussolutioninthepresenceofcarbonate anions[7,8].ThisprocessleadsgenerallytotheformationofB-type CAP [7] but A-B-type CAPmay be also formed[9]. In the dry process,A-typeCAPcouldbegenerated byheatingpureCa-HA underCO2fluxathightemperatureinordertoreplacehydroxyl
groupsbycarbonategroups[10].CAPcouldbealsoobtainedbythe commonmechano-chemicalprocessasdemonstratedby Sucha-neket al.[11],whereinanaqueous suspensionof Ca(OH)2 and
CaCO3(orNa2CO3)and (NH4)2HPO4 aretreatedina
laboratory-scalemill.Butthislastwayrequiresseverereactionconditions. We recently developed a new one-step synthesis process of Ca-HAfromCaCO3andorthophosphoricacid(H3PO4)aseconomical
starting materials at moderate synthesis conditions [12]. The advantageofthisprocesscomparedto classicalCa-HA synthesis pathways remainsin the factthat pure Ca-HA canbeobtained withoutanyfurtherpurificationstep,thankstotheabsenceofall counterionsinthereactionmedium[12].Amodificationofthis processwasthendevelopedfortheone-stepsynthesisofCAPunder moderatehydrothermalconditions[13].CAPcontaining4.2wt.%of carbonatecouldbeobtainedfromthereactionofCaCO3andH3PO4in
aclosebatchreactorunderCO2pressure.CaCO3waschosenbecause
itisthesourceofbothcalciumcationsandcarbonateanionsforthe formationofCAP,andalsobecauseithasconsiderableindustrial importanceandabundantdiversityasabiomineral[14].Inthiswork, we firstlycompared thereaction of calcite with different alkali orthophosphates and orthophosphoric acid in orderto selectthe best orthophosphate.Then,weinvestigatedtheinfluenceofdifferent
* Correspondingauthor.Tel.:+33563493258;fax:+33563493043. E-mailaddress:doan.phamminh@mines-albi.fr(D.PhamMinh).
parameters includingthecontentofwaterintheinitialsuspensionof CaCO3, the reaction temperature and the reaction time, on the
contentofcarbonateinserted inthefinalsolidCAP. Theinvitro biomineralizationactivityofthesolidCAPproductcontainingthe highestcarbonatecontentwasalsoinvestigatedbeforeandafter thermaltreatmentatdifferenttemperatures.
2. Materialsandmethods
Orthophosphoricacid(H3PO4,85wt.%inwater)waspurchased
from Merck. Calcitepowder (CaCO3, d50of 23.6
m
m), and alkaliorthophosphates(NaH2PO4,KH2PO4,NH4H2PO4)ofanalyticalpurity
from Fisher Scientific were used as received without further purification.The reaction of CaCO3 with these orthophosphates
was carried out in a 250mL stainless steel close batchreactor (TopIndustry,France) whichwasequippedwithanelectricalheating jacketandamagneticstirrer.Whenorthophosphoricacidwasused, 10gof calcite powderand45–135gof water were fedintothe reactor. Thereactorwasclosedandthen6.92gH3PO4(85wt.%)wasquickly
injectedintothereactorviaasyringe.Whenalkaliorthophosphates wereused,10gofwaterwassetintothereactor.Thislastonewas closedand135gofanaqueoussolutioncontaining60mmolofan alkaliorthophosphatewasquicklyinjectedintothereactor.Inall
case,themolarratioofcalciumtophosphorusintheinitialreaction mixturewas1.67.Thereactionmixturewaskeptat60or80!Cunder
stirringof800rpmfor48–168hofreaction.Theconditionsforthe synthesisofdifferentCAPweresummarizedinTable1.Duringthe reaction, the pressure in thebatch reactor was measured by a manometer.Whenthereactionreachedthedesiredreactiontime, thereactor was freelycooled down to room temperature.Solid products were separated from liquid phase by filtration using 0.45
m
mfilterpaper.Thentheywereair-driedat50!Cfor48hbeforefurtheranalysesandcharacterizations.
Elemental analysis was carried out by inductively coupled plasma atomic emission spectroscopy (ICP-AES, HORIBA Jobin YvonUltima2apparatus).Infraredspectroscopy(IR)wascarried outusingaShimadzuFTIR8400Sspectrometerinthewavenumber range 4000–500cm"1. X-ray diffraction (XRD) measurements
were carried out using a Phillips Panalytical X’pert Pro MPD diffractometer.Scanningelectronmicroscopy(SEM)observation wasperformedonaPhilipsXL30ESEMapparatus. Thermogravim-etry analysis (TG) was carried out with a SDTQ600 analyzer (TAInstruments)underairflow(100mLmin"1)withtheheating
rateof20!Cmin"1.TheamountofremainingCaCO
3inthefinal
solidwascalculatedcorrespondingtotheobservedweightlossof CO2from610to720!C.
3. Results
3.1.Orthophosphatecomparison
Fig.1presentsXRDresultsofthesolidproductsobtainedfrom thereactionofcalcitewithfourorthophosphatesundersimilar reaction conditions. Inall cases, Ca-HA appeared as the main crystallinecalciumphosphate.Ontheotherhand,the decompo-sitionofcalcite wasdifferent fromeachotherorthophosphate. Under the reaction conditions used, orthophosphoric acid alloweddecomposingcompletelytheintroducedcalcite,because the signal of themain diffractionpeakof calcite at 29.4! was
negligible (CAP–H pattern). For three alkali orthophosphates used,diffractionpeaksofremainingcalcitewereclearlyobserved, which indicates that the reaction of calcite and these
Table1
SynthesisconditionsofCAPfromCaCO3(100mmolor10g),alkaliorthophosphates,
andH3PO4 (66.7mmol);D isthemass ratioofwater tocalciteintheinitial
suspension.Thefinalpressureinthebatchreactorvariedintherangeof9–16bar. Orthophosphate T (!C) t (h) D Water (g) Solidproduct designation Note NaH2PO4 80 48 13.5 135 CAP–Na Comparisonof
orthophosphate KH2PO4 80 48 13.5 135 CAP–K NH4H2PO4 80 48 13.5 135 CAP–N H3PO4 80 48 13.5 135 CAP–H H3PO4 60 48 4.5 45 T60/t48/D4.5 Parametric study H3PO4 60 48 9 90 T60/t48/D9 H3PO4 80 48 4.5 45 T80/t48/D4.5 H3PO4 80 48 9 90 T80/t48/D9 H3PO4 80 168 9 90 T80/t168/D9
orthophosphateswasnotcompleted.Thisresultwasconfirmed byTGanalysis(resultsnotshown).Theremainingcalcitecontent presentinthefinalsolidswas4.4,1.4,0.5and<0.06wt.%forCAP– Na,CAP–K,CAP–NandCAP–H,respectively.Probably, orthophos-phoric acid with higher acidity compared to other alkali orthophosphates was more favorable for the dissolution of calcite particles,which is crucial for the formation of calcium phosphatephases [15].Whencalciteparticleswerecoveredby solid calcium phosphate layers, the contact between these covered calcite particlesandorthophosphatessourcesmust be preventedandsothereactionisstoppedorsloweddown[15,16]. Inadditiontothishigherperformanceforthedecompositionof calcite, nowashingstepwasneededfor thepurificationofthe solidproduct startingfromorthophosphoricacidthanks tothe absenceofcounter-ionssuchasalkalis.Soforthenextparametric
study,CAPsynthesiswasperformedonlywithorthophosphoric acid.
3.2.CAPfromcalciteandorthophosphoricacid–XRD characterization
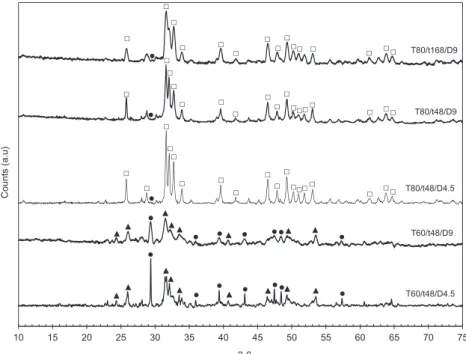
Fig.2presentsXRDpatternsofthesolidproductsobtainedat different synthesis conditions. Both productsobtained at 60!C
(T60/t48/D4.5andT60/t48/D9)containedoctocalciumphosphate (OCP, Ca8(HPO4)2(PO4)4
#
5H2O) as the main crystalline calciumphosphate, and high contents of remaining calcite. So the temperatureof60!Cwasnotenoughforthecompletedissolution
ofcalciteparticles.Probablythistemperatureledtotheformation ofcore–shellparticlesinwhichcalcitecorewascoveredbysolid calciumphosphatelayers(shell)[17].Thisstructurepreventedthe
Fig.2.XRDpatternsofthesolidproductsobtainedatdifferentsynthesisconditions;(&)Ca-HA(JCPDSstandardNo.01-072-1243);(~)OCP(JCPDSstandardNo. 00-026-1056);(*)calcite(JCPDSstandardNo.00-047-1743).
500 1000 1500 2000 2500 3000 3500 4000 Tr ansm ittance, a. u Wavenumber, cm-1 T80/t168/D9 T80/t48/D9 T80/t48/D4.5 T60/t48/D9 T60/t48/D4.5 OH Phosphate CO32 Phosphate CO32
contact of remaining calcite core with phosphate species and sloweddownor stopped thereaction at this temperature. The increaseofthedilutionofcalcitesuspensiondidnotimprovethe dissolutionofcalciteat60!C.
Onthe other hand, whenthe reaction temperature rose to 80!C,calcitecouldbecompletelydissolvedasindicateditsvery
weak signal of the principal XRD peak at 29.4!. At this
temperatureandforthesamereactiontimeof48h,thedilution ofCaCO3suspensionhadnoremarkableinfluenceontheformed
product.Themaincrystallinecalciumphosphatephaseidentified fortheproductssynthesizedat80!CwasCa-HA.However,CAP
(JCPDS standard No. 96-900-3550, 96-900-3551, 96-900-3552) mightbealsopresentintheseproductsbuttheirXRDdiffractions areusuallysuperposedonthoseofCa-HA.SothepresenceofCAP needstobeconfirmedbyotheranalyses.
3.3.CAPfromcalciteandorthophosphoricacid–IRcharacterization IRspectraofallthesolidproductsarepresentedinFig.3.Inall cases,characteristicpeaksofphosphategroupscouldbefoundin thewavelengthrangesof650–550cm"1and1300–910cm"1[13].
Fortwosolidsobtainedat60!C,nosignalofcarbonategroupswas
observed, despite remarkable contents of remaining calcite as shown by XRD analysis (Fig. 2). As mentioned above, calcite remnants must be covered by calcium phosphate layers and thereforecouldnotbedetectedbydiffusereflectanceIRanalysis, whichrecordsonlysurfaceinformationforsolidsamples.
For the solids synthesized at 80!C, absorption bands of
carbonategroupscouldbeclearlyobservedat1545,1450,1415, 880,and870cm"1.Alltheseabsorptionbandscouldbeattributed
tocarbonate groups inserted in theapatite structure of Ca-HA
80
85
90
95
100
105
0
500
1000
1500
TG
(w
t.
%
)
Temperature,
oC
T60/t48/D4.5
T60/t48/D9
(A)
0
0
500
100
0
150
0
DT
G
(w
t.
%
/
oC),
a
u
Temperature,
oC
T60/t48/D4.5
T60/t48/D9
(B)
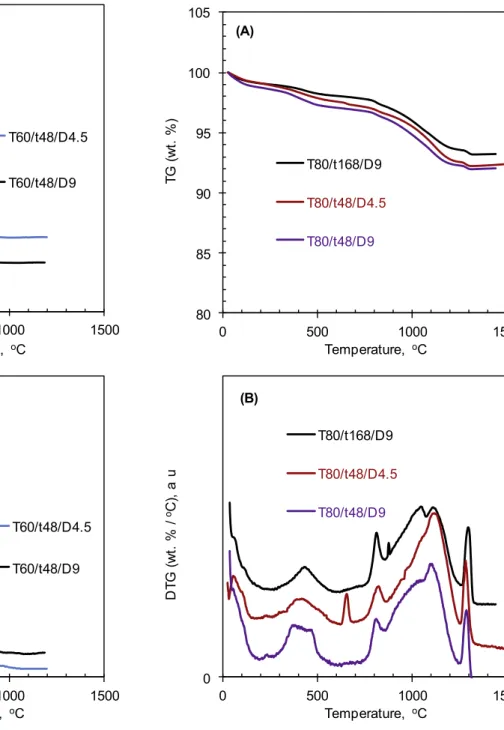
Fig.4.TGanalysisofthesolidproductssynthesizedat60!C.
80
85
90
95
100
105
0
500
100
0
150
0
TG
(w
t.
%
)
Temperature,
oC
T80/t168/D9
T80/t48/D4.5
T80/t48/D9
(A)
0
0
500
100
0
150
0
DT
G
(w
t.
%
/
oC),
a
u
Temperature,
oC
T80/t168/D9
T80/t48/D4.5
T80/t48/D9
(B)
[18–20].Carbonategroupsreplacedbothphosphateandhydroxyl groupstoformA-B-typeCAP,asillustratedbytheircharacteristic absorption bands at 1545 and 880cm"1 (A-type) and 1450,
1415and870cm"1(B-type)[18,19].
Theelementalanalysisofthreesolidproductsobtainedat80!C
showedthatthemolarratioofcalciumtophosphorusvariedin 1.83–1.90 range, which was higher than the value of the stoichiometricCa-HA. Thiscanbeexplainedbytheinsertion of carbonategroupsatthepositionofphosphategroups.
3.4.CAPfromcalciteandorthophosphoricacid–TGcharacterization Fortheuseasbiomaterials,calciumphosphatebasedproducts areusuallytreatedbyasinteringstep[21].Thermalbehaviorofthe preparedsolidswasinvestigatedinlargerangeoftemperatureas showninFigs.4and5.Forthesolidpreparedat60!C(Fig.4),after
thefirstweightlossabout100!Cduetotheremovalofsurface
humidity, different weight losses in the temperature range of 100–600!CwereobservedforthedehydrationofOCP[18,22–25],
which wasthemaincalciumphosphatepresentinthesesolids. Remainingcalciteshowedanintenseweightlossatabout610!C,
followedbythedecarbonationofCAPat750!C.Nofurtherweight
losswasobservedupto1200!Cforthesetwosolids.
For the solids synthesizedat 80!C, afterthedehydration of
surface moisturearound 100!C, thedehydrationof
hydrogeno-phosphate groupstook placein therangeof 100–600!C. These
weightlossesweremuchlowercomparedtothesolidssynthesized at 60!C (Fig. 4). As expected, no notable signal of calcite
decomposition was detectedaround 610!C. On theotherhand,
thedecarbonationofCAPtookplacewithimportantweightlosses. Thisdecarbonationstartedatabout750!Candfinishedatabout
1200!C. The last weight loss around 1250!C was due to the
dehydration of hydroxyl group of apatitic structure to form oxyhydroxyapatite[5].So,whenthesynthesistemperaturewasset at80!C, thereactiontimeand thedilution oftheinitialcalcite
suspensionhadnonotableinfluenceonthefinalCAP.Threesolid productssynthesizedat80!Cseemedalsotohavesimilarthermal
behavior.FromTGanalysis,thecontentofcarbonateinsertedinthe apatiticstructurecouldbecalculated,supposingthatweightloss in thetemperature range of750–1250 was due tothethermal decarbonation. Thus, three solid products synthesized at 80!C
fromcalciteandorthophosphoricacidhadthecarbonatecontent intherangeof4.2–4.6wt.%.
3.5.CAPfromcalciteandorthophosphoricacid–thermaltreatment andbiomineralizationactivityinSBFsolution
For use as biomaterials, hydroxyapatite based products, includingCAP,mayneedstabilizationstepbythermaltreatment. InthecaseofCAP,thethermaltreatmentshouldnotleadtodeep decarbonation, in order tomaintain apatitic carbonate content similartothatofbonemineral[6].Inthissection,thermalstability ofCAPsynthesizedat80!C(T80/t48/D4.5)isdiscussed.Thissolid
T80/t48/D4.5 was chosen because of its lowest content of remainingCaCO3.ThereactivityofsolidproductsinSBFtest,as
the biomineralization activity, is also reported. This test was carriedoutwithT80/t48/D4.5thermallytreatedatthe tempera-turerangeof105–1400!Cunderairatmosphere.
ThermaltreatmentofT80/t48/D4.5powderwascarriedoutin the TG–DSC analyzer under air atmosphere. The powder was heatedat10!Cmin"1uptodesiredfinaltemperatureandwaskept
atthis temperaturefor5h.The finaltemperature variedin the range600–1400!C.After,theovenwasfreelycooleddowntoroom
temperature. 500 700 900 1100 1300 1500 1700 Tr an sm itt an ce (a . u .) Wavenumber (cm-1) 105oC 600 oC 800 oC 1000 oC 1545 1450 1415 1200 oC 1400 oC
IR spectrawere rapidlyrecorded onsinteredproducts, and are presented in Fig.6.Althoughthe decarbonationof CAPstarted around750!C(TGanalysis,Fig.5),thethermaltreatmentinthe
temperaturerangeof600–800!Cseemedtohavenoimpactonthe
carbonatecontentofT80/t48/D4.5solid.Thismaybeexplainedby alowkineticdecarbonationofT80/t48/D4.5at600–800!C.When
thesinteringtemperatureroseto1000!C,apartialdecarbonation
couldbeobserved,leadingtoreductionofIRintensityofapatitic carbonategroups.At1200–1400!C,absorptionbandsofcarbonate
groups disappeared, indicating that the decarbonation was complete. Thecalcinationat1200!CledtothecharacteristicIR
spectra ofCa-HA,while thecalcinationat 1400!C decomposed
Ca-HA intotetracalciumphosphate(TTCP)andtricalcium phos-phate(TCP)[18,26].Accordingtopreviousreports[18,19,26],the thermal treatmentunderairatmosphereisexpected toincrease the crystallinities of Ca-HA and CAP, and decrease the apatitic carbonatecontentofCAP,asconfirmedbyIRresults(Fig.6).Butat the very high temperature of 1200–1400!C, Ca-HA and CAP
decomposedintoTCPandTTCP.
Biomineralization activityof sinteredsolids wasinvestigated usingsimulatedbodyfluid(SBF)solution.SyntheticSBFsolution waspreparedaccordingtopreviouswork[27].Forthistest,asolid sinteredataspecifictemperaturewassubmergedintoSBFsolution andtheobtainedsuspensionwasthermostatedat37!Cfor7days.
SEM studywas thenperformedonthepowdersrecoveredafter risingwithwateranddryingatroomtemperature.
Ontheleft-hand-sideof Fig.7,thesinteringat600–1000!C
hadnosignificantinfluenceontheapparentmorphologyofCAP particles.Particlesofflat-needle-likemorphologydominatedCAP powders, driedat105!Corsintered at600–1000!C.Whenthe
temperature rose to 1200–1400!C (left-hand-side, Fig. 8),
sintering took place with the increase of particle size, true density and the decrease of porosityandspecific surfacearea [28,29].
ForCAPpowderstreatedat105–1000!C,despitethedecrease
ofcarbonatecontentwhenthecalcinationtemperatureincreased as showed by TGanalysis, CAP particles werecovered by new calcium phosphate layers after contact with SBF solution (Fig. 7, right-hand-side). Previous reports showed that Ca-HA basedmaterialshadappropriatebiomineralizationactivityforthe growthofcalciumphosphatelayersinSBFsolution[27,30,31].SBF solutionwasalsofoundasfavorablemediumfortheprecipitation ofcalcium andphosphate toformCa-HA [32,33].For oursolid synthesizedat80!C,thepresenceoftraceamountof remained
calcitemustnotinfluencethegrowthofCa-HAbecausecalcitehas beenshownasbiomineralanditsgrowthtookplaceinSBFsolution [14,33]. For the solids calcined at 1200–1400!C, although the
thermaldecarbonationwascomplete,thegrowthofnewcalcium phosphatelayersalsotookplace(Fig.8,right-hand-side).Infact, Ca-HA,andnon-apatiticcalciumphosphatesincludingTTCPand TCP, which resulted from thermal treatment of CAP at 1200–1400!C, are also known as active biomineralization
materialsinSBFsolution[26,31,34,35]. 4. Conclusions
The reaction of calcite and different orthophosphates was investigated.Alkaliphosphatesdidnotallowacomplete decom-positionofcalciteat80!Cinaclosebatchreactor.
Ontheotherhand,orthophosphoricacidledtothecomplete decompositionofcalcitepowder,andcarbonatedhydroxyapatite (CAP)wasformed.Thereactiontemperatureof80!Cwasfoundto
be crucial for the total decomposition of calcite and thus the formationofapatiticphase.Thereactiontimeandthedilutionof calcitesuspensionhadnosignificantinfluenceonthereaction.
CAPpowderstartingfromorthophosphoricacidandcalcitewas thermally stable at 600 and 800!C under air atmosphere. At
1000!C for 5h, partial decarbonation was observed. The
decarbonationwastotalat1200orabove.AllCAPanditsthermal decompositionproductswerefoundtobereactiveinSBFsolution, promotingcrystallizationofbiomimeticapatites.
Acknowledgment
Theauthorsgratefullyacknowledgecolleaguesat RAPSODEE Centerfortechnicalhelp.
References
[1]E.D.M.D.Pellegrino,R.M.B.S.Biltz,Medicine44(1965)397–418.
[2]K.J.Quelch,R.A.Melick,P.J.Bingham,S.M.Mercuri,Archs.Oral.Biol.28(1983)
665–674.
[3]J.P.Bilezikian,L.G.Raisz,T.J.Martin,PrinciplesofBoneBiology,3rded.,Elsevier,
2008.
[4]E.S.Thian,Z.Ahmad,J.Huang,M.J.Edirisinghe,S.N.Jayasinghe,D.C.Ireland,R.A.
Brooks,N.Rushton,W.Bonfield,S.M.Best,Biomaterials29(2008)1833–1843.
[5]D.PhamMinh,N.D.Tran,A.Nzihou,P.Sharrock,Mater.Sci.Eng.C33(2013)
2971–2980.
[6]E.Landia,G.Celottia,G.Logroscinob,A.Tampieria,J.Eur.Ceram.Soc.23(2003)
2931–2937.
[7]R.M. Wilson,J.C.Elliott,S.E.P.Dowker,R.I.Smith,Biomaterials25(2004)
2205–2213.
[8]S.Padilla,I.Izquierdo-Barba,M.Vallet-Regí,Chem.Mater.20(2008)5942–5944.
[9]H.E.Mason,A.Kozlowski,B.L.Phillips,Chem.Mater.20(2008)294–302.
[10]T. Tonegawa,T. Ikoma,Y.Suetsugu,N.Igawa,Y.Matsushita,T. Yoshioka,
N.Hanagata,J.Tanaka,Mater.Sci.Eng.B173(2010)171–175.
[11]W.L.Suchanek,P.Shuk,K.Byrappa,R.E.Riman,K.S.TenHuisen,V.F.Janas,
Biomaterials23(2002)699–710.
[12]D.PhamMinh,N.D.Tran,A.Nzihou,P.Sharrock,Ind.Eng.Chem.Res.52(2013)
1439–1447.
[13]D.PhamMinh,N.D.Tran,A.Nzihou,P.Sharrock,Mater.Res.Bull.51(2014)
236–243.
[14]M.Mozafari,F.Moztarzadeh,Challengesofspontaneousformationofcalcite
duringbioactivityassessmentonbioactivematerialsinterfaces,in:J.Dobrev,P.
Markovic (Eds.), Calcite: Formation, Properties and Applications, NOVA
SciencePubInc.,NewYork,2011,pp.95–122.
[15]D.PhamMinh,N.Lyczko,H.Sebei,A.Nzihou,P.Sharrock,Mater.Sci.Eng.B177
(2012)1080–1089.
[16]C.Verwilghen,M.Chkir,S.Rio,A.Nzihou,P.Sharrock,G.Depelsenaire,Mater.
Sci.Eng.C29(2009)771–773.
[17]D.PhamMinh,H.Sebei,A.Nzihou,P.Sharrock,Chem.Eng.J.198–199(2012)
180–190.
[18]J.C.Elliott,StudiesinInorganicChemistry18:StructureandChemistryofthe
ApatitesandOtherCalciumOrthophosphates,Elsevier,Amsterdam–London–
NewYork–Tokyo,1994.
[19]M.E.Fleet,X.Liu,Biomaterials28(2007)916–926.
[20]M.E.Fleet,Biomaterials30(2009)1473–1481.
[21]M.Jarcho,Clin.Orthop.Relat.Res.157(1981)259–278.
[22]A.I. Mitsionis, T.C. Vaimakis, C.C. Trapalis, Ceram. Int. 36 (2010)
623–634.
[23]H.Monma,Y.Nishimura,T.Okura,Phosph.Res.Bull.18(2005)127–134.
[24]A.Lebugle,E.Zahidi,G.Bonel,React.Solids2(1986)151–161.
[25]A.Bigi,G.Cojazzi,M.Gazzano,A.Ripamonti,N.Roveri,J.Inorg.Biochem.40
(1990)293–299.
[26]C.J.Liao,F.H.Lin,K.S.Chen,J.S.Sun,Biomaterials20(1999)1807–1813.
[27]S.Jalota,S.B.Bhaduri,A.C.Tas,Mater.Sci.Eng.C28(2008)129–140.
[28]S.Bailliez,A.Nzihou,Chem.Eng.J.98(2004)141–152.
[29]D.PhamMinh,M.GaleraMartinez,A.Nzihou,P.Sharrock,J.Therm.Anal.
Calorim.112(2013)1145–1155.
[30]T.Kokubo,S.Ito,M.Shigematsu,S.Sakka,T.Yamamuro,J.Mater.Sci.22(1987)
4067–4070.
[31]T.Kokubo,H.Takadama,Biomaterials27(2006)2907–2915.
[32]A.C.Tas,Biomaterials21(2000)1429–1438.
[33]M.Mozafari, F.Moztarzadeh, M.Tahriri, J. Non-Cryst.Solids 356 (2010)
1470–1478.
[34]K.Kurashina,H.Kurita,M.Hirano,J.M.A.deBlieck,C.P.A.T.Klein,K.deGroot,
J.Mater.Sci.:Mater.Med.6(1995)340–347.
[35]L.A.DosSantos,L.C.DeOliveira,E.C.S.Rigo,R.G.Carrodeguas,A.O.Boschi,