© Gratien Adoumandjali, 2020
Variabilité génétique, moléculaire et quantitative du
Tilapia du Nil (Oreochromis niloticus, Linnaeus, 1758)
dans le bassin du Congo
Thèse
Gratien Adoumandjali
Doctorat en biologie
Philosophiæ doctor (Ph. D.)
Variabilité génétique, moléculaire et quantitative du
Tilapia du Nil (Oreochromis niloticus, Linnaeus, 1758)
dans le bassin du Congo
Thèse de doctorat en biologie
Gratien Adoumandjali
Sous la direction de :
Louis Bernatchez, directeur de recherche
Damase Khasa, codirecteur de recherche
ii
Résumé
Dans ce travail de thèse de doctorat, deux objectifs ont été poursuivis à savoir : i) évaluer la structure génétique des populations du tilapia du Nil (Oreochromis niloticus, Linnaeus, 1758) pour orienter les mesures de gestion à l’échelle de la sous-région du bassin du Congo; ii) et évaluer l’héritabilité des traits de croissance afin d’envisager une amélioration génétique de ladite espèce.
Dans la perspective de déterminer la structuration génétique du tilapia du Nil dans le bassin du Congo à l'aide de récents outils génomiques de type GBS, 13,792 SNPs neutres étaient identifiés à partir de 438 tilapias du Nil collectés au Cameroun, en République Centrafricaine et en République Démocratique de Congo. La présence de groupements génétiques distincts a été élucidée par la méthode de regroupement bayésien implémentée dans le programme Admixture exécuté en utilisant 2000 bootstraps avec un nombre de groupes (K) variant de 1 à 14. L’estimation du flux génique potentiel, entre les sites, a été réalisée à l’aide du logiciel Treemix et l’évaluation de degré d’apparentement par paire entre deux individus d’un même site ou écosystème (utilisant les indices de relation de parenté ou relatedness, AJK), par vcftools.
L’estimation de l’héritabilité des traits de croissance du tilapia du Nil a été réalisée en utilisant les données de 660 poissons cultivés dans les hapas placés en étang entre 185 et 209 jours d’expérience. Les mesures de masse corporelle et de longueur standard ont été prises in situ tandis que le facteur de condition a été déduit en utilisant la formule de Fulton. La détermination de données liées à la morphologie a été obtenue par analyse Procruste généralisée. Une analyse utilisant le modèle animal mixte a été appliquée à ces traits morphométriques pour estimer les différentes composantes de variance génétique et environnementale à partir desquelles l'héritabilité de chaque caractère, les effets des facteurs environnementaux et les corrélations génétiques entre les traits ont été déduits.
Les résultats de l’analyse de structure sur les 13,792 marqueurs neutres ont révélé l’existence d'une structuration génétique mise en évidence avec l’identification de 5 populations génétiquement différentes (moyenne Fst = 0.079; CI: 0.073 à 0.086, P-valeur = 0.001). Il ressort de cette étude qu’il y a eu flux de gènes entre certains sites et qu’il y a moins d’individus apparentés dans les rivières et lacs par rapport aux piscicultures.
On a démontré que l’héritabilité au sens strict (h2) de la masse corporelle était élevée (0.67±0.30), modérée pour la morphologie (0.30 ± 0.12), faible pour la longueur standard (0.10 ± 0.10) et le facteur
iii
de condition (0.03 ± 0.04). L’héritabilité au sens large (H2) a varié de 0.22 ± 0.13 à 0.93 ± 0.10 pour tous les traits. La corrélation génétique entre la masse corporelle et la morphologie (0.91) était élevée mais moyenne entre la longueur standard et la morphologie (0.21) ainsi qu’entre la masse corporelle et la longueur standard (0.20).
La corrélation phénotypique entre la masse corporelle et la longueur standard (0.95) était élevée tandis qu’elle était moyenne entre la longueur standard et la morphologie (0.21) ainsi qu’entre la masse corporelle et la morphologie (0.27).
Au vu de ces résultats, on peut conclure qu’il y a existence d'une structuration génétique pouvant permettre une gestion et utilisation durables de l’espèce tilapia du Nil dans le cadre des activités piscicoles et d’ensemencement. L’existence d’une bonne diversité́ génétique associée à l’héritabilité des caractères de croissance augure de bonnes perspectives pour des programmes d’amélioration génétique de l’espèce dans la sous-région du bassin du Congo.
iv
Abstract
In this PhD thesis work, two objectives were pursued: i) to assess the genetic structure of Nile tilapia populations (Oreochromis niloticus, Linnaeus, 1758) in order to guide management strategies at the sub-regional scale; region; ii) and assess the heritability of the growth traits in order to consider a genetic improvement of the species.
In order to determine the genetic structuring of Nile tilapia in the Congo Basin using recent GBS-type genomic tools, 13,792 neutral SNPs were identified from 438 Nile tilapias collected in Cameroon, Central African Republic and Democratic Republic of Congo. The presence of distinct genetic groupings was elucidated by the Bayesian clustering method implemented in the Admixture program run using 2000 bootstraps with a number of groups (K) ranging from 1 to 14. The estimation of potential gene flow between sites, was performed using Treemix software and peer paired degree evaluation between two individuals from the same site or ecosystem (using kinship or relatedness indices, AJK), by vcftools.
The assesment of the heritability of Nile tilapia growth traits was performed using data from 660 fish grown in hapas placed in a pond between 185 and 209 days of experience. The body mass and standard-length measurements were taken in situ while the condition factor was deduced using the Fulton formula. Determining morphology-related data was obtained by generalized Procrustes analysis. An analysis using the mixed animal model was applied to these morphometric traits to estimate the different components of genetic and environmental variances from which the heritability of each trait, the effects of environmental factors and the genetic correlations between traits were derived.
The results of the structural analysis on the 13,792 neutral markers revealed the existence of a genetic structure highlighted with the identification of 5 genetically different populations (average Fst = 0.079, CI: 0.073 to 0.086, P-value = 0.001). This study shows that there has been gene flow between some sites and that there are fewer inbred individuals in the rivers and lakes while fish farmers have been more consanguineous.
Strict heritability (h2) of body mass was shown to be high (0.67 ± 0.30), moderate for morphology (0.30 ± 0.12), low for standard length (0.10 ± 0.10), and condition factor (0.03 ± 0.04). Heritability in the
v
broad sense (H2) ranged from 0.22 ± 0.13 to 0.93 ± 0.10 for all traits. The genetic correlation between body mass and morphology (0.91) was high but average between standard length and morphology (0.21) and between body weight and standard length (0.20).
The phenotypic correlation between body mass and standard length (0.95) was high while it was average between standard length and morphology (0.21) as well as between body mass and morphology (0.27).
In view of these results, it can be concluded that there is a genetic structure that can allow sustainable management and use of the Nile tilapia species in the context of fish farming and fish seed production. The existence of a good genetic diversity associated with the heritability of growth traits is promising for the development of genetic improvement programs of the of Nile tilapia species in the Congo Basin sub region.
vi
Table des matières
Résumé ... ii
Abstract ... iv
Table des matières ... vi
Liste des figures ... xi
Liste des tableaux ... xiv
Liste des abréviations, sigles, acronymes ... xv
Remerciements ... xvii
Introduction ... 1
Bref historique de la structure génétique de populations ... 1
Structure génétique de populations au service de la conservation
... 3
Mécanismes évolutifs et biologie évolutive ... 5
Nouvelles technologies de séquençage et implications ... 6
Variabilité intrapopulation : héritabilité et corrélations génétiques 7
Définition et historique de l'héritabilité ... 8Héritabilité et partitionnement de la variance totale : Paramètres de population ... 9
Applications ... 14
Héritabilité et génétique évolutive ... 15
Variabilité de l'héritabilité ... 17
Héritabilité à l'ère de la génomique ... 18
Héritabilité avec pedigrees inconnus. ... 19
Exploiter la variation de la parenté. ... 19
Héritabilité de l'expression des gènes. ... 20
Tilapia du Nil (Oreochromis niloticus, Linnaeus, 1758) ... 21
vii
Production mondiale de tilapias et pays producteurs ... 22
Systématique ... 25
Habitat et distribution géographique ... 26
Régime alimentaire et nutritionnel ... 28
Biologie de la reproduction ... 28
Comportement reproductif ... 28
Maturité sexuelle et longévité ... 30
Fécondité ... 31
Croissance ... 31
Caractéristiques morphologiques de l'espèce ... 32
Exigences écologiques ... 34
Risques pathologiques ... 35
Problématique ... 38
Objectifs ... 40
Chapitre 1 Matériels et méthodes ... 41
Structure génétique de populations de tilapia du Nil dans l’espace
du bassin hydrographique du Congo en vue d’une gestion éclairée
... 41
Échantillonnage ... 41
Techniques moléculaires ... 43
Bio-informatique et génotypage ... 44
Diversité et différenciation génétique ... 45
Détection des SNP sous sélection ... 46
Regroupement individuel et de population ... 46
Évaluation de degré de consanguinité dans les différents sites et écosystèmes (rivières, lacs et piscicultures) ... 47
Variabilité génétique : héritabilité des traits phénotypiques de
croissance chez le tilapia du Nil (Oreochromis niloticus, Linnaeus,
1758) dans le bassin du Congo ... 48
Zone d’étude ... 48
Dispositif expérimental ... 48
Protocole d’étude ... 49
Acquisition des données ... 50
viii
Facteur de condition K ... 51
Morphologie ... 51
Analyses statistiques ... 54
Modèle statistique ... 54
Estimation des valeurs d’héritabilités, d’effets environnementaux et corrélations ... 57
Chapitre 2 Résultats ... 58
Structure génétique des populations ... 58
Résultats de génotypage ... 58
Diversité génétique ... 59
Différenciation de la population ... 59
Regroupement de populations ... 62
Identification de possibles flux de gènes entre les différentes populations ... 63
Évaluation de degré de consanguinité dans les différents sites et écosystèmes (rivières, lacs et piscicultures) ... 65
Variabilité génétique : héritabilité des traits phénotypiques de
croissance chez le tilapia du Nil (Oreochromis niloticus, Linnaeus,
1758) dans le bassin du Congo ... 66
Variation de forme du corps ... 66
Statistiques descriptives ... 69
Héritabilité et effets des facteurs environnementaux ... 71
Corrélations génétiques et phénotypiques ... 72
Chapitre 3 Discussion ... 74
Structure génétique des populations ... 74
Diversité génétique intra population de tilapia du Nil ... 74
Différenciation génétique et regroupement de population de tilapia du Nil ... 76
Estimation de la relation de parenté par paires à l'aide de données génomiques de population ... 78
Variabilité génétique : héritabilité des traits phénotypiques de
croissance chez le tilapia du Nil (Oreochromis niloticus, Linnaeus,
1758) dans le bassin du Congo ... 79
Héritabilité ... 79
ix
Corrélations génétiques et phénotypiques. ... 83
Limites de l’étude ... 85
Implications de l’étude ... 85
Conclusion ... 86
Structure génétique des populations ... 86
Analyses génomiques ... 86
Implications pour la gestion du tilapia du Nil ... 87
Variabilité génétique : héritabilité des traits phénotypiques de
croissance chez le tilapia du Nil (O. niloticus, Linnaeus, 1758) dans
le bassin du Congo ... 88
Forces et limites de l’étude ... 89
Bibliographie ... 90
Annexes ... 126
Annexe 1 : Les estimations de la littérature sur l'héritabilité et les
effets environnementaux sur la masse corporelle et longueur
standard à différents âges chez le tilapia du Nil ; SD : écart-type,
H : héritabilité, C : effets environnementaux ; N : nombre individus,
FS : pleins-frères, HS : demi-frères, S : mâles, D : femelles, REML
: Méthode de vraisemblance maximale restreinte et MCMC :
Méthodes de Monte-Carlo par chaînes de Markov. ... 126
Annexe 2 : Protocole d’extraction ADN par la méthode aux sels
... 128
Annexe 3 : Protocole PicoGreen ... 129
Annexe 4 : Script bash pour préparer les données d’entrée
TreeMix ... 132
Annexe 5 : Recherche des individus très apparentés avec vcftools
... 133
Annexe 6 : Différentes étapes du génotypage par séquençage
... 133
Annexe 7 : Tableau de valeurs de FST pour toutes les paires de
populations des 348 individus et 18099 loci ; Koupa-Matapite
(KOU); Lac Magba (MAG); Lac Bamendjin (BAM); Lac Lagdo
(LAG); Lac Boali (BOA); Lac Boukoko (BOU); Rivière Oubangui
(OUB); Wango (WAN); Rivière Bandoro (BAN); Fleuve Congo
x
(CON); Ferme Amani (FAM); Ferme Bumaki (FBU); Ferme Futuka
(FFU). Les FST moyens ont été de 0.043 au Cameroun, 0.028 en
République Centrafrique et 0.082 en République Démocratique du
Congo. Toutes les p-valeur sont hautement significatives (P =
0.001 = ***) ... 135
xi
Liste des figures
Figure 1: Production aquacole mondiale de tilapia (Fao, 2012; Fitzsimmons, 2017,
2015) ... 23
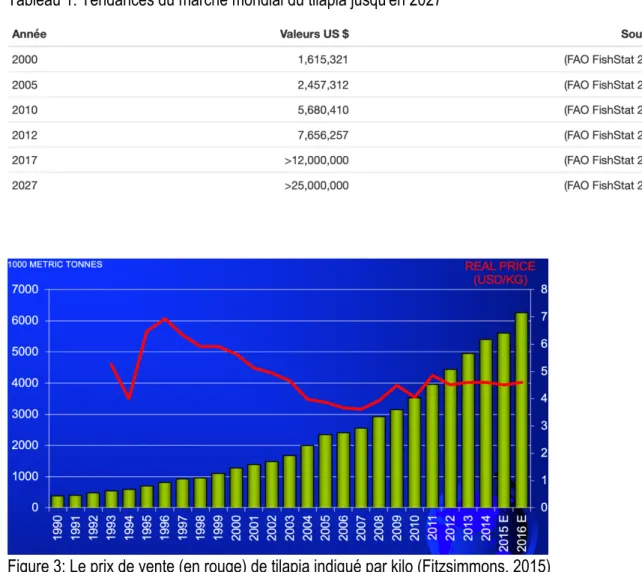
Figure 2: Principaux pays de production de tilapia (6,265,800 mt) en 2016 ... 23 Figure 3: Le prix de vente (en rouge) de tilapia indiqué par kilo (Fitzsimmons, 2015)
... 24
Figure 4: Aire de répartition naturelle du tilapia du Nil O. niloticus (en gris), avec les
localisations d’échantillonnage de 6 populations sauvages étudiés pour tester leur détermination sexuelle : les lacs Manzala (Egypte), Rudolph (Kenya), Volta (Ghana), Koka & Metahara (Éthiopie). Les symboles correspondent aux références suivantes : ★Altena et Hörstgen-Schwark, 2002; ● Tessema et al., 2006; ■ Bezault et al., 2007. ... 27
Figure 5: La photographie de Oreochromis niloticus mâle prise lors de
l’échantillonnage ... 33
Figure 6: Carte montrant les lieux d'échantillonnage du tilapia du Nile : A
(Cameroun), B (Centrafrique), C (République Démocratique du Congo). ... 42
Figure 7: Photographie montrant les prélèvements des échantillons du Lac Lagdo
... 42
Figure 8: a) Le dispositif expérimental en U présentant les 30 hapas dans un étang
de 800 m2 de l’amont vers l’aval et un couple de géniteurs (gauche); b) un couple
de géniteur (droite) ... 50
Figure 9: a) les œufs (gauche) ; b) les larves (droite) ... 50 Figure 10: a) Photographie des poissons; b) berceau de maille sur lequel est
poisson est étalé. ... 52
Figure 11: Dix points de repères pour décrire la variation morphométrique. 1)
museau, 2) premier rayon dur nageoire dorsale, 3) dernier rayon mou nageoire dorsale, 4) limite postérieur supérieur du pédoncule, 5) dernière insertion de la ligne latérale, 6) limite postérieur inférieur du pédoncule, 7) rayon postérieur de la nageoire anale, 8) rayon antérieur de la nageoire anale, 9) rayon postérieur de la nageoire pelvienne, 10) point antérieur du maxillaire. ... 53
Figure 12: Dendrogramme de population de Fst et heatmap basés sur les valeurs
de Fst parmi 13 lieux d'échantillonnage de tilapia du Nil. Le code de couleur de heatmap illustre la matrice Fst en considérant quatre groupes Fst différents délimités de la distribution par paire de Fst: faible Fst (inférieur au 5ème centile, Fst <0,0377), Fst moyen faible (du 5ème au 25ème centile, 0,0377 ≤ Fst <0,0616), intermédiaire (25ème au 75ème percentile, 0,0616 ≤ Fst < 0.113), Fst élevé (supérieur au 75ème centile, FST ≥ 0,113). ... 61
Figure 13: Ce graphique montre une nette diminution de la valeur de l’erreur de la
validation croisée jusqu’à k = 5 groupes, après quoi elle augmente. Dans ce cas, le coude de la courbe correspond également à la plus petite erreur de la validation croisée et indique clairement que 5 groupes peuvent être conservés ... 62
Figure 14: Structure de la population ; L’assignation individuelle bayésienne utilisant
xii
structure génétique de la population. Le nombre de groupes génétiques (K = 5), a été choisi. Les différentes couleurs représentent les différents groupes génétiques. Pisciculture-Koupa-Matapite (KOU); Lac Magba (MAG); Lac Bamendjin (BAM); Lac Lagdo (LAG); Lac Boali (BOA); Lac Boukoko (BOU); Rivière Oubangui (OUB); Pisciculture Wango (WAN); Rivière Bandoro (BAN); Fleuve Congo (CON); Ferme Amani (FAM); Ferme Bumaki (FBU); Ferme Futuka (FFU); La carte représente les trois pays d’échantillonnage, A (Cameroun), B (Centrafrique), C (République Démocratique du Congo) et les couleurs les populations distinctes. ... 63
Figure 15 : Variation génétique ancestrale partagée entre les populations de tilapia
de Nil et les mélanges entre paires des sites échantillonnés. La longueur des branches est proportionnelle à la quantité de dérive génétique dans chaque branche, et la barre d'échelle indique 10 fois l'erreur type (s.e) moyenne des entrées de la matrice de covariance entre des paires de populations. L'échelle de couleur indique le poids des événements de migration déduite, le rouge indiquant une forte probabilité de migration ou de lien ancestral. KOU (Pisciculture-Koupa-Matapite); MAG (Lac Magba); BAM (Lac Bamendjin); LAG (Lac Lagdo); BOA (Lac Boali); BOU (Lac Boukoko); OUB (Rivière Oubangui); WAN (Pisciculture Wango); BAN (Rivière Bandoro); CON (Fleuve Congo); Ferme Amani (FAM); Ferme Bumaki (FBU); Ferme Futuka (FFU). La carte représente les trois pays d’échantillonnage, A (Cameroun), B (Centrafrique), C (République Démocratique du Congo) et les couleurs les populations distinctes. ... 64
Figure 16: Relation de parenté consanguine entre les individus de différents sites
échantillonnés (Lac, Pisciculture et Rivière). La couleur rouge indique les lacs, verte, les piscicultures et bleu les rivières. ... 66
Figure 17: Superposition des moindres carrés des 420 spécimens, montrant les
dispersions observées (variation totale) autour de chaque point de repère. ... 67
Figure 18: Graphique de l’analyse bâton brisé présentant les pcs représentant de
variance de conformation : ligne rouge : conformation consensus ; noir, conformation de chaque individu produit par R. Les 4 PCs représentent 70.8 % de la variance de la conformation: PC1 (29.8 %), PC2 (20 %), PC3 (10.9 %) et PC4 (10.1%). ... 68
Figure 19: Graphique de dispersion de scores selon les quatre premiers
composants principaux d'un PCA basé sur tous les individus utilisés dans la présente étude. Chaque individu est représenté dans le graphique à deux axes, par un point unique. Les points ronds représentent les progénitures. Les quelques points couleurs pour les progénitures représentent ceux dont les scores de variation de forme ont le plus contribué dans les PC par rapport à ceux en petit gris. Chaque couleur de triangle correspond à un couple de famille. Le nuage, constitué de points et triangles obtenus, correspond à l’ensemble des configurations des individus étudiés. Deux formes diffèrent entre elles lorsque les deux points les représentant sont éloignés dans l’espace. A l’inverse, une distance nulle indique que les deux formes sont identiques. ... 68
Figure 20: Changement de forme produit par le logiciel MorphoJ. La ligne grise
pointillée représente la configuration moyenne de la forme du corps parmi les 420 individus utilisés comme configuration de référence ou tangente, la ligne pleine et noire représente la déformation par rapport à la forme moyenne vers la direction positive ou négative sur les composantes principales (PC). ... 69
xiii
Figure 21: représentation des poids du corps (a), longueur standard(b), coefficient
de condition(c) et morphologie(d) en fonction du sexe. ... 70
Figure 22 : Corrélations génétiques à gauche et phénotypiques à droite entre les
xiv
Liste des tableaux
Tableau 1: Tendances du marché mondial du tilapia jusqu'en 2027 ... 24 Tableau 2: Les principales maladies affectant le tilapia du Nil sont citées dans le tableau ci-dessous
(FAO, 2005) ... 36
Tableau 3: Sites d’échantillons: latitude et longitude, date d'échantillonnage et nombre d'individus
échantillonnés ... 43
Tableau 4: Description des facteurs qui ont été considérés susceptibles d’influencer les phénotypes
des progénitures dans l’expérience. ... 56
Tableau 5: Modèle mixte animal : dam représente les femelles ; kdam, le facteur de condition des dam
; âges, la durée de l’expérience (185-209 jours); pha, la position des hapa dans l’étang en fonction de l’entrée d’eau ; densité, le nombre d’individus par hapa à la fin de l’expérience ; bwdam, poids du corps des dam ; sldam, longueur standard des dam. ... 56
Tableau 6: Nombre de SNP supprimés après chaque étape de filtrage. ... 58 Tableau 7: Indices de diversité génétique par population: Hétérozygotie observée (Ho); Hétérozygotie
attendue (He); Coefficient de consanguinité (GIS). ... 60
Tableau 8: Analyse de la variance moléculaire (AMOVA) sur 381 individus et 18109 SNP ... 61 Tableau 9 : Nombre d'enregistrements (N), moyenne globale par sexe, écart-type (SD) pour les
mesures du corps chez le tilapia du Nil (O. niloticus) ... 70
Tableau 10: Modèle mixte ou animal : dam représente les femelles ; kdam, le facteur de condition des
dam ; âges, la durée de l’expérience (185-209 jours); pha, la position des hapa dans l’étang en fonction de l’entrée d’eau ; densité, le nombre d’individus à la fin de l’expérience ; bwdam, poids du corps des dam ; sldam, longueur standard des dam; les critères d’information de déviance (DIC). ... 71
Tableau 11: Estimations de l'héritabilité au sens large (H2 = s2G /s2p), l'héritabilité au sens strict (h2 = s2A /s2p) et des effets environnementaux (E2 = s2E/s2p) de la distribution postérieure avec les intervalles de confiance (CI) à 95% pour le poids, la longueur standard, le facteur de condition et la morphologie du tilapia du Nil. s2p désigne la somme de toutes les composantes de variance (s2G + s2E + s2r); dam, densité, âges, pha sont des covariables, dans le modèle, pour expliquer la variance des traits des progénitures ; n désigne le nombre d’individus. ... 72
xv
Liste des abréviations, sigles, acronymes
ADN: Acide désoxyribonucléique
AMOVA : Analyse de la variance moléculaire (Analysis of molecular variance) BAM: Bamendjin
BAN: Bandoro BOA: Boali BOU: Boukoko CON: Congo FAM: Ferme Amani
FAO: Food and Agriculture Organization FBU : Ferme Bumaki
FFU : Ferme Futuka
FSG : Faculté des Sciences et Génie
FST: Indice de différentiation génétique (Index of genetic differentiation)
GBS : Génotypage par séquençage, ou (de l’anglais « genotyping by sequencing ») IBIS : Institut de Biologie Intégrative et des Systèmes
IBD : Identité par Descendant
IRAD : Institut de Recherche Agricole pour le Développement KOU : Koupa-Matapite
LAG: Lagdo MAG: Magba
NGS : Séquençage de Nouvelle Génération OUB : Oubangui
PCR = Polymerase Chain Reaction. Réaction en chaîne de polymérisation
PFOGRN-BC : Programme Élargi de Formation en Gestion des Ressources Naturelles dans Le Bassin
du Congo
BAD : Banque africaine de développement
P-value : Valeur de probabilité associée à un test statistique (probability value associated to a statistical
test)
RCA: République Centrafricaine
RDC: République Démocratique du Congo
RIFFEAC : Réseau des Institutions de formation forestière et environnementale d’Afrique centrale SNP : Polymorphisme à un seul nucléotide (Single Nucleotide Polymorphism)
UL: Université Laval VCF: Variant Call Format WAN: Wango
xvi
Je dédie ce travail de thèse de doctorat à mon défunt papa Albert Akapa et à ma maman Madeleine Ambélo
xvii
Remerciements
Plusieurs personnes ont contribué à l’élaboration de cet ouvrage sans pour autant y figurer. Grâce à elles aujourd’hui, je réalise un rêve incroyable et j’espère pouvoir un jour les aider autant qu’elles ne l’ont fait pour moi. Je formule l’espoir de ne manquer personne et surtout avoir les mots justes pour exprimer ma profonde gratitude et l’estime que j’aurai toujours envers tous.
J’ai le plaisir de me rappeler de nos premiers échanges que je présente dans un petit dialogue utilisant nos prénoms:
§ Gratien : Bonjour Professeur,
Je suis bénéficiaire de la bourse doctorale du Programme Élargi de Formation en Gestion des Ressources Naturelles dans le Bassin du Congo (PEFOGRN-BC) pour réaliser une thèse de doctorat à l’Université Laval. J’ai discuté avec le Professeur André Desrochers lors de la formation organisée à Yaoundé dans le cadre de la sélection des candidats à cette bourse. Étant donné que mon thème est lié à votre spécialité, il m'a été conseillé de vous contacter pour la direction du travail. J'ai donc parcouru certaines de vos publications qui m’ont beaucoup impressionné et je voudrais savoir si vous accepteriez d’encadrer ma thèse de doctorat. Le thème que j'ai retenu, pour le moment, est: "Caractérisation génétique des populations de tilapia du Nil, Oreochromis niloticus (Linnaeus, 1758) en République Centrafricaine". Cela me ferait plaisir si vous vouliez bien réorienter le sujet pour lui donner le sens qui convienne. Je sollicite votre accord écrit pour me permettre de commencer la procédure d’inscription à l’Université Laval. Je vais vous faire parvenir dans un bref délai, le DRAFT de la proposition de recherche pour orientations.
Veuillez agréer, Monsieur le Professeur, mes considérations les plus distinguées.
§ Louis : Bonjour Gratien,
Merci beaucoup pour votre message.
Malheureusement, mon laboratoire est à pleine capacité avec 30 personnes et il ne m'est présentement impossible de recruter de nouveaux étudiants dans un avenir rapproché.
Bonne chance pour la suite des choses.
§ Gratien : Bonjour Professeur,
xviii
Si vous connaissez d'autres Professeurs que je pourrais contacter, je serai heureux de recevoir leurs contacts.
§ Louis : Bonjour Gratien,
Le professeur Grant Vanderberg pourrait être intéressé car je crois qu’il a travaillé sur les tilapias en aquaculture.
§ Louis : Bonjour Gratien,
J'ai changé d'idée et accepterai en principe de te diriger.
Mais pour donner mon approbation finale, je vais discuter avec mon ami Damase des détails entourant le contexte qui ne m'a pas du tout été expliqué par le Dr Desrochers avant qu'il te recommande à moi, ce qui est un peu dommage et non orthodoxe comme procédure.
Bien à toi.
§ Gratien : Bonjour Professeur,
Je suis très heureux de savoir que vous accepteriez de diriger mon travail. Pour ma part, je ferai tout ce qui est possible pour mériter votre confiance.
Mes respects les plus distingués.
§ Louis : Bonjour Gratien.
Cependant, je devrai auparavant avoir plus d'informations sur le contexte précis, ton financement etc. Je contacterai Damase à cet effet. Tu commencerais quand, à l'automne 2012 ou en 2013?
Cordialement,
Et bien mon cher Louis, tu as été un modèle pour moi et je n’ai pas d’autres mots qui soient plus forts que merci pour être généreux comme toi. Cette thèse résume l’essentiel de mon cheminement et des travaux de recherches que j’ai réalisés depuis 2013 aussi bien dans ton laboratoire à l’Institut de Biologie Intégrative et des Systèmes (IBIS) de l’Université Laval et qu’au Cameroun, en République Centrafricaine et en République Démocratique du Congo. Grace au solide soutien que tu m’as toujours accordé, j’ai réussi à rassembler ce document constitué d’une introduction faisant la synthèse des principaux arguments théoriques et des travaux antérieurs, d’une méthodologie montrant la démarche ayant permis d’acquérir les données ainsi que leurs analyses. Je voudrais aussi en profiter pour te remercier pour tes soutiens financiers qui m’ont permis de joindre les deux bouts.
xix
Cette thèse a reçu un soutien financier du Programme élargi de formation en gestion des ressources naturelles dans le Bassin du Congo (PFOGRN-BC). Je te remercie mon co-directeur, Damase. Une thèse sans financement n’est pas possible. Tu t’es impliqué dans le Programme Élargi de Formation en Gestion des Ressources Naturelles dans le Bassin du Congo (PEFOGRN-BC) financé par le fonds pour les forêts du bassin du Congo géré par la Banque africaine de développement (BAD) et grâce à ta contribution dans ce programme, j’ai bénéficié du financement qui m’a permis de réussir ma formation à l’Université Laval.
Je profite de cette exceptionnelle occasion pour remercier les membres de mon comité de thèse : Julie Turgeon et Grant Vanderberg. Merci à Julie pour m’avoir suivi depuis le début de mon doctorat jusqu’à la fin. Tu es une scientifique modèle et j’espère un jour avoir une carrière aussi épanouie que la tienne. Tu as été pour moi comme un co-directeur avec tes réflexions, commentaires, critiques, conseils, encouragement et disponibilité qui m’ont aidé tout au long de mon parcours. Tu es une dame magnifique. Merci pour tous ces moments ensembles.
C’est plus tard, que je me suis rendu compte que j’avais déjà pris contact avec Grant pour une possible thèse quand j’étais encore en Belgique. Même si je n’avais pas reçu de réponse, la nature nous a encore réuni autrement. Merci pour ton soutien et tes conseils Grant.
Je remercie Caroline Côté pour son soutien qui s’est ajouté à celui de son mari Louis. Tu as été d’une compréhension exceptionnelle. Merci pour la botte d’hiver que tu m’avais offert et pour le petit job qui m’avais dépanné.
À mon arrivée au laboratoire Bernatchez, j’ai trouvé des personnes exceptionnelles qui m’ont tout de suite mis en confiance. Jean-Sébastien Moore, Anne-Marie Dion-Cté, Scott Pavey, Anne Danziel, Thierry Gosselin, Fabien Lamaze, Charles Perrier, Anaïs Lacoursière-Roussel.
Mes remerciements particuliers vont à l’endroit de Jean-Sébastien et Anne-Marie qui ont été formidables dans le laboratoire Bernatchez. Vous avez été à mes côtés quand mes enfants étaient en danger dans mon pays. Vous avez organisé des collectes de fonds avec la croix rouge pour venir en aide aux Centrafricains qui en avaient besoin. Votre geste est resté vivace dans mes pensées. Un remerciement particulier à Eric Normandeau qui a toujours été disponible pour apporter l’aide nécessaire, à Martin Laporte, Clément Rougeux, Bérénice Bougas, Alysse Pérault-Payette, Vincent
xx
Bourret, Guillaume Côté, Cécilia Hernandez, Anne Carrier, Maëlle Sevellec, Ben Sutherland, Simon, Charles, Damien, Gaétan, Lucie Papillon et Jérôme pour les soutiens multiples.
Je remercie toutes les personnes qui m’ont aidé et soutenu au Cameroun avec toutes les tracasseries des forces de l’ordre dans ce pays. Mes remerciements vont à l’endroit de Victor et Charlotte Pouomogne qui m’ont accueilli dans leur maison comme un membre de la famille. Cela m’a permis de prendre des repères. Merci à leurs enfants Marie, Yolande, Esther, Laurence, Ruth, Daniel et Joseph Pouomogne. Une pensée particulière à Marie qui s’est impliquée dans la collecte de mes données à des endroits difficiles d’accès. Quand mes papiers ont été retenus par les forces de l’ordre camerounaise, elle a décidé de m’accompagner pendant toute la durée de mon séjour en informant régulièrement ses parents qui étaient déjà inquiets de ma situation. Il était arrivé que je sois à court d’argent car aucune banque pour faire le retrait. Elle appelait régulièrement sa mère pour me dépanner. Mes remerciements aux personnels de l'IRAD dont la collaboration a été un apport significatif. Il s’agit de Rostand, Salifou, Arouna, Aurélien, Mariama, Jean, Abiba et Ousmanou. Si j’ai réussi à collecter quelques-uns de mes échantillons qui étaient régulièrement vandalisés, c’est grâce à Ousmanou. Remarquant que le surveillant du dispositif expérimental ne faisait pas bien son travail, Ousmanou a pris l’engagement de le faire comme il le pouvait en me proposant des orientations.
En République Centrafricaine, mes remerciements vont à l’endroit du couple Eddy Fourier et Ida Séverine Gbondele. Ida dont l’aide a été précieuse dans les contacts avec certains exploitants piscicoles, a été remarquable. Merci Eddy car je me souviens encore au moment où le gros véhicule se dirigeait droit sur nous qui étions à moto et tu as fait de ton mieux pour qu’on s’enfonce dans la végétation ne sachant pas sur quel terrain on s’aventurait. Tu aurais pu laisser ta vie rien que pour m’aider à avoir les nageoires de poissons pour ma thèse. Trouve ici ma pleine gratitude.
Merci à mes parents, Albert Akapa et Madeleine Ambélo. À mes grandes sœurs Honorine Nzangué, Cathérine Natoua, Marie Claire Gbélébele et Brigitte Kokoti qui ont toujours cru en moi et m’ont soutenu dans mes études. Je ne t’oublierais jamais ma défunte cadette Médibobongui Vianeige. Tu as toujours ta place dans mon cœur. Les meilleures motivations de ma vie Victor Alex Adoumandjali, Victoire Josiana Medibobongui Adoumandjali, Wenceslas Alpha Adoumandjali et Roy Christopherson Akapa Adoumandjali, prenez la relève. Je vous aime.
1
Introduction
L’étude de la structure génétique de populations et de l’héritabilité de caractères de croissance est d’une grande importance pour une gestion durable, responsable et une amélioration génétique pour supporter une production soutenue d’une espèce d’importance économique telle que le tilapia du Nil (Oreochromis niloticus). Cette introduction se propose de faire un bref état des lieux, entre autres, des problématiques liées à l’impact de l’activité humaine sur les populations de tilapia du Nil. Elle se structure selon les différents angles abordés et approches utilisées dans ce travail dans le contexte général de la biologie et de l’évolution des populations de tilapia du Nil.
Bref historique de la structure génétique de populations
Il y a un siècle, deux articles fondamentaux ont inauguré le domaine de la génétique des populations (Casillas et Barbadilla 2017). Les premières mesures de la variation génétique chez l’espèce
Drosophila pseudoobscura (Lewontin et Hubby 1966) et chez l’homme (Harris 1966) ont été fournies,
par application de la technique d’électrophorèse sur gel de protéines pour détecter les loci d’allozymes. À cette époque, la génétique des populations tablait sur une base théorique étendue intégrant les principes de l'héritage mendélien avec les forces affectant les changements de fréquences d'allèles dans les populations, cherchant à formaliser la vision darwinienne selon laquelle l'évolution biologique est un processus de population par lequel la variation génétique au sein d'une espèce se transforme en variation génétique entre espèces (Mayr 1963). Cependant, en raison de l’incapacité technique de mesurer la variation génétique pour presque tous les loci, cet exercice formel exhaustif s’est déroulé dans un vide factuel virtuel. Avec peu de données, les modèles demeuraient généraux, non restreints par le monde contingent (Lewontin 1974). Après des décennies de difficultés à mesurer la variation génétique, de nombreuses données sur la variation électrophorétique ont enfin ouvert le dialogue nécessaire entre données et théorie. Depuis lors, ce dialogue a continué de catalyser les principales avancées en génétique des populations.
De nos jours, la révolution génomique a généré des données génétiques détaillées sur les populations, dépassant de loin les rêves de tout généticien pré-moléculaire. Des ensembles volumineux de données
2
incluant des séquences génomiques complètes de nombreux individus issus de populations naturelles appartenant à de nombreuses espèces ont transformé les inférences génétiques de population d’échantillons de loci en génomique de populations (analyse des profils de variation de l’ADN à l’échelle du génome au sein d’une espèce et entre espèces). Des catalogues de presque tous les variants polymorphes sont actuellement disponibles pour les espèces modèles telles que D. melanogaster (Grenier et al. 2015; Huang et al. 2014; Lack et al. 2015; Langley et al. 2012; Mackay et al. 2012), levures (Liti et al. 2009; Strope et al. 2015), Arabidopsis thaliana (Cao et al. 2011; Gan et al. 2011; The 1000 Genomes Project Consortium et al. 2015), Caenorhabditis elegans (Andersen et al. 2012) et les humains (Sudmant et al. 2015; The 1000 Genomes Project Consortium et al. 2010, 2012, 2015). Dans les années à venir, les données génomiques de populations continueront de croître, à la fois en nombres de séquences et en nombre d'espèces (Ellegren 2014; Tyler-Smith et al. 2015).
Dans un livre influent, intitulé « La base génétique du changement évolutif » (Lewontin 1974), la question était de savoir si le mécanisme de la génétique des populations était empiriquement insuffisant, non plus par manque de données, mais par manque de précision des paramètres théoriques qui l'ont rendu incapable de rendre compte des observations. Les progrès de la génétique moléculaire évolutive ont ensuite enrichi le domaine avec de nombreux nouveaux concepts, termes, processus, techniques moléculaires et méthodes statistiques et informatiques. Mais remarquablement, les forces fondamentales de l'évolution établies par les pères fondateurs du domaine (Fisher 1931; Haldane 1990; Kimura 1955; Wright 1931), à savoir la sélection naturelle, la dérive génétique, la mutation, la recombinaison et le flux de gènes, constituent toujours les facteurs explicatifs essentiels utilisés pour comprendre la base génétique du changement évolutif dans une population (Charlesworth 2010; Lynch et Walsh 2007).
3
Structure génétique de populations au service de la conservation
Les impacts possibles du stockage comprennent une diminution significative de la taille effective de la population, en raison du faible nombre de reproducteurs utilisés pour effectuer une reproduction de soutien (Laikre et al. 2010), une perte de diversité génétique dans les populations ensemencées (Eldridge, Myers, et Naish 2009) et une perte de différenciation génétique entre les populations (Lamaze et al. 2012; Marie, Bernatchez, et Garant 2010; Perrier et al. 2013). Un objectif de longue date de la biologie de la conservation a été de délimiter et de hiérarchiser les unités de conservation intraspécifiques qui devraient être préservées en raison de leur importance écologique ou évolutive (Fraser et Bernatchez 2001; Waples 1991). Le rôle de la variation génétique dans le contexte évolutif a été inventé par le concept d'unités évolutives significatives (ESU) (Ryder 1986), lignées génétiques intraspécifiques résultant de l'isolement historique en raison de barrières persistantes au flux génétique. Ces unités génétiquement uniques sont des cibles hautement prioritaires pour la conservation (Crandall et al. 2000). Bien que de nombreuses définitions de l'ESU aient été proposées, elles sont généralement définies comme des populations qui présentent un isolement reproductif en raison de leur divergence évolutive à long terme et en tant que telles représentent une composante évolutive importante de l'espèce (Crandall et al. 2000; Fenster et al. 2018; Frankham, Briscoe, et Ballou 2002; Fraser et Bernatchez 2001; Funk et al. 2012). D'un autre côté, une forte variation génétique au sein de la population est un autre critère important pour prioriser les populations à protéger. Outre sa contribution à la diversité génétique totale des espèces, une variation génétique suffisamment élevée au sein de la population est cruciale pour la survie à long terme d'une population d'espèces et sa capacité à répondre et à s'adapter aux changements environnementaux changeants (Neale 2012).
La structure génétique des populations apparait donc comme un outil de gestion des ressources naturelles car il a été montré que la documentation des modèles spatiaux de ladite structure et des caractéristiques qui façonnent la connectivité marine ont été essentielles pour affiner les efforts de conservation tels que la conception de
4
réseaux de réserves marines à des échelles appropriées (Cros et al. 2017) et la gestion des pêches (Bernatchez et al. 2017). La connaissance de l'étendue et de la structure de la diversité génétique des espèces est essentielle pour l'établissement d'une stratégie de conservation efficace, car les facteurs génétiques contribuent au risque d'extinction des espèces par la dépression de consanguinité, la perte de diversité génétique et la perte de potentiel évolutif (Frankham 2012; Frankham, Bradshaw, et Brook 2014). Donner la priorité à des populations particulières dans les décisions de conservation est généralement basé sur leur contribution à la diversité génétique totale des espèces. Pour cette raison, il est important de connaître le niveau de divergence d'une population par rapport aux autres populations et sa variation intra-population.
Le développement des outils moléculaires a donné la possibilité d’analyser la variabilité et les différences génétiques entre les individus et les populations d’une même espèce. La structure génétique des populations est un domaine de recherche qui vise à décrire ces différences génétiques au niveau spatial et temporel et, est particulièrement utilisée pour comprendre les patrons de colonisation historique d’une espèce qui occupe un territoire donné (Bernatchez et Wilson 1998; Boulet et Gibbs 2006; Turgeon et Bernatchez 2001). Elle permet de comprendre les traits d’histoire de vie associés à une espèce (Garant, Dodson, et Bernatchez 2000; Sinclair 1988), détecter l’influence de la sélection naturelle sur les gènes fonctionnels (Prugnolle et al. 2005; Westerdahl et al. 2004). Elle permet aussi de mieux comprendre les impacts des activités humaines sur les populations naturelles, telle l’influence des ensemencements sur l’évolution des populations de poissons (Eldridge et Naish 2007; Hansen 2002; Ruzzante et al. 2004) et de détecter les immigrants et estimer le flux génique entre populations (Castric et Bernatchez 2004; Manel, Gaggiotti, et Waples 2005; Rannala et Mountain 1997; Tallman et Healey 1994).
Les études qui caractérisent la structure génétique des populations naturelles peuvent fournir des indications relatives aux flux géniques et à la dérive génétique (Loiselle et al. 1995). De manière générale, la présence de populations
5
génétiquement distinctes peut révéler l'influence de barrières aux flux géniques et de la dérive génétique importante (Hoffman et al. 2011). Des populations plus homogènes reflètent des flux de gènes historiques et contemporains et des dérives génétiques plus faibles (Holsinger et Weir 2009). De plus, les facteurs extrinsèques (événements climatiques et géologiques, hétérogénéité de l'habitat) et intrinsèques (capacité de dispersion, système d'accouplement et préférence d'habitat) liés à la structuration génétique jouent un rôle dans le processus de diversification et de spéciation (Schluter 2001). Des études ont révélé chez les cichlidés, l'impact des fluctuations des niveaux d’eau paléo-historiques sur la diversité génétique (Egger et al. 2007), mais aussi l’importance de l'hétérogénéité de l'habitat, de l'écologie et de la capacité de dispersion sur la différenciation de la population (Brawand et al. 2014; Wagner et al. 2013). La structure génétique des populations est également de plus en plus utilisée pour déterminer des populations génétiquement distinctes permettant d’établir des unités de gestion appropriées pour orienter les mesures de gestion et de conservation (Palsbøll, Berube, et Allendorf 2007). Chez les salmonidés de la côte ouest américaine, la structure génétique des populations a permis de déterminer des unités de conservation et de définir l’échelle spatiale à laquelle une population est identifiée (Waples et Gaggiotti 2006).
Mécanismes évolutifs et biologie évolutive
Les organismes (unicellulaires et pluricellulaires) constituent une biodiversité façonnée par l'influence des quatre forces évolutives qui sont la mutation, la dérive génétique, la migration et la sélection. La mutation est la source fondamentale des variations génétiques. Elle agit sur une échelle temporellement longue et dépend des taux de mutation généralement faibles. La mutation et la dérive génétique tendent à différencier aléatoirement les populations isolées d'une même espèce tandis que la migration tend à homogénéiser les populations isolées en apportant de matériel génétique neuf via des individus migrants. Ces trois forces sont dites « neutres » car elles agissent aléatoirement sur l’ensemble du génome. Par opposition aux forces neutres, la sélection agit de manière spécifique sur certains loci en réponse aux pressions environnementales (Cavalli-Sforza 1966; Frankham et al. 2004).
6
Depuis les travaux sur l’origine des espèces, revues historiques (Darlington 1950; Darwin et Peckham 2010), jusqu’à nos jours, les fondements de la compréhension de l’évolution du monde vivant, ont graduellement été établis en considérant une espèce comme un groupe d’individus se reproduisant et interagissant avec les autres membres du même groupe dans un espace donné (Waples et Gaggiotti 2006). Cette compréhension de l’évolution du vivant est éclairée par la biologie évolutive qui est l’une des branches de la biologie qui vise à comprendre l’influence des mécanismes évolutifs sur l’évolution du vivant, à étudier les forces évolutives (sélection naturelle, mutation, recombinaison, dérive, migration) qui font changer les êtres vivants au cours du temps. Tout en abordant la question de la compréhension des processus responsables de la diversification des populations et des espèces, la biologie évolutive mène des études sur les bases génomiques sous-tendant des traits phénotypiques (complexes, variables et potentiellement adaptatifs). L’interaction entre le génome d'un organisme et l'hétérogénéité des environnements dans lequel évolue ce dernier est considérée comme l’une des causes de la différentiation. Ainsi, on parle de divergence adaptative lorsque des populations d’une même espèce s’adaptent à des conditions environnementales différentes.
Nouvelles technologies de séquençage et implications
Le domaine de la génétique a été révolutionné, grâce aux avancées technologiques, par l’augmentation considérable des informations pouvant être obtenues (Narum et al. 2013; Pool et al. 2010). L’arrivée du séquençage de nouvelle génération (NGS) (Metzker 2010) a permis d’augmenter la résolution des études génétiques avec l’obtention, à moindre coût, de milliers de marqueurs SNPs distribués sur l’ensemble du génome. Les techniques de génotypage par séquençage (GBS, Annexe 6) permettent de réduire la complexité des génomes, de découvrir des loci polymorphes et de simultanément génotyper des individus. Brièvement, le tampon de restriction (NEB4) et deux enzymes de restriction (PstI et MspI) ont été ajoutés à chaque échantillon. La digestion a été complétée par incubation à 37°C pendant deux heures et les enzymes ont été inactivées par incubation à 65°C pendant 20 minutes. Deux adaptateurs (un unique pour chaque échantillon et le second commun) ont été ajoutés à chaque échantillon et la ligature a été réalisée en utilisant un « Master Mix » suivi de l'addition de ligase T4. La réaction de ligature a été complétée à 22°C pendant 2 heures puis à 65°C pendant 20 minutes pour inactiver les enzymes. Les échantillons ont été regroupés dans du 48-plex et
7
nettoyés en utilisant des kits de purification QIAquick PCR. La banque a ensuite été amplifiée par PCR et séquencée sur la puce Ion Torrent Proton P1v2 (Université Laval, IBIS, Québec, Canada). Ces avancées ont permis d’identifier des loci potentiellement impliqués dans la divergence adaptative (Narum et al. 2013; Stapley et al. 2010). L’investigation des bases génomiques d’un plus grand nombre de traits phénotypiques sans informations génétiques préalables est devenue possible et les traits phénotypiques, souvent polygéniques, impliquant de nombreux loci à faibles effets (Atwell et al. 2010; Davies et al. 2011) ont donc vu leur capacité d’être étudiés accrue par les NGS. Les marqueurs tels que les SNPs, générés par les NGS, ont l’avantage de se situer soit dans des régions neutres, dans des gènes d’intérêt ou encore d’être liés à ceux-ci. L’utilisation de ces marqueurs peut favoriser la caractérisation de la divergence aussi bien neutre entre populations en déduisant la structure populationnelle que potentiellement adaptative entre populations (Allendorf, Hohenlohe, et Luikart 2010; Helyar et al. 2011; Pool et al. 2010; Stapley et al. 2010).
Variabilité intrapopulation : héritabilité et corrélations génétiques
La variabilité génétique des caractères à l’intérieur des populations est d’un plus haut intérêt pour le sélectionneur, puisque c’est un paramètre qui va lui permettre de diriger l’évolution de sa population d’élevage vers la combinaison de caractères la plus souhaitable à un moment donné, dans un contexte donné. À l’opposé du choix de la meilleure souche qui, malgré son importance, la connaissance et l’exploitation de variabilité génétique intra population permettent, à petits pas mais de façon continue, d’améliorer les caractéristiques d’une souche, avec peu de limites théoriques sur les niveaux que peuvent atteindre les caractères d’intérêt. En particulier, on peut dans certains cas porter sans difficulté, en quelques générations, la moyenne de la population à un niveau supérieur à celui du meilleur individu dans la population de départ. Pour mettre en œuvre la sélection de façon efficace, il est nécessaire de connaitre l’héritabilité des caractères, ainsi que les corrélations génétiques qui les relient entre eux.
Une question centrale en biologie est de savoir si la variation observée d'un trait particulier est due à des facteurs environnementaux ou à des facteurs génétiques. Des multiples définitions techniques de l'héritabilité (considérée comme étant un terme technique en génétique et un paramètre de population
8
avec des définitions spécifiques) et leurs nombreuses applications en biologie évolutive, médecine et agriculture ont été abordées (Visscher, Hill, et Wray 2008).
Définition et historique de l'héritabilité
Le paramètre qui indique le potentiel évolutif d'un trait dans une population est l'héritabilité. L'héritabilité est définie comme la proportion de variation phénotypique due à la variation génétique interindividuelle déterminant l'ampleur et la vitesse du changement phénotypique en réponse à la sélection (Falconer and Mackay 1996, Gurevitch, Scheiner, and Fox 2002). Une autre définition de l'héritabilité est la proportion de variation d'un trait ayant une base génétique avec une capacité à répondre à la sélection par un changement de la valeur moyenne du caractère dans la population ou bien le degré de ressemblance, entre les parents, due à des gènes partagés.
L'héritabilité étant un rapport de variances, le numérateur et le dénominateur doivent être examinés de près. Le dénominateur contient la variation totale observée, excluant généralement la variation due à des facteurs fixes et à des co-variables connus, tels que le sexe, l'âge et la cohorte. Le numérateur de l’héritabilité contient une variation due à des valeurs génétiques additives dans la population. Ces valeurs, appelées « valeurs d’élevage » dans la littérature (Falconer et Mackay 1996), sont définies comme la somme des effets moyens des gènes des parents qui donnent la valeur génotypique moyenne de leur descendance. La définition de l'héritabilité a pour conséquence d’être liée au concept de de population, car la variation des facteurs génétiques additifs et non additifs, ainsi que la variance environnementale, sont spécifiques à la population. La variance génétique dépend de la ségrégation, dans une population, d’allèles influençant le trait, des fréquences des allèles, de l’effet des variants et du mode d’action des gènes, toutes ces variables peuvent différer d'une population à l'autre. Comme la variance environnementale peut varier d'une population à l'autre, en théorie, l'héritabilité dans une population ne permet pas de prédire l'héritabilité du même trait dans une autre population (Visscher, Hill, et Wray 2008).
Du point de vue historique, il est devenu courant d'utiliser le symbole h2 pour l'héritabilité, car h (pour hérédité) a été utilisé pour désigner la corrélation entre génotype et phénotype (Wright 1920). Le carré de cette corrélation (c’est-à-dire h2) est, par définition, la proportion de la variation du phénotype qui est attribuable au passage du génotype au phénotype. Dans un article classique de 1919, la
9
ressemblance entre parents en termes de coefficients de corrélation et de régression a été paramétrée, tout en donnant également un exemple du pourcentage de la variance totale de la taille chez l'homme pouvant être attribuée aux génotypes et aux "génotypes essentiels" (Fisher 1919). Ces pourcentages correspondent à ce que l’on appelle maintenant l'héritabilité au sens large et au sens étroit (Visscher et al. 2008). On pense que J. L. Lush a été le premier à utiliser officiellement le terme «héritabilité» pour décrire la proportion de variation due à des facteurs héréditaires (Lush 1940).
Héritabilité et partitionnement de la variance totale : Paramètres de population
Les phénotypes observés (P) d’un trait d’intérêt peuvent être divisés, selon les modèles de nature-culture biologiquement plausibles, en un modèle statistique représentant la contribution du génotype non observé (G) et de facteurs environnementaux non observés (E) :
Phénotype (P) = Génotype (G) + Environnement (E)
La variance des phénotypes observables (σ2P) peut être exprimée par la somme des variances sous-jacentes non observées (σ2G et σ2E): σ2P = σ2G + σ2E.
L'héritabilité est définie comme un rapport de variance, exprimant la proportion de la variance phénotypique pouvant être attribuée à la variance des valeurs génotypiques :
• Héritabilité (sens large) = H2 = σ2G / σ2P
La variance génétique peut être divisée en variance d'effets génétiques additifs (valeurs de sélection; σ2A), de dominance (interactions entre allèles au même locus, σ2D) et d'épistatique (interactions entre allèles à différents loci, σ2I): σ2G = σ2A + σ2D + σ2I,
• Héritabilité (sens étroit ou strict) = h2 = σ2A / σ2P
En général, σ2E peut être décomposé en un nombre quelconque de facteurs contributifs identifiables, mais aléatoires, pouvant être spécifiques au phénotype. Les exemples incluent la variance environnementale commune à certains groupes, par exemple les frères et sœurs et les portées (σ2CE), et la variance non génétique commune aux mesures répétées d'individus (σ2PE).
Le reste de la variance environnementale, qui ne peut être attribuée à d'autres facteurs, est défini comme la variance résiduelle dans l'environnement, qui inclut la variance d'erreur stochastique individuelle et l'erreur de mesure (σ2RE): σ2E = σ2CE + σ2PE + σ2RE
10
Dans le partitionnement le plus simple, aucun facteur spécifique contribuant à σ2E n'est identifié et σ2RE
= σ2E. Les variances génétiques et environnementales peuvent être divisées davantage pour un trait tel que la masse corporelle à la naissance de la progéniture afin d'inclure les effets génétiques et environnementaux maternels attribuables à la mère (Willham 1963).
La partition de la variance phénotypique suppose l'absence de covariance de génotype par environnement (σG,E). Un autre terme, souvent ignoré, est l’interaction entre génotype et environnement (G * E), lorsque l’effet du génotype dépend de l’environnement. L’exemple le plus étudié et le moins controversé de G * E chez l’homme est l’interaction entre des événements stressants de la vie (l’environnement) et le polymorphisme de longueur du gène transporteur de la sérotonine (le génotype) et leurs effets sur la dépression majeure (le phénotype) (Caspi et al. 2003). Si G * E existe, P = G + E + G * E, une partition plus complète de la variance phénotypique est donc:
σ2P = σ2G + σ2E + 2 σG,E + σ2G *E
Les co variations G et E et les interactions G * E sont souvent ignorées, généralement parce qu’elles ne peuvent pas être estimées. Si l'un des deux est présent, ignorer le premier gonflera les estimations de σ2G et ignorer le second gonflera les estimations de σ2E (Falconer et Mackay 1996).
Méthodes d’estimation de l'héritabilité
L'estimation de l'héritabilité dans les populations dépend de la répartition de la vaiation observée en facteurs génétiques et environnementaux non observés. La méthodologie statistique, utilisée pour répartir la variation et estimer l'héritabilité, est bien développée et elle a plus ou moins convergé entre les espèces et les disciplines (Lynch et Walsh 1998). Cependant, le modèle d'analyse repose sur des hypothèses et souvent non vérifiables, ce qui rend l'inférence statistique difficile et parfois controversée, en particulier pour les phénotypes comportementaux chez l'homme. Dans ces circonstances, l'héritabilité peut changer et son utilité à l'ère de la génomique est capitale. L'importance de l'héritabilité reste centrale et les nouvelles opportunités qu'elle offre (par exemple, pour mesurer l'expression des gènes, la méthylation et les métabolites) permettront de disséquer davantage la variation phénotypique et l'interaction entre les gènes et l'environnement.
Les héritabilités et les variances qui y contribuent sont des paramètres d'une population. En réalité, la seule compréhension de ces paramètres est leur estimation (Jacquard 1983). L'héritabilité peut être
11
estimée à partir de données empiriques sur la ressemblance observée et attendue entre parents. La ressemblance attendue entre parents dépend d'hypothèses concernant ses causes environnementales et génétiques sous-jacentes. Lorsque la sélection est appliquée, le rapport entre la réponse de sélection observée (R, le changement du phénotype moyen entre générations) et le différentiel de sélection observé (S, la différence de phénotype moyen entre les parents sélectionnés pour la reproduction et la moyenne globale de leurs progénitures) peut être utilisé pour estimer l'héritabilité. Cette relation est résumée dans l’équation de l’éleveur, R = h2S (Lynch et Walsh 1998). Pour une expérience qui s'étend sur plusieurs générations, l'héritabilité réalisée a été définie comme le rapport ou la régression de la réponse de sélection cumulative (la somme de toutes les réponses) au différentiel de sélection cumulé (la somme de tous les différentiels de sélection) (Falconer et Mackay 1996).
Traditionnellement, l'héritabilité était estimée à partir de conceptions simples et souvent équilibrées, telles que les fonctions simples de la régression de la progéniture sur les phénotypes parentaux, la corrélation des frères et demi-frères et la différence de corrélation entre les paires de jumeaux monozygotiques (MZ) et dizygotiques (DZ) (Falconer et Mackay 1996). Lorsque des mesures phénotypiques sont disponibles pour les individus avec une combinaison de relations, à la fois au sein de générations différentes, ou en général lorsque le design est déséquilibré, les estimations de la variance génétique additive et des composantes environnementales sont estimées plus efficacement à partir d'un modèle mixte linéaire. Le «modèle animal» est notamment devenu le modèle de choix en génétique d’élevage (Lynch et Walsh 1998), en génétique évolutive (Kruuk 2004) et dans certaines applications en génétique humaine (Almasy et Blangero 1998). Le modèle animal a été dérivé à l'origine pour des applications en génétique du bétail (Quaas et Pollak 1980) et un effet génétique additif aléatoire, valeur de reproduction (Falconer et Mackay 1996; Lynch et Walsh 1998) est ajusté pour chaque individu dans l'arbre généalogique. Dans ce modèle, toutes les relations génétiques additives par paires de l'ensemble du pedigree sont utilisées et, aux fins d'analyse, toutes les sources d'informations sont pondérées de manière appropriée par leur variance d'échantillonnage. Les méthodes d'estimation basées sur le modèle animal sont itératives et sont donc plus intensives en calcul que les estimations d'héritabilité basées sur des coefficients de régression ou de corrélation. Le maximum de vraisemblance résiduel (Patterson et Thompson 1971) est la méthode standard d'estimation des composantes de la variance génétique dans les arbres généalogiques complexes. Il
12
est utilisé pour le bétail, les populations naturelles et les populations humaines (Koerhuis et Thompson 1997; Kruuk et al. 2000; Lynch et Walsh 1998; Macgregor et al. 2006; McRae et al. 2007; Meyer 1985). La précision d'une estimation de l'héritabilité dépend de son erreur d'échantillonnage, qui est fonction de la taille de l'échantillon et de la structure de l'arbre généalogique, ainsi que d'un biais, qui peut provenir d'une confusion. Des biais peuvent apparaître, par exemple, lors de l’appariement et de la sélection de l’assortiment. La variance d'échantillonnage de l'estimation de l'héritabilité est inversement proportionnelle à la relation des individus au carré, au nombre de familles et, dans une moindre mesure, au nombre d'individus dans une famille (Falconer et Mackay 1996). Par conséquent, des centaines d'observations sont nécessaires pour obtenir une erreur type inférieure à 0,1, et des milliers sont nécessaires pour obtenir des estimations très précises.
L'estimabilité fait référence au nombre de paramètres pouvant être estimés à partir de données et dépend de la mesure dans laquelle l'expérience visant à estimer l'héritabilité permet de partitionner la variance totale observée en sources causales présumées. Parfois, un certain nombre de ces sources sont inévitablement confondues dans le plan expérimental, de sorte que leur contribution à la variance globale ne peut être séparée. Par exemple, les estimations de la variance de dominance et de la variance environnementale commune sont confondues lorsque la partition de la variance est réalisée à l'aide d'observations sur des pleins frères. Dans la conception classique des paires jumelles MZ et DZ, il n’existe que trois statistiques essentielles pouvant être estimées à partir de leurs phénotypes, à savoir la ressemblance MZ (par exemple, la covariance ou la corrélation), la ressemblance DZ et la variation phénotypique globale dans l’échantillon. Par conséquent, seules trois composantes de la variance peuvent être estimées, bien que de nombreuses autres composantes causales de la variance, génétiques et non génétiques, peuvent être postulées pour influer sur la ressemblance de la ZM et de la ZZ (Falconer et Mackay 1996; Lynch et Walsh 1998). La confusion pourrait conduire à un biais grave dans l'estimation de l'héritabilité. Par exemple, si la ressemblance des parents et de la progéniture est en partie due à des effets environnementaux courants, alors l'héritabilité basée sur leur ressemblance pourrait être surestimée. Bien que les corrélations de parents éloignés soient moins susceptibles d'être biaisées par des facteurs non génétiques, leur relation est si faible que les estimations d'héritabilité basées sur des parents éloignés présentent une erreur d'échantillonnage élevée.
Le dénominateur de l'héritabilité est la variance phénotypique totale, estimée comme étant la variance du trait après correction des effets fixes connus tels que le sexe, l'âge ou la cohorte. Si les
13
identificateurs de ces facteurs sont inconnus, les estimations de la variance phénotypique seront plus grandes (et l'estimation de l'héritabilité moins). Par exemple, chez l’homme, si la différence de hauteur moyenne de 15 cm entre les hommes et les femmes était ignorée, l’estimation de l’héritabilité serait réduite à 0,6 contre 0,8 quand cette différence est prise en compte. La variance phénotypique totale doit-elle être ajustée pour tenir compte des effets fixes connus lors de l'estimation de la fraction de variance due à des facteurs génétiques? Les obtenteurs de végétaux et d’animaux diraient oui, car ils obtiennent la meilleure prédiction de la performance future en travaillant avec la quantité de variation qui n'est pas expliquée par les effets connus. De même, les généticiens humains diraient que oui, car l'héritabilité est utilisée pour comprendre la composante génétique du risque de maladie, indépendamment des facteurs de risque environnementaux connus. Les généticiens évolutionnistes pourraient dire non, car la matière première de la sélection naturelle est la variation totale entre les individus. La prédiction de la réponse à la sélection naturelle dépend du fait que la sélection ait lieu entre ou parmi les facteurs à l'origine de la variation. Par exemple, si les fluctuations climatiques d’une année à l’autre ont un effet important sur la viabilité moyenne d’une plante annuelle mais que la sélection naturelle opère au cours des années, la meilleure prédiction de la réponse serait alors fondée sur une héritabilité estimée en ajustant entre variation d'une année.
Des mesures répétées sur un individu peuvent être prises pour certains caractères, temporellement, par exemple, la masse corporelle à différents âges ou spatialement, par exemple, le nombre de soies des côtés gauche et droit de Drosophila melanogaster. La variation qui en résulte peut-être partitionnée entre et au sein des individus. Si l'on suppose que ces mesures répétées sont des expressions du même génotype, la variation au sein des individus est alors causée par des erreurs de mesure et d'autres facteurs environnementaux aléatoires. La corrélation est alors appelée répétabilité et constitue une limite supérieure pour l'héritabilité au sens large (Falconer et Mackay 1996). L'héritabilité peut être inférieure à la répétabilité car la corrélation des mesures répétées d'un individu peut être d'origine environnementale aussi bien que génétique. Si le phénotype d'un trait est estimé à partir de la moyenne de plusieurs observations, il convient de définir soigneusement l'héritabilité, car l'héritabilité dépend du nombre d'enregistrements utilisés et n'est pas identique à l'héritabilité d'une observation individuelle. L'héritabilité de la moyenne peut être beaucoup plus grande si l'héritabilité d'une seule observation et la répétabilité sont faibles, par exemple, lorsque l'héritabilité d'une observation est égale à 0,10 et que la corrélation de mesures répétées n'a pas de composante environnementale, l'héritabilité de la