MARIE-KRYSTEL GAUTHIER
GENETIQUE MOLECULAIRE DE
LA MALADIE DE STARGARDT
Etude des mutations du gène ABCA4 dans
la population canadienne-française
Mémoire présenté
à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de maîtrise en biologie cellulaire et moléculaire pour l'obtention du grade de Maître es sciences (M.Sc.)
FACULTE DE MEDECINE UNIVERSITÉ LAVAL
QUÉBEC
2010
II
RÉSUMÉ
La maladie de Stargardt est la dégénérescence maculaire la plus fréquente chez les enfants de 6 à 12 ans. La maladie se caractérise par une perte graduelle et irréversible de la vision centrale. La forme récessive du Stargardt (90% des cas) est causée par le gène ABCA4. Le but de notre étude était d'identifier la nature et la prévalence des mutations retrouvées dans ABC A4 chez la population canadienne-française souffrant de Stargardt. Parmi les 19 familles recrutées, 29 patients souffraient de Stargardt et se partageaient 39 haplotypes distincts. Sur les 20 variations détectées, 15 étaient de réelles mutations alors que les cinq autres étaient des polymorphismes sans conséquence sur la maladie. 45% des 58 chromosomes étudiés ont été associés à une mutation. Sept nouvelles mutations ont été confirmées, mais aucun effet fondateur n'a été observé. Ceci est dû au gène ABCA4 qui est extrêmement polymorphique et sujet aux réarrangements géniques.
m
ABSTRACT
Stargardt disease is the most prevalent macular degeneration in children from 6 to 12 years old. This disease is characterized by a gradual and non reversible loss of central vision. The recessive form of Stargardt (90% of all cases) is caused by the ABCA4 gene. The objective of our project was to study the prevalence and nature of mutations found in ABCA4 in the French-Canadian population suffering from Stargardt disease. From 19 recruited families, 29 patients were diagnosed with Stargardt and were sharing 39 distinct haplotypes. On the 20 detected variations, 15 were known mutations and the other five were polymorphisms with no consequences on the disease. 45% of all 58 studied disease chromosomes were associated with a mutation. Seven novel mutations were confirmed, but no founder effect was observed. This could be because ABC A4 is a highly polymorphic gene and subject to DNA rearrangements.
IV
REMERCIEMENTS
J'aimerais tout d'abord remercier mon directeur de recherche, Dr Vincent Raymond, de m'avoir chaleureusement accueillie dans son équipe, où j'ai eu l'occasion d'apprendre énormément. J'ai eu le privilège de présenter mes travaux à de nombreux congrès et journées de recherche, notamment avec l'American Society of Human Genetics, un congrès d'envergure internationale qui a eu lieu à Hawaii. Je suis consciente et très fière de la confiance qu'il m'a accordée.
Un merci très spécial à Stéphane Dubois, sans qui le laboratoire ne pourrait tout simplement pas fonctionner. Ses conseils avisés et sa patience sans limite m'ont été d'un grand secours en de nombreuses occasions. Grâce à lui, j'ai pu approfondir mes connaissances de laboratoire de façon extraordinaire; c'est un professeur exceptionnel. Merci aussi aux autres étudiants du laboratoire : Mathieu Lanthier-Veilleux, pour ses idées et recommandations très utiles, ainsi que Pascal Belleau, sans qui mon savoir bioinformatique ne serait pas ce qu'il est aujourd'hui. Merci à Rose Arseneault, l'infirmière de notre équipe, pour la gestion efficace des dossiers des patients, en plus d'avoir participé à la relecture de ce mémoire. J'aimerais aussi remercier Marc-André Rodrigue et toute l'équipe de la plate-forme de séquençage, Sylvie Desjardins, Annie-Claude Collin-Deschesnes, Guy Reimnitz, Nathalie Gaudreault et Angèle Gareau-Pagé, pour leur assistance lors du séquençage. Merci aussi à Edith Gagnon pour son beau sourire à tous les matins et ses judicieux conseils.
J'aimerais aussi remercier tous les organismes subventionnaires qui ont grandement aidé à la réalisation de ce projet, tout particulièrement le CRSNG, le Réseau Vision du FRSQ et la Fondation des maladies de l'œil. Merci également aux nombreuses familles qui ont rendu cette étude possible grâce à leur participation active.
Enfin, j'aimerais remercier tout spécialement mes parents, Brigitte et Yves, mes sœurs, Arianne et Charlène, ainsi que mon copain Philippe Simon, pour leur soutien moral sans faille et leur compréhension tout au long de mes études graduées. Philippe a particulièrement fait preuve d'une patience incroyable lors des moments plus difficiles, et je dois avouer que son sens de l'humour a sauvé plus d'une fois la situation...
AVANT-PROPOS
Je suis la principale personne ayant travaillé sur la partie expérimentale de ce projet de recherche. J'ai effectué le génotypage des patients et de leur famille, préparé le séquençage des individus atteints de Stargardt et de leurs parents, analysé les résultats et confirmé par essais spécifiques les nouvelles mutations détectées. Pourtant, tout ceci n'aurait pas été possible sans l'aide précieuse de mes nombreux collègues et collaborateurs.
Elie Deilhes a commencé ce projet lors de ses études de doctorat et avait ainsi débuté le génotypage de quelques familles. Stéphane Dubois et Marc-André Rodrigue, professionnels de recherche, ont respectivement réalisé l'extraction d'ADN des patients et le séquençage, en plus de superviser étroitement mon travail en m'assistant dans l'analyse des résultats. Pascal Belleau, étudiant au doctorat dans mon laboratoire, a créé une base de données permettant de classer toutes les familles et individus souffrant non seulement de Stargardt, mais également de toutes les maladies oculaires étudiées au laboratoire. Rose Arseneault, l'infirmière de recherche de notre équipe, était responsable de la révision des dossiers ophtalmologiques ainsi que du contact avec les patients et leur famille. Elle a également complété la généalogie des familles québécoises afin d'identifier des ancêtres communs.
Les Drs Mario Malenfant, Jean-Paul Lachance et Yvon Tardif sont les principaux ophtalmologistes ayant participé activement au recrutement des patients pour cette étude. De nombreux optométristes à travers le Québec et le Canada ont également contribué au recrutement. Le Dr Vincent Raymond, mon directeur de recherche, est professeur au Département de médecine moléculaire à la Faculté de médecine de l'Université Laval et chercheur au Centre de recherche du CHUL. Il est directeur du laboratoire de génétique moléculaire des systèmes sensoriels ainsi que fondateur de la plateforme de séquençage et de génotypage des génomes du centre de recherche du CHUL.
VI
A mes parents,
Brigitte Beaulieu et Yves Gauthier,
qui m'ont encouragée sans relâche tout au long de mes études.
Sans eux, je ne serais pas devenue ce que je suis aujourd'hui.
VII
TABLE DES MATIÈRES
RESUME II ABSTRACT Ill REMERCIEMENTS IV
AVANT-PROPOS V TABLE DES MATIÈRES VII
LISTE DES FIGURES X LISTE DES TABLEAUX XI CHAPITRE 1. INTRODUCTION 1
1.1. La maladie de Stargardt 1
1.1.1. Description 1 1.2. Le gène délétère : ABCA4 3
1.2.1. Superfamille des transporteurs de type ABC 3
1.2.2. Caractéristiques du gène ABCA4 3 1.2.3. Caractéristiques de la protéine ABCA4 4
1.2.4. Rôle d'ABCA4 7 1.2.4.1. Photoréception, phototransduction et cycle visuel 7
1.2.4.2. Accumulation de dépôts de lipofuscine 7 1.2.5. Mutations et polymorphismes dans ABCA4 11
1.2.5.1. Un gène polymorphique 11 1.2.5.2. Distinction entre mutations et polymorphismes 11
1.2.5.3. Combinaisons de mutations dans ABCA4 12
1.2.5.4. Une micropuce d'ABCA4 12
1.3. Association d,ABCA4 avec d'autres maladies oculaires 13
1.4. Traitements en vue 13 1.4.1. Protection physique 13 1.4.2. Pharmacologie 14 1.4.3. Interventions chirurgicales 14
1.4.4. Thérapie génique 15 1.5. Étude de la population québécoise 15
1.5.1. L'effet fondateur 15 1.5.1.1. Histoire de la population québécoise 15
1.5.1.2. Le Stargardt et l'effet fondateur 16
1.5.2. Recrutement des familles 16 1.5.3. Impact du dépistage génétique 17
1.6. Hypothèses et objectifs 17 1.6.1. Problématique et hypothèses 17
VIII CHAPITRE 2. Identification de nouvelles mutations dans le gène ABCA4 chez la
population canadienne-française 20 2.1. Le Stargardt dans la population canadienne-française 20
2.2. Objectif du projet 20 2.3. Matériel et méthodes 20
2.3.1. Recrutement des familles 20 2.3.2. Extraction d'ADN 21 2.3.3. Génotypage de microsatellites 21
2.3.4. Séquençage d'ABCA4 21 2.3.5. Culture de cellule et extraction d'ARN 22
2.4. Résultats 22 2.4.1. Les patients et leur famille 22
2.4.2. Ségrégation des haplotypes 24 2.4.3. Caractérisation génétique 24 2.4.4. Confirmation des nouvelles mutations 37
2.4.4.1. Étude des changements d'acides aminés 37
2.4.4.2. Étude des mutations d'épissage 39 2.4.4.3. Mutation dans le promoteur du gène ABCA4 41
2.5. Discussion 41 2.5.1. L'éventail des degrés de sévérité du Stargardt 41
2.5.2. Une ségrégation des haplotypes parfois singulière 42 2.5.3. Le séquençage d'ABCA4 ne révèle pas toutes les variations 44
2.5.4. Les nouvelles variations détectées sont des mutations associées au Stargardt ...44 2.5.4.1. Les changements d'acides aminés sont de nouvelles mutations causant la
maladie de Stargardt 44 2.5.4.2. Les variations d'épissage sont des mutations causant la maladie de Stargardt
45 2.5.4.3. La variation localisée dans le promoteur d'ABCA4 serait une mutation
causative du Stargardt 46
CHAPITRE 3. Absence d'effet fondateur pour ABCA4 dans la population
canadienne-française 48 3.1. L'effet fondateur au Québec 48
3.2. Objectif du projet 48 3.3. Matériel et méthodes 49
3.3.1. Établissement des liens interfamiliaux 49 3.3.2. Généalogie des familles concernées 49
3.4. Résultats 49 3.4.1. Localisation géographique des cas de Stargardt au Québec et dans les environs49
3.4.2. Relations entre les familles étudiées 51 3.4.3. Généalogie des familles à l'étude 52
IX
3.5. Discussion 52 3.5.1. Absence d'effet fondateur pour ABCA4 chez les Canadiens-Français 52
CHAPITRE 4. CONCLUSION 54 4.1. Les nouvelles mutations dans le gène ABCA4 sont spécifiques à la population
canadienne-française 54 4.2. Absence d'effet fondateur pour ABCA4 dans la population canadienne-française.55
4.3. ABCA4 et la dégénérescence maculaire liée à l'âge 55
4.4. Un autre gène qu,ABCA4? 56
4.5. Perspectives de recherche 56 4.5.1. Confirmation approfondie des nouvelles mutations 56
4.5.2. Approches alternatives afin de déterminer les mutations non identifiées 57
4.5.3. Études de corrélation génotype-phénotype 57 ANNEXE 1 : Vérification de la mutation IVS37-2AA par PCR 60
LISTE DES FIGURES
Figure 1. Photographies de la région centrale de fonds d'œil 2 Figure 2. Localisation du gène ABCA4 sur le chromosome 1 4
Figure 3. Anatomie de la partie centrale de la rétine 5 Figure 4. Bâtonnet en association avec une cellule de l'EPR 6
Figure 5. Cycle visuel, photoréception, phototransduction 8 Figure 6. Réaction chimique de la conversion du tout-trans-rétinal en A2PE 10
Figure 7. Photographies de fonds d'œil 23 Figure 8. Génotypage des familles de Stargardt étudiées 25
Figure 9. Positionnement des nouvelles mutations sur la protéine ABC A4 40
Figure 10. Analyse de la mutation IVS38-2AA 45 Figure 11. Provenance régionale des patients souffrant de Stargardt 50
XI
LISTE DES TABLEAUX
Tableau 1. Liste des variations détectées chez les 29 patients souffrant de Stargardt 38
Tableau 2. Liste des nouvelles mutations détectées 39 Tableau 3. Les mutations de changements d'acides aminés sont entourées de mutations
déjà connues 41 Tableau 4. Comparaison du site de liaison de FOXC1 et de la séquence du promoteur
d'ABCA4 47 Tableau 5. Représentation des haplotypes communs entre les familles de Stargardt 51
CHAPITRE 1. INTRODUCTION
1.1. La maladie de Stargardt
La vue constitue le sens le plus développé chez l'être humain. En effet, les yeux comprennent environ 70% des récepteurs sensoriels de l'organisme, et près de la moitié du cortex cérébral est lié au processus de la vision [1]. Il est ainsi normal que la détérioration ou la perte de ce sens soit catastrophique. Plusieurs chercheurs et scientifiques travaillent avec acharnement sur une multitude de maladies affectant la vue afin de mieux les comprendre et éventuellement, trouver des traitements efficaces. Ce mémoire traitera d'une cause de cécité en particulier, la maladie de Stargardt, qui se développe surtout chez les enfants.
1.1.1. Description
La maladie de Stargardt est une forme de dégénérescence maculaire juvénile. La maladie a été décrite pour la première fois en 1909 par Karl Stargardt [2]. La prévalence de cette maladie se situe entre 1/8000 et 1/10 000 en Amérique du Nord, ce qui en fait la dystrophie maculaire héréditaire la plus répandue chez les enfants. Elle se développe habituellement entre 6 et 12 ans [3]. Le Stargardt se caractérise par une perte graduelle et bilatérale de la vision centrale, une atrophie de l'épithélium pigmentaire rétinien (EPR), une dégénérescence des photorécepteurs et l'apparition de « mouchetures » (ou flecks, en anglais) autour de la macula (Figure 1) [4-7]. La vision des couleurs est souvent également altérée [5]. Dans 90% des cas, le Stargardt ségrége selon un mode autosomal récessif. Dans les 10% restant, cette maladie se transmet selon un mode autosomal dominant [8]. Le sujet principal de ce projet de recherche est la forme récessive de la maladie de Stargardt, causée par des mutations dans le gène ABCA4 [9]. Des mutations dans les gènes ELOVL4 ou PROML1 entraînent la forme dominante de la maladie, plus rare [10-11]. Il n'existe présentement aucun traitement pour cette maladie.
Œil droit
Œil gauche
B
Figure 1. Photographies de la région centrale de fonds d'œil. A) Patient sain présentant un fond d'œil normal, clair et uniforme.
B) Patient atteint de Stargardt où des flecks jaunâtres autour de la macula sont identifiés par une flèche. Il s'agit de dépôts visibles fluorescents d'A2E, un composé de la lipofuscine.
1.2. Le gène délétère : ABCA4
1.2.1. Superfamille des transporteurs de type ABC
Il existe 48 gènes codant pour des protéines ABC (ATP binding cassette) dans le génome humain. Ces protéines sont divisées en sept sous-familles distinctes, selon la similitude de leur séquence d'acides aminés et la phylogénie les reliant [12-13]. ABCA4 fait partie de la superfamille des transporteurs de type ABC, sous-famille ABCA (12 membres), et sa structure est très caractéristique de ce groupe. Les transporteurs de la famille ABC sont très conservés parmi tous les vertébrés [5]. Ils lient et hydrolysent l'ATP afin d'utiliser cette énergie pour transporter des composés dont la nature varie beaucoup : vitamines, ions, peptides, glucides, lipides, en passant par diverses drogues [12, 14]. En plus de jouer un rôle en tant que transporteurs, les protéines de la famille ABC agissent comme récepteurs et canaux protéiques [15]. Des défauts dans ces protéines sont associés à de nombreuses maladies héréditaires, comme la maladie de Tangier et la déficience familiale en HDL (ABCAl), la fibrose kystique (ABCC7) et des problèmes majeurs au foie (ABCB11) [16-17]. 1.2.2. Caractéristiques du gène ABCA4
Le gène ABC A4, à l'origine appelé ABCR pour retina-specific ABC transporter, a été identifié en 1997 par Allikmets et al. comme le gène délétère de la forme autosomale récessive de la maladie de Stargardt [9]. Ce gène couvre environ 150 kb d'ADN génomique, et comprend 50 exons, pour un total de 2273 acides aminés. Il est situé sur le chromosome 1, au locus lp22.1 (Figure 2) [17-19]. ABCA4 est exprimé presqu'exclusivement dans les photorécepteurs de la rétine [5, 9]. Une étude récente démontre que son ARNm a aussi été retrouvé dans certaines régions du cerveau de rats. La signification fonctionnelle de la présence d,ABCA4 dans le cerveau n'est pas encore déterminée. [9, 20-21]. Quoiqu'il en soit, ABCA4 est exprimé chez tous les vertébrés, dans la membrane des disques des segments externes des cônes et des bâtonnets. Fonctionnant à de faibles intensités de lumière, les bâtonnets sont plus sensibles que les cônes, et sont donc adaptés à la vision nocturne et périphérique. Les cônes, eux, réagissent à une lumière très intense et sont donc responsables de la distinction des couleurs de même que de l'acuité visuelle (Figure 3) [1, 22].
ABCA4: Ip22.1-p21 u on
■d
B
lOOf, 20011 ' ' )I l I M 11 i
TTTTTT
< < 9 * 359293 Cf»1 IA8CA41 GenelD:24 |1_53313*p |93_>4* 94230981 << NC 0000019 " H - h - H hHII I I II I l l l l l l l llll l l l l II H I l l l l l NM.0003502Figure 2. A) Localisation du gène ABCA4 sur le chromosome 1, au locus Ip22.1-p21. B) Représentation du gène ABCA4 avec ses 50 exons. Source http://refgene. com/gene/24
1.2.3. Caractéristiques de la protéine ABCA4
ABCA4 est une glycoprotéine de 256 kDa qui possède deux domaines transmembranaires de six boucles chacun, deux domaines extracellulaires glycosylés ainsi que deux domaines intracellulaires très conservés liant l'ATP. Ces derniers comprennent les motifs Walker A et B ainsi que le motif « signature » typique ABC [5, 17, 23]. ABC A4 est insérée dans la membrane des disques du segment externe des photorécepteurs, en association étroite avec les cellules de l'EPR (Figure 4) [18]. Ces dernières sont responsables de recycler et de phagocyter les disques des photorécepteurs qui se renouvellent à tous les jours, en plus de leur apporter des nutriments essentiels [1, 14]. Les protéines de type ABC étant très grosses, il est difficile de les exprimer ou de les purifier en quantité suffisante pour les cristalliser afin d'étudier leur structure en 3 dimensions [12]. Néanmoins, le dernier modèle proposé suite à la compilation de nombreuses études est présenté à la figure 4.
Lorsque ABCA4 lie l'ATP, elle subit un changement de conformation et agit comme une flipase afin de transporter un dérivé du rétinol à travers la membrane des disques des cônes et bâtonnets [5, 18]. Molday et al. ont démontré en 2009 l'importance de l'extrémité C-terminale de la protéine, où un motif d'acides aminés très conservés, VFVNFA, participerait au repliement correct d'ABCA4 et à son bon fonctionnement [24].
B
Cellules pigmentaire. de 1 epithelium rétinien (EPR1 Cônes Bâtonnets Cellules horizontales Cellules bipolaires Cellules amacrines Cellules ganglionnaires Fibres nerveuses - * Trajet de la lumière
Figure 3. Anatomie de la partie centrale de la rétine. A) Coupe transversale d'un œil. Les dégénérescences maculaires touchent une zone de la rétine dénomée « macula », un léger renfoncement mesurant à peine quelques millimètres de diamètre. Cet endroit est dépourvu de vaisseaux sanguins afin d'optimiser la vision centrale. Source : Vetopsy.fr. http://www, vetopsy. fr/ sens/visu/images/oeil ret, gif,
B) Coupe transversale de la rétine. La lumière traverse les différentes couches de cellules en commençant par les fibres nerveuses, les cellules ganglionnaires, amacrines, bipolaires et horizontales, et les photorécepteurs (cônes et bâtonnets). Figure adaptée de : Elena Kaye, Maryam Etezadi Amoli, Sharareh Noorbaloochi et Khuram Zia. Rhodopsin Based Vision Prosthesis Model.
http://scien.stanford.edu/class/psvch221/proiects/07/channel_rhodopsin/finalProie ct.htm.
A
Phagosome Grannie de mélanine C ellule de l'épithélium pigmentaire rétinien Zone externe Segment externe du bâtonnet Membra nt da disque Segment interne du bâtonnet Zone interne Corps cellulaire Régionsynaptique Vésicule» syniptiques Photorécepteur : bâtonnet
Figure 4. Bâtonnet en association avec une cellule de l'épithélium pigmentaire rétinien (EPR). A) Le bâtonnet comprend un segment externe, une tige de connexion ciliée, un segment interne, un corps cellulaire et une région synaptique. Le segment externe est composé d'un empilement de disques entourés d'une membrane plasmique. La cellule de l'EPR adjacente au photorécepteur lui fournit les nutriments nécessaires, participe au cycle visuel et phagocyte des parties du segment externe afin de régénérer et recycler les disques du photorécepteur.
B) Agrandissement de la membrane d'un disque où est situé ABCA4.
C) Modèle topologique d'ABCA4, comportant deux domaines exocytoplasmiques (ECD), deux domaines transmembranaires ainsi que deux domaines liant l'ATP (NBD). La protéine ABCA4 transporte certains dérivés du rétinol à travers la membrane du disque du photorécepteur, du lumen vers le côté cytoplasmique. Image traduite tirée de : Molday, R.S. (2007). ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J Bioenerg Biomembr 39, 507-517.
1.2.4. Rôle d'ABC A4
1.2.4.1. Photoréception, phototransduction et cycle visuel
La photoréception est le processus par lequel l'œil détecte l'énergie lumineuse. Les segments externes des photorécepteurs contiennent des pigments visuels, composés d'une molécule photosensible appelée rétinal (dérivée de la vitamine A), combinée à une protéine, l'opsine. Le pigment visuel des bâtonnets est la rhodopsine. Lorsque la rhodopsine absorbe la lumière, le rétinal, alors sous la forme de rétinal-11-cis, se transforme en son isomère tout-trans-rétinal et se détache de l'opsine. L'opsine libérée agit alors comme une enzyme et catalyse une cascade de signalisation menant à l'hyperpolarisation des membranes des photorécepteurs, et subséquemment, au processus de la vision. C'est ce qu'on appelle la phototransduction : l'énergie lumineuse est transformée en signal électrique, propagé jusqu'au lobe occipital via les neurones pour permettre de voir (Figure 5a) [1].
Pendant ce temps, le tout-trans-rétinal doit être recyclé en rétinal-11-cis pour se lier de nouveau à une opsine et ainsi régénérer la rhodopsine. C'est ici qu'intervient ABCA4. La fonction principale de cette protéine est de transporter le tout-trans-rétinal, libéré dans le lumen et sur la paroi interne des membranes, jusque dans le cytoplasme (Figure 5a). Une fois passé dans le cytoplasme, le tout-trans-rétinal est transformé en vitamine A, pour ensuite être transféré dans les cellules de l'EPR et être recyclé en rétinal-11-cis. C'est ce qu'on appelle le cycle visuel [14, 17-18]. Le tout-trans-rétinal libéré suite à la photoexcitation peut également réagir avec le phosphatidyléthanolamine (PE) pour former le N-rétinylidène-phosphatidyléthanolamine (N-Ret-PE), un composé également transporté dans le cytoplasme par ABC A4 [17]. Certaines équipes suggèrent même que c'est sous la forme de N-Ret-PE qu'ABCA4 évacue le plus efficacement les dérivés du rétinol des disques des photorécepteurs [17, 23].
1.2.4.2. Accumulation de dépôts de lipofuscine
Lorsqu'ABC A4 est mutée, sa fonction de transporteur est altérée, voire complètement inhibée. ABCA4 ne peut donc pas évacuer efficacement le tout-trans-rétinal, le N-Ret-PE, ni aucun de leurs dérivés, qui s'accumulent alors dans le lumen et sur la face interne des membranes des disques des bâtonnets (Figure 5b). [23]. Le N-Ret-PE peut alors
Cellule EPR
Fennetiire des canaux sodium et hyperpolarisation
Disques du segment externe d'un bâtonnet
Phagocytose
. Disques du segment ' externe d"un bâtonnet
LEGENDE ! ! Rhodopsine Opane
1
Retm_l-1 1-cis T out- U _ ns-1 - Una 1A ■
•
TrMWctucme et Ç »n«l à V lUm me A phosphochestetase sodiumFigure 5. Cycle visuel, photoréception, phototransduction et rôle joué par ABCA4 dans ces processus (page précédente). A) Situation normale, où la protéine ABCA4 est fonctionnelle. Un photon de lumière excite la rhodopsine. Le rétinal-11-cis est transformé en tout-trans-rétinal, libérant l'opsine. C'est la photoréception. L'opsine libérée active une transducine et une phosphodiesterase, ce qui ferme les canaux à sodium par une cascade de signalisation. L' hyperpolarisation de la membrane du photorécepteur est le signal transmis aux cellules des couches sous-jacentes afin de permettre la vision. C'est la phototransduction. ABCA4 fait alors passer le tout-trans-rétinal du côté du lumen vers le côté cytoplasmique du bâtonnet pour régénérer la rhodopsine. Le tout-trans-rétinal est transformé en plusieurs intermédiaires, comme la vitamine A, pour finalement être converti en rétinal-11-cis dans les cellules de l'EPR. C'est le cycle visuel.
B) Situation où la protéine ABCA4 est mutée et non fonctionnelle. La photoréception et la phototransduction durent un certain temps, mais puisqu'ABCA4 ne peut transporter le tout-trans-rétinal, celui-ci s'accumule dans le lumen du disque. Il forme alors, avec le N-ret-PE, un composé intermédiaire, l'A2PE. Celui-ci est phagocyté par les cellules de l'EPR lors du recyclage des photorécepteurs, et est métabolisé en A2E toxique dans le lysosome des cellules de l'EPR. Les dépôts de lipofùscine deviennent alors visibles, et les cellules de l'EPR, de même que les photorécepteurs associés, dégénèrent progressivement.
Image inspirée de : Molday, R.S. (2007). ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J Bioenerg Biomembr 39, 507-517.
réagir avec une molécule de tout-trans-rétinal et former du bis-rétinoïde-pyridinium-phosphatidyléthanolamine, ou A2PE, qui s'accumule à son tour dans les disques [25-26]. Lors de la phagocytose des disques des bâtonnets par les cellules adjacentes de l'EPR, les composantes du segment externe sont métabolisées dans les lysosomes de ces cellules; A2PE est alors hydrolyse en bis-rétinoide-N-rétinylidine-N-rétinyléthanolamine, ou A2E (Figure 6) [27]. L'A2E est une composante fluorescente majeure de la lipofùscine, qui s'accumule dans les cellules de l'EPR sous forme de dépôts fluorescents, créant ainsi les flecks si caractéristiques du Stargardt [27-29]. L'A2E agit comme un détergent et peut aussi
être converti en époxydes très toxiques, causant l'apoptose des cellules de l'EPR [30-32]. La mort des cellules de l'EPR causerait ensuite la dégénérescence des photorécepteurs qui y sont associés, ce qui explique la perte de vision chez les gens souffrant de Stargardt [14,
10
( J ^ ^ ^ o ♦ PE
Tout (ran s vét in a 1 Phosphatidyléthauolamine N-rétinvlidène-PE Hvdrolvse Condensation, réarrangement et oxydation
E
H.
o q j a î ,
r
A2E A2PEFigure 6. Réaction chimique de la conversion de tout-trans-rétinal et du phosphatidyléthanolamine (PE) en A2PE, un composé bisrétinoïde formé dans les segments externes des photorécepteurs, et en A2E, un composé fluorescent toxique formé par l'hydrolyse de A2PE dans les cellules de l'épithélium pigmentaire rétinien.
Figure adaptée de : Molday, R.S., ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J Bioenerg Biomembr, 2007.
39(5-6): p. 507-17
Il est à noter qu'une faible accumulation d'A2E dans les cellules de l'EPR est normale en vieillissant, car il s'agit d'un produit dérivé de la dégradation des disques des photorécepteurs [33]. Par contre, lorsque cette concentration dépasse un certain seuil critique (comme dans le cas du Stargardt, où les niveaux d'A2E sont de 2 à 5 fois trop élevés [34]), la lumière bleue est alors fortement absorbée par le surplus d'A2E qui initie l'apoptose des cellules de l'EPR [35]. Cette accumulation d'A2E est plus grave au niveau de la macula, région responsable de l'acuité visuelle, puisque c'est là où la concentration de photorécepteurs est la plus grande [5].
11 1.2.5. Mutations et polymorphismes dans ABC A4
1.2.5.1. Un gène polymorphique
ABCA4 est un très gros gène : ses 50 exons et 2273 acides aminés couvrent un grand nombre d'acides nucléiques [17-18]. À ce jour, plus de 500 mutations ont été répertoriées dans ce gène, ce qui démontre une grande hétérogénéité génotypique. De nouvelles variations sont d'ailleurs découvertes constamment [6]. L'éventail de mutations varie de simples substitutions d'une base nucléique à des deletions de plusieurs exons [36-37]. De plus, la fréquence de porteurs hétérozygotes dans la population générale est particulièrement élevée, ce qui entraîne parfois un mode de transmission pseudodominant de la maladie [38-40]. ABCA4 est l'un des gènes les plus polymorphiques connus à ce jour [41].
1.2.5.2. Distinction entre mutations et polymorphismes
Puisque le gène est très polymorphique, les variations retrouvées dans ABCA4 sont composées de mutations causant la maladie, mais également de nombreux polymorphismes. Un polymorphisme génétique est la coexistence de plusieurs alleles différents pour un même gène, mais qui ne sont pas associés à une maladie. Quelques fois, il est difficile de déterminer si une variation dans l'ADN est une mutation ou un polymorphisme sans conséquence sur la maladie. Plusieurs mutations sont considérées comme des polymorphismes dans certaines études, alors que quelques années plus tard, une autre équipe les considère comme des mutations, et vice versa [42]. Ceci rend l'analyse des variations plutôt complexe. La majorité d'entre elles sont aussi des mutations faux-sens qui demeurent rares autant chez les malades que chez les individus sains [6, 43]. Des cas où des porteurs de deux mutations dans ABCA4 restaient sains ont même déjà été rapportés [42, 44]. Ceci impliquerait la présence de facteurs externes, comme un gène modificateur (gène affectant l'expression de certains autres gènes), ou peut-être une penetrance incomplète de la maladie [41,44-45].
La classification d'une variation en une mutation ou un polymorphisme requiert avant tout un test biochimique adéquat, comme l'étude de protéines recombinantes in vitro [43, 46] et un essai ATPase. Ce dernier test consiste à purifier la protéine recombinante
12
ABCA4 et à la reconstituer en l'insérant dans une membrane de liposome. Par la suite, différents dérivés du rétinol (le tout-trans-rétinal et le N-ret-PE par exemple) sont soumis à ABCA4, dont l'activité d'hydrolyse de l'ATP est quantifiée [18, 31]. Cette méthode constitue en fait le seul moyen disponible pour tester la fonctionnalité d'ABCA4, mais ne permet malheureusement pas de quantifier la quantité de substrat transporté, ce qui serait une donnée complémentaire primordiale à obtenir dans l'étude de cette protéine. Le fait que le rétinal soit extrêmement hydrophobe et que la protéine, très grosse, soit difficile à purifier, compliquent les recherches [47].
1.2.5.3. Combinaisons de mutations dans ABC A4
Plusieurs équipes de recherche se concentrent également sur le fait que différentes combinaisons de mutations engendrent des phenotypes de Stargardt différents. En effet, cela influence grandement le degré de sévérité de la maladie, affectant l'âge d'apparition des symptômes, l'agressivité de la progression, l'acuité visuelle, le patron de dépôts de lipofùscine ainsi que les résultats d'électro-rétinogramme (ERG) [39, 48]. D'autres équipes ont aussi émis l'hypothèse que les mutations faux-sens causeraient plutôt une forme modérée de la maladie de Stargardt, et que la nature de ces mutations, autant que leur position dans le gène ABCA4 (domaine de liaison d'un facteur de transcription par rapport à une région non-fonctionnelle), influenceraient l'âge d'apparition des symptômes ainsi que les caractéristiques ophtalmoscopiques [42, 45, 49].
1.2.5.4. Une micropuce d,ABCA4
La compagnie Asper Ophthalmics Biotech a récemment développé une micropuce commerciale détectant 558 variations connues dans ABCA4 (http://www.asperbio.com/ index.php?nid=88&pid=82&rid=37). Cependant, son utilité est limitée puisqu'elle ne détecte pas les nombreuses nouvelles mutations dans le gène. Son taux de détection se compare alors à celui des autres méthodes d'analyse de l'ADN, moins dispendieuses, comme le séquençage direct ou la chromatographic dHPLC [50-53]. Ces techniques sont d'ailleurs souvent utilisées afin de compléter les résultats obtenus avec la micropuce d'Asper qui elle, constitue plutôt un outil de détection et de diagnostic de base.
13 1.3. Association d'ABCA4 avec d'autres maladies oculaires
Il est également connu que différentes combinaisons de mutations dans ABCA4 causent tout un éventail de maladies oculaires [40, 54-55]. En effet, le Fundus Flavimaculatus [56], la rétinite pigmentaire [57], la dystrophic cônes-bâtonnets [58] et la dégénérescence maculaire liée à l'âge (DMLA), sont toutes reliées à des mutations à ce locus, quoiqu'une association avec la DMLA reste toutefois controversée [59]. Un modèle basé sur l'activité résiduelle de la protéine due à des mutations dans ABCA4 a été proposé ainsi : la sévérité du phénotype observé serait inversement proportionnelle à l'activité résiduelle de la protéine [42, 60-62]. Des patients porteurs de deux alleles nuls seraient alors atteints de rétinite pigmentaire, alors que ceux possédant un allele nul et une mutation moyennement sévère auraient une dystrophie cônes-bâtonnets. Une mutation nulle avec une mutation légère, ou alors deux mutations moyennement sévères causeraient le Stargardt, alors que des individus porteurs d'un allele sauvage et d'un allele mutant quelconque auraient de plus grands risques de développer la DMLA [41, 60, 63]. Il est également fréquent que certaines combinaisons de mutations entraînent des formes intermédiaires et/ou chevauchantes de maladies oculaires, et ce parfois dans la même famille [54, 64].
1.4. Traitements en vue
Il n'existe actuellement aucun traitement pour la maladie de Stargardt. Cependant, les recherches progressent dans de nombreuses directions, et le modèle de souris knockout ABCA4'1' s'avère un outil très efficace pour tester les nouvelles avenues thérapeutiques [32].
1.4.1. Protection physique
La rétine est la seule partie du système nerveux central à être directement exposée aux rayons UV. Il est d'ailleurs connu depuis des millénaires qu'une exposition excessive à la lumière cause des dommages importants à la rétine [65]. C'est notamment le cas lorsqu'une éclipse solaire est regardée directement, ou lorsque l'on fixe la lumière du soleil sans lunettes de protection [66]. L'activation de la rhodopsine par la lumière forme le
14 composé intermédiaire tout-trans-rétinal qui est directement impliqué dans la biosynthèse d'A2E, tel que discuté dans la section 1.2.4 [67]. En l'absence d'une protéine ABCA4 fonctionnelle, l'A2E s'accumule donc rapidement. Certains chercheurs ont ainsi démontré que des souris ABC A4'1' accumulaient ce composé beaucoup plus vite en présence de lumière que celles ayant été élevées dans la pénombre [68]. Ils en ont conclu que le port de lunettes filtrant 100% des rayons UV et des rayons bleus du soleil pourrait s'avérer une option intéressante de protection pour les patients souffrant de Stargardt.
1.4.2. Pharmacologie
Au niveau pharmacologique, l'administration d'isotrétinoïde, aussi connu sous le nom commercial d'Accutane, bloque la formation et l'accumulation d'A2E chez des souris ABC A4 ~'~ en réduisant le niveau de N-Ret-PE, un précurseur d'A2E [69]. Le Fenretinide, un autre composé commercial connu pour ses effets anticancéreux, semble avoir le même effet, mais en causant une déficience en vitamine A dans l'œil [70]. D'ailleurs, un groupe de chercheur a pu déterminer qu'une déficience en vitamine A protégeait contre l'accumulation d'A2E chez la souris ABCA4", alors qu'une diète supplémentée en vitamine A augmentait la présence d'A2E et de ses précurseurs chez ces souris [71]. Il a aussi été découvert qu'un supplément oral diététique de lutéine permet d'augmenter la pigmentation de la macula chez les patients, sans toutefois restaurer leur vision centrale, et ce après 6 mois de traitement. La lutéine est un pigment maculaire primordial dans le processus de vision normale ainsi qu'un facteur clé dans la protection des photorécepteurs et des cellules de l'EPR contre les dommages oxydatifs [72]. Ceci corrobore le fait que l'acuité visuelle de patients atteints de Stargardt est proportionnelle à la quantité de pigments présents dans la macula [73]. L'augmentation d'AMPc par des moyens pharmacologiques restaure aussi le pH des lysosomes des cellules de l'EPR et augmente ainsi leur pouvoir de dégradation et de recyclage des photorécepteurs même en présence d'A2E [74].
1.4.3. Interventions chirurgicales
Il est possible de prévenir la maladie de Stargardt chez les individus atteints de cette pathologie grâce à une fécondation in vitro suivie d'un diagnostic génétique de
15 préimplantation. Le cas d'un homme atteint de Stargardt désirant concevoir un enfant sain avec sa conjointe, elle-même porteuse d'une mutation dans ABC A4, a été rapporté en 2010 [75]. Une fécondation in vitro a eu lieu, et un embryon porteur d'un allele mutant du père et de 1'allele sauvage de la mère a été implanté avec succès dans l'utérus de la mère. Une petite fille porteuse mais en santé est venue au monde 9 mois plus tard.
1.4.4. Thérapie génique
La thérapie génique constitue aussi une voie très prometteuse. Une construction de la forme sauvage du gène ABCA4 humain dans un vecteur lentiviral a déjà été transfectée avec succès dans l'espace subrétinien de souris ABCA4 ", permettant ainsi une diminution significative de l'accumulation d'A2E grâce à une expression quasi normale d,ABCA4 [76]. Ce traitement pourrait éventuellement être envisagé, avec précaution, pour soigner toutes les formes de dégénérescences maculaire associées à la défectuosité d,ABCA4 chez l'humain.
1.5. Étude de la population québécoise 1.5.1. L'effet fondateur
1.5.1.1. Histoire de la population québécoise
Un effet fondateur se produit lors de l'établissement d'une nouvelle population sur un territoire neuf, où la notion de goulot d'étranglement, ou d'entonnoir démographique, est caractéristique. On observe une importante réduction de la taille de la population fondatrice par rapport à la population mère, ainsi qu'un changement du profil des fréquences alléliques dans cette nouvelle population [77]. Ainsi, des alleles de fréquence plus rare associés à certaines maladies mendéliennes se retrouvent beaucoup plus répandus dans cette population, menant ainsi à une prévalence accrue de ces maladies.
Au Québec, 6 millions de Canadiens-Français descendent d'environ 8500 immigrants Français, arrivés entre 1608 et 1759. L'établissement de la population de la Nouvelle-France a par la suite entraîné une série d'effets fondateurs régionaux dans la
16 province [78]. Certaines maladies mendéliennes y sont aussi plus fréquentes qu'ailleurs au monde, comme par exemple dans les régions de Charlevoix et Saguenay-Lac-St-Jean. La fibrose kystique, la tyrosinémie héréditaire de type 1 et la dystrophic musculaire oculo-pharyngée constituent de bons exemples de maladies possédant une prévalence supérieure dans certaines régions de la province [79]. L'effet fondateur au Québec est donc un concept bien connu, qui caractérise de nombreuses maladies monogéniques.
1.5.1.2. Le Stargardt et l'effet fondateur
Certaines mutations associées à la maladie de Stargardt sont plus fréquentes, telles que G863A, A1038V et G1961E, et pourraient avoir des fréquences différentes dans des populations distinctes, ceci dû à la présence d'un effet fondateur [40, 63]. Il est d'ailleurs possible que chez des groupes ethniques différents, un allele puisse avoir des effets beaucoup plus drastiques, dépendant de la prévalence de gènes modificateurs encore inconnus, d'effets environnementaux ou de la fréquence d'autres mutations dans ABCA4 chez cette population [80].
Le Stargardt étant le plus souvent une maladie héréditaire récessive et monogénique, il est possible de supposer qu'un effet fondateur serait impliqué dans sa fréquence et sa distribution chez la population canadienne-française, comme bon nombre d'autres maladies génétiques. Un effet fondateur pour le Stargardt, ou du moins une fréquence allélique plus élevée pour certaines mutations, ont en effet été observés dans d'autres pays, comme en Allemagne [81], en Afrique du Sud [82], au Portugal [53], en Hongrie [83], en Europe de l'Est [63] ainsi qu'au Danemark [84].
1.5.2. Recrutement des familles
Au Québec, l'incidence et la prévalence du Stargardt restent encore inconnues. Notre étude réunit 19 familles répandues à travers la province, allant d'un noyau de trois individus prélevés à 43 par famille, pour un total de 29 patients atteints de Stargardt récessif. Ces derniers ont tous été examinés par un ophtalmologiste qualifié et ont donné leur consentement afin de participer à cette étude.
17 1.5.3. Impact du dépistage génétique
Le dépistage génétique possède des avantages non négligeables. En effet, il permet de confirmer un diagnostic au niveau moléculaire lorsque deux dystrophies se chevauchent ou possèdent des symptômes semblables. Le dépistage fournit également un pronostic de l'état de la vision pour l'avenir du patient. Il permet d'établir un système de corrélation génotype-phénotype, de trouver de nouveaux gènes et voies biochimiques, d'identifier les porteurs et de conseiller les couples désirant avoir des enfants, et enfin, de guider les futures stratégies thérapeutiques [85].
Dans le cas du Stargardt, la situation est encore plus complexe, car le taux de détection de mutations varie entre 66-80% seulement [86]. De plus, pour des raisons encore obscures mais tout de même relativement fréquentes en génétique humaine, certains porteurs de deux mutations ne développeront pas la maladie, alors que d'autres, qui ne possèdent aucune variation dans la séquence codante du gène, seront atteints de Stargardt. Il faut ainsi être très prudent lors d'entrevues de conseil génétique avec des familles, même si ce genre de rencontre leur permet de mieux comprendre la maladie. Le dépistage permet donc de faire avancer la recherche, en plus de fournir une aide précieuse aux familles atteintes de la maladie de Stargardt.
1.6. Hypothèses et objectifs 1.6.1. Problématique et hypothèses
Puisque le gène ABCA4 est le seul à être associé à ce jour à la forme récessive du Stargardt, nous en avons fait l'objet d'une étude sur des familles canadiennes-françaises souffrant de cette maladie. Il s'agit de la première étude de cette envergure au Québec et dans les environs, regroupant un nombre considérable de familles.
Comme il a été mentionné dans la section 1.5.1, le Stargardt est également une maladie qui, dans certaines régions du monde, est influencée par un effet fondateur. L'établissement particulier de la population québécoise en Nouvelle-France rend ce phénomène fréquent dans la province pour plusieurs autres maladies aussi.
KS Ainsi, notre hypothèse de départ était que l'étude d'ABCA4, un gène très polymorphique, révélerait beaucoup de variations dans la population canadienne-française, autant des mutations que des polymorphismes. Il était également plausible que de nouvelles variations soient répertoriées. Ces dernières avaient le potentiel d'être spécifiques à la population canadienne-française.
De plus, comme bon nombre d'autres maladies génétiques, nous pensions qu'un effet fondateur influencerait l'incidence du Stargardt dans la province et les environs.
1.6.2. Objectifs
L'objectif de ce projet était d'effectuer une vaste étude sur ABCA4 dans la population canadienne-française.
Nous voulions tout d'abord étudier ce gène chez les familles souffrant de la forme récessive du Stargardt, afin de répertorier le plus de mutations possible. Nous voulions également évaluer la présence d'un effet fondateur au sein de la population étudiée.
Objectifs spécifiques :
1. Génotyper des marqueurs microsatellites entourant le locus ABC A4 chez les patients et les familles recrutés.
- Confirmer la ségrégation du gène selon le mode de transmission récessif de la maladie de Stargardt.
2. Sequencer les 50 exons du gène, les jonctions introns-exons ainsi que la région promotrice chez tous les patients.
3. Analyser les variations observées.
- Comparer les séquences entre tous les individus afin d'identifier les variations présentes.
19 - Classer les variations comme étant nouvelles ou déjà répertoriées selon la littérature et les différentes bases de données disponibles.
- Déterminer si ce sont des mutations ou des polymorphismes selon leur classement dans la littérature et les bases de données.
- Confirmer les nouvelles mutations détectées par des méthodes alternatives (analyse sur gel, programme bioinformatique, comparaison avec la littérature, etc.). - Établir le taux de détection des mutations dans ABCA4 chez la population canadienne-française.
4. Évaluer la présence ou l'absence d'un effet fondateur.
- Comparer les haplotypes associés à des mutations partagées par plus d'une famille. - Étudier la généalogie des familles possédant des haplotypes communs.
20
CHAPITRE 2. Identification de nouvelles mutations dans le gène ABCA4
chez la population canadienne-française
Ce chapitre détaille le premier objectif à l'origine de mon projet de maîtrise, soit l'analyse des variations dans le gène ABCA4 chez la population canadienne-française.
2.1. Le Stargardt dans la population canadienne-française
À ce jour, aucune étude d'envergure sur la maladie de Stargardt n'a été menée au Québec. On parle en général d'une incidence de 1/8000 à 1/10 000 en Amérique du Nord [3], bien que cette étude ait été réalisée aux États-Unis, et non spécifiquement au Québec. La prévalence actuelle constitue donc une estimation grossière.
2.2. Objectif du projet
Mon projet de maîtrise visait donc l'étude du gène ABCA4 dans la population canadienne-française. Un projet regroupant un si grand nombre de familles n'avait jamais été rapporté auparavant. Le gène ABCA4, tel que discuté au chapitre précédent, est très polymorphique, ce qui portait à croire que nous trouverions beaucoup de variations, autant des mutations que des polymorphismes, dans la population étudiée. Nous espérions également détecter de nouvelles variations, potentiellement spécifiques à la population de la province de Québec et des environs.
2.3. Matériel et méthodes 2.3.1. Recrutement des familles
Ce projet a été approuvé par le comité d'éthique du centre de recherche du CHUL et respecte les principes de la déclaration d'Helsinski. Un formulaire de consentement a été
21 signé pour chacun des patients et les membres de leur famille participants. Les implications du projet leur ont été expliquées clairement.
Vingt-neuf patients provenant de 19 familles ont été recrutés parmi la population canadienne-française. Ils ont tous été examinés par des ophtalmologistes et optométristes certifiés. L'examen de base comprenait une mesure de l'acuité visuelle, une angiographie, un ERG et dans certains cas un test des couleurs. Soixante-cinq individus normaux sans lien avec les familles participantes ont également été recrutés en tant que contrôles.
2.3.2. Extraction d'ADN
Chaque patient ainsi que les membres de sa famille participant ont ensuite donné un échantillon de sang. L'ADN a été extrait avec la trousse commerciale Gentra PureGene DNA isolation (Qiagen). Chaque échantillon était par la suite codé et dosé au spectrophotomètre (Ultrospec 2100 pro), et finalement dilué à 10 pg/pl pour utilisation. 2.3.3. Génotypage de microsatellites
Quatre marqueurs microsatellites couvrant 6,9 Mb ont été génotypes (D1S435, D1S2804, D1S2819 et D1S497) grâce à des amorces couplées à quatre fluorochromes. Les réactions de génotypage ont été effectuées sur un PCR Perkin Elmer 9700 avec 15 ng d'ADN génomique. Les conditions de PCR ont été décrites précédemment [87].
Après la réaction de PCR, les échantillons étaient dilués entre 1/750 et 1/2000, puis combinés, pour les quatre marqueurs, dans 10 pi de formamide et 0,25 pi du standard de poids moléculaire GeneScan-500 LIZ (Applied Biosystems). Les échantillons ont été analysés à l'aide d'un appareil de séquençage ABI 3130XL (Applied Biosystems). Le logiciel Gene Mapper (version 4.0, Applied Biosystem) a été utilisé pour analyser les résultats de génotypage.
2.3.4. Séquençage d'ABCA4
Le gène ABCA4 a ensuite été analysé dans son ensemble par séquençage direct, selon la méthode de Sanger, pour tous les individus atteints de Stargardt. Les 50 exons, dont les jonctions exon-introns, ont été amplifiés par des amorces spécifiques et des
22
conditions utilisées auparavant [9]. La région promotrice a été amplifiée (de -833 à +35) par les amorces 5'-CC ATCC AGGTCTCAAGGGTCT-3' et 5'-GAGCCAGAG GCGCTCTTAAC-3'. Les conditions de PCR étaient 5 minutes à 95°C, 35 cycles de (20 secondes à 94°C, 40 secondes à 58°C et 1 minute à 72°C) suivi de 10 minutes à 72°C.
Les réactions de séquençage ont été effectuées par la plateforme de séquençage du CHUL, sur un appareil de séquençage ABI 3130XL (Applied Biosystems). Les régions où des mutations avaient été détectées ont également été séquencées chez les parents des patients, afin de les associer au bon haplotype. Les nouvelles variations ont aussi été vérifiées par séquençage direct de 130 chromosomes contrôles normaux. Le logiciel Automated Splicing mutation a permis de confirmer les nouvelles mutations d'épissage alors que Matlnspector a permis d'identifier la mutation dans la région du promoteur.
2.3.5. Culture de cellules et extraction d'ARN
Les cellules de lymphoblastes des patients souffrant de Stargardt ont été immortalisées avec le virus Epstein Bar. L'ARN a été extrait des cellules en culture avec une solution de Trizol (Invitrogene) et l'ADNc a été préparé avec la SS-III (Invitrogene). Les réactions de PCR et de nested PCR ont nécessité des amorces spécifiques et des conditions optimales qui sont décrites en annexe 1. Les amplicons d'ADN ont été visualisés sur gel d'agarose 2%. Les contrôles de rétine ont été obtenus de donneurs consentants sains, et l'ARN en a été extrait avec la solution de Trizol (Invitrogene).
2.4. Résultats
2.4.1. Les patients et leur famille
Le diagnostic de Stargardt a été confirmé sans équivoque par plusieurs ophtalmologistes chez les 29 patients de cette étude. Malgré cela, différents degrés de sévérité ont pu être observés, faisant varier l'âge d'apparition des symptômes chez certains, alors que d'autres montraient une progression plus lente ou plus rapide de la maladie, selon le cas. Certains présentaient un fond d'œil très caractéristique de la maladie de Stargardt, avec des flecks jaunâtres entourant la macula. Cette caractéristique était par contre moins
23
B
Patient FL-005
Patient MX-006
Patient PI-008
Figure 7. Photographies de fonds d'œil. A) Patient présentant des flecks jaunâtres caractéristiques. B) Patients ne présentant pas de flecks de façon
24
évidente dans d'autres cas. La figure 7 illustre un exemple des différences perçues lors de l'examen de fond d'œil chez quelques patients. La plupart des membres de la famille des patients étaient également examinés par un ophtalmologiste afin de confirmer leur état normal. Certaines familles présentent aussi d'autres cas de maladies de l'œil, telle que la rétinite pigmentaire, la DMLA et la sclérose choroïdienne.
2.4.2. Ségrégation des haplotypes
Le génotypage des familles visait à confirmer le mode de ségrégation autosomal récessif de la maladie. Il a été estimé qu'un total de quatre marqueurs microsatellites était suffisant pour étudier la ségrégation des haplotypes dans les 19 familles. Les familles participantes comprenaient 29 patients atteints de Stargardt qui se partageaient un total de 39 haplotypes distincts ségrégant avec la maladie.
Le mode autosomal récessif était confirmé lorsque les parents du patient étaient sains et que l'enfant était atteint. De plus, cet enfant devait posséder un allele du père et un allele de la mère, et ce génotype devait être associé exclusivement à la maladie, c'est-à-dire que quiconque possédant la même combinaison d'allèles devait être malade aussi.
Pour 13 de ces familles, le mode autosomal récessif a bien été confirmé. Par contre, pour les 6 familles restantes, soient BR, DI, MR, MX, SM et S W, il existe des discordances dans le patron de ségrégations des alleles (Figure 8). Les familles DI, MR et SW présentent un patient atteint de Stargardt possédant le même génotype qu'un frère ou une sœur normal(e). Les familles BR, MX et SM comprennent des patients se partageant trois alleles potentiellement associés à la maladie, ce qui implique qu'un des parents aurait dû présenter des symptômes aussi, ce qui n'est pas le cas.
2.4.3. Caractérisation génétique
Le séquençage direct a permis d'identifier des variations dans le gène ABCA4 chez de nombreux patients. Ces variations, lorsque déjà répertoriées dans la littérature, ont été classées en mutation ou en polymorphisme. Les nouvelles variations ont été vérifiées par différents essais spécifiques qui seront décrits plus loin.
25
Figure 8. Génotypage des familles de Stargardt étudiées, où les mutations détectées sont couplées aux haplotypes associés avec la maladie A) Familles dont la ségrégation des haplotypes est normale. B) Familles dont la ségrégation des haplotypes n'est pas typique du mode autosomal récessif. Les alleles de couleurs sont associés à la maladie de Stargardt, alors que les alleles blancs ne sont pas associés à la maladie. Les mutations ayant été associées avec un haplotype sont inscrites à côté de ce dernier.
LEGENDE
Q Femme ^ Rétinite pigmentaire | | Homme ^ ) Rétinopatie centrale séreuse y \ Patient décédé ^ Glaucome
# 0 Patient prélevé ( \ Dystrophic fovéo-maculaire
| Stargardt Q Sclérose choroïdienne 0 DMLA Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille # 19 Haplotypes et mutations correspondantes I L1_50P I G8.3A
o
^ ^ « • 0 0 9 ".-CCSi~5=
_.-___. . - - C C * 0 " HZf[
-à à o
• • • • vC-(D3 JC-CCT ot -010 ' . : ■ , . X-OS] ] ] [ m
à&
ô ô ù 6 0 b a ô
jT~*
u î >
■JC-CÙS uc-ooe C6~&
c77)
J: <___ x - o n X-G2* X-Olt i_ -OIS jr-Olî X-OI * X-CMS * *
X - O I t X-OI 2 ■C - j i. X-OIT X-CCO .'. - ' i t
m
li
Confirmation du mode autosomal récessif (AR)
26
LEGENDE
O Femme __\ Rétinite pigmentaire J Homme >lj) Rétinopatie centrale séreuse \__\ Patient décédé ^_\ Glaucome
Q Patient prélevé f"fc Dystrophie fovéo-maculaire | Stargardt \ _ \ Sclérose choroïdienne Q DMLA Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille #17
0-TS-011■0
TS-012S
TS-016ùTJ
TS-018 TS-008 TS-017 TS-013 TS-007 A1773ET A1773E I A 1 7 7 3 E |Confirmation AR pour les trois patients Une mutation détectée (1/2) Famille #15 TE-003 N96H ffl
à
TE-002Û
TE-004 TE-005 . • TE-001- j ]
Confirmation AR Une mutation détectée (1/2).0
TS-0021
27 LÉGENDE Ç j Femme J Homme \ / \ Patient décédé Ç j Patient prélevé | Stargardt 0 DMLA __\ Rétinite pigmentaire ^ J Rétinopatie centrale séreuse M Glaucome >f"fc Dystrophic fovéo-maculaire \ _ \ Sclérose choroïdienne Famille #2 CM-002
B
5
CM-003D
CM-001 I73T I R2107H «r CM-0047>
CM-005 Confirmation AR Une mutation détectée (1/2) H [73T D R2107H Famille #7 D-LQ-007 LQ-007a
G1961EI Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Confirmation AR 537C Deux mutations détectées (2/2)P P .o
LQ-007 LQ-007 LQ-007 ■R537C BR537C B R 5 3 7 C28 LÉGENDE Ç j Femme J Homme ï / \ Patient décédé Ç j Patient prélevé | Stargardt 0 DMLA
> ^ Rétinite pigmentaire microsatellites Marqueurs ^ J Rétinopatie centrale séreuse génotypes
PB Glaucome D1S435 Çh Dystrophic fovéo-maculaire | _\ Sclérose choroïdienne D1S2804 ABCA4 D1S2819 D1S497 Famille #4
D
FH-009 FH-004 «T ffl.<T7>
FH-OU FH-010H
n
0-
FH-001HO
FH-002E
D .D
FH-003 Confirmation AR Aucune mutation détectée (0/2) FH-005E
29 LÉGENDE Ç j Femme I | Homme 1/1 Patient décédé Ç j Patient prélevé I Stargardt 0 DMLA Famille #5 __\ Rétinite pigmentaire •jj) Rétinopatie centrale séreuse JPB Glaucome Ç^ Dystrophic fovéo-maculaire | _\ Scléroese choroïdienne M a r q u e u r s microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497
.©
FL-002 W821R (promoteur) - 439 A>GB
a
FL-001a .□
FL-003 FL-004 FL-005 W821R (promoteur)- 439 A>GH
I T ] W821R| I (promoteur»ni
43?A>G
i
Confirmation AR Une mutation détectée (1/2) Famille #8 R537C IVS19+1 G>C R537CB IIVS19 R537cl ly;,G>c
Confirmation AR VS19+1 G>C £>eux mutations détectées (2/2)30 LÉGENDE Ç j Femme I I Homme \ / \ Patient décédé Ç j Patient prélevé | Stargardt Q DMLA __\ Rétinite pigmentaire ^ J Rétinopatie centrale séreuse
Marqueurs microsatellites génotypes P B Glaucome Ç ^ Dystrophic fovéo-maculaire D1S435 D1S2804
| _\ Sclérose choroïdienne ABCA4
D1S2819 D1S497 Famille #11 Famille #12
.o-NG-002[
■
NG-003]
Confirmation AR Aucune mutation détectée (0/2)-0
NG-001 _ _ NG-004]
D
PI-008I
£>
PI-003E
Confirmation AR Aucune mutation détectée (0/2)31
LEGENDE
Ç j Femme __\ Rétinite pigmentaire | | Homme ^ J Rétinopatie centrale séreuse \ / \ Patient décédé JPB Glaucome
Ç j Patient prélevé Ç% Dystrophic fovéo-maculaire | Stargardt \_\ Sclérose choroïdienne Q DMLA
Famille #6
I
Confirmation AR0
T
Deux mutations G196]E | |L]25op détectées (2/2) Famille #16 TH-001
I
O
I
TH-002 1863AI L1250PE
Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 GY-002 GY-003 #GY G1961EI
TH-003 TH-0041
863A Confirmation AR Une mutation détectée (1/2)32 LÉGENDE ^ J Femme J Homme \ / \ Patient décédé Ç j Patient prélevé I Stargardt 0 DMLA >^| Rétinite pigmentaire ^ J Rétinopatie centrale séreuse JPB Glaucome >£fc Dystrophic fovéo-maculaire | _\ Sclérose choroïdienne Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille #18
O-TU-011.Ô
TU-010 TU-0021
TU-001 G1961EE
- .6 .6 .à
U-003 TU-004 TU-005 TU-006
B
G1961E H G 1 9 6 1 ELU LI
□ .n .n
TU-007 TU-008f l
■G1961EConfirmation A R pour les deux patients Une mutation détectée (1/2)
. TU-009
33
B
Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille #9 GH ^ n SE HSHhl Oi?, ShH °3SE Oag -r_Jr. ■ 0 . 8 ■0.5 ■O.s HShtl."l
r q . s O 2 3 U s -O.s -O.» ■O.sn
-■D On -n K • «"0,8
-O.SIo.si
■q..,i -o.gi 4111 On?; Confirmation AR Deux mutations détectées (2/2)■q.1
□Mn _
rP. Confirmation AR Une mutation détectée (1/2) -D -E O J ; ! ^ □ n 1 Q nit
l;:6sAfc.r..______3
ation AR-n "' '
i l i t o t i /-_«-_ c '—'__ ri _■ Pas de confirmation AR Deux mutations détectées (2/2)-o.
34 LÉGENDE Ç j Femme J Homme X/\ Patient décédé Ç j Patient prélevé I Stargardt Q DMLA __\ Rétinite pigmentaire ^ J Rétinopatie centrale séreuse JPB Glaucome Çh Dystrophic fovéo-maculaire \_\ Sclérose choroïdienne Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille #14
D
SM-003h
0
SM-001 SM-004 SM-005 # SM-002 SM-006.n n n
SM-007 SM-008 SM-009Famille #10 f 3 ~
MX-001 MX-003 ra£À
MX-004 MX-002B
MX-005I
MX-006 Pas de confirmation AR pour les quatre patientsAucune mutation détectée (0/2)
35 LÉGENDE Ç j Femme J Homme X/\ Patient décédé Ç j Patient prélevé | Stargardt Q DMLA >^l Rétinite pigmentaire ^ J Rétinopatie centrale séreuse JPB Glaucome Çh Dystrophic fovéo-maculaire | _\ Sclérose choroïdienne Marqueurs microsatellites génotypes D1S435 D1S2804 ABCA4 D1S2819 D1S497 Famille #3
D
_ _6
>I-006I
Famille #13.o
SW-006 A595vlÂ
SW-007D
û
•DI-005 * DI-003 *DI-004
1
I E
?w
n
-005P
SW-0086
SW-009o
DI-007B
O
o û
ù
6ù
Pas de confirmation AR Aucune mutation détectée (0/2) A595V A595vl L A595V-1 A595V Pas de confirmation ARA595v|| |A.595V Deux mutations
36 LÉGENDE Ç j Femme J Homme \ / \ Patient décédé Ç j Patient prélevé | Stargardt Q DMLA
__\ Rétinite pigmentaire microsatellites Marqueurs ^ j Rétinopatie centrale séreuse génotypes
PO Glaucome
D1S435
Çh Dystrophic fovéo-maculaire D1S2804
\_\ Sclérose choroïdienne ABCA4
D1S2819 D1S497 Famille #1
D
BR-005[
O
BR-006I
BR-007 BR-008 BR-009.□
BR-010.6 . 6 <
Pas de confirmation AR Aucune mutation détectée (0/2)O
BR-011 BR-012 BR-013
37
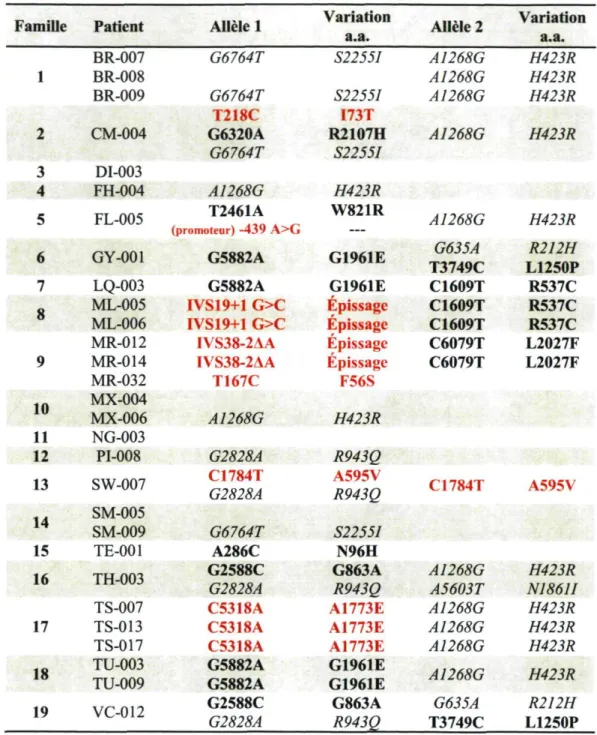
Seulement 26 des 58 chromosomes étudiés ont pu être associés à une mutation dans ABCA4 causant le Stargardt, ce qui donne un taux de détection de 45%. Un total de 15 mutations ont été répertoriées, soient 12 changements d'acides aminés, deux mutations altérant l'épissage du gène ainsi qu'une mutation dans le promoteur. Huit de ces mutations étaient déjà connues : N96H, R537C, W821R, G863A, L1250P, G1961E, L2027F et R2107H. Sept étaient de nouvelles variations qui n'avaient jamais été répertoriées dans la littérature, soit F56S, I73T, A595V, A1773E, IVS19+1 G>C, IVS38-2AA et une dans le promoteur, à -439 A>G. Cinq polymorphismes fréquents ont également été répertoriés :
R212H, H423R, R943Q NI86II et S2255I. Le tableau 1 illustre le classement des variations en mutation ou en polymorphisme pour chacun des 29 patients.
2.4.4. Confirmation des nouvelles mutations
Les exons porteurs des sept nouvelles mutations ont été analysés par séquençage direct chez 130 chromosomes normaux, et aucune n'a été retrouvée chez ces individus contrôles. Le tableau 2 dresse une liste des nouvelles mutations, de leur fréquence dans la population étudiée ici ainsi que chez les individus contrôles.
2.4.4.1. Étude des changements d'acides aminés
La figure 9 représente une protéine ABCA4 schématisée où ont été positionnées toutes les mutations répertoriées dans cette étude, sauf celle étant localisée dans le promoteur du gène. Les nouvelles mutations de changements d'acides aminés (F56S, I73T, A595V, A1773E) se trouvent toutes à proximité d'un domaine transmembranaire, ce qui indique qu'elles sont probablement critiques au bon repliement ainsi qu'au fonctionnement de la protéine. De plus, de part et d'autres de chacune de ces quatre nouvelles mutations se trouvent des mutations déjà bien répertoriées dans la littérature (Tableau 3), ce qui renforce l'idée que ces quatre nouvelles variations seraient de réelles mutations.
38
Famille Patient Allele 1 Variation a.a. Allele 2 Variation a.a.
BR-007 G6764T S2255I A1268G H423R 1 BR-008 A1268G H423R BR-009 G6764T S2255I I73T A1268G H423R T218C S2255I I73T A1268G 2 CM-004 G6320A G6764T R2107H S2255I A1268G H423R 3 DI-003 4 FH-004 A1268G H423R W821R A1268G 5 FL-005 T2461A (promoteur) -439 A>G H423R W821R A1268G H423R 6 GY-001 G5882A G5882A G1961E G1961E G635A T3749C C1609T R212H L1250P 7 LQ-003 G5882A G5882A G1961E G1961E G635A T3749C C1609T R537C 8 ML-005 IVS19+1 G>C Épissage C1609T R537C 8 ML-006 IVS19+1 G>C Épissage C1609T R537C MR-012 IVS38-2AA Épissage C6079T L2027F 9 MR-014 IVS38-2AA Épissage C6079T L2027F MR-032 T167C F56S 10 MX-004 T167C F56S 10 MX-006 A1268G H423R 11 NG-003 13 PI-008 SW-007 G2828A C1784T G2828A R943Q A595V R943Q C1784T A595V 14 SM-005 14 SM-009 G6764T A286C S2255I N96H 15 TE-001 G6764T A286C S2255I N96H 16 TH-003 TS-007 G2588C G2828A C5318A G863A
R943Q A1268G A5603T N1861I H423R TH-003
TS-007
G2588C G2828A
C5318A A1773E A1268G H423R
17 TS-013 C5318A A1773E A1268G H423R
TS-017 C5318A A1773E A1268G H423R
18 TU-003 TU-009 G5882A G5882A G1961E G1961E A1268G H423R
19 VC-012 G2588C G2828A G863A R943Q G635A T3749C R212H L1250P
Tableau 1. Liste des variations détectées chez les 29 patients souffrant de Stargardt. Les variations ont été classées selon la littérature en mutation ou en
polymorphisme. Les nouvelles mutations sont indiquées en rouge, alors que les
espaces blancs indiquent qu'aucune mutation n'a pu être détectée dans ABCA4 pour ces patients, (a.a. : acides aminés)
39
,, Changement de ... Nombre de patients Individus nucleotides porteurs contrôles
3 3 13 (promoteur) -439 A>G T167C T218C C1784T ND F56S I73T A595V 1 1 1 1 2 3 2 0/65 0/65 0/65 0/65 0/65 0/65 0/65 19 IVS19+1 G>C C5318A IVS38-2AA Épissage 1 1 1 1 2 3 2 0/65 0/65 0/65 0/65 0/65 0/65 0/65 38 38 IVS19+1 G>C C5318A IVS38-2AA A1773E Épissage 1 1 1 1 2 3 2 0/65 0/65 0/65 0/65 0/65 0/65 0/65
Tableau 2. Liste des nouvelles mutations détectées. La fréquence des nouvelles mutations dans la population étudiée ainsi que chez les individus contrôles sont indiquées. ND : non déterminé.
2.4.4.2. Etude des mutations d'épissage
Le programme de prédiction d'épissage Automated splicing mutation (ASM) a démontré que les deux mutations d'épissage, IVS19+1 G>C et IVS38-2AA, causeraient le saut d'un exon lors de l'épissage de l'ARNm d,ABCA4. Les deux mutations d'épissage ont également été positionnées sur la protéine ABCA4 à la figure 9.
Dans le cas de IVS19+1 G>C, c'est l'exon 19 qui est sauté, puisque le site donneur est altéré. Cela entraîne un changement du cadre de lecture et l'apparition d'un codon STOP, donc d'une protéine tronquée. Cette mutation, présente uniquement dans la famille ML, a également été vérifiée aux Pays-Bas par le laboratoire du Nederlands Institute for Neuroscience. Cette famille avait en effet consulté ce laboratoire, et nous avons confirmé les résultats qu'ils avaient obtenus. Ils ont eux aussi affirmé que cette variation n'avait jamais été répertoriée dans la littérature, et qu'elle n'était pas présente chez 120 de leurs
chromosomes normaux.
La mutation IVS38-2AA entraîne le saut de l'exon 38, toujours selon le programme de prédiction d'épissage ASM. Le site accepteur serait délété en partie, ce qui pourrait altérer l'épissage de l'ARNm. Les cellules de lymphoblastes des patients porteurs de cette mutation avaient été immortalisées dans notre laboratoire avant mon arrivée. L'ARN a été
40 extrait de ces cellules mises en culture et l'ADNc obtenu a pu être étudié. La figure 10 montre un gel d'agarose illustrant la présence de l'allèle sauvage (ARNm complet) et de l'allèle muté (sans exon 38) chez le même patient. Une forme intermédiaire d'ARNm possédant uniquement la deuxième moitié de l'exon 38 est également visible chez un autre patient. Cette forme alternative d'épissage tronque seulement une partie de l'exon 38 sans pour autant changer le cadre de lecture subséquent. L'abondance relative de ces deux formes d'ARNm reste toujours à déterminer, de même que si la deletion d'une partie de l'exon 38, située dans la région transmembranaire, affecte ou non la fonction d'ABCA4.
R537C W821R N96H I73T 1VS37-2AA A1773E G863A IVS19+1G> L2027F R2107H
Figure 9. Positionnement des nouvelles mutations sur la protéine ABCA4. Les
nouvelles mutations sont positionnées près des domaines transmembranaires et d'un domaine de liaison à l'ATP, ce qui indique leur rôle potentiellement important dans le bon fonctionnement de la protéine. Les mutations déjà répertoriées dans la littérature sont également indiquées.