HAL Id: hal-02663287
https://hal.archives-ouvertes.fr/hal-02663287
Submitted on 31 May 2020
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Screening potential repurposed COVID-2019 3C-like
protease inhibitors
Clifford Fong, South Australia
To cite this version:
Clifford Fong, South Australia. Screening potential repurposed COVID-2019 3C-like protease in-hibitors. [Research Report] Eigenenergy. 2020. �hal-02663287�
Screening potential repurposed COVID-2019 3C-like protease inhibitors
Clifford W. Fong
Eigenenergy, Adelaide, South Australia, Australia. Email: [email protected]
Keywords: COVID-2019 or SARS-CoV-2; 3C-like protease, or 3CLpro, or Mpro; inhibition; ACE2 inhibitor; HOMO-LUMO; quantum mechanics;
Abbreviations: Structure activity relationships SAR, ΔGdesolv,CDSfree energy of water desolvation, ΔGlipo,CDS
lipophilicity free energy, CDS cavity dispersion solvent structure of the first solvation shell, Dipole moment DM, Molecular Volume Vol, HOMO highest occupied molecular orbital, LUMO lowest unoccupied molecular orbital, HOMO-LUMO energy gap, RBD receptor binding domain.
Abstract
This study describes a linear free energy quantum mechanical method for relatively ranking how well a wide range of repurposed drugs might inhibit the SARS-CoV-2 protease. Inhibitors that are predominantly charged at physiological pH, such as Hydroxychloroquine and Chloroquine, are unlikely to be effective inhibitors as charged species are not easily transported across host cell membranes. Small lipophilic neutral with low water desolvation properties inhibitors are likely to be most effective, all else being equal, with Favipiravir being an example. An examination of Remdesivir and its metabolites suggest that GS441524 is likely to be most effective inhibitor. An examination of Remdesivir and its metabolites suggest that GS441524 is likely to be most effective inhibitor. It is predicted that the neutral form of Imatinib binds better to the ACE2 receptor than the neutral Arbidol, with both charged species showing much reduced binding.
Introduction
The search for therapeutic drugs that can ameliorate the effects of COVID-2019 (SARS-CoV-2) infections is being vigorously pursued. Screening studies to evaluate the potential efficacy of repurposed and novel 3C-like SARS-CoV-2 protease inhibitors typically include docking binding energies to the protease using the x-ray protease structure PDB6LU7 combined with molecular dynamics studies. [1-18] One study uses a Deep Docking quantitative structure-activity relationship (QSAR) model trained on docking scores of database subsets to approximate in an iterative manner the docking of over one billion drugs to identify potential new inhibitors of the 3C-like protease. [3] Another study has also used a similar deep learning approach to identify new inhibitors. [4] Docking studies are approximate and dependent upon the particular docking method use, and molecular dynamics studies are intensive and reliant on the application of force fields. The methods can give “ab initio” binding properties that may relate to in vivo biological inhibition of the 3C-like protease.
Alternately, QSAR studies rely on experimentally determined in vitro protease inhibitory data (IC50, pIC50 etc) that apply to a known structurally similar series of inhibitors, and can then be
used to predict other likely more effective inhibitors. Typically physiochemical descriptors used in QSAR studies include molecular weight, number of rotatable bonds, number of aromatic rings, number of stereocenters, number of hydrogen bond acceptors, number of hydrogen bond donors, Lipinski rule of five indicators, etc. [4]
Both the “ab initio” and QSAR approaches do not often translate to in vivo clinical success in the history of therapeutic drug development. Failures mainly occur in targeting efficacy and
undesirable side effects. Clearly another important factor in determining drug efficacy at the desired target is the transport of the drug and how well the drug can be transported across the cell membrane to the target receptor.
We have recently developed a quantum mechanically based structure activity model for the inhibition of the 3C-like protease from the HKU4 (HKU4-CoV) [18] for a particular series of inhibitors. HKU4 (HKU4-CoV) belongs to the same 2c lineage as MERS-CoV and shows high sequence similarity with MERS-CoV. [19] It has been shown that equation 1 accurately describes the inhibition of the protease:
Eq 1 Inhibition of 3C-like protease pIC50 for 40 compounds from [19] was:
pIC50= 0.05ΔGdesolv,CDS - 0.11ΔGlipo,CDS - 0.08Dipole Moment – 0.23(HOMO-LUMO) + 6.63
where ΔGdesolv,CDS is the free energy of water desolvation, ΔGlipo,CDS is the lipophilicity free
energy, the dipole moment in water, and HOMO-LUMO is the energy gap in water.
The important finding is that pIC50 is dominantly related to the HOMO-LUMO energy gap of the
inhibitors. The HOMO-LUMO gap is an inherent descriptor of the innate reactivity of the inhibitor, and is related to how the inhibitor binds to the protease. In particular, how the HOMO of the protease (HOMOprot) interacts with the LUMO of the inhibitor (LUMOinhib), and how the
HOMO of the inhibitor (HOMOinhib) interacts with the LUMO of the protease (LUMOprot). These
molecular interactions fundamentally define the inhibitor-protease binding interaction.
Unfortunately calculating the HOMOprotand LUMOprot is prohibitive, and other molecular factors
are also involved determining the binding process in the protease binding pocket and eventually the binding energy. [23]
One advantage in finding relationships like eq 1 is that the HOMO-LUMO gap of the inhibitor also gives some indication of the likely side effects of the inhibitors, since the HOMO-LUMO gap reflects the inherent chemical reactivity of the inhibitors to other non-desirable biological targets and processes. [20-23]
This study seeks to evaluate whether the HOMO-LUMO gap may be useful for screening potential inhibitors of the 3C-like protease of SARS-CoV-2, extrapolating from the relationship previously discovered in eq 1. [19] This methodology could be a useful adjunct to more detailed docking and molecular dynamics studies which do not use electron molecular orbitals, which are fundamental descriptors of chemical or physical properties and processes.
A critical factor determining the efficacy of potential 3C-like protease inhibitors is how such drugs can prevent viral replication after infection of the host cell. SARS-CoV-2 shares 79.5% sequence identity with SARS-CoV, and uses the same angiotensin converting enzyme 2 (ACE2) receptor as SARS-CoV as a mechanism of cell entry. ACE2 is a trans-membrane
metallocarboxypeptidase known to be highly concentrated in airway epithelial cells. The envelope-anchored spike protein promotes coronavirus entry into host cells by binding to the host ACE2 receptor, fusing viral and host membranes. Coronaviruses, including SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), and infectious bronchitis virus (IBV), fuse at the plasma membrane or use receptor-mediated endocytosis and fuse with endosomes, depending on the cell or tissue type. This virus-receptor interaction promotes both cross-species and human-to-human transmission of the virus, allowing the viral genome to be transported to the host cell cytoplasm for replication.
Viral spread and pathogenesis in the infected host requires spike protein binding to the ACE2 receptor, and spike protein priming by host cellular proteases resulting in fusion of viral and cellular membranes using endosomal proteases. Genomic RNA is then released into the cytoplasm of the host cell and translated into polyproteins and processed by two proteases, the main protease (3C-like protease or Mpro) and the papain-like protease (PLpro). Protease inhibitors block the viral life cycle by selectively preventing such proteolytic cleavage.
Zhang et al [24] determined that the crystal structure of Mpro(or 3CLpro) of SARS-CoV-2 is very similar to the structure of the SARS CoV Mpro, as expected from the 96% sequence identity. The Mpro of SARS-CoV-2 forms a tight dimer, through a salt-bridge interaction. In particular, they
studied the inhibitory efficacy of some α-ketoamide compounds (see Figure 1) with SARS CoV Mpro, and found that compound 13b inhibited the purified recombinant SARS-CoV-2 Mpro with IC50 = 0.67 μM, whereas the corresponding IC50 values for inhibition of the SARS-CoV Mpro
and the MERS-CoV Mpro were 0.90 and 0.58 μM respectively. These IC50 values are equivalent
within experimental error, so these data and other studies indicate that the protease has a very close identity in the SARS-CoV-2, SARS-CoV and MERS-CoV replicating process. [24-26] Lan et al [27] have determined the crystal structure of the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the cell receptor ACE2. The overall
ACE2-binding mode of the SARS-CoV-2 RBD is nearly identical to the RBD of SARS-CoV, which also uses ACE2 as the cell receptor. A notable feature of both RBD-ACE2 interfaces was the networks of hydrophilic interactions: 13 hydrogen bonds and 2 salt bridges at the
SARS-CoV-2 ACE2 interface, and 13 hydrogen bonds and 3 salt bridges at the SARS-CoV RBD-ACE2 interface. The structure contains residues Thr333-Gly526 of the SARS-CoV-2 RBD, residues Ser19-Asp615 of the ACE2 N-terminal peptidase domain, one zinc ion, four N-acetyl-β-glucosaminide (NAG) glycans linked to ACE2 Asn90, Asn322 and Asn546 and to RBD Asn343, as well as 80 water molecules. It is clear that desolvation and resolvation processes must be important for drugs that act as inhibitors of ACE2, such as Arbidol and Imatinib.
Arbidol is an antiviral agent that blocks viral fusion with target membranes, prohibiting viral entry into cells. It is known that Arbidol can effectively inhibit SARS-CoV-2 infection at a concentration of 10-30 µM in vitro. [16] Blocking virus-host fusion through inhibiting the Abl kinase pathway is also a promising target for the development of antiviral therapies, since this kinase activity is required for entry of coronaviruses. Abl kinase inhibitors such as Imatinib may prevent the coronavirus membrane fusion step. A high-throughput screen identified Imatinib as an inhibitor of SARS-CoV and MERS-CoV. Imatinib has previously been demonstrated to inhibit both SARS-CoV and MERS-CoV, as well as infectious bronchitis virus entry to cells. Both SARS-CoV and SARS-CoV-2 viruses use ACE2 as a receptor. [2] A clinical trial will test whether Imatinib can block the first round of cell to cell virus infection and therefore stop or prevent SARS-CoV-2 infection in humans. [17]
We have previously shown that the general equation 2 can successfully describe transport processes of drugs across the cell membrane. [28-34]
Eq 2
Drug Transport = ΔGdesolv,CDS + ΔGlipo,CDS + Dipole Moment + Molecular Volume
This study will examine whether the four independent molecular specifiers in eq 2 can be useful in screening how drugs might be transported across the host cellular membrane or block the virus-host membrane interaction that is critical for progressing COVID-2019 infection. The study will also examine if eq 1 can be applicable to the inhibition of the protease involved in the SARS-CoV-2, SARS-CoV and MERS-CoV replication process. It is proposed that the molecular specifiers used in eq 1 and eq 2 can be useful screening tools to evaluate the potential efficacy of therapeutic drugs that may be active against the SARS-CoV-2 virus.
Results
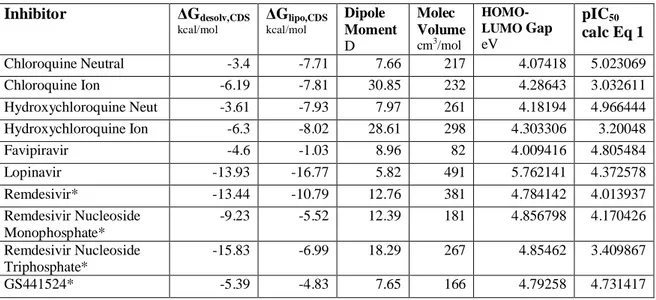
Table 1 shows the molecular specifiers of a diverse range of repurposed inhibitors that are being considered as therapeutic agents against the COVID-2019 virus. Most of these drugs are
currently used for other diseases and have the advantage of being clinically or near clinically available in terms of human safety considerations.
On the assumption that eq 1 for the 3C-like protease from the HKU4 (HKU4-CoV) is a good surrogate for the SARS-CoV-2 protease, the HOMO-LUMO gap is the critical decider of likely
efficacy against the COVID-2019 virus. The smaller the gap of a particular inhibitor, the more reactive is the inhibitor towards the SARS-CoV-2 protease, with other molecular specifiers such as lipophilicity, dipole moment and water desolvation being of lesser importance. [19] The column titled pIC50calc Eq 1 in Table 1 shows the calculated inhibition of the 3C-like protease
of COVID-2019 from eq 1. This assumes that eq 1 derived for the HKU-CoV applies to the SARS-CoV-2 protease, consistent with Zhang’s and others findings. [24-26]
However, the efficacy of the inhibitors also depends on how well the drug can enter the host cell by being transported across the cell membrane before inhibiting the protease. As eq 2 shows, drug transport is dependent upon water desolvation, lipophilicity, dipole moment and molecular volume.
We have shown that water desolvation and lipophilicity are critical factors in determining how drugs can cross cell membranes by diffusion, facilitated diffusion or other active transport processes. [28-34] Desolvation of drugs becomes critical as drugs leave the bulk aqueous environment of the blood plasma and interacts with the cell membrane to initiate the transport process: in particular charged drugs have high desolvation requirements, as can be seen for the ΔGdesolv,CDS values of Chloroquine, Hydroxychloroquine, Remdesivir and Remdesivir
triphosphate, Nafamostat, Galidesivir, Arbidol, Imatinib. The lipophilicity ΔGlipo,CDSvalues do
not change significantly from the neutral to the charged species (since most drugs are relatively large organic molecules with just one or two charged moieties) but the dipole moment changes are large for the charged species. Table 1 shows the calculated ΔGdesolv,CDS, ΔGlipo,CDS, and
molecular volumes for the various drugs.
It is clear that inhibitors that are predominantly charged at physiological pH levels such as Chloroquine, Hydroxychloroquine, Remdesivir and Remdesivir triphosphate, Nafamostat, Galidesivir, Arbidol, Imatinib will not pass through cell membranes as fast as their neutral counterparts, due to unfavorable larger ΔGdesolv,CDS values. Hence their overall efficacy as
inhibitors of the SARS-CoV-2 protease will be diminished. The same conclusions apply to Arbidol and Imatinib which are known to interact with the ACE2 receptor, [16,17] since the charged forms of the drugs will require some larger degree of desolvation of their primary solvation shells before interacting with the ACE2 receptor than would the neutral forms. [28-34] The HOMO-LUMO gap for the neutral Imatinib is lower than that of the neutral Arbidol, indicating that it binds more effectively to ACE2 than Arbidol.
It is known that increased drug lipophilicity increases cell membrane transport, while increased water desolvation and molecular volume of the drugs decrease the ease of cell membrane transport processes. [28-34] Since viral infectivity depends on interactions between the host cell plasma membrane and the virus envelope, lipid dependent attachment to human host cells is a critical factor. It may be feasible to reduce the infectivity of some coronaviruses, possibly by
inhibiting viral lipid-dependent attachment to host cells. [35] While the charged Arbidol and Imatinib suffer significant desolvation penalties compared to the neutral species, the lipophilic values are comparable.
Inspection of Table 1 pIC50 calc values suggest that Ruxolitinib, GS441524, Saquinavir,
Ledipasvir, Baricitinib, Carfilzomib, Favipiravir and Efavirenz are the strongest inhibitors of the protease after excluding any drugs that may be substantially charged at physiological pH levels. However using the raw HOMO-LUMO gap values, Saquinavir, Ledipasvir, Velpatasvir,
Efavirenz and Nitazoxanide are the inhibitors likely to be most active. Favipiravir shows high inhibitory properties as per eq 1, and would be expected to show high membrane transport properties based on its eq 2 molecular specifier properties.
Comparison of Remdesivir and its metabolites GS441524, Remdesivir nucleoside
monophosphate and Remdesivir nucleoside triphosphate (all as neutral species) [36] shows that GS441524 has a lower desolvation energy, and higher lipophilicity of all the Remdesivir family (see Figure 2). Eq 1 predicts that GS4412514 has the greatest inhibitory effect and would be expected to have an enhanced membrane transport compared to other members of the Remdesivir family based on the molecular specifiers in eq 2.
Examination of the inhibitors studied by Zhang [24] shows that compound Z11 is predicted to have a higher pIC50 than Z13A or Z13B, in agreement with Zhang’s findings. However Zhang
found that Z14 had almost nil inhibitory effect on Mpro(or 3CLpro) of SARS-CoV-2, and concluded that the bulky t-Butylcarboxy group was required to pass through the cellular membrane of the human Calu-3 cells infected with SARS-CoV-2. However Table 1 shows the predicted pIC50 is higher than Z11, Z13A or Z13B. It appears that Z14B exists as the protonated
species at physiological pH levels, and subsequently has a much reduced pIC50 value.
Discussion
This study seeks to find approximate screening methods that can allow the relative ranking of various inhibitors that can bind with the Mpro(or 3CLpro)of SARS-CoV-2, as discussed in the Results section. The rankings are based on the assumption that eq 1 holds for the SARS-CoV-2 protease. It is also assumed that the structure activity relationship eq 1 also hold for other structurally diverse inhibitors than the series for which eq 1 was applicable. There is good literature evidence that the Mpro(or 3CLpro)of SARS-CoV-2, CARS-CoV and MERS are very similar.[24-26]
We have previously discussed the relationship amongst the frontier molecular orbitals of the protein and inhibitors that bind to that protein, ie the HOMOprot, LUMOprot, and the HOMOlig,
LUMOlig. [23] Where the HOMO and LUMO of the ligand and the protein are close together,
Abuhammad et al [37] also examined bat HKU4 coronavirus 3CLpro inhibitors as antivirals against the MERS coronavirus. However attempts to derive successful QSAR
models failed, prompting the use of ligand efficiency [LE = −log(IC50)/heavy atom count] as an
alternative response variable instead of -log(IC50). The best QSAR showed a reliance on number
of aromatic bonds, X,Y and Z dipoles, LUMO and various fitting values derived from a statistical training analysis. Eq 1 appears to be a simpler and more informative QSAR model, that does not require statistical artifacts such as training sets, but uses the raw experimental data. The molecular specifiers in eq 2 also may be used to give a qualitative insight into how well the inhibitors can access various cell membrane transport processes. It is noted that traditionally docking studies and sometimes molecular mechanics studies have been used to screen for
improved inhibitors, the present method is simpler and also gives information about how well the drugs might pass through cell membranes.
Conclusions
This study describes a linear free energy quantum mechanical method based on eqs 1 and 2 for relatively ranking how well a wide range of repurposed drugs might inhibit the SARS-CoV-2 protease. Inhibitors that are predominantly charged at physiological pH, such as
Hydroxychloroquine and Chloroquine, are unlikely to be effective inhibitors as charged species are not easily transported across host cell membranes. Small lipophilic neutral with low water desolvation properties inhibitors are likely to be most effective, all else being equal, with Favipiravir being an example. An examination of Remdesivir and its metabolites suggest that GS441524 is likely to be most effective inhibitor. It is predicted that the neutral form of Imatinib binds better to the ACE2 receptor than the neutral Arbidol, with both charged species showing much reduced binding.
Experimental Methods
All calculations were carried out using the Gaussian 09 package. Energy optimizations were at the DFT/B3LYP/6-31G(d,p) (6d, 7f) level of theory for all atoms in water. Selected
optimizations at the DFT/B3LYP/6-311+G(d,p) (6d, 7f) level of theory gave very similar results to those at the lower level. Optimized structures were checked to ensure energy minima were located, with no imaginary frequencies. Energy calculations were conducted at the
DFT/B3LYP/6-31G(d,p) (6d, 7f) for neutral and cationic compounds with optimized geometries in water, using the IEFPCM/SMD solvent model. With the 6-31G* basis set, the SMD model achieves mean unsigned errors of 0.6 - 1.0 kcal/mol in the solvation free energies of tested neutrals and mean unsigned errors of 4 kcal/mol on average for ions. [38] The 6-31G** basis set has been used to calculate absolute free energies of solvation and compare these data with experimental results for more than 500 neutral and charged compounds. The calculated values were in good agreement with experimental results across a wide range of compounds. [39,40] Adding diffuse functions to the 6-31G* basis set (ie 6-31+G**) had no significant effect on the
solvation energies with a difference of less than 1% observed in solvents, which is within the literature error range for the IEFPCM/SMD solvent model. HOMO and LUMO calculations included both delocalized and localized orbitals (NBO).
It is noted that high computational accuracy for each species in different environments is not the focus of this study, but comparative differences between various species is the aim of the study.
References
[1] C Liu, Q Zhou, Y Li, LV Garner, SP Watkins, LJ Carter, J Smoot, AC Gregg, AD Daniels, S Jervey, D Albaiu, Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases, ACS Cent. Sci. 2020, 6, 315-331
[2] L Dong, S Hu, J Gao, Discovering drugs to treat coronavirus disease 2019 (COVID-19), Drug Discov Therapeutics, 2020, 14, 58-60.
[3] AT Ton, F Gentile,[M Hsing, F Ban,[ A Cherkasov, Rapid Identification of Potential Inhibitors
of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds, Mol. Inf. 2020, 39, 2000028-2000036
[4] A Zhavoronkov, V Aladinskiy, A Zhebrak, B Zagribelnyy, V Terentiev, DS Bezrukov, D Polykovskiy, R Shayakhmetov, A Filimonov, P Orekhov, Y Yan, O Popova, Q Vanhaelen, A Aliper, Ivanenkov, Potential COVID-2019 3C-like Protease Inhibitors Designed Using Generative Deep Learning Approaches, doi.org/10.26434/chemrxiv.11829102.v2
[5] M Macchiagoden, M Pagliai, P Procacci Inhibition of the Main Protease 3CLPro of the Coronavirus Disease 19 via Structure-Based Ligand Design and Molecular Modeling, ArXiv:2020.09937v2, 2 Mar 2020
[6] V Kumar, KP Tan, YM Wang, S Wei Lin, PH Liang, Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors, Bioorg. Med. Chem. 2016,
http://dx.doi.org/10.1016/j.bmc.2016.05.013
[7] BR Beck, B Shin, Y Choi, S Park, K Kang, Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model, Comp Struct Biotech J, 2020, 18, 784–790
[8] YW Chen , C-P Yiu , K-Y Wong, Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates, F1000Research 2020, 9, 129-146
[9] CJ Gordon, EP Tchesnokov, JY Feng, DP Porter, M Götte, The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus, 2020, https://www.jbc.org/cgi/doi/10.1074/jbc.AC120.013056
[10] ES Amirian, JK Levy,
Current knowledge about the antivirals remdesivir (GS-5734)
and GS-441524 as therapeutic options for coronaviruses,
One Health, 2020, 9,100128-100135
[11] M Wang, R Cao, L Zhang, X Yang, J Liu, M Xu, Z Shi, Z Hu, W Zhong, G Xiao,
Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro, Cell Research, 2020, 0, 1–3; https://doi.org/10.1038/s41422-020-0282-0 [12] KM Kapoor, A Kapoor, Role of Chloroquine and Hydroxychloroquine in the Treatment of COVID-19 Infection-A Systematic Literature Review, medRxiv preprint, 2020, doi:
[13] Z Xu, C Peng, Y Shi, Z Zhu, K Mu, X Wang, W Zhu, Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation, 2020,
bioRxiv preprint, https://doi.org/10.1101/2020.01.27.921627
[14] S Khaerunnisa, H Kurniawan, R Awaluddin, S Suhartati, S Soetjipto Potential Inhibitor of COVID-19 Main Protease (Mpro) from Several Medicinal Plant Compounds by
Molecular Docking Study, www.preprints.org, 2020, doi:10.20944/preprints202003.0226.v1 [15] J Stebbing, A Phelan, I Griffin, C Tucker, O Oechsle, D Smith, P Richardson, COVID-19: combining antiviral and anti-inflammatory treatments, www.thelancet.com/infection, 2020, 20, https://doi.org/10.1016/S1473-3099(20)30132-8
[16] S Weston, CM Coleman, R Haupt, J Logue, K Matthews, MB Frieman, Broad anti-coronaviral activity of FDA approved drugs against SARS-CoV-2 in vitro and SARS-CoV in vivo, BioRxiv, 2020, doi: https://doi.org/10.1101/2020.03.25.008482
[17] P Rousselot, Versailles Hospital, Imatinib in COVID-19 disease in aged patients, https://clinicaltrials.gov/ct2/show/NCT04357613
[18] F Lin, XM Fu, C Wang, SY Jiang, JH Wang, SW Zhang, L Yang, Y Li,
QSAR, Molecular Docking and Molecular Dynamics of 3C-like Protease Inhibitors, Acta Phys. Chim. Sin. 2016, 32, 2693-2708
[19] CW Fong, Inhibition of COVID-2019 3C-like protease: structure activity relationship using quantum mechanics, 2020, https://hal.archives-ouvertes.fr/hal-02529030
[20] CW Fong, Screening anti-colorectal cancer drugs: free radical chemotherapy, HAL Archives, 2019,https://hal.archives-ouvertes.fr/hal-02271521v1
[21] CW Fong, Role of stable free radicals in conjugated antioxidant and cytotoxicity treatment of triple negative breast cancer, HAL archives 2018,https://hal.archives-ouvertes.fr/hal-01803297
[22] CW Fong, Toxicology of platinum anticancer drugs: oxidative stress and antioxidant effect of stable free radical Pt-nitroxides, HAL Archives, 2019,hal-01999011, version 1,
https://hal.archives-ouvertes.fr/hal-01999011v1
[23] CW Fong, The antioxidant properties of Edaravone analogs and Amyloid β induced oxidative stress and neurotoxicity in Alzheimer’s disease – a quantum mechanical study, 2020, https://hal.archives-ouvertes.fr/hal-02428379v1
[24] L Zhang, D Lin, X Sun, U Curth, C Drosten, L Sauerhering, S Becker, K Rox, R Hilgenfeld,
Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors, Science, 2020, 368, 409–412.
[25] F Wang, C Chen, W Tan, K Yang, H Yang, Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Sci Rep. 2016, 6, 22677 https://doi.org/10.1038/srep22677.
[26] H Yang, M Bartlam, Z Rao, Drug design targeting the main protease, the Achilles' heel of coronaviruses, Curr Pharm Des. 2006, 12, 4573‐4590.
[27] J Lan, J Ge, J Yu, et al, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature, 2020, https://doi.org/10.1038/s41586-020-2180-5
[28] CW Fong, Permeability of the Blood–Brain Barrier: Molecular Mechanism of Transport of Drugs and Physiologically Important Compounds, J Membr Biol. 2015, 248,651-69.
[29] CW Fong, The extravascular penetration of tirapazamine into tumours: a predictive model of the transport and efficacy of hypoxia specific cytotoxic analogues and the potential use of cucurbiturils to facilitate delivery, Int J Comput Biol Drug Design. 2017, 10, 343-373
[30] CW Fong, Statins in therapy: Understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur J Med Chem. 2014, 85, 661-674 [31] CW Fong, Predicting PARP inhibitory activity – A novel quantum mechanical based model. HAL Archives. 2016, https://hal.archives-ouvertes.fr/hal-01367894v1.
[32] CW Fong, A novel predictive model for the anti-bacterial, anti-malarial and hERG cardiac QT prolongation properties of fluoroquinolones, HAL Archives. 2016, https://hal.archives-ouvertes.fr/hal-01363812v1.
[33] CW Fong, Statins in therapy: Cellular transport, side effects, drug-drug interactions and cytotoxicity - the unrecognized role of lactones, HAL Archives, 2016, https://hal.archives-ouvertes.fr/hal-01185910v1.
[34] CW Fong, Drug discovery model using molecular orbital computations: tyrosine kinase inhibitors. HAL Archives, 2016, https://hal.archives-ouvertes.fr/hal-01350862v1
[35] M Baglivo, M Baronio, G Natalini, et al, Natural small molecules as inhibitors of coronavirus lipid dependent attachment to host cells: a possible strategy for reducing SARS-COV-2 infectivity? Acta Biomed 2020, 91, 161-164
[36]TK Warren, R Jordan et al, Therapeutic efficacy of the small molecule GS-5734
against Ebola virus in rhesus monkeys, Nature, 2016, 531, 381-385
[37] A Abuhammad, RA Al-Aqtash,BJ Anson,AD Mesecar,MO Taha, Computational modeling
of the bat HKU4 coronavirus 3CLpro inhibitors as a tool for the development of antivirals against the emerging Middle East respiratory syndrome (MERS) Coronavirus, J Mol Recognit. 2017, 30, e2644, https://doi.org/10.1002/jmr.2644
[38] AV Marenich, CJ Cramer, DJ Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J Phys Chem B, 2009, 113, 6378 -96
[39] S Rayne, K Forest, Accuracy of computational solvation free energies for neutral and ionic compounds: Dependence on level of theory and solvent model, Nature Proceedings, 2010, http://dx.doi.org/10.1038/npre.2010.4864.1.
[40] RC Rizzo, T Aynechi, DA Case, ID Kuntz, Estimation of Absolute Free Energies of Hydration Using Continuum Methods: Accuracy of Partial Charge Models and Optimization of Nonpolar Contributions, J Chem Theory Comput. 2006, 2, 128-139
Figure 1. α-ketoamide inhibitors from Zhang [24]
Figure 2. (A) Remdesivir, (B) GS441524, (C) Remdesivir nucleoside triphosphate Table 1. Inhibitor ΔGdesolv,CDS kcal/mol ΔGlipo,CDS kcal/mol Dipole Moment D Molec Volume cm3/mol HOMO-LUMO Gap eV pIC50 calc Eq 1 Chloroquine Neutral -3.4 -7.71 7.66 217 4.07418 5.023069 Chloroquine Ion -6.19 -7.81 30.85 232 4.28643 3.032611 Hydroxychloroquine Neut -3.61 -7.93 7.97 261 4.18194 4.966444 Hydroxychloroquine Ion -6.3 -8.02 28.61 298 4.303306 3.20048 Favipiravir -4.6 -1.03 8.96 82 4.009416 4.805484 Lopinavir -13.93 -16.77 5.82 491 5.762141 4.372578 Remdesivir* -13.44 -10.79 12.76 381 4.784142 4.013937 Remdesivir Nucleoside Monophosphate* -9.23 -5.52 12.39 181 4.856798 4.170426 Remdesivir Nucleoside Triphosphate* -15.83 -6.99 18.29 267 4.85462 3.409867 GS441524* -5.39 -4.83 7.65 166 4.79258 4.731417
Saquinavir -11.96 -14.84 10.08 501 3.892677 4.543604 Invermectin B1A -18.95 -15.67 16.56 547 5.060344 3.443211 Ritonavir -14.09 -14.74 8.81 644 5.223616 4.233208 Atazanavir -18.24 -12.86 6.16 544 4.496239 4.387325 Nelfinavir -11.77 -13.78 11.8 390 4.77026 4.20506 Ledipasvir -17.12 -17.67 2.11 666 3.58953 5.018438 Velpatasvir -14.99 -18.09 15.9 521 3.84887 3.99003 Nitazoxanide -9.08 -6.79 13.18 172 3.85376 4.360445 Ruxolitinib -4.35 -7.47 8.55 226 4.5512 4.795694 Baricitinib -3.54 -7.06 6.86 259 4.53243 4.966201 Carfilzomib -14.54 -15.94 5.38 580 4.46168 4.667594 Nafamostat Neutral -6.86 -9.19 15.39 296 3.86302 4.318895 Nafamostat Diion -9.51 -9.38 19.85 228 4.06112 3.800342 Ribavirin -4.63 -5.52 9.85 174 5.95208 4.337202 Darunavir -10.78 -10.68 8.72 331 5.00619 4.404456 Sofusbuvir -14.37 -9.51 7.61 397 5.37546 4.220914 Galidesivir Neutral -2.55 -5.49 12.09 170 4.84244 4.517209 Galidesivir Ion -3.53 -5.62 14.51 179 5.23695 4.192102 Dolutegravir -8.9 -8.43 17.07 306 4.45515 3.945386 Efavirenz -8.24 -5.37 10.3 199 1.82348 5.07675 Grazoprevir -13.53 -13.61 12.7 521 4.11908 4.198282 Arbidol neutral -6.99 -10.45 10.84 263 4.13269 4.619391 Arbidol Ion -9.4 -10.56 17.9 282 4.49733 3.870374 Imatinib Neutral -2.4 -13.91 6 404 3.46789 5.408195 Imatinib Ion -5.47 -14.05 48.94 400 3.4747 1.911489 Z11 -12.44 -15.14 5.17 474 3.97785 4.887255 Z13A -13.45 -12.49 10.29 437 3.62627 4.491128 Z13B -14.41 -13.19 8.68 439 3.73757 4.552729 Z14B Neutral -11.93 -11.63 5.68 354 2.81644 5.100469 Z14B Ion -12.9 -11.8 21.07 389 4.25079 3.525458