THÈSE DE DOCTORAT
Présentée par
Youssef RAMLI
Discipline : Chimie
Spécialité: Chimie organique / Pharmacochimie
3-METHYLQUINOXALIN-2-ONES : SYNTHÈSE, RÉACTIVITÉ ET EVALUATION DE LEURS
PROPRIETES ANTIBACTERIENNES ET LEUR EFFICACITE INHIBITRICE DE CORROSION
Soutenue le : 10 décembre 2011, devant le jury :
Président :
El Mokhtar ESSASSI Professeur à la Faculté des Sciences, Université Mohammed V- Agdal, Rabat
Examinateur :
Youssef KANDRI RODI Professeur à la Faculté des Sciences et Technique de Fés, Université Sidi Mohamed Ben Abdllah
Said LAZAR Professeur à la Faculté des Sciences et Technique de Mohammedia, Université Hassan II Mohammedia–Casablanca
Mohammed BENCHIDMI Professeur à la Faculté des Sciences, Université Mohammed V- Agdal, Rabat
Patrick MARTIN Professeur à l’Institut Universitaire de Technologie, Université d’Artois, Béthune, France
Invité :
Mohamed MASSOUI Docteur d’Etat ès Sciences Physiques
Ahmed MOUSSAIF Docteur en Chimie (CNESTEN)
UNIVERSITÉ MOHAMMED V – AGDAL
FACULTÉ DES SCIENCES
Rabat
UNIVERSITÉ MOHAMMED V – AGDAL
FACULTÉ DES SCIENCES
A mes chers parents ;
A ma femme ;
A ma fille ;
A mes frères ;
A mes soeurs ;
A mes beaux parents
A toute ma famille ;
A mes amis ;
A tous ceux qui me sont chers ;
Pour leur présence de tous les instants,
Pour le soutien qu’ils m’ont apporté,
AVANT PROPOS
Ce travail a été réalisé au Laboratoire de Chimie Organique Hétérocyclique de la Faculté des Sciences de l’Université Mohammed V-Agdal, sous la direction de Monsieur le Professeur El Mokhtar ESSASSI.
Tout d’abord, j’adresse mes plus vifs remerciements à Monsieur le Professeur El Mokhtar ESSASSI, de m’avoir accueilli dans son laboratoire, pour sa gentillesse, sa disponibilité et sa rigueur scientifique. Ses compétences et son efficacité ont fortement contribué à la réalisation de ce travail.
Je tiens également à exprimer mes sincères remerciements à Monsieur Mohammed Benchidmi Professeur à la Faculté des Sciences de Rabat, pour l’honneur qu’il m’a fait en acceptant de siéger dans ce jury et pour ses encouragements.
Je tiens encore à lui exprimer ma profonde gratitude pour son aide et tous ses précieux conseils.
Je tiens à adresser mes remerciements à Monsieur Youssef KANDRI RODI Professeur à la Faculté des Sciences et Techniques de Fés, qui a porté un intérêt considérable à ce travail et qui n’a ménagé aucun effort pour siéger au jury de cette thèse.
Je remercier très vivement Monsieur Said LAZAR, Professeur à la Faculté des Sciences et Techniques de Mohammedia, pour avoir bien voulu juger cette thèse en tant que rapporteur et membre de jury.
Je tiens à remercie Monsieur Mohamed MASSOUI, Ancien Professeur à la Faculté des sciences de Kénitra pour l’honneur qu’il m’a fait de juger mon travail.
Je remercie Monsieur le Professeur Patrick Martin à l’Institut Universitaire de Technologie de l’Université d’Artois à Béthune d’avoir accepté de faire partie de ce jury de thèse.
Je tiens à remercier Monsieur Ahmed MOUSSAIF, Cadre Superieur au Centre National
d’Etudes Scientifiques et Techniques d’Energie Nucléaire, d’avoir accepté de faire partie de ce jury et à qui j’adresse mes plus vifs remerciements.
Je tiens également à remercier tous les respectables Professeurs de l’UFR « Pharmacochimie » pour la qualité de leurs enseignements. Qu’ils soient tous honorés par ce travail.
Bien que la thèse soit fondamentalement un travail individuel, elle n’aurait pu être menée à bien sans une équipe de collègues qui contribuent au bon fonctionnement du laboratoire, avec lesquels il est possible d’échanger conseils et suggestions. Je remercie pour cela tous mes anciens et actuels collègues de laboratoire de Chimie Organique Hétérocyclique, dans le désordre,D. AZIANE, M. EL KARBANE, R. ALAM, K. MOUDEN, F. SBAI, M. BENLFILALI, S. FADEL A. ALSUBARI, Z. BOUHARASS, R. BOUHFID, H. GHAYATI, A. MRABTI, A. RHIOUI, H. BENZEID, I. CHAKIB, C. JARMOUMI, S. AZIANE.
Je suis particulièrement reconnaissant à Monsieur EL MANSOURI Mohammed Préparateur au Département de Chimie pour l’aide morale. Je lui exprime mes sincères remerciements.
Introduction générale ... 3
Références bibliographiques ... 6
PARTIE I
Chapitre I : Mise au point sur la synthèse, la réactivité et l’activité biologique
de la quinoxaline
Introduction ... 10I. Synthèse de la quinoxalin-2-one ... 10
1. Condensation de l’o-phénylènediamine avec les acides α-cétocarboxyliques et les cétoesters ... 10
2. À partir de quinoxalines intermédiaires ... 12
3. À partir des dérivés de l’aniline ... 12
4. À partir de systèmes hétérocycliques ... 13
4.1. Par extension de cycle……….………..………..13
4.1.1. À partir de l’indolinone ... 13
4.1.2. À partir de la diazétidone ... 14
4.1.3. À partir de la 4-arylidène-2-méthyl-1,3-oxazolidin-5-one ... 14
4.1.4. À partir de l’acide déhydroascorbique ... 14
4.1.5. À partir de composés à cinq chaînons dicarbonylés. ... 15
4.1.6. À partir d’hétérocycles azotés polycarbonylés ... 16
4.2. Par rétrécissement de cycle……..……….16
4.2.1. À partir de la 1,5-benzodiazépin-2-one ... 16
4.2.2. À partir de la 1,4-benzodiazépin-2-one ... 17
II. Réactivité de la quinoxalin-2-one ... 18
1. Alkylation ... 18
2. Substitution nucléophile en position 2... 21
3. Réactivité du groupement alkyle en position 3 ... 22
4. Réactions de cyclisation impliquant les positions 2 et 3 de la quinoxaline ... 24
5. Réactions de cycloaddition 1,3-dipolaire sur les quinoxalines ... 28
III. Propriétés biologiques des dérivés de la quinoxaline ... 31
Conclusion ... 34
Chapitre II : Synthèse et réactivité de la quinoxalin-2-one
Introduction ... 41
1. Synthèse de la 3-alkylquinoxalin-2-one ... 41
2. Synthése des styrylquinoxalines à partir de la alkylquinoxaline-2-one ... 45
Rappel bibliographique ... 45
Fusion de la 6,7-dichloro-3-méthylquinoxaline avec les aldéhydes ... 48
3. Alkylation des quinoxalines dans les condition des la catalyse par transfer de phase ... 51
3.1. Alkylation de la 3-méthylquinoxaline ... 53
3.2.Alkylation de la 3-(4-méthoxystyryl)quinoxalin-2(1H)-one ... 58
4. Sulfuration da la 3-méthylquinoxalin-2-one et ses dérivés ... 61
5. Alkylation de la 3-méthylquinoxalin-2-thione ... 66
6. Synthése des bis-quinoxalines ... 75
Conclusion ... 82
Partie expérimentale ... 83
Références bibliographiques ... 98
Chapitre III : Synthèse de nouvelles molécules polyhétérocycliques dérivées
de la 3-méthylquinoxalin-2-one par cycloaddition dipolaire-1,3
Introduction ... 1011. Élaboration et réactivité des dipôles ... 102
1.1. La diphénylnitrilimine (DPNI) ... 102
1.2. Les nitriloxydes (oximes) ... 106
2. Réactions de cycloaddition dipolaire-1,3 sur la 3-méthylquinoxalin-2-one ... 109
2 .1. Réactions de cycloaddition dipolaire-1,3 de la 3-méthylquinoxalin-2-(1H)-one : action de la diphénylnitrilimine ... 110
2 .2. Réactions de cycloaddition dipolaire-1,3 de la 3-méthylquinoxalin-2-(1H)-one : action de la N-aryl-C-éthoxycarbonylnitrilimine. ... 114
2. 3. Réaction de cycloaddition dipolaire-1,3 de la 3-styrylquinoxalin-2(1H)-one : action de la diphénylnitrilimine ... 117
2. 4. Réaction de cycloaddition dipolaire-1,3 du 1-propargyl 3-méthylquinoxalin-2-one : action de de nitriloxyde ... 120
Conclusion ... 123
Partie expérimentale ... 124
Chapitre IV : Synthèse des thiènoquinoxalines
Introduction ... 132
1. Rappels bibliographiques ... 132
2. Synthèse des thiènoquinoxalines ... 135
Conclusion ... 143
Partie expérimentale ... 144
Références bibliographiques ... 148
PARTIE II
Chapitre I :
Evaluation de l’activité antibactérienne de quelques
dérivés de la 3-méthylquinoxaline
Introduction ... 151Les bactéries ... 151
Résistances des bactéries aux antibiotiques ... 153
Résistance naturelle ou intrinsèque ... 153
Résistance acquise ... 155
Les Antibiotiques ... 156
1. Paramètres d’activité d’un antibiotique ... 156
2. Mode d’action d’un antibiotique ... 156
Méthodes d’étude de la sensibilité bactérienne aux antibiotiques ... 157
1. La méthode de dilution en milieu liquide ... 157
2. La méthode de dilution en milieu solide ... 157
3. La méthode de diffusion sur gélosé (antibiogramme) ... 158
Partie experimentale ... 159
Matériel et Méthodes ... 159
Matériel ... 159
1. Souches bactériennes ... 159
2. Produits à tester ... 160
3.Milieux de culture et Solvants ... 161
B. Méthodes ... 162
2. Préparation des solutions mères des produits ... 162
3. Préparation de différentes dilutions ... 162
4. Préparation de l’inoculum ... 164 5. Ensemencement ... 164 6. Lecture ... 164 Résultats ... 166 Discussion ... 170 Conclusion ... 171 Références bibliographiques ... 172
Chapitre II : Etude du pouvoir inhibiteur de quelques dérivés de la
3-méthylquinoxalin-2-one
Introduction ... 174Méthodes d’estimation de l’efficacité des inhibiteurs ... 174
Méthodes permettant d’apprécier l’efficacité des inhibiteurs ... 175
1. la gravimétrie ... 175
2. Exploitation des courbes de polarisation ... 175
Aperçu sur l’inhibition de la corrosion du Bronze en milieu NaCl ... 176
Aperçu sur l’inhibition de la corrosion de l’acier en milieu HCl ... 177
Etude du pouvoir inhibiteur de quelques dérivés de la 3-méthylquinoxalin-2-one sur la corrosion du bronze, dans un milieu NaCl 3%. ... 178
Etude du pouvoir inhibiteur de quelques dérivés de la 3-méthylquinoxalin-2-one sur la corrosion de l’acier, dans un milieu HCl. ... 187
Etude de l’efficacité inhibitrice de la 2-phenylthieno[3,2-b]quinoxaline ... 193
Conclusion ... 195
Références bibliographiques ... 196
Conclusion générale……… ………199
Abréviations et symboles
AcOEt Acétate d’éthyle m Multiplet
MeOH Méthanol MeCN Acétonitrile
CDCl3 Chloroforme deutérié CTP Catalyse par transfert de phase
D Doublet q Quadruplet
DCM Dichlorométhane dd Doublet dédoublé
Rdt Rendement RMN Résonance magnétique
nucléaire
DMF Diméthylformamide s Singulet
DMSO-d6 Diméthylsulfoxyde deutérié t Triplet
DPNI Diphénylnitrilimine IR Infrarouge
EtOH Éthanol TEA Triéthylamine
H Heure TA Température ambiante
TMS Tétraméthylsilane THF Tétrahydrofurane
IE Impact électronique VIH Virus d’Immunodéficience
Humain
F° Point de fusion HAr Proton aromatique
ppm Partie par million Ph Phényle
J Constante de couplage δ Déplacement chimique
FAB fast atoms Bombardment ESI Électrospray ionization
INH Inhibiteur k kelvin
CMI La concentration minimale inhibitrice
CMB La concentration minimale bactéricide
NaCl Chlorure de soduim HCl Acidechloridrique
Introduction
Générale
3
Introduction générale
L’importance des dérivés de la quinoxaline dans différents domaine et, en particulier, en chimie, en biologie et en pharmacologie, a incité les chercheurs à développer de nombreuses méthodes de synthèse pour leurs préparations et à trouver de nouveaux domaines d’application.
En effet, la quinoxaline est parmi les plus anciens systèmes hétérocycliques dont la chimie de synthèse n’a cessé d’évoluer depuis des siècles, et son exploitation fut variée de l’agrochimie et la biologie, à la médecine et la thérapie.
En agrochimie, notons l’existence d’herbicides [1], d’insecticides [2] et de pesticides [3] à base de quinoxaline. L’Oxythioquinox connu par son nom commercial : Morestan, est parmi les meilleurs insecticides utilisés de nos jours.[4-7] (Figure 1)
Figure 1 : Morestan
La chimie de la quinoxaline a contribué concrètement à l’avancement des recherches dans la microbiologie par la découverte de plusieurs agents antibactériens[8,10] ou antifongiques.[11] Et récemment, de nouvelles quinoxalines ayant à la fois des activités antibactériennes et antifongiques ont été mises au point.[12,13]
En médecine, la découverte d’antibiotiques à base de quinoxaline a fait une vraie révolution dans le monde des médicaments, citons l’exemple des antibiotiques suivants : L’échinomycin,[14-16] et la triostin A.[17] (Figure 2), et depuis cette découverte, les recherches n’ont cessé de progresser pour essayer de trouver d’autres antibiotiques de structures proches de celle de l’échinomycin .[18]
4
Figure 2
D’autres quinoxalines ne sont pas encore en phase de commercialisation, mais ont montré des activités antivirales, citons l’exemple le plus révolutionnaire, celui des quinoxalines actives contre le VIH-1.[19-21]
Certains chercheurs ont pu découvrir une multitude d’activités biologiques chez une seule molécule. Par exemple certaines quinoxalines peuvent avoir à la fois des activités antibactériennes, antifongiques et anti VIH.[22] D’autres possèdent des activités à la fois antibactérienne, antifongique, antimycobactérienne, anticancéreuse et anti VIH.[23]
Vu la richesse de la chimie des quinoxalines, et la diversité de leurs applications, nous nous sommes intéressés dans le présent travail à préparer de nouvelles séries de quinoxalines, qui feront l’objet d’autres réactions chimiques, ainsi que de tests biologiques et de test de l’inhibition de corrosion. Ce travail se répartit, ainsi, en deux volets :
La première partie traitera la synthèse de nouvelles quinoxalines et se présentera en quatre chapitres.
Le premier chapitre est un rappel bibliographique sous forme d’une mise au point qui rapporte les différentes voies de synthèse des quinoxalines et leurs dérivés ainsi qu’un étalement sur leurs activités biologiques.
Le deuxième chapitre discutera la synthèse de quelques quinoxalin-2-ones, la sulfuration, la condensation avec plusieurs aldehydes et l’alkylation par transfert de phase solide-liquide de la 3-méthylquinoxalin-2-one et ses dérivés.
5
Nous consacrerons le troisième chapitre à la condensation de la 3-méthylquinoxalin-2-one et ses dérivés synthétisés avec les dipôles, dans des réactions de cycloaddition dipolaire-1,3 pour préparer des systèmes polyhétérocycliques.
Le dernier chapitre, de la première partie, fera l’objet de la synthèse de systèmes polyhétérocycliques, appelés thiènoquinoxalines, à partir d’un ensemble de styrylquinoxalines ,déjà préparés dans le deuxième chapitre. Cet ensemble de molécules est doté de propriétés inhibitrices de la corrosion d’acier dans un milieu HCl, une caractéristique qui sera étudiée dans la 2ème partie de ce travail.
La deuxième partie traitera diveres propriétés de nouvelles quinoxalines et se présentera en deux chapitres.
Le premier chapitre de cette partie sera consacré à l’évaluation de l’activité antibacterinne de quelques dérivés de la 3-méthylquinoxalin-2-one, préparées dans la première partie.
Le deuxième chapitre présentera une étude d’effet inhibiteur de quinoxalines. Nous montrerons que quelques dérivés de la 3-méthylquinoxalin-2-one présentent des propriétés antibactériennes importantes et que d’autres dérivés de la même molécule sont de térs bon inhibiteur de corrosion.
6
Références bibliographiques
1. Cerecetto H., Dias E., Di Maio R., González M., Pacce S., Saenz P., Seoane G., Suescun L., Mombrú A., Fernández G., Lema M., Villalba J., J. Agric. Food Chem., 2000, 48, 2995. 2. Sasse K., Weger R., Untersten HoeferG., Grewe F., Angew. Chem., 1960, 72, 973. 3. Knowles O. C., Environ. Health Persp., 1976, 14, 93.
4. Harding W.C., Pesticide profiles, part I, Univ. Maryland, Coop. Ext. Serv. Bull., 1979, 267, 30.
5. Thomson W. T., Agricultural chemicals - book 1, Revised ed. Thomson Publ., Indianapolis,
IN, 1976, 232.
6. Meister R. T., Berg G. L., Sine C., Meister S., Poplyk J., Farm Chemicals Handbook, 1984, 70.
7. Worthing C. R., The Pesticide Manual: A World Compendium, 7eédition. British Crop Protection Council, 1983, 7, 695.
8. Khan S. A., Saleem K., Khan Z., Eur. J. Med. Chem., 2007, 42, 103. 9. Khan S. A., Saleem K., Khan Z., Eur. J. Med. Chem., 2008, 43, 2257. 10. Sharaf El-Din N., Chem. Heterocycl. Comp., 2000, 36, 449.
11. Petrova G. M., Charushin V. N., Shcherbakova N. G., Golod M. S., Chupakhin O. N.,
Pharma. Chem. J., 1987, 21, 518.
12. Tandon V. K., Yadav D. B., Maurya H. K., Chaturvedi A. K., Shukla P. K., Bioorg. Med.
Chem., 2006, 14, 6120.
13. Sandeep A. K., Devanand B. S., Bioorg. Med. Chem. Lett., 2006, 16, 6181.
14. Kim Y. B., Kim Y. H., Park J. Y., Kim S. K., Bioorg. Med. Chem. Lett., 2004, 14, 541. 15. Waring M., Makoff A., Mol. Pharmacol., 1974, 10, 214.
16. (a) Waring M. J., Wakelin L. P. G., Nature, 1974, 252, 653. (b) Wakelin L. P. G., Waring M. J., Biochem. J., 1976, 157, 721. 17. (a) Kuroya M., Ishida N., J. Antibiotics Ser. A., 1961, 14, 324.
(b) Mutssuura S., J. Antibiotics Ser. A, 1965, 18, 335.
18. Kim Y. B., Kim Y. H., Park J. Y., Kim S. K., Bioorg. Med. Chem. Lett., 2004, 14, 541. 19. Campiani G., Fabbrini M., Morelli E., Nacci V., Greco G., Novellino E., Maga G., Spadari
S., Bergamini A., Faggioli E., Uccella I., Bolacchi F., Marini S., Coletta M., Fracasso C., Caccia S., Antivir. Chem. Chemoth., 2000, 11, 141.
7
20. Kleim J. P., Bender R., Billhardt U. M., Meichsner C., Riess G., Rosner M., Winkler I., Paessens A., Antimicrob. Agents Chemother., 1993, 37, 1659.
21. Campiani G., Aiello F., Fabbrini M., Morelli E., Ramunno A., Armaroli S., Nacci V., Garofalo A., Greco G., Novellino E., Maga G., Spadari S., Bergamini A., Ventura L., Bongiovanni B., Capozzi M., Bolacchi F., Marini S., Coletta M., Guiso G., Caccia S., J.
Med. Chem., 2001, 44, 305.
22. Carta A., Sanna P., Loriga M., Setzu M. G., La Colla P., Loddo R., Il Farmaco, 2002, 57, 19.
8
9
Chapitre I : Mise au point sur la
synthèse, la réactivité et
l’activité biologique de la
quinoxaline
10
Introduction
Les quinoxalines constituent une classe de composés hétérocycliques présentant diverses applications dans différents domaines; que ce soit en pharmacologie,[1-5] en agrochimie,[6,7] ou encore en industrie chimique où plusieurs brevets ont été déposés.[8-11] Ainsi plusieurs nouvelles méthodes de synthèse ont été décrites dans la littérature. Nous présentons dans cette mise au point la synthèse, la réactivité et les propriétés biologiques de systèmes hétérocycliques dérivés de la quinoxaline.
I. Synthèse de la quinoxalin-2-one
Les principales méthodes de synthèse proposées, peuvent être divisées en deux catégories. La première met en jeu des réactions de cyclocondensation entre des o-phénylènediamines et des composés aliphatiques électrophiles, dans des conditions conventionnelles, ou en présence de sels métalliques en solution ou sur support solide sous micro-ondes.
Le noyau pyrazine peut être également formé en utilisant des dérivés de l’aniline o-substitués.
Une autre classe de réactions, met en jeu des hétérocycles azotés à différents chaînons susceptibles de subir des réactions d’ouverture ou de réarrangements dans différentes conditions, conduisant à des intermédiaires ouverts qui se cyclisent ultérieurement pour donner les dérivés de la quinoxaline.
1. Condensation de l’o-phénylènediamine avec les acides α-cétocarboxyliques et les cétoesters
Les quinoxalin-2-ones et leurs dérivés substitués en position 3, ont été obtenus par la condensation de l'o-phénylènediamine 1 avec les acides α-cétocarboxyliques ou avec leurs esters correspondants selon la méthode de Hinsberg[12-16] (Schéma 1).
11
Schéma 1
Il est à noter que lorsque la réaction met en jeu les o-phénylènediamines monosubstituées, il a été possible d’obtenir un mélange de deux isomères [17-24]
5 et 6 (Schéma 2).
Schéma 2
La condensation de l’o-phénylènediamine avec l’acétylène dicarboxylate d’éthyle 7 dans l’éthanol, permet de préparer le composé 8 de structure quinoxaline pouvant se présenter sous deux formes tautomères (schéma 3).[25]
12
2. À partir de quinoxalines intermédiaires
King et al.[26] ont réalisé la synthèse de la quinoxaline 3 en condensant des α-halogénoesters 10 avec l’o-phénylènediamine 1 et en effectuant une oxydation à l’aide de l’eau oxygénée (Schéma 4).
Schéma 4
Plusieurs exemples ont été décrits dans la littérature concernant l’hydrolyse des 2- aminoquinoxalines qui conduit aux quinoxalin-2-ones. Ainsi, la 2,3-diaminoquinoxaline est hydrolysée par l’acide chlorhydrique (2.5 M) à 100°C, pendant 5 minutes pour donner la 3-aminoquinoxalin-2-one.[27] De même le traitement de la 2-amino-3-phénylquinoxaline par l’acide nitrique, donne la 3-phénylquinoxalin-2-one avec un excellent rendement.[28]
En portant l’oxime quinoxalinylglyoxal au reflux de la diméthylaniline, il a été possible de préparer la quinoxaline 3 provenant d’un intermédiaire spiro 13.[29] (Schéma 5)
Schéma 5
3. À partir des dérivés de l’aniline
La 2-bromo-N-tert-butyl-6-(2-chloroacétamido)aniline 14 conduit, au reflux de l’acétonitrile, pendant 24 heures, à la 5-bromo-4-tertiobutylquinoxalin-2-one 15 avec un rendement de 79% (schéma 6).[30]
13
Schéma 6
Ultérieurement, le rendement de la réaction a été amélioré à 97% en faisant subir à la
N,N-dibenzyl-2-(éthoxycarbonylméthyl)amino-4-(trifluorométhyl)aniline 16 une réduction sous
une pression de 3 atmosphères, qui induit une cyclisation spontanée, donnant la 6-trifluorométhylquinoxalin-2-one 17 (schéma 7).[31]
Schéma 7
4. À partir de systèmes hétérocycliques
4.1. Par extension de cycle 4.1.1. À partir de l’indolinone
La 3-méthylquinoxaline 3apeut être également préparée par une extension de cycle.[32] Ainsi la 3-azido-3-méthyl-2-indolinone 18 est transformée en quinoxaline au reflux du xylène (schéma 8).
14
4.1.2. À partir de la diazétidone
La quinoxaline 20 est obtenue avec un rendement de 34% en chauffant la diazétidone 19 au reflux du méthanol (schéma 9).[33-34]
Schéma 9
4.1.3. À partir de la 4-arylidène-2-méthyl-1,3-oxazolidin-5-one
Jellal et al.[35] ont réalisé la condensation, dans le n-butanol à reflux, de l’o-phénylènediamine 1 avec l’arylidène-2-méthyloxazolin-5-one 22, permettant d’accéder à la 3-arylméthylquinoxalin-2-one 23 (Schéma 10).
Schéma 10
4.1.4. À partir de l’acide déhydroascorbique
De même, la condensation de l’acide déhydroascorbique 24 avec l’o-phénylènediamine 1, en présence des arylhydrazines, conduit au composé 25 qui se transforme en pyrazolylquinoxalinone 26, (Schéma 11).[36]
15
Schéma 11
4.1.5. À partir de composés à cinq chaînons dicarbonylés D’une manière similaire les auteurs[37-39]
ont obtenu les quinoxalines 31-34 en condensant l’o-phénylènediamine avec les composés hétérocycliques dicarbonylés 27-30 (Schéma 12).
NH2 NH2 O O O CH3 CH3 N H N O HO CH3 CH3 + 28 1 32
16
Schéma 12
4.1.6. À partir d’hétérocycles azotés polycarbonylés
D’autres composés renfermant des fonctions lactames 35 et 36 ont été également utilisés, comme agents de cyclisation, pour la préparation des quinoxalines[40-43] 37 et 38 (Schéma 13).
Schéma 13
4.2. Par rétrécissement de cycle
4.2.1. À partir de la 1,5-benzodiazépin-2-one
Ahabchane et al.[44] ont pu préparer la quinoxaline 41 à partir de la benzodiazépine-2-thione 39 en deux étapes. D’abord ils ont procédé à une ouverture du cycle à sept éléments, en faisant réagir l’hydrazine sur la benzodiazépine-2-thione, obtenue par sulfuration de la 1,5-benzodiazépin-2-one. L’o-aminophénylaminopyrazole 40, ainsi obtenu, subit ultérieurement une condensation avec l’acétylène dicarboxylate d’éthyle, pour conduire, à côté de la benzimidazoline 43, à un nouveau dérivé de la quinoxaline qui se présente sous deux formes tautomères (schéma 14).
17
Schéma 14
4.2.2. À partir de la 1,4-benzodiazépin-2-one
L’irradiation de la 1,4-benzodiazépine 44, donne la quinoxaline 45 dans le cas où (X = Cl), dans le cas où (X = SMe), on note également la formation de l’oxadiazocine 46[45] (Schéma 15).
18
II. Réactivité de la quinoxalin-2-one
1. Alkylation :
La réaction d’alkylation de la quinoxalin-2-one donne un mélange de dérivés O-alkylé et
N-alkylé. Ainsi la méthylation de la quinoxaline par le diazométhane[46,47] permet d’engendrer l’alkylation des deux centres de la fonction lactame (schéma 16).
Schéma 16
Cette réaction a été généralisée à d’autres agents alkylants, dans les conditions de la catalyse par transfert de phase, conduisant aux composés N- et O-alkylés.
L’ORTEP de la éthyl-3-méthylquinoxalin-2-one confirme la structure du composé N-alkylé (Fig 3).[48]
Figure 3
Les réactions d’alkylation ont été exploitées pour préparer des quinoxalines différemment fonctionnalisées en positions 1 et 2.
19
Ainsi, Ali et al.[49], ont adopté un protocole qui consiste à utiliser du bromure d’allyle comme agent d’alkylation et d’hydrure de sodium dans le diméthylformamide à 100°C. La dihydroxylation des dérivés N- et O-allylés 51 et 52, a été réalisée avec l’AD-mixβ dans un mélange t-butanol-eau (Schéma 17).
Schéma 17
Benksim et al.[50] ont mis au point une méthode de synthèse efficace permettant d’obtenir des analogues de nucléosides57 et 58, à partir de la quinoxalinone en faisant réagir les dérivés 1,2-O-sulfonyles de structure gluco- 55 et arabino- 56 avec la 3-méthylquinoxalin-2-one 3 en présence d’une base faible K2CO3 dans le DMF à 80 °C (Schéma 18).
20
Schéma 18
La condensation des quinoxalinones 3 avec le 5,6-anhydro-1,2-O-isopropylidène- -D-glucofuranose 59, conduit aux O-glucoquinoxalines 60a-b, selon une réaction mettant en jeu le réarrangement du 5,6-anhydro-1,2-O-isopropylidène--D-glucofuranose en 3,6-anhydro correspondant, qui réagit préférentiellement avec l’atome d’oxygène de la fonction lactame de la quinoxaline. Les O-glucoquinoxalines 60a-b obtenues ont été identifiées par les données spectroscopiques et confirmées par la diffraction des rayons X (figure 4).[51]
21
Figure 4
Il est à noter, que lorsque le groupe hydroxyle du 5,6-anhydro-1,2-O-isopropylidène- -D-glucofuranose, est protégé par un groupe alkyle, la même réaction permet d’isoler le composé de N-alkylation à côté du composé de l’O-alkylation (Schéma 20).
Schéma 20
2. Substitution nucléophile en position 2
La substitution nucléophile en position 2 de la quinoxalin-2-one 3 a permis aux auteurs
[52-54]
d’isoler les produits chlorés 62 par action du POCl3 ou PCl5 (Schéma 21).
N H N N N R O R Cl 3 62 Schéma 21
22
Dans le cas où R=H,[55] la réaction de chloration donne la 2,3-dichloroquinoxaline.
La réaction de thionation a été réalisée par action du pentasulfure de phosphore dans la pyridine (schéma 22).[56,57]
Schéma 22
3. Réactivité du groupement alkyle en position 3
Le groupement alkyle, en position 3, de la quinoxalin-2-one est très réactif vis-à-vis de quelques agents électrophiles. Ainsi, Leese et al.[47] ont facilement réalisé la bromation de la 3-méthylquinoxalin-2-one 3a
Baranov[58] a étudié l’oxydation de la quinoxaline 3a par l’oxyde de sélénium pour obtenir la 3-formyl-quinoxaline 65 (Schéma 23).
Schéma 23
Selon une réaction d’oxydation, Romanenko[59]
a synthétisé la 3-acylquinoxalin-2-one (Schéma 24).
23
Schéma 24
Kurasawa et al.[60] ont réalisé l’action du sel d’aryldiazonium sur la 3-méthylquinoxalin-2-one 3 (Schéma 25).
Schéma 25
La condensation de la 3-méthylquinoxalin-2-one 3a avec les aldéhydes aromatiques donne les composés 68 (Schéma 26).[61]
Schéma 26
Récemment Vakhid et al.[62] ont préparé le 2-(indolizin-2-yl) bezimidazole 71, en condensant la 3-(arylchlorométhyl)quinoxalin-2-one 69, avec l’-picoline 70 (schéma 27).
24
Schéma 27
4. Réactions de cyclisation impliquant les positions 2 et 3 de la quinoxaline
Les réactions de cyclisation conduisant aux systèmes hétérocycliques condensés sont connues depuis longtemps; elles permettent d’obtenir des hétérocycles oxygénés ou soufrés accolés à la quinoxaline. La réaction de Marchlewski et Sosnowski,[63] réalisée en présence de l’acide chlorhydrique, en constitue un très ancien exemple, (schéma 28).
Schéma 28
Andrejčikov et al.[64]
ont réalisé la cyclisation de la 3-aroylméthylquinoxalin-2-one 74, en mettant en jeu le groupe carbonyle de la fonction lactame, en présence de l’acide polyphosphorique. (Schéma 29)
25
Dans les mêmes conditions que précédemment, les auteurs[65-68] ont réalisé la cyclisation des oximes 76 (Schéma 30).
Schéma 30
La cyclisation des dérivés soufrés insaturés 78 peut donner, selon les conditions opératoires, un cycle furane 79 ou thiophène 80 (Schéma 31).[69]
Schéma 31
Le chauffage, en présence de l’oxychlorure de phosphore, de la 3-(1,3,4-oxadiazol-2-yl)-méthylquinoxalin-2-one 81, permet de préparer la furoquinoxaline 82[70] (Schéma 32).
26
Essassi et al. ont décrit la synthèse de la 2-(thiènyl)-2,dihydrofurano[2,b]quinoxaline 84 en condensant,à fusion, à 150°C, la méthylquinoxalin-2-one 3a et le 3-formyl-thiophène 83 (schéma 33).[71]
Schéma 33
La littérature rapporte d’autres travaux concernant l’obtention de quinoxalines accolées à des hétérocycles azotés tels que l’indole et le pyrazole. En effet, Schunk,[72]
ainsi que Wiendermanova,[73] ont pu cycliser la 3-(o-aminophényl)quinoxalin-2-one 72 (Schéma 34), au reflux de l’acide acétique ou de l’acide chlorhydrique.
Schéma 34
D’autres auteurs[74-76]
ont réalisé la cyclisation de l’hydrazone 86 pour obtenir les dérivés de la pyrazolo[3,4-b]quinoxaline 87 appelés «Flavazoles» (Schéma 35).
27
La cyclisation s’effectue dans une solution alcaline[77-78]
ou au reflux de l’acide acétique.[79] Plusieurs flavazoles ont été, ainsi préparés, avec R = aryle ou sucre. Dans le cas où R = H, la cyclisation n’a pu avoir lieu, ni en milieu alcalin, ni par chauffage en milieu acide.
Un système tricyclique renfermant la quinoxaline accolée au thiazole a été obtenu en exploitant la présence du synthon -énaminoester.
Ainsi, la bromation de la 3-éthoxycarbonylméthylidène-quinoxalin-2-one 88 par, le brome en milieu acide acétique en présence de l’acétate de sodium, conduit au composé bromé 89 qui subit ultérieurement l’action du thiocyanate de potassium pour donner la thiazoloquinoxalin-2-one 90 (schéma 36).[80]
Schéma 36
La structure du composé 88 a été confirmée par une étude cristallographique qui met en évidence, en particulier, la présence d’une liaison hydrogène entre le groupe NH et le carbonyle de la fonction ester (Figure 5).[81]
28
Des résultats similaires ont été observés en condensant le 7-amino-indazole 91 avec l’acétylène dicarboxylate d’éthyle 7. La pyrazoloquinoxaline 92 obtenue, après une réaction de bromation suivie de l’action du thiocyanate de potassium, conduit à la pyrazolothiazolo-quinoxaline 93 (schéma 37).[82]
Schéma 37
5. Réactions de cycloaddition 1,3-dipolaire sur les quinoxalinones
La littérature rapporte peu de travaux concernant la synthèse de systèmes hétérocycliques renfermant la quinoxaline liée à divers noyaux pentagonaux de type pyrazole, isoxazole, imidazole et 1,2,3-triazole.
Ainsi, Ferfra et al.[83] ont étudié l’action de l’hydrazono-α-bromoglyoxylate d’éthyle 95 sur la quinoxaline 94 en présence de la triéthylamine; la réaction de cycloaddition 1,3-dipolaire, conduit à un mélange de trois produits provenant de deux réactions compétitives; une réaction de cycloaddition 1,3-dipolaire mettant en jeu la double liaison du groupe allyle, conduisant à la pyrazoline; ainsi qu’une réaction de cyclocondensation engageant le synthon énaminoester donnant le pyrazole. (Schéma 38)
29
Schéma 38
Dans les mêmes conditions opératoires que précédemment, la condensation du bromure d’hydrazonoyle 95 sur la 1-éthoxycarbonylméthyl-3-(éthoxycarbonylméthylène)-2-oxoquinoxaline 99, conduit exclusivement à la 1-(éthoxycarboxyméthyl)-5-hydroxypyrazolin-4’-yl)-2-oxoquinoxaline 100 (Schéma 39).[84]
Schéma 39
Dans la même série de réactions, le traitement de la quinoxaline 94 par l’α-chloro-phényl phénylhydrazone 101 pendant 48 heures, conduit à un mélange de quatre produits issus comme
30
précédemment, de réactions de cycloaddition 1,3-dipolaire et de cyclocondensation, ainsi que d’une réaction d’alkylation de l’atome d’azote en position 4 de la quinoxaline. (Schéma 40)
Schéma 40
Les auteurs ont également étudié l’action du chlorobenzaldoxime 106 sur le composé 94, celui-ci contient plusieurs sites dipolarophiles. La réaction conduit à un mélange de trois produits 107-109 (Schéma 41).
31
Schéma 41
III. Propriétés biologiques des dérivés de la quinoxaline
Les quinoxalines, ont marqué ces dernières années un véritable exploit dans les domaines biologique et pharmacologique.
Les études de la toxicité aigue et de l’activité psychotrope ont été réalisées sur les 1-alkyl-3-(éthoxycarbonylméthylène)-2-oxoquinoxaline. Les résultats obtenus montrent que ces produits, ne sont pas toxiques aux doses thérapeutiques et présentent des propriétés sédatives, myrolaxantes et anxiolytiques.[85]
32
Figure 6
De même lors de l’étude de l’activité anti-inflammatoire, Li et al. ont évalué l’activité de la quinoxaline 111 en faisant varier les motifs R et R’. Les composés A et B se sont avérés être des antagonistes non peptidiques de récepteurs de la molécule interleucine-8, qui est impliquée dans plusieurs maladies inflammatoires et cancéreuses. [86]
Avec A :
Et B :
Figure 7
De même les quinoxalines possèdent des propriétés antivirales.[87,88] Plusieurs travaux ont montré l’activité de certaines quinoxalines vis-à-vis du virus de l’Immunodéficience Humaine (VIH-1) qui est l’agent responsable du SIDA (Syndrome de l’Immunodéficience Acquise), parmi lesquels la 6,7-diméthyl-2-(pent-4-ènyloxy)quinoxaline 112,[89] et le S-2720 113, qui, non seulement inhibent le VIH-1 RT, mais empêchent sa réplication au niveau du tissu cellulaire.[90]
33
Figure 8
Une autre activité marquante des quinoxalines, est l’activité antimicrobienne. En effet, Carta et al.[91] ont démontré que les quinoxalines 114, 115 et 116, possèdent une activité à la fois antibactérienne et antifongique.
Figure 9
Le composé 117[92] est un nouveau macrocycle moléculaire dérivé de la quinoxalin-2-one inhibiteur des kinases dépendantes de cyclines CDK1, 2, 4 et 6. Cependant le composé 118[93] inhibe le glycogène de phosphorylase qui est l’enzyme responsable du métabolisme du glycogène au glucose, étant donné que le glucose est surproduit chez les patients souffrant de diabète.
34
Il y a un grand nombre de dérivés de la quinoxaline qui ont montré une activité antitumorale, Corona et al.[94] ont montré que la 5,7-diamino-3-phényl-2-[(3,5-diméthoxy)phénoxy]quinoxaline 119 possède une activité antitumorale, in vitro, vis-à-vis de plusieurs types de tumeurs.
Figure 11
Pour clore cette partie de l’activité biologique, citons quelques brevets montrant les diverses activités de la quinoxaline, comme inhibiteur de la protéine Kinase,[95] ou comme antagonistes de Bradykinin qui est un peptide responsable de la dilatation des vaisseaux sanguins et conduisant donc à l’abaissement de la tension artérielle.[96]
Conclusion
Les dérivés de la quinoxalinone occupent une place appréciable dans plusieurs domaines, notamment dans le domaine pharmaceutique. Aussi les modifications de structure de base de la quinoxalin-2-one, ont permis l’apparition de nouveaux dérivés présentant un large spectre d’activités biologiques.
La chimie des dérivés de la quinoxalin-2-one présente une richesse très importante due à la présence de différents sites réactifs engagés dans des réactions d’alkylation, d’amination, de chloration, de sulfuration et de cycloaddition 1,3-dipolaire.
Les études effectuées sur ces dérivés ont montré que la modification structurale permet d’améliorer son profil pharmacologique lui conférant des propriétés antibactériennes, anticancéreuses, anti VIH, tranquillisantes et sédatives.
35
Références bibliographiques
1. Khan S. A., Saleem K., Khan Z., Eur. J. Med. Chem., 2008, 43, 2257. 2. Kotharkar S. A., Shinde D. B., Bioorg. Med. Chem. Lett., 2006, 16, 6181.
3. Vicente E., Lima L. M., Bongard E., Charnaud S., Villar R., Solano B., Burguete A.,
Perez-Silanes S., Aldana I., Vivas L., Monge A., Eur. J. Med. Chem., 2008, 43, 1903.
4. Undevia S. D., Innocenti F., Ramirez J., House L., Desai A. A., Skoog L. A., Singh D. A.,
Karrison T., Kindler H. L., Ratain M. J., Eur. J. Cancer, 2008, 44, 1684.
5. Desplat V., Geneste A., Begorre M. A., Fabre S. B., Brajot S., Massip S., Thiolat D.,
Mossalayi D., Jarry C., Guillon J., J. Enzym. Inhib. Med. Chem., 2008, 23, 648.
6. Zheng H., Jiang C., Chiu M. H., Covey J. M., Chan K. K., Drug. Metab. Dispos., 2002, 30, 344.
7. Cerecetto H., Dias E., Di Maio R., González M., Pacce S., Saenz P., Seoane G., Suescun L., Mombrú A., Fernández G., Lema M., Villalba J., J. Agr. Food. Chem., 2000, 48, 2995. 8. Chen Y., Cushing T. D., Hao X., Reichelt A. H. X., Rzasa R. M., Seganish J., Shin Y.,
Zhang D., 2008, Pub. No.: WO/2008/118455.
9. Suzuki T., Seo S., Kawakami S., 2008, Pub. No.: WO/2008/102713
10. Egawa M., Kawakami S., Nakashima H., Ohsawa N., Seo S., Nomura R., 2007, WO/2007/108403.
11. Gawa M., Kawakami S., Ohsawa N., Inoue H., Seo S., Nomura R., 2007, WO/2007/032258.
12. Hinsberg O., Liebigs Ann. Chem., 1887, 237, 1228. 13. Hinsberg O., Liebigs Ann. Chem., 1887, 237, 327. 14. Hinsberg O., Liebigs Ann. Chem., 1896, 292, 245.
15. Andrejčikov Ju. S., Saraeva R. F., Fridman A. L., Khim. Geter. Soed., 1973, 259. 16. Wolf F. J., Beutel R. H., Stevens J. R., J. Am. Chem. Soc., 1948, 70, 2572.
17. Kurasawa Y., Satoh J., Ogura M., Okamoto Y., Takada A., Heterocycles, 1984, 22, 1531. 18. Platt B. C., J. Chem. Soc., 1948, 1310.
19. Otomasu H., Yoshida K., Chem. Pharm. Bull., 1960, 5, 475.
20. Wolf F. J., Pfister K., Beutel R. H., Wilson R. M., Robinson C. A., Stevens J. R., J. Am.
Chem. Soc., 1949, 71, 6.
21. Horner L., Schwenk U., Liebigs Ann. Chem., 1953, 579, 212.
36
23. Hockenhull D. J. D., Floodgate G. D., Biochem. J., 1952, 52, 38.
24. Ali M. M., Ismail M. M. F., El-Gaby M. S. A., Zahran M. A., Ammar Y. A., Molecules, 2000, 5, 864.
25. Iwanami Y., J. Chem. Soc. Japen, 1961, 82, 788.
26. King F. E., Clark-Lewis J. W., J. Chem. Soc., 1951, 3379. 27. Schipper E., Day A. R., J. Am. Chem. Soc., 1951, 73, 5672. 28. Krönke F., Leister H., Chem. Ber., 1958, 91, 1479.
29. Titov V. V., Kozhokina L. P., Tetrahedron Lett., 1973, 1105.
30. Mickelson J. W., Jacobsen E. J., Carter D. B., Im H. K., Im W. B., Schreur P. J. K. D., Sethy V. H., Tang A. H., McGee J. E., Petke J. D., J. Med. Chem., 1996, 39, 4654.
31. Jacobsen E. J., Stelzer L. S., TenBrink R. E., Belonga K. L., Carter D. B., Im H. K., Im W. B., Sethy V. H., Tang A. H., VonVoigtlander P. F., Petke J. D., Zhong W.-Z, Mickelson J. W., J. Med. Chem., 1999, 42, 1123.
32. Tamura Y., Chun M. W., Nishida H., Kwon S., Ikeda M., Chem. Pharm. Bull., 1978, 26, 2866.
33. Fischer W., Fahr E., Angew. Chem. Int. Ed., 1967, 6, 630. 34. Morrow D. F., Regan L. A., J. Org. Chem., 1971, 36, 27.
35. Jellal M., Ramli Y., Moussaif A., Kandri Rodi Y., Fifani J., Essassi E.M., Pierrot M., J.
Soc. Chim. Tun., 2005, 7, 19.
36. Gris J., Glisoni R., Fabian L., Fernandez B., Albertina G., Tetrahedron Letters, 2008, 49, 1053.
37. Ohle H., Gross W., Ber. Deutch.Chem.Ges., 1935,68,2262 38. Fries K., Barten K., Liebigs Ann.Chem., 1925,442,257
39. Terpetschning E., Ott W., Kollene G., Peters E.M., Monatch.Chem., 1988, 119, 367. 40. Barlaw R., Ing H., Lewis I., J. Chem. Soc., 1951, 3242
41. King F.B., Clark-Lewis J.W., J. Chem. Soc.,1951,3379
42. Schunk E., Marchelewski L., Ber. Deutch. Chem. Ges., 1895, 28, 2525. 43. Schunk E., Marchelewski L., Ber. Deutch. Chem. Ges., 1896, 29, 194.
44. Ferfra S., Ahabchane N. H., Garrigues B., Essassi E. M., C. R. Acad. Sci. Paris, Chimie, 2001, 4, 905.
45. Ning R. Y., Field G. F., Sternbach L. H., J. Heterocycl. Chem., 1970, 7, 475. 46. Jones R. G., Kornfeld E. C., McLaughlin K. C., J. Am. Chem. Soc., 1950, 72, 3539. 47. Leese C. L., Rydon H. N., J. Chem. Soc., 1955, 303.
37
48. Benzeid H., Vendier L., Ramli Y., Garrigues B., Essassi, E. M., Acta Cryst. 2008, E64, o2234.
49. Ali I. A. I., Fathalla W., Heteroatom. Chem., 2006, 17, 280. 50. Benksim A., Thèse doctorat Amiens, France 2006.
51. Jarmoumi C., Lakhrissi B., Mondieig D., Négrier P., Léger J. M., Massip S., Lazar Z., Benali B., Massoui M., Essassi E. M., J. Phy. Org. Chem., 2009, 22, 585.
52. Westphal G., Wasicki H., Zielinski V., Weberr F. G., Tonew M., Tonew E., Pharmazie, 1977, 32, 570.
53. Borkovec J., Michalský J., Podpěrová A., Chem. Listy, 1955, 49, 1405. 54. Platt B. C., Sharp T. M., J. Chem. Soc., 1948, 2129.
55. Motylewski S., Ber. Deutsch. Chem. Ges., 1908, 41, 800.
56. Badr M. Z. A., El-Naggar G. M., El-Sherief H. A. H., Abdel-Rahman A. E.-S., Aly M. F.,
Bull. Chem. Soc. Jpn., 1983, 56, 326.
57. Moustafa O. S., J. Chin. Chem. Soc., 2000, 47, 351.
58. Baranov S.N., Plachuk-Tarnavsya N.E., Ukr. Zh., 1983, 29, 82. 59. Romaneko V.D., Burmistrov S.I., Khim. Geter. Soed., 1973, 852.
60. Kurasawa Y., Yamazaki K., Tajima S., J. Heterocycl. Chem., 1986, 23, 957.
61. Badr M. ZA., El-Naggar G.M., El-Sherif H.A., Bull. Chem. Soc. Jpn., 1983, 56, 326. 62. Mamedov V. A., Saifina D. F., Gubaidullin A. T., Saifina A. F., Rizvanov I. k.,
Tetrahedron Letters, 2008, 49, 6231.
63. Marchlewski L., Sosnowski J., Ber. Deutsch. Chem. Ges., 1901, 34, 1108. 64. Andrejčikov Ju. S., Saraeva R. F., Fridman A. L., Khim. Geter. Soed., 1973, 259. 65. Dahn H., Nussbaum J., Helv. Chim. Acta, 1969, 52, 1661.
66. Kurasawa Y., Suzuki K., Nakamura S., Moriyama K., Takada A., Heterocycles, 1984, 22, 695.
67. Kurasawa Y., Shimabukuro S., Okamoto Y., Takada A., Heterocycles, 1985, 23, 65.
68. Kurasawa Y., Ichikawa M., Kamata I., Okamoto Y., Takada A., Heterocycles, 1985, 23, 281.
69. Terpetschnig E., Ott W., Kollenz G., Peters K., Peters E. M., Von Schnering G. H.,
Monatsh. Chem., 1988, 119, 367.
70. Kurasawa Y., Moritaki Y., Ebukuro T., Takada A., Chem. Pharm. Bull., 1983, 31, 3897. 71. Anothane C., Bouhfid R.,Essassi, E. M., Molbank, 2007, M536.
38
73. Wiedermannová I., Jirovský D., Hlaváč J., Slouka J., Acta Universitatis Palackianae
Olomucensis, Facultas Rerum Naturalium, 2001, 40, 79.
74. Romanenko V. D., Burmistrov S. I., Khim. Geter. Soed., 1973, 852.
75. Ortega M. A., Sainz Y., Montoya M. E., Lopéz De Ceráin A., Monge A., Pharmazie, 1999, 54, 24.
76. Henseke G., Dittrich K., Chem. Ber., 1959, 92, 1550. 77. Somogyi L., Carbohyd. Res., 1992, 229, 89.
78. Mousaad A., Awad L., Shimy N., El Ashry E-S. H., J. Carbohyd. Chem., 1989, 8773. 79. Sumoto K., Irie M., Mibu N., Miyano S., Nakashima Y., Watanabe K., Yamaguchi T.,
Chem. Pharm. Bull., 1991, 39, 792.
80. Benchidmi M., Essassi E.M., Ferfra S., Fifani J., Bull. Soc. Chim. Belg., 1993, 102, 679. 81. Ferfra S., Essassi E.M., El-Bali B., Bolte, M. Acta Cryst. 1999, C55, IUC9900021.
82. Boutayeb M., El Imadi S., Benchidmi M., Essassi E. M., El Ammari L., Synthetic
Communications, sous presse.
83. Ferfra S., Ahabchane N. H., Essassi E. M., Garrigues B., J. Mar. Chim. Heterocycl., 2002, 1, 12.
84. Ferfra S., Ahabchane N. H. Essassi E. M., Molbank, 2006, M472.
85. Ferfra S., Zellou A., Essassi E.M., Cherrah Y., Hassar M. Ann. Pharm. Fr., 2002, 60, 341. 86. Li J. J., Carson K. G., Trivedi B. K., Yue W. S., Ye Q., Glynn R. A., Miller S. R., Connor
D. T., Roth B. D., Luly J. R., Low J. E., Heilig D. J., Yang W., Qin S., Hunt S., Bioorg.
Med. Chem., 2003, 11, 3777.
87. Rübsamen-Waigmann H., Huguenel E., Shah A., Paessens A., Ruoff H.-J., Briesen H V., Immelmann A., Dietrich U., Wainberg M. A., Antivir. Res., 1999, 42, 15.
88. Patel M., McHugh R. J., Cordova C. B., Klabe R. M., Erickson-Viitanen S., Trainor G. L., Rodgers J. D., Bioorg. Med. Chem. Lett. 2000, 10, 1729.
89. Ali I. A. I., Al-Masoudi I. A., Hassan H. Gh., Al-Masoudi N. A., Chem. Heterocycl. Comp., 2007, 43, 1052.
90. Kleim J. P., Bender R., Billhardt U. M., Meichsner C., Riess G., Rösner M., Winkler I., Paessens A., Antimicrob. Agents Ch., 1993, 37, 1659.
91. Carta A., Loriga M., Zanetti S., Sechi L. A., Il Farmaco, 2003, 58, 1251.
92. Kalinski C., Umkehrer M., Ross G., Kolb J., Burdack C., Hiller W., Tetrahedron Lett. 2006, 47, 3423.
39
93. Dudash Jr. J., Zhang Y., Moore J. B., Look R., Liang Y., Beavers M. P., Conway B. R., Rybczynski P. J., Demarest K. T., Bioorg. Med. Chem. Lett., 2005, 15, 4790.
94. Corona P., Carta A., Loriga M., Vitale G., Paglietti G., Eur. J. Med. Chem., 2008, 44, 1579. 95. Bemis G. W., Duffy J. P., 2005, WO 2005/ 056547 A2.
96. Grant F., Bartulis S., Brogley L., Dappan M., Kasar R., Khan A., Neitzel M., Pleiss M. A., Thorsett E. D., Tucker J., Ye M., Hawkinson J., 2003, WO 03/093245.
40
Chapitre II: Synthèse et
réactivité de la
41
Introduction
La littérature rapporte un certain nombre de travaux concernant la synthèse et la réactivité de dérivés de la quinoxaline renfermant plusieurs sites réactionnels (Voir chapitre précédent).
Dans ce chapitre, nous rapportons les divers travaux réalisés sur la synthèse et la réactivité de la quinoxaline, en se focalisant sur quelques dérivés de la 3-méthylquinoxalin-2-one.
1. Synthèse de la 3-alkylquinoxalin-2-one :
Différentes réactions de synthèse de la quinoxalin-2-one ont été rapportées par la littérature ; nous nous contentons dans ce paragraphe de citer quelques synthèses de la quinoxaline étudiée, la 3-méthylquinoxalin-2-one.
Ainsi, Burger et al.[1] ont obtenu une série de quinoxalines différemment substituées en position 3, en faisant condenser l’o-phénylènediamine 1 avec la 3,3-bis-(trifluorométhyl)-5-oxazolinone (R = H) 120 dans l’acétate d’éthyle en présence de l’acide acétique comme catalyseur à température ambiante (Schéma 42).
Schéma 42
Dans un autre travail, King et al.[2] ont également préparé des dérivés de la quinoxaline, d’abord en condensant l’o-phénylènediamine 1 avec les α-halogénoesters 10 ,puis par une déshydrogénation dans l’eau oxygénée (Schéma 43).
42
Schéma 43
La quinoxalin-2-one 3 est obtenue par cyclisation de l’o-nitroaniline de type 121. En effet, la cyclisation se fait dans l’hydroxyde de sodium en présence d’un mélange (eau-éthanol) ; cette réaction donne l’intermédiaire quinoxalin-2-on-4-oxyde 122 qui est ensuite réduit par le dithionite de sodium.[3] (Schéma 44).
Schéma 44
Après avoir rapporté des exemples qui illustrent la synthèse de la 3-alkylquinoxaline, nous indiquons qu’il existe d’autres travaux où la 3-méthylquinoxaline[4-6]
et ses dérivés ont été préparés.
Pour notre part, nous avons choisi la méthode de Krishnan[7] pour préparer la 3-méthylquinoxaline et ses différents dérivés étudiés, et ceci en suivant deux voies :
Voie 1 : faire réagir l’o-phénylènediamine 1 avec l’acide pyruvique 2 dissous dans l’eau à température ambiante pendant 10 minutes, pour obtenir la 3-méthylquinoxaline.
Voie 2 : faire réagir l’o-phénylènediamine 1 avec le pyruvate d’éthyle 2 dissous dans un mélange H2O-HCl à température ambiante, pendant 15 minutes,pour obtenir la
43 NH2 NH2 R1 R2 + R1 R2 H N N CH3 O R O OR' O R=Me R'=H,Et 1a:R1=R2=H 1b:R1=CH3,R2=H 1c:R1=Cl,R2=Cl 1d:R1=CO2H,R2=H 1e:R1=NO2,R2=H 1f :R1=Cl,R2=H 3a:R1=R2=H 3b:R1=CH3,R2=H 3c:R1=Cl,R2=Cl 3d:R1=CO2H,R2=H 3e:R1=NO2,R2=H 3f:R1=Cl,R2=H 2 Schéma 45
D’autre part, nous avons mis au point une nouvelle voie de synthèse de la 3-méthylquinoxalin-2-one, qui consiste à faire réagir l’o-phénylènediamine avec des acides aminés : la serine et la cystéine en milieu acide chlorhydrique 5.5N à chaud. (Schéma 46)
R1 R2 NH2 NH2 HX H2 C COOH NH2 + N H N O CH3 R1 R2 HCl 5.5N 1a:R1=H=H 11c:R1=R2=Cl X=O,S 3a:R1=H=H 3c:R1=R2=Cl 123 Schéma 46
Ainsi la réaction de l’o-phénylènediamine avec la thréonine dans les même conditions que
précédement,conduit à la formation de la 3-éthylquinoxalin-2-one.( Schéma 47) NH2 NH2 HO HC CH3 CO OH NH2 + N H N O Et HCl 5.5N 1a 124 3g Schéma 47
44
La structures du produit 3g a été identifiée grâce aux données spectrales (RMN ¹ H, ¹³C et masse.
Le spectre RMN ¹H du composé 3g dans le CDCl3, montre en particulier, un quartet et un
triplet relatifs au groupement éthyle, en plus un signal vers 11ppm attribuable au groupe NH. Le spectres de masse (IE) présente le pic moléculaire relatif à l’ ion moléculaires à m/z = 174 confirmant ainsi la structure proposée.
La formation de ces produits peut être expliquée par le mécanisme réactionnel suivant :
H C HC NH2 CO2H OH R H -H2O R C H C NH2 COOH RH2C C NH,H COOH NH2 NH2 R N H N NH2 CH2R O H R N H N CH2R O R -NH3 Schéma 48
Il est à noter que la condensation de l’o-phénylènediamine dans les conditions de la haute dilution, avec un large excès de la serine, nous a permis d’obtenir en plus du composé 3, le produit 125 de type
45 NH2 NH2 HO HC COO H NH2 + N N HCl 5.5N H N N CH3 1 123 125 + N H N O CH3 3a Schéma 49
46
2. Synthèse des styrylquinoxalines à partir de la 3-alkylquinoxalin-2-one : Rappels bibliographiques :
La littérature rapporte différentes méthodes pour préparer les styrylquinoxalines.[7,8] Ainsi, Taylor et al.[9] ont préparé d’abord la quinoxaline 126 chlorée en position 2, qu’ils ont engagé dans une réaction de Wittig, conduisant à la 2-styrylquinoxaline 128, (Schéma 51)
N N Cl CH2=PPh3 N N CH=PPh3 PhCHO N N CH=CHPh 126 127 128 Schéma 51
De même Kaupp et al.[10] ont préparé selon un procédé similaire la 2-chloro-3-styrylquinoxaline 130 ,à partir de la 2,3-dichloroquinoxaline 129. (Schéma 52).
N N Cl Cl N N CH=PPh3 Cl 1) Ph3P=CH2, THF, 20°C, 2 h 2) PhCHO, 20°C, 30 min 129 130 Schéma 52
Dans des conditions proches de celles décrites précédemment, Kim et al.[11] ont obtenu un mélange de cis- et trans-6-styrylquinoxaline 132 à partir de la 6-quinoxalinecarbaldéhyde 131, (Schéma 53). N N OHC N N PhHC=HC PhCH2PPh3Cl, BuLi THF, 20°C, 6 h 131 132 Schéma 53
Pour notre part ,nous avons suggéré une autre voie de synthèse qui consiste à faire réagir à fusion la 3-méthtylquinoxaline-2-one 3a avec une série d’aldéhydes aromatiques 133a-f, Cette
47
méthode, réalisée en l’absence de solvant, nous a permis d’isoler les composés attendus avec d’excellents rendements, variant entre 76% et 93% (Schéma 54)
N H N CH3 O O H N H N O 2h R2 R2 133a- R1=R2=H 133b- R1=Cl, R2=H 133c- R1=H, R2=Cl 133d- R1=H, R2=COOH 133e- R1=H, R2=N(CH3)2 133f- R1=H, R2=OCH3 R1 134a- R1=R2=H 134b- R1=Cl, R2=H 134c- R1=H, R2=Cl 134d- R1=H, R2=COOH 134e- R1=H, R2=N(CH3)2 134f- -R1=H, R2=OCH3 R1 3a Schéma 54
Dans ce type de réactions, la quinoxaline 3a peut se présenter sous la forme tautomère 3a’ de structure énamine. Cette dernière réagit sur le carbonyle de l’aldéhyde selon une réaction de type aldol. L’intermédiaire [A], ainsi formé, subit ultérieurement une réaction de déshydratation, conduisant à la 3-styrylquinoxaline (Schéma 55).
Schéma 55
Une étude de la spectrométrie de RMN1H, 13C et masse a permis d’identifier les structures des styrylquinoxalines (tableau 1 ).
48
Fusion de la 6,7-dichloro-3-méthylquinoxaline avec les aldéhydes
De même, nous avons procédé à la condensation de la 6,7-dichloro-3-méthylquinoxaline 3c avec le p-méthoxy le p-chloro et l’o-chlorobenzaldéhyde dans les mêmes conditions que précédemment (Schéma 56). Notons que la double liaison C=C, adopte exclusivement la
configuration trans, attestée par la grande valeur des constantes de couplage observées dans les spectres de RMN1H des produits 135b-d , (voir tableau 1 ).
N H N CH3 O O H N H N O 2h R2 R2 133b- R1=Cl, R2=H 133c- R1=H, R2=Cl 133d -R1=H, R2=OCH3 R1 135b- R1=Cl, R2=H 135c- R1=H, R2=Cl 135d- -R1=H, R2=OCH3 Cl Cl R1 3c Cl Cl Schéma 56
Les structures des styrylquinoxalines 135b-d ont été définies grâce aux données spectrales RMN1H, 13C et masse.
Ainsi, la RMN1H des styrylquinoxalines, nous a permis de constater que la double liaison C=C formée adopte une configuration trans grâce à la constante de couplage J3 qui est comprise entre 16 et 16.5 Hz.
Le tableau suivant indique les valeurs en ppm des déplacements chimiques des protons éthylèniques des produits 134a-e et 135b-d:
49
Tableau 1 : Valeurs des déplacements chimiques des protons éthylèniques des styrylquinoxalines préparés.
Produits Déplacements chimiques des protons éthylèniques DMSO-d6 134a 7.63 (d, 1H, CHéthylènique, 3J = 16.5 Hz) et 8.08 (d, 1H, CHéthylènique, 3J = 16.5 Hz) 134b 7.64 (d, 1H, CHéthylènique, 3J = 16.2 Hz) et 8.45 (d, 1H, CHéthylènique, 3J = 16.2 Hz) 134c 7.63 (d, 1H, CHéthylènique, 3J = 16.2 Hz) et 8.05 (d, 1H, CHéthylènique, 3J = 16.2 Hz), 134d 8.11 (d, 1H, CHéthylènique, 3J = 16.2 Hz) et 8.75 (d, 1H, CHéthylènique, 3J = 16.2 Hz) 134e 7.38 (d, 1H, CHéthylènique, 3J = 16 Hz) et 7.99 (d, 1H, CHéthylènique, 3J = 16 Hz) 135b 7.60 (d, 1H, CHéthylènique, 3J = 16,2 Hz) et 8.45 (d, 1H, CHéthylènique, 3J = 16,2 Hz) 135c 7.60 (d, 1H, CHéthylènique, 3J = 16.2 Hz) et 8.06 (d, 1H, CHéthylènique, 3J = 16.2 Hz) 135d 7.61 (d, 1H, CHéthylènique, 3J = 16.2 Hz) et 8.36 (d, 1H, CHéthylènique, 3J = 16.2 Hz)
Nous représentons sur la figure suivante, comme exemple des styrylquinoxalines, les spectres RMN1H et RMN13C du produit 134c.
50
51
3. Alkylation des quinoxalines dans les conditions de la catalyse par transfert de phase
Dans notre laboratoire, nous avons rapporté plusieurs alkylations de différentes quinoxalines; citons l’exemple de l’alkylation de la 3-(éthoxycarbonylméthylène)-2-oxo-quinoxaline 136 établie par Ferfra et al.[12] La réaction effectuée dans les conditions de la catalyse par transfert de phase, solide-liquide, montre que le résultat dépend de la nature de l’agent d’alkylation utilisé. Ainsi, si les halogénures d’alkyle réagissent préférentiellement sur le carbone énaminique, le sulfate de méthyle affecte exclusivement l’azote amidique (Schéma 57). N H N O OEt O N H N O OEt O N H N O OEt O N H N O OEt O R R H R CH3 CTP RX (CH3)2SO4 136 137 138 139 Schéma 57
La quinoxaline-2,3-dione 140 a été alkylée par Mustaphi[13] à l’aide de différents agents alkylants dans les conditions de la catalyse par transfert de phase solide-liquide (Schéma 58).
N H H N O O RX N N O O R R CTP sol-liq DMF, K2CO3 140 141 X= Br ou Cl R= Méthyle, éthyle, propargyle et acétate d'éthyle Schéma 59
52
Dans le cas où R = Allyle, l’auteur a obtenu un mélange de quinoxalines mono et dialkylée au niveau des azotes.
Dans un travail récent, Jarmoumi[14] a pu greffer des sucres sur la 3-méthylquinoxaline, et obtient deux produits : le N- et le O-alkylé (Figure13).
O O O O N N O R1 H H 142 R1= CH3,H N N CH3 O O OR2 O O HO H3C 143 R2= CH3,C3H5,C4H9, C8H17,C10H21,C12H25 N N CH3 O O OR3 O O HO Cl 144 R3= CH3,C3H5,C8H17,C12H25 Figure 13
Une façon d’augmenter la séléctivité de la réaction pour avoir un seul produit d’alkylation, consiste en la condensation de la o-méthylaminoaniline avec l’acide pyruvique dans l’éthanol à 50°C,[15] (Schéma 59). NH2 NH CH3 O HO O N N CH3 CH3 O CH3 145 146 EtOH 2 Schéma 59
53
Pour notre part nous avons choisi l’alkylation de la 3-méthylquinoxaline-2-one et de ses dérivés, en catalyse par transfert de phase liquide –solide, dans le diméthylformamide en présence du carbonate de potassium comme base.
3.1. Alkylation de la 3-méthylquinoxalin-2-one
Nous avons procédé à l’alkylation de la 3-méthylquinoxalin-2-one par différents agents alkylants dans les conditions de la catalyse par transfert de phase liquide-solide dans le diméthyl formamide en présence de carbonate de potassium comme base et du bromure de tetra-n-butylammonium comme catalyseur.La réaction conduit aux N-alkylquinoxalines 146, 148, 150, 152, 154 ,et O-alkylquinoxalines 147, 149, 151,153. (Schéma 60)
N H N CH3 O + RX K2CO3/BTBA DMF N N CH3 OR N R N CH3 O + 3 147,149,151,153 146,148,150,152,154 RX= I-CH3 146: R=CH3 Cl-CH2-Ph 147, 148: R= -CH2-Ph Br-C3H3 149, 150: R= -C3H3 Br-C3H5 151, 152: R= -C3H5 Br-C2H5 153, 154: R= -C2H5 Schéma 60
Les structures de ces composés alkylés ont été identifiées grâce aux données spectrales (RMN ¹ H, ¹³C et masse). Les spectres RMN ¹ H, pris dans le CDCl 3,montrent,en particulier,les signaux relatifs aux groupes alkyles,mettant en évidence la disparition des signaux attribuables au groupe NH,ce qui atteste leur engagement dans la réaction.
54
Outre l’analyse spectroscopique de RMN, une étude de diffraction aux rayons-X a confirmé les structures attribuées aux composés 148, 150 , 152 et 154.
Figure 14 : ORTEP du composé 1-ethyl-3-methylquinoxalin-2(1H)-one 154 Tableau 2: Données cristallographiques du composé 154
Empirical formula C11 H12 N2 O
Formula weight 188.23
Temperature 180 K
Wavelength 0.71073 Å
Crystal system, space group Triclinic, P 1
Unit cell dimensions a = 7.4101(6) Å , = 84.976(7)° b = 9.1405(8) Å, =78.717(7)°. c = 14.2969(12) Å, = 88.137(7)° Volume 945.82(14) Å3 Z, Calculated density 1.322 Mg/m3 Absorption coefficient 0.09mm-1 F(000) 400 Crystal size 0.18 x 0.13 x 0. 07 mm3 Theta range for data collection 2.8 to 26.4°.
Limiting indices -9≤h≤8
-11≤k≤11 -14≤l≤17
Reflections collected 3865
55
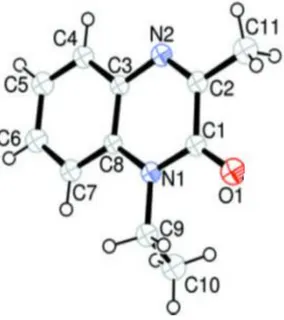
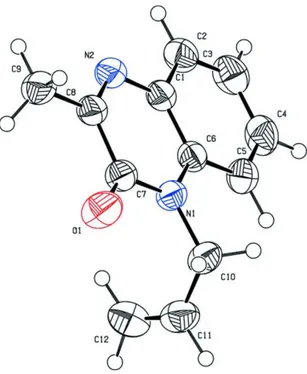
Figure 15 : ORTEP du composé 3-méthyl-1-(prop-2-ynyl)quinoxalin-2(1H)-one 150
Tableau 1: Données cristallographiques du composé 150
Empirical formula C12 H10 N2 O
Formula weight 198.22
Temperature 180 K
Wavelength 0.71073 Å
Crystal system, space group Monoclinic, P2/n Unit cell dimensions a = 21.1241(1) Å ,
b = 9.1405(8) Å, =105.354(6)°. c = 22.246(1) Å, Volume 1980(2) Å3 Z, Calculated density 1.329 Mg/m3 Absorption coefficient 0.09mm-1 F(000) 832 Crystal size 0.20 x 0.15 x 0. 08 mm3 Theta range for data collection 2.7 to 32.2°.
Limiting indices -22≤h≤26
-5≤k≤5 -26≤l≤27
Reflections collected 4058
56
Figure 16 : ORTEP du composé 1-allyl-3-méthylquinoxalin-2(1H)-one 152
Tableau 4 : Données cristallographiques du composé 152
Empirical formula C12 H12 N2 O
Formula weight 200.24
Temperature 1486 K
Wavelength 0.71073 Å
Crystal system, space group Monoclinic, P2/n Unit cell dimensions a = 5.O722(5) Å ,
b = 13.4707(13) Å, =95.082(5)°. c = 15.057(5) Volume 1024.31(17) Å3 Z, Calculated density 1.298 Mg/m3 Absorption coefficient 0.09mm-1 F(000) 424 Crystal size 0.32 x 0.31 x 0. 13 mm3 Theta range for data collection 2.7 to 28.3°.
Limiting indices -6≤h≤6
0≤k≤17 0≤l≤20
Reflections collected 2546
57
ORTEP du composé 1-allyl-3-méthylquinoxalin-2(1H)-one 152
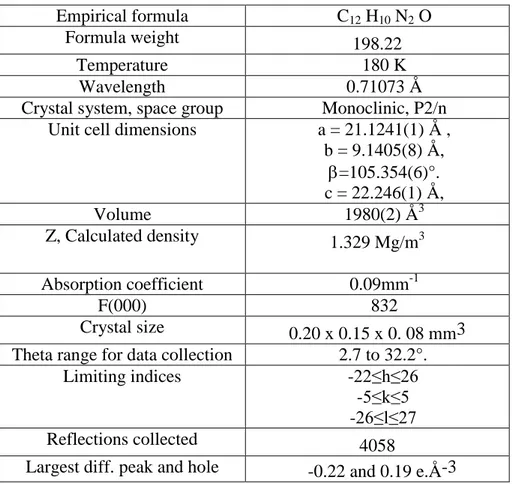
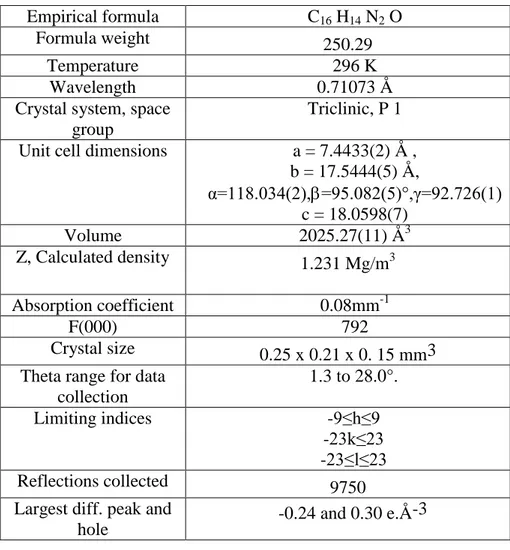
Figure 17 : ORTEP du composé 1-benzyl-3-méthylquinoxalin-2(1H)-one 148
Tableau 5 : Données cristallographiques du composé 148
Empirical formula C16 H14 N2 O
Formula weight 250.29
Temperature 296 K
Wavelength 0.71073 Å
Crystal system, space group
Triclinic, P 1 Unit cell dimensions a = 7.4433(2) Å ,
b = 17.5444(5) Å, α=118.034(2),=95.082(5)°,γ=92.726(1) c = 18.0598(7) Volume 2025.27(11) Å3 Z, Calculated density 1.231 Mg/m3 Absorption coefficient 0.08mm-1 F(000) 792 Crystal size 0.25 x 0.21 x 0. 15 mm3 Theta range for data
collection 1.3 to 28.0°. Limiting indices -9≤h≤9 -23k≤23 -23≤l≤23 Reflections collected 9750
Largest diff. peak and