© Myriam Tremblay, 2019
Caractérisation de l'expression des isoformes du DCIR
dans l'infection par le VIH-1
Mémoire
Myriam Tremblay
Maîtrise en microbiologie-immunologie - avec mémoire
Maître ès sciences (M. Sc.)
Caractérisation de l’expression des isoformes du DCIR
dans l'infection par le VIH-1
Mémoire
Maîtrise en microbiologie-immunologie
Maître ès sciences (M. Sc.)
Myriam Tremblay
Sous la direction de :
Caroline Gilbert, directrice de recherche
Frédéric Barabé, codirecteur de recherche
ii
Résumé
Le DCIR, une lectine de type C, a été identifié comme un facteur facilitant le transfert des virus des cellules dendritiques vers les lymphocytes TCD4 lors de l’infection au VIH-1. Cinq isoformes différentes du DCIR existent. L’expression de certaines isoformes peut être modulée de façon indépendante dans certaines pathologies. L’hypothèse qui a été posée est que les isoformes du DCIR sont modulées au cours de l’infection au VIH-1 et qu’il est possible de générer des cellules déficientes en DCIR par la technique de CRISPR/Cas9. Les objectifs étaient de développer une PCR quantitative spécifique pour chaque isoforme du DCIR, de quantifier l’expression des isoformes du DCIR dans des cellules immunitaires de patients infectés par le VIH-1, de déterminer s’il existe des corrélations entre le patron d’expression de chaque isoforme et les données cliniques des patients VIH-1 et, finalement, de développer un outil CRISPR/Cas9 permettant le knock-out du gène du DCIR dans des cellules souches hématopoïétiques. Les résultats montrent que l’expression des isoformes 1 à 4 n’est pas modulée par l’infection au VIH-1. Cependant, une corrélation existe entre le ratio CD4/CD8 des patients traités et l’expression de l’isoforme 1 du DCIR dans les cellules polynucléées. De plus, dans les cellules mononucléées sanguines périphériques, les isoformes 1 et 3 du DCIR sont les plus exprimées et, dans les cellules sanguines polynucléées, l’isoforme 1 du DCIR est la plus exprimée. Finalement, un outil CRISPR/Cas9, permettant d’inactiver le gène du DCIR dans des cellules souches hématopoïétiques par infection lentivirale, a été développé. Ces données permettront de mieux caractériser les rôles des isoformes du DCIR, contribuant ainsi au développement de stratégies thérapeutiques ciblant cette lectine.
iii
Abstract
The C type lectin DCIR was identified as viral transfer factor from dendritic cells to CD4 T cells during HIV-1 infection. There are five known isoforms of the DCIR protein. The expression of some of these isoforms can be modulated independently in some pathologies. The hypothesis of this project is that DCIR isoforms can be modulated during HIV-1 infection and that it is possible to generate DCIR deficient cells with CRISPR/Cas9 technology. Our objectives were: to develop a specific quantitative PCR for each isoforms of DCIR; to quantify the expression of DCIR isoforms in immune cells of HIV-1 infected patients; to determine if there are correlations between the expression pattern of each isoform and the patients’ clinical data and; to develop a CRISPR/Cas9 tool allowing the knock out of the DCIR gene in hematopoietic stem cells. The results show that the expression of DCIR isoforms 1 to 4 is not modulated by HIV-1 infection. However, a positive correlation exists between the CD4/CD8 T cell ratio of treated HIV-1 patients and the expression of DCIR isoform 1 in polymorphonuclear cells. Furthermore, the DCIR isoforms 1 and 3 are the most expressed isoforms in the patients’ peripheral blood mononuclear cells, while the DCIR isoform 1 is the most expressed isoform in the patients’ polymorphonuclear cells. Finally, our CRISPR/Cas9 tool, allowing the inactivation of the DCIR gene in hematopoietic stem cells by lentiviral infection, has been developed. These results will allow us to better characterize the roles of DCIR isoforms, contributing so to the development of therapeutic strategies targeting this lectin.
iv
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... vi
Liste des tableaux ... viii
Liste des abréviations ... ix
Remerciements ... xii
Introduction ... 1
1. Le virus de l’immunodéficience humaine 1 ... 1
1.1. Génome viral ... 1
1.2. Cycle de réplication ... 2
1.3. Mécanismes d’infection ... 4
1.4. Cures contre le VIH-1... 4
2. Dérèglement du système immunitaire ... 5
2.1. Lymphocytes T CD4... 5
2.2. Lymphocytes T régulateurs ... 6
2.3. Lymphocytes T CD8... 7
2.4. Lymphocytes T auxiliaires folliculaires ... 7
2.5. Lymphocytes B ... 8
3. Les cellules dendritiques ... 9
3.1. Altération des cellules dendritiques par le VIH-1... 9
3.2. Classes de cellules dendritiques ... 10
3.3. Interaction des cellules dendritiques avec le VIH-1... 12
3.4. Facteurs de restriction des cellules dendritiques à l’infection par le VIH-1 ... 13
4. Thérapies en développement contre le VIH-1 ... 14
5. Les lectines et les lectines de type C ... 16
6. Le DCIR ... 16
6.1. Gène et protéine ... 18
6.2. DCIR murin ... 21
6.3. Les ligands du DCIR ... 22
6.4. Signalisation des récepteurs inhibiteurs ... 23
6.5. Rôles du DCIR dans l’homéostasie immunitaire ... 27
6.6. Modulation des isoformes du DCIR ... 31
Chapitre 1 : Hypothèse et objectifs ... 33
1. Problématique ... 33
2. Hypothèse ... 34
3. Objectifs ... 34
Chapitre 2 : Matériels et méthodes ... 35
1. Développer une PCR quantitative spécifique pour chaque isoforme du DCIR ... 35
1.1. Choix des amorces ... 35
1.2. Détermination des conditions optimales ... 37
1.3. Conception des courbes standards ... 40
2. Quantifier l’expression des isoformes du DCIR dans des cellules immunitaires de patients infectés par le VIH-1 ... 41
2.1. Cohorte de patients ... 42
2.2. Extraction d’ARN et qPCR ... 43
3. Déterminer les corrélations entre le patron d’expression de chaque isoforme et les données cliniques des patients VIH-1 ... 43
v
4. Développer un outil CRISPR/Cas9 permettant le knock-out du gène du DCIR dans des cellules souches
hématopoïétiques ... 44
4.1. Fabrication de l’outil CRISPR/Cas9 ... 46
4.2. Transfection de cellules HEK 293T avec les plasmides ... 49
4.3. Extraction de l’ADNg des cellules HEK 293T transfectées ... 50
4.4. Amplification des régions cibles par réaction en chaîne par polymérase (PCR) ... 51
4.5. Validation par surveyor assay ... 53
4.6. Production de lentivirus ... 54
4.7. Infection des cellules souches hématopoïétiques ... 55
4.8. Tri cellulaire et différenciation en cellules dendritiques immatures ... 56
4.9. Coloration et microscopie ... 56
Chapitre 3 : Résultats ... 58
1. Développer une PCR quantitative spécifique pour chaque isoforme du DCIR ... 58
1.1. Validation des séquences amplifiées par les amorces ... 58
1.2. Élaboration des courbes standards ... 60
2. Quantifier l’expression des isoformes du DCIR dans des cellules immunitaires de patients infectés par le VIH-1 ... 61
2.1. Expression des isoformes dans les cellules mononucléées sanguines périphériques ... 63
2.2. Expression des isoformes dans les cellules sanguines polynucléées ... 64
3. Déterminer les corrélations entre le patron d’expression de chaque isoforme et les données cliniques des patients VIH-1 ... 65
3.1. Corrélations dans les cellules mononucléées sanguines périphériques ... 65
3.2. Corrélations dans les cellules sanguines polynucléées ... 68
4. Développer un outil CRISPR/Cas9 permettant le knock-out du gène du DCIR dans des cellules souches hématopoïétiques ... 70
4.1. Validation des plasmides ... 71
4.2. Production de lentivirus ... 74
4.3. Infection des cellules souches hématopoïétiques ... 75
4.4. Prolifération des cellules souches hématopoïétiques ... 77
4.5. Tri cellulaire ... 78
4.6. Différenciation en cellules dendritiques immatures ... 80
4.7. Validation par microscopie ... 82
Conclusion ... 84
5. Quantification de l’expression des isoformes du DCIR ... 84
5.1. Mise au point d’une technique de qPCR ... 84
5.2. Patron d’expression des isoformes du DCIR dans des cellules immunitaires de patients infectés par le VIH-1 ... 85
6. Corrélations entre le patron d’expression des isoformes et les données cliniques des patients VIH-1 87 6.1. Dans les cellules mononucléées sanguines périphériques ... 87
6.2. Dans les cellules sanguines polynucléées ... 88
7. Développement d’un outil CRISPR/Cas9 permettant le knock-out du gène du DCIR dans des cellules souches hématopoïétiques ... 90
8. Perspectives ... 92
vi
Liste des figures
Figure 1 : Schéma de la structure d’une particule du VIH-1. ... 1
Figure 2 : Structure du génome du VIH-1 (9200 à 9600 nucléotides). ... 2
Figure 3 : Vue d’ensemble du cycle de réplication du VIH-1. ... 3
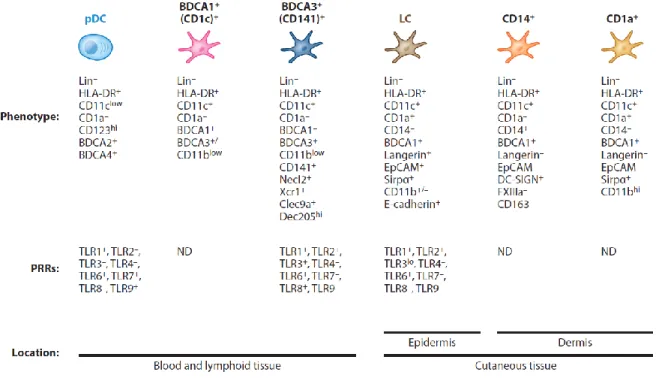
Figure 4 : Phénotypes, expression de récepteurs de pathogènes et localisation des différents groupes de cellules dendritiques chez l’humain. ... 12
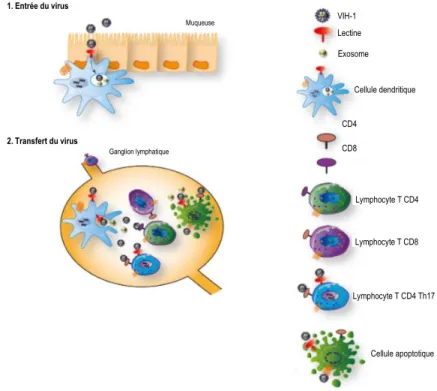
Figure 5 : Rôle des cellules dendritiques dans la transmission du VIH-1. ... 13
Figure 6 : Expression du DCIR dans différents types cellulaires et différentes conditions... 17
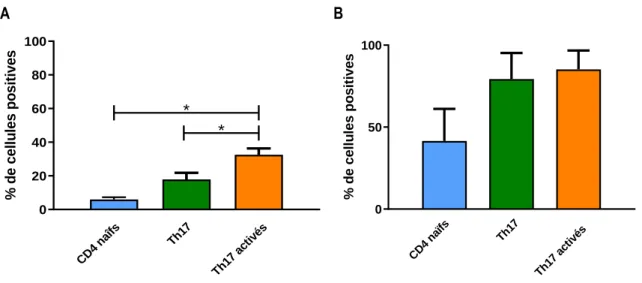
Figure 7 : Expression du DCIR dans des lymphocytes T CD4 naïfs et des lymphocytes T CD4 polarisés en Th17. ... 18
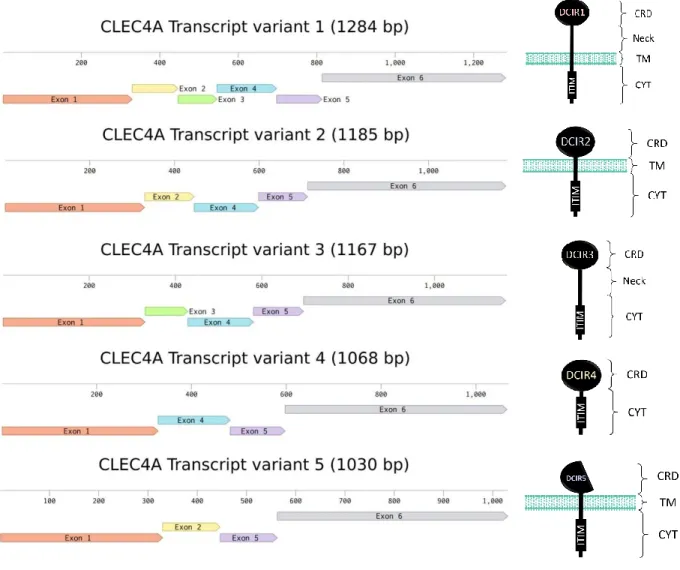
Figure 8 : Séquence ADN codante et séquence d’acides aminés du gène et de la protéine du DCIR humain. 20 Figure 9 : Exons des différentes isoformes du DCIR et leur structure protéique. ... 21
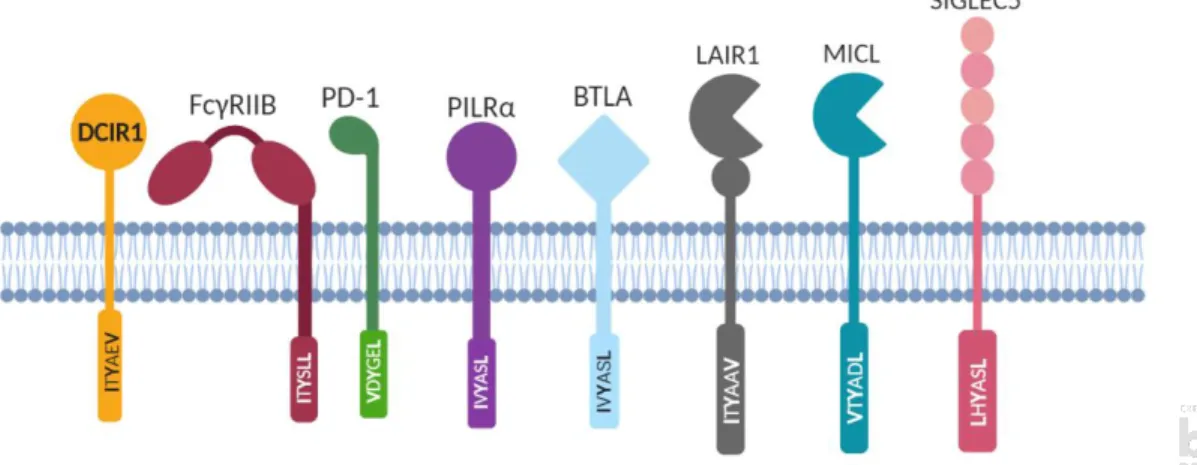
Figure 10 : Différents récepteurs de l’immunité possédant un motif ITIM et la séquence de leur ITIM. ... 24
Figure 11 : Voie de signalisation de l’activation du récepteur DCIR. ... 26
Figure 12 : Rôles du DCIR dans les cellules dendritiques. ... 29
Figure 13 : Position des amorces sur l’ADNc des différentes isoformes du DCIR. ... 37
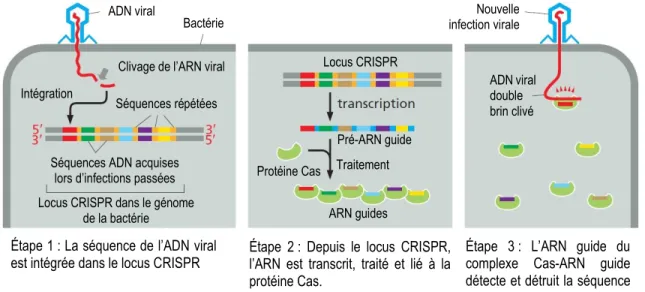
Figure 14 : Mécanisme de défense procaryotique CRISPR. ... 44
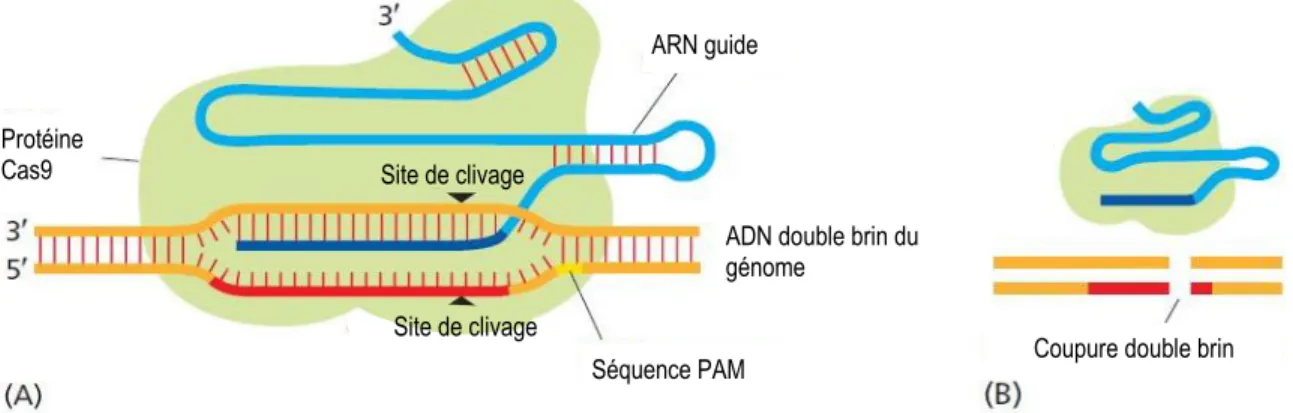
Figure 15 : Complexe protéique de l’endonucléase Cas9 de la technique de CRISPR/Cas9. ... 45
Figure 16 : Schéma expérimental des différentes étapes de l’objectif 5. ... 46
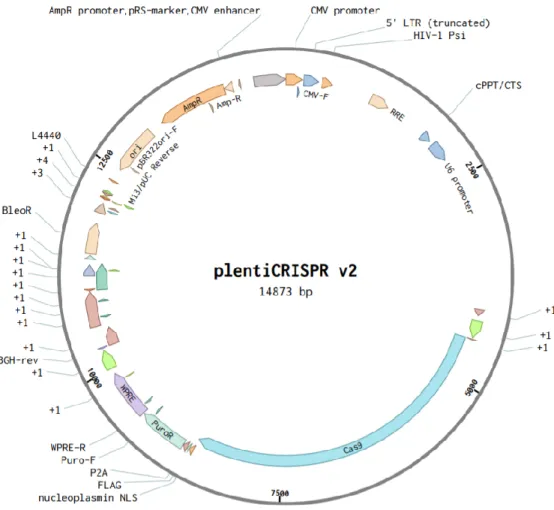
Figure 17 : Plasmide utilisé pour fabriquer les plasmides témoins, DCIR1, DCIR2 et DCIR3. ... 47
Figure 18 : Positions des amorces des ARN guides des plasmides DCIR1, DCIR2 et DCIR3 sur le gène du DCIR. ... 48
Figure 19 : Construction du plasmide DCIR1, aussi nommé pLentiCRISPRv2_2a_GFP DCIR sgRNA1 (cl.1). 49 Figure 20 : Positions des amorces de la PCR sur le gène du DCIR et la région en amont du gène. ... 52
Figure 21 : Séquençage des amplicons obtenus avec les paires d’amorces des isoformes 1 à 4 du DCIR. .... 59
Figure 22 : Courbes standards des qPCR du CT par rapport au nombre de copies d’ADNc par µL pour les pairs d’amorces des isoformes du DCIR. ... 60
Figure 23 : Quantification de l’expression des isoformes du DCIR dans les cellules immunitaires des patients VIH-1. ... 62
Figure 24 : Quantification de l’expression des isoformes du DCIR dans les cellules mononucléées sanguines périphériques des patients VIH-1. ... 63
Figure 25 : Quantification de l’expression des isoformes du DCIR dans les cellules sanguines polynucléées des patients VIH-1. ... 64
Figure 26 : Cellules HEK 293T transfectées avec les différents plasmides. ... 71
Figure 27 : Cytométrie en flux des cellules HEK 293T transfectées ou non avec les plasmides témoin, DCIR1, DCIR2 et DCIR3. ... 72
Figure 28 : Migration sur gel des amplicons des régions des exons 1 et 2 du DCIR ciblé par la Cas9 dans nos cellules HEK 293T transfectées avec nos quatre plasmides. ... 73
Figure 29 : Migration des amplicons du PCR du surveyor assay... 74
Figure 30 : Cellules HEK 293 48 heures après leur transfection avec les plasmides RRE, REV, VSV-G et le plasmide de transfert correspondant à leur condition. ... 75
Figure 31 : Photographies des précurseurs de cellules dendritiques infectées. ... 76
Figure 32 : Résultats de l’analyse de cytométrie en flux des échantillons de précurseurs de cellules dendritiques infectées. ... 77
Figure 33 : Prolifération des précurseurs de cellules dendritiques... 78
Figure 34 : Résultats du tri des précurseurs de cellules dendritiques infectées par les lentivirus... 79
Figure 35 : Photographies des précurseurs de cellules dendritiques infectés et triés positifs à la GFP. ... 80
Figure 36 : Résultats de l’analyse de cytométrie en flux des cellules dendritiques immatures triées positives à la GFP et cultivées en présence de TNFα. ... 81
vii
Figure 37 : Cellules dendritiques immatures infectées par les lentivirus et triées positives à la GFP après 6 jours en culture avec du TNFα. ... 83 Figure 38 : Expression des isoformes du DCIR dans les PMN de patients de différents groupes d’âge. ... 89
viii
Liste des tableaux
Tableau 1 : Différents noms du DCIR chez l’humain et leur abréviation. ... 17
Tableau 2 : Les différents ligands du DCIR et de DC-SIGN (180, 225, 230, 248-250, 267, 269-275). ... 23
Tableau 3 : Séquence d’acides aminés des ITIM de différents récepteurs de l’immunité. ... 25
Tableau 4 : Caractéristiques des différentes amorces du projet. ... 36
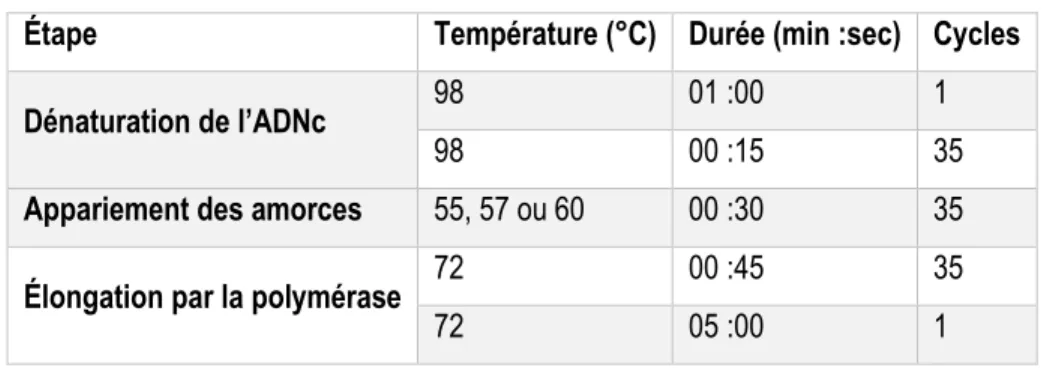
Tableau 5 : Caractéristiques des différentes étapes de la PCR. ... 39
Tableau 6 : Caractéristiques des différentes étapes du qPCR. ... 40
Tableau 7 : Données cliniques des patients VIH-1 des différents groupes de la cohorte. ... 43
Tableau 8 : Caractéristiques des amorces sélectionnées pour les ARN guides pour l’outil CRISPR/Cas9. ... 48
Tableau 9 : Informations sur les paires d’amorces utilisées pour la PCR. ... 51
Tableau 10 : Programme de la PCR. ... 52
Tableau 11 : Programme du surveyor assay. ... 53
Tableau 12 : Données obtenues pour la qPCR avec les amorces des isoformes du DCIR. ... 61
Tableau 13 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 1 du DCIR dans les cellules mononucléées sanguines périphériques des patients VIH-1. ... 65
Tableau 14 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 2 du DCIR dans les cellules mononucléées sanguines périphériques des patients VIH-1. ... 66
Tableau 15 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 3 du DCIR dans les cellules mononucléées sanguines périphériques des patients VIH-1. ... 67
Tableau 16 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 4 du DCIR dans les cellules mononucléées sanguines périphériques des patients VIH-1. ... 67
Tableau 17 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 1 du DCIR dans les cellules sanguines polynucléées des patients VIH-1. ... 68
Tableau 18 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 2 du DCIR dans les cellules sanguines polynucléées des patients VIH-1. ... 69
Tableau 19 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 3 du DCIR dans les cellules sanguines polynucléées des patients VIH-1. ... 69
Tableau 20 : Corrélations de Pearson entre les données cliniques des patients des différents groupes de la cohorte et l’expression de l’isoforme 4 du DCIR dans les cellules sanguines polynucléées des patients VIH-1. ... 70
Tableau 21 : Résumé des corrélations établies de l’expression des isoformes du DCIR dans les cellules mononucléées sanguines périphériques avec les données cliniques des patients VIH-1. ... 87
Tableau 22 : Résumé des corrélations établies de l’expression des isoformes du DCIR dans les cellules sanguines polynucléées avec les données cliniques des patients VIH-1. ... 88
ix
Liste des abréviations
ADN : Acide désoxyribonucléique ADNg : ADN génomique
ANOVA : Analyse de variance
APOBEC3G/3F : Apoplipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G or 3F ARN : Acide ribonucléique
BAFF : Facteur d’activation des cellules B BDCA -1 : Blood dendritic cell antigen 1 BDCA-3 : Blood dendritic cell antigen 3 BLAST : Basic Local Alignment Search Tool BLIMP : PR domain zinc finger protein 1
BMC : Cellules de moelle osseuse de souris (bone marrow cells) BTLA: B- and T-lymphocyte attenuator
Cas9 : CRISPR associated protein 9 CCR : Récepteur à C-C chimiokine
CD : Récepteur de différenciation (cluster of différenciation) CLEC4A : C-type lectin domain family 4 member A
CLEC9A : C-type lectin domain family 9 member A CLECSF6 : C-type lectin superfamily 6
CMH-II : Complexe majeur d’histocompatibilité II CpG : Dinucléotide cytosine-phosphate-guanine
CRD : Domaine de reconnaissance des hydrates de carbone
CRISPR : Clustered Regularly Interspaced Short Palindromic Repeats CSH : Cellules souches hématopoïétiques
CX3CR1 : CX3C chemokine receptor 1 CXCR : Récepteur à C-X-C chimiokine DC : Cellules dendritiques
DCAR : Dendritic cell immunoactivating receptor f DCIR : Dendritic cell immunoreceptor
DC-SIGN : Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin dNTP : Désoxyribonucléotides triphosphate
EDTA : Acide éthylène diamine tétra-acétique EFV : Efavirenz
Env : Enveloppe
EpCAM : Epithelial cell adhesion molecule FcγRIIB : Récepteur Fc γRIIB
FcεR1 : Récepteur d’immunoglobuline E à haute affinité FSC : Taille relative des cellules (foward-scatter) FXIIIa : Facteur IIIa
Gag : Antigène spécifique de groupe ou group-specific antigen GFP : Protéine fluorescente verte (green Fluorescent Protein) Gp : Glycoprotéine
HCV : Virus de l’hépatite C
HLA-DR : Human Leukocyte Antigen – DR isotype IDT : Integrated DNA technologies
IFNα : Interféron α IL : Interleukine
IM-MDDC : Cellules dendritiques immatures dérivées de monocytes INDEL : Insertions et délétions
x
ITAM : Motif activateur à base de tyrosine (Immunoreceptor tyrosine-based activation motif) ITIM : Motif inhibiteur à base de tyrosine (Immunoreceptor tyrosine-based inhibitory motif) LAIR1 : Leukocyte-associated immunoglobulin-like receptor 1
LC : Cellules de Langerhans LLIR : Lectin-like Immunoreceptor LPS : Lipopolysaccharide bactérien
LTR : Longues répétitions terminales (long terminal repeat) MMR : Macrophage mannose receptor
MR : Mannose receptor
NCBI : National Center for Biotechnology Information NEB : New England BioLabs inc.
NK : Cellules tueuses naturelles
NNRTI : Inhibiteurs non-nucléosides de la transcriptase inverse ou non-nucleoside reverse transcriptase
inhibitors
NRTI : Inhibiteurs nucléosides de la transcriptase inverse ou nucleoside reverse transcriptase inhibitors P : Protéine
PAM : Protospacer adjacent motif
PCR : Réaction en chaîne par polymérase (polymerase chain reaction) PD-1 : Programmed-death 1
pDC : Cellules dendritiques plasmacytoïdes
PIC : Complexe de préintégration ou pre-integration complex PILRα : Paired immunoglobulin-like type 2 receptor α
PMA : Acétate de phorbol myristate Pol : Polymérase
PréDC : Précurseurs de cellules dendritiques PRRs : Pattern-recognition receptors PSGlu : Pénicilline-Streptomycine-Glutamine PTP : Protéines tyrosine phosphatases
qPCR : Réaction en chaîne par polymérase quantitative (quantitative polymerase chain reaction) RNase : Ribonucléase
SAMHD1 : Sterile alpha motif and HD domain 1 SD : Erreur standard
SDS : Dodécylsulfate de sodium SH-2 : SRC homology-2 domain
SHP-1 : Src homology region 2 domain-containing phosphatase-1 SHP-2 : Src homology region 2 domain-containing phosphatase-2 SIDA : Syndrome de l’immunodéficience acquise
SIGLEC5 : Sialic acid binding immunoglobulin-like lectin 5 siRNA : Petits ARN interférents
SSC : Granularité relative (sidescatter) SSG : Sodium stibogluconate
T-bet : T-box transcription factor TBX21 Tbr2 : éomésodermine ou T-box brain protein 2 tetherin/CD317 : Bone marrow stromal cell antigen-2 TLR : Récepteurs de type toll (toll-like receptors) Tm : Températures de demi-dénaturation TNF : Facteur de nécrose tumorale
UHRESS : Unité Hospitalière de recherche, d’enseignement et de soins sur le SIDA VIH-1 : Virus de l’immunodéficience humain 1
xi
Ordinary people with commitment can make an extraordinary impact on their world. -John C. Maxwell
xii
Remerciements
Ce mémoire marque la fin de nombreux mois de recherche de littérature scientifique, d’expériences de laboratoire, de rédaction de documents et, soyons honnêtes, d’impasses et de moments plus difficiles. Je n’aurais jamais pu écrire ce mémoire et terminer mon projet sans l’aide de ma directrice de recherche, de mes collègues et de ma famille.
Tout d’abord, je souhaite remercier ma directrice de recherche Dre Caroline Gilbert pour son soutien, sa patience et son aide constructive. Si Dre Gilbert ne m’avait pas offert l’opportunité de travailler dans son laboratoire pour mes nombreux stages et pour ma maîtrise, je ne serais pas rendue où j’en suis aujourd’hui. Je la remercie aussi pour tout ce que j’ai appris avec elle, pour sa disponibilité et aussi pour sa confiance en moi, qui m’ont d’autant plus aidé durant ces dernières années. Merci aussi au Dr Frédéric Barabé d’avoir accepté d’être mon codirecteur de maîtrise et pour son expertise.
Ensuite, je voudrais remercier tous les membres de mon équipe de recherche, anciens et actuels, pour leur aide considérable et leur soutien moral. Merci aux étudiants Julien Boucher, Lindsay Grimard-Letellier, Wilfried Bazié, Gabriel Pépin, Camille Contant, Émilie Côté et Julien Roy pour leur support technique et moral ainsi que pour leur camaraderie. Chacun d’eux m’a appris de nombreuses choses et a contribué à la réussite de mon projet. Merci aussi à Dominique Ouellet, ancienne professionnelle de recherche de l’équipe du Dr Jacques P. Tremblay, et à Benjamin Goyer, notre professionnel de recherche, pour leur patience, leur professionnalisme, leur support et leur participation dans mon projet de maîtrise. Je souhaite aussi remercier les membres des autres équipes de recherche de l’axe des maladies infectieuses et immunitaires du CHU ainsi que les techniciens et les professionnels de recherche des plateformes de génomique, d’imagerie et de cytométrie du CHU de Québec, qui ont répondu à mes questions et qui ont participé de près ou de loin à mon projet par leur collaboration. Finalement, je remercie grandement et je dédie ce mémoire à ma mère Maryse Beaupré, à mon père Bruno Tremblay ainsi qu’à mon conjoint Jordan Woodbury pour leur immense soutien moral. Ils ont cru en moi durant tout mon parcours et m’ont soutenu dans mes moments plus difficiles. Un petit merci aussi à mon perroquet, qui m’a accompagné durant ma rédaction et qui m’a aussi soutenu, à sa manière.
1
Introduction
1. Le virus de l’immunodéficience humaine 1
Le virus de l’immunodéficience humaine 1 (VIH-1) est responsable du développement du syndrome de l’immunodéficience acquise (SIDA) (1). Ce rétrovirus, faisant partie de la famille des lentivirus, a été isolé pour la première fois en 1983 à l’Institut Pasteur en France et possède une structure sphérique ayant un diamètre d’environ 120 nanomètres (1-5). La particule virale mature possède une double membrane externe lipidique qui contient des trimères de protéine Env à sa surface. La protéine Env est formée de trimères de la glycoprotéine 120 (gp120), qui sont ancrés à la membrane externe par des trimères de la protéine transmembranaire gp41 (4, 5). La capside virale possède une forme conique et est formée par la protéine p24 (6). La Figure 1 illustre la structure d’une particule du VIH-1.
Figure 1 : Schéma de la structure d’une particule du VIH-1.
Le VIH-1 est un rétrovirus à ARN. gp = Glycoprotéine; p = protéine. Figure inspirée de la figure 3 de la revue de la German
Advisory Committee Blood, 2016 (7) et conçue avec la plateforme BioRender.
1.1. Génome viral
Le génome du VIH, contenu dans la capside virale, comprend deux fragments identiques d’acide ribonucléique (ARN) simple brin. L’acide désoxyribonucléique (ADN) double brin du provirus est produit par transcription inverse pour ensuite être intégré dans le génome de la cellule hôte. Cet ADN est flanqué à chaque extrémité par une séquence de longues répétitions terminales (long terminal repeat ou LTR), qui n’est pas transcrite. Vient
2
ensuite le gène Gag, qui code pour des protéines de la membrane externe du noyau viral, pour des protéines de la capside, de la nucléocapside et pour une protéine stabilisante d’acides nucléiques. Le gène Gag est suivi par le gène Pol codant pour la protéase, la transcriptase inverse et l’intégrase du virus. Le gène Env, situé en aval du gène Pol, code pour les deux glycoprotéines de l’enveloppe virale, gp120 et gp41. Des gènes régulateurs sont aussi présents : Tat et Rev, qui sont nécessaires à l’initiation de la réplication virale, ainsi que Nef, Vif, Vpr et Vpu, qui agissent sur la réplication virale, le bourgeonnement et la pathogenèse du virus (8, 9) La Figure 2 illustre le génome complet du VIH-1 (7).
Figure 2 : Structure du génome du VIH-1 (9200 à 9600 nucléotides).
Les cadres de lectures des gènes du VIH-1 sont illustrés. LTR = long terminal repeat; gag = antigène spécifique de groupe ou group-specific antigen; pol = polymérase; env = enveloppe. Figure inspirée de la figure 1 de la revue de la German
Advisory Committee Blood, 2016 (7) et conçue avec la plateforme BioRender.
1.2. Cycle de réplication
Pour infecter une cellule immune, la glycoprotéine gp120 du VIH-1 mature se lie au récepteur de différenciation (CD ou cluster of differenciation) 4 de la cellule hôte. Ainsi, toutes les cellules possédant le CD4, notamment les lymphocytes T auxiliaires, les macrophages, les cellules dendritiques et les astrocytes, sont susceptibles d’être infectées par le VIH-1. Par la suite, un changement de conformation du CD4 et de la gp120 se produit, permettant l’ouverture d’un site de liaison entre la gp120 et le récepteur à C-C chimiokine (CCR) de type 5 ou le récepteur à C-X-C chimiokine (CXCR) de type 4, présents à la surface de la cellule hôte (10, 11). La liaison simultanée de gp120 au CD4 et au corécepteur mène de nouveau à un changement conformationnel de gp120 et gp41 (3, 12). Gp41 prend alors la forme d’un canal qui s’insère dans la membrane plasmique de la cellule hôte. C’est ainsi que l’enveloppe virale se fusionne à la membrane cellulaire de l’hôte. À la suite de la fusion, la capside virale est transloquée dans le cytoplasme et est décapsidée, permettant la libération de son contenu dans le cytoplasme. C’est dans le cytoplasme que la transcriptase inverse du VIH-1 est activée et peut alors transcrire l’ARN simple brin viral en ADN complémentaire (ADNc). L’ARN est ensuite dégradé par une ribonucléase (RNase) de l’hôte, puis l’ADN complémentaire simple brin est converti en ADN double brin, que
3
l’on nomme l’ADN proviral. L’ADN proviral est transporté dans le noyau de la cellule hôte par un pore nucléaire. L’intégrase qui accompagne alors l’ADN proviral insère au hasard ce dernier dans le génome de la cellule hôte, ce qui marque l’établissement ainsi que la persistance de l’infection virale. Le génome viral peut alors être répliqué lors de la division cellulaire ou lors de l’activation de la cellule hôte. Ensuite, les nouveaux ARN produits par transcription sont transloqués dans le cytoplasme et sont traduits en protéines virales, permettant l’assemblage de nouvelles particules virales immatures par bourgeonnement de la membrane plasmide. Les particules virales vont finalement devenir matures par le clivage de protéines structurales menant à la formation d’une protéine Gag mature. Au terme de ces étapes, les virions sont infectieux. Six à huit heures s’écoulent entre l’attachement du virus à la cellule hôte (cette étape prenant de trente minutes à deux heures) et la formation de l’ADN proviral. L’intégration de l’ADN proviral au génome de l’hôte prend place environ douze à quatorze heures après le début de l’infection. Vingt-quatre heures après le premier contact avec le virus, les premières particules virales formées de novo sont détectables. Le cycle de réplication complet du VIH-1 est illustré à la Figure 3 (13).
Figure 3 : Vue d’ensemble du cycle de réplication du VIH-1.
Les étapes principales du cycle sont: la liaison du récepteur CD4 et du corécepteur au virus; la fusion du virus avec la membrane de la cellule hôte; la décapsidation; la libération de l’ARN et des protéines virales dans le cytoplasme; la transcription inverse de l’ARN viral en ADN; la formation du complexe de préintégration (pre-integration complex ou PIC); la translocation de l’ADN viral dans le noyau; l’intégration, la transcription et la traduction de l’ADN viral dans le génome de l’hôte, menant à la formation de nouveaux ARN et protéines virales; la translocation des ARN et des protéines nouvellement formées à la surface de la cellule hôte; l’assemblage et la libération d’une nouvelle particule virale immature et la maturation du virus. Les différentes familles de traitements antirétroviraux sont mentionnées dans les encadrés verts aux étapes du cycle de réplication qu’elles bloquent. Des facteurs de restriction du VIH-1 sont illustrés en rouge ainsi que leur antagoniste
4
viral correspondant en bleu. LTR = long terminal repeat; NRTI = nucleoside reverse transcriptase inhibitors; NNRTI =
non-nucleoside reverse transcriptase inhibitors. Figure et légende tirées de l’article de Barré-Sinoussi, 2013 (13).
1.3. Mécanismes d’infection
Le virus peut se propager de cellule en cellule de plusieurs façons, selon qu’il est associé à une cellule ou sous forme de particule libre. Lorsque le virus est transmis d’une cellule infectée à une autre directement, il se crée un lieu de forte proximité entre les deux cellules. On nomme cette zone la synapse virologique. Dans la synapse virologique, des particules virales formées de novo sont présentes en grande quantité, permettant ainsi une transmission virale efficace par la fusion ou l’endocytose des particules virales dans la cellule. Pour qu’il y ait la formation d’une synapse virologique, une infection productive de la cellule est nécessaire. En fait, l’infection productive de la cellule mène à l’expression de la protéine Env à sa surface. Env peut ensuite se lier au récepteur CD4 d’autres cellules avoisinantes (14).
1.4. Cures contre le VIH-1
La principale conséquence de l’infection par le VIH-1 est une dérégulation du système immunitaire. Dès les premières heures de l’infection, le virus se transmet d’une cellule à l’autre et se réplique si rapidement qu’une charge d’ARN virale allant jusqu’à 1 million de copies par millilitre de plasma est atteinte avant même que les lymphocytes B, les lymphocytes T CD4 et les lymphocytes T CD8 aient eu le temps de déclencher une réponse anti-VIH efficace (15, 16). Une fois que le virus a pris le dessus, le dérèglement de l’immunité de l’hôte est irréversible. Malgré que des traitements antirétroviraux existent pour contrôler l’infection, les patients infectés devront vivre toute leur vie avec cette infection chronique (14). De plus, les traitements ne permettent pas la restauration de la réponse immunitaire et apportent leur lot d’effets secondaires. C’est pourquoi le développement d’un traitement fonctionnel, c’est-à-dire d’un traitement qui permet de contrôler de façon permanente l’infection sans nécessairement éradiquer le virus, est souhaitable (13).
Selon la revue de Lui en 2015, on parle de deux catégories de cures du VIH-1. Il y a la cure stérilisante, qui désigne que toute trace de virus dans le corps d’un patient a été éliminée (17), tandis que la cure fonctionnelle désigne un contrôle à long terme de la réplication virale par le système immunitaire du patient, en absence de thérapie antirétrovirale, ainsi que l’élimination des symptômes de la maladie. La cure fonctionnelle n’éliminerait pas totalement le virus, qui continuerait à se répliquer, mais sous le contrôle du système immunitaire de son hôte (18). Le système immunitaire de certains patients, les contrôleurs du VIH-1, parvient spontanément à contrôler la réplication virale du VIH-1. Par conséquent, ces patients ne développent pas le SIDA (19-21). Les
5
mécanismes sous-jacents de ce contrôle de la réplication virale ont inspiré de nombreuses équipes de recherche afin de développer une thérapie immunomodulatrice qui mènerait à ce genre de cure fonctionnelle. En effet, le développement d’une cure stérilisante pour l’infection au VIH-1 s’avère laborieux en raison, entre autres, de l’établissement d’un réservoir viral dans des cellules immunitaires en latence. Cela suggère d’autant plus que le développement d’une cure fonctionnelle du VIH-1 pour les patients infectés devrait être une priorité. Afin de pouvoir un jour atteindre ce but, il est nécessaire de bien comprendre l’effet qu’a l’infection par le VIH-1 sur le système immunitaire, et, plus particulièrement, sur chaque type cellulaire impliqué.
2. Dérèglement du système immunitaire
Le dérèglement du système immunitaire survenant lors de l’infection par le VIH-1 est caractérisé par une hyperactivation généralisée qui atteint, entre autres, les lymphocytes T CD4. En effet, les lymphocytes T CD4 infectés ont une durée de vie allant de 2 à 4 jours, puisqu’ils seront éliminés par la lyse des cellules productrices de virus ou par des lymphocytes T cytotoxiques du système immunitaire de l’hôte (7, 22, 23). La diminution marquée du nombre de lymphocytes T CD4, accompagnée par l’inhibition de la production de nouveaux lymphocytes, mène au déclin rapide de leur population. Ce déclin mène, à long terme, à l’immunodéficience à laquelle on associe le VIH-1 (7). Puisque le virus est intégré dans le génome de nombreuses cellules immunes comme les macrophages, les astrocytes ou les lymphocytes mémoires, il peut demeurer dans un état de latence pendant de nombreuses années. Les virus sortent de leur latence lors de l’activation de ces dernières cellules, entamant la production de nouvelles particules virales (7).
2.1. Lymphocytes T CD4
Lors d’une infection par le VIH-1, les lymphocytes T CD4 spécifiques au VIH-1 supportent la réponse des lymphocytes T CD8. Cependant, puisque l’infection mène rapidement à la déplétion de la population de lymphocytes T CD4 et puisque la plupart des lymphocytes T CD4 restants ne sont pas totalement fonctionnels, ce support est absent de la réponse. Le dysfonctionnement des lymphocytes T CD4 serait entre autres lié à l’expression du récepteur inhibiteur programmed-death 1 (PD-1) (24-26). PD-1 est un récepteur immunitaire inhibiteur qui permet de réduire l’activation des lymphocytes T CD4. Dans le contexte de l’infection au VIH-1 et dans le contexte d’autres infections chroniques, PD-1 est surexprimé sur les lymphocytes T CD4, ce qui mène à l’inhibition de la prolifération de ces derniers ainsi qu’à leur apoptose (27, 28). Cette surexpression a aussi été associée à l’altération des fonctions antivirales des lymphocytes T (27). De plus, un répresseur transcriptionnel de la différenciation des lymphocytes T et des lymphocytes B, nommé PR domain zinc finger protein 1 (BLIMP), serait hautement surexprimé sur les cellules PD-1 positives (29, 30). Plusieurs études suggèrent qu’une réponse
6
anti-VIH rapide par les lymphocytes T CD4, avant que ces derniers soient atteints et détruits par le virus, empêcherait la progression de l’infection (31, 32).
Un sous-groupe de lymphocytes T CD4, les lymphocytes T CD4 Th17, sont particulièrement affectés par l’infection au VIH-1. Ces cellules se situent à la surface des muqueuses et protègent l’intégrité de ces dernières contre des organismes pathogènes (33, 34). On dénote une déplétion marquée des lymphocytes T CD4 Th17, proportionnelle à la progression de l’infection, dans la muqueuse intestinale et génitale lors de l’infection au VIH-1 (35-37). Ces cellules seraient particulièrement permissives à l’infection, puisqu’elles expriment les deux corécepteurs du virus (38). Malgré leur fragilité à l’infection, ce type de lymphocyte serait important dans la défense contre le virus, puisqu’il a été rapporté chez les macaques que les individus ayant un plus grand nombre de lymphocytes T CD4 Th17 avant l’infection avaient une charge virale maximale moindre que les macaques avec un plus petit nombre de lymphocytes T CD4 Th17 (15, 39). Un ratio lymphocytes T CD4 Th17/lymphocytes T régulateurs inférieur à 1 chez un individu serait directement lié à sa tendance de progression vers la maladie (40, 41).
2.2. Lymphocytes T régulateurs
Les lymphocytes T régulateurs, eux, ont un rôle plus mitigé dans l’infection au VIH-1. En général, la quantité de lymphocytes T régulateurs, comparativement à celle des cellules effectrices, augmente avec la progression de l’infection et avec l’augmentation de la charge virale (42). Cette augmentation du nombre de lymphocytes T régulateurs serait renversée par les traitements antirétroviraux (43). De plus, il a été rapporté que le compte de lymphocytes T régulateurs est plus bas dans le sang périphérique et les muqueuses des patients contrôleurs de VIH-1 (44, 45). Certaines études rapportent que les lymphocytes T régulateurs ralentissent la progression de l’infection en diminuant l’activation immune chronique (46, 47). D’autres études suggèrent que les lymphocytes T régulateurs interfèrent avec la réponse immune antivirale des lymphocytes T CD4 et CD8 (48, 49). Ceci s’explique par le fait que les différentes études utilisent des définitions différentes pour décrire la population de lymphocytes T régulateurs qu’ils ont étudiée (15, 50). Par exemple, certaines études ne considèrent qu’une population particulière plus stricte de lymphocytes T régulateurs (51-53). D’autres utilisent le compte absolu ou le compte relatif de lymphocytes T régulateurs et d’autres ne se concentrent que sur la spécificité antigénique ou la capacité à éliminer l’activation des lymphocytes T (54, 55). Par exemple, l’expansion de la population de lymphocytes T régulateurs possédant le CD39 serait associée avec la progression de l’infection, avec la déplétion de lymphocytes T CD4 et avec l’activation immunitaire (52, 53). Cependant, il a été rapporté que l’activité du CD39 permettrait de médier l’élimination de la réplication du VIH-1 dans les lymphocytes T CD4 in vitro (56).
7
2.3. Lymphocytes T CD8
En plus du déclin de la population de lymphocytes T CD4, on remarque une augmentation du nombre et de la cytotoxicité des lymphocytes T CD8 (15, 57). En fait, les lymphocytes T CD8 spécifiques au VIH font leur apparition dans la circulation sanguine tôt avant la séroconversion d’un patient, c’est-à-dire avant la détection d’anticorps anti-VIH plasmatiques (58). Leur population augmente rapidement et serait associée avec une diminution de la charge virale (15, 59-61). La première réponse des lymphocytes T CD8 ne cible cependant que certains épitopes immunodominants du virus, ce qui permet au virus ne les possédant pas de s’échapper de la réponse cytotoxique (62). De plus, malgré la présence d’une réponse cytotoxique anti-VIH de grande envergure, l’activation chronique du système immunitaire mène ultimement à l’épuisement des lymphocytes T CD8, rendant avec le temps cette réponse immunitaire inefficace (63, 64). En effet, comme pour les lymphocytes T CD4, la stimulation antigénique chronique des lymphocytes T CD8 mène à l’expression du récepteur PD-1 et, par conséquent, à leur épuisement (65, 66). C’est la qualité et la rapidité plutôt que la quantité des lymphocytes T CD8 qui importent dans la réponse anti-VIH. Il a été rapporté qu’une dysfonction de l’expression de certains facteurs de transcription contrôlant le développement, la différenciation ainsi que le fonctionnement des lymphocytes T CD8, comme le T-box transcription factor TBX21 (T-bet) et l’éomésodermine (ou T-box brain
protein 2, Tbr2), soit la source de l’inefficacité de la réponse des lymphocytes T CD8 durant l’infection (67, 68).
2.4. Lymphocytes T auxiliaires folliculaires
Un autre sous-groupe de lymphocytes T, les lymphocytes T auxiliaires folliculaires, participe aussi dans le dérèglement immunitaire causé par l’infection par le VIH-1 (15). Ces lymphocytes T auxiliaires folliculaires résident dans les centres germinatifs des organes lymphoïdes secondaires et sont caractérisés par l’expression de CXCR5 et de PD-1 (69, 70). Ces lymphocytes T contribuent à la production d’anticorps spécifiques de haute affinité par les lymphocytes B ainsi qu’à la maturation des lymphocytes B en cellules plasmatiques et en lymphocytes B mémoires (71, 72). La population de lymphocytes T auxiliaires folliculaires augmente durant l’infection au VIH-1 (73-76). Puisque ces cellules n’expriment pas beaucoup le récepteur CCR5 à leur surface, on croyait qu’elles seraient protégées contre le VIH-1, mais ce n’est pas le cas (75, 76). En fait, les lymphocytes T auxiliaires folliculaires peuvent être infectés par le VIH-1 à une vitesse comparable à celle de l’infection des lymphocytes T CD4 mémoires (76, 77). La cause de l’expansion de leur population durant l’infection demeure incertaine (75).
8
2.5. Lymphocytes B
Les lymphocytes B sont aussi affectés par l’infection par le VIH-1, contribuant d’autant plus à l’épuisement de la réponse immunitaire. En fait, la population de lymphocytes B mémoires diminuent progressivement lors de l’infection, menant ultimement au développement du SIDA (78-80). De plus, l’expression du marqueur d’épuisement PD-1 est aussi augmentée chez les lymphocytes B (15, 80, 81). Il a été rapporté que le VIH-1 peut se lier aux lymphocytes B in vivo par le CD21 (exprimé sur les lymphocytes B matures et modulant leur activation), par le dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN, une lectine de type C) et par des immunoglobulines de surface, ce qui faciliterait la transmission du virus par des contacts cellule à cellule et, dans une moindre mesure, stimulerait les lymphocytes B (82-86). Ce contact direct entre le virus et les lymphocytes B n’est cependant que de petite envergure en comparaison avec les relations indirectes qu’ils partagent.
Le VIH-1 cause, de façon indirecte, une hyperactivation marquée des lymphocytes B, qui se caractérise par la production d’une quantité fulgurante d’anticorps non spécifiques (de l’hypergammaglobulinémie) (87-90), une augmentation de l’activation de lymphocytes B polyclonaux (87, 88, 91), une augmentation du taux de renouvellement des cellules (92, 93), une augmentation de l’expression de marqueurs d’activation (CD70, CD71, CD80 et CD86) (94-97), une augmentation de la différenciation des lymphocytes B en plasmablastes (88, 92, 98, 99), une augmentation de la production d’auto-anticorps (91, 100, 101) et, finalement, une augmentation de la fréquence de tumeurs malignes des lymphocytes B (102). La réponse anti-VIH par les anticorps à la suite de l’infection par le VIH-1 n’est pas efficace, puisqu’elle n’a pour cible qu’une large gamme d’épitopes non neutralisants de l’enveloppe du virus. De plus, le virus évolue et se diversifie rapidement, empêchant le rétablissement de la réponse des lymphocytes B (103, 104). Il a été rapporté que la réponse par les immunoglobulines A, spécifique au VIH-1, est faible au site muqueux de l’infection comparativement à la réponse par les autres classes d’immunoglobulines (105, 106). La destruction précoce des tissus lymphoïdes associés à la muqueuse intestinale et l’inhibition de la commutation de classe des immunoglobulines par le virus pourraient être la cause de l’affaiblissement de cette réponse (107).
On ne connait pas encore clairement quels facteurs contribueraient à l’hyperactivation des lymphocytes B durant l’infection au VIH-1. Il a été suggéré que l’interféron α (IFNα), le facteur de nécrose tumorale (TNF), l’interleukine (IL) 6, l’IL-10, le ligand CD40 ainsi que le facteur d’activation des cellules B (BAFF) participeraient de près ou de loin dans cette hyperactivation (86, 108-111). En fait, leur concentration dans le sérum des patients atteints par le VIH-1 est augmentée durant l’infection. L’IFNα en particulier serait libéré par des cellules dendritiques plasmacytoïdes après que ces dernières aient été activées par du lipopolysaccharide bactérien (LPS) libéré
9
dans la circulation sanguine par la destruction des tissus lymphoïdes intestinaux (108, 112). En effet, le LPS est libéré lorsque l’intégrité de la muqueuse intestinale est altérée par la déplétion marquée de lymphocytes T CD4 Th17 dans l’intestin (80, 113). Ce seraient donc ces facteurs, produits par d’autres cellules, qui activeraient indirectement les lymphocytes B.
En résumé, les fonctions de l’immunité acquise sont altérées par le VIH-1 dans l’infection. Ces altérations mènent au dérèglement de l’immunité qui caractérise le SIDA. En plus de l’immunité adaptative, l’immunité innée est aussi altérée, et ce, très tôt dans l’infection. On parle alors de la phase précoce de l’infection. Cette dernière met en jeu les cellules dendritiques, une autre cible du VIH-1, qui ont un rôle dans la communication entre l’immunité innée et l’immunité acquise au cours des premières étapes de l’infection et, plus précisément, dans le transfert viral.
3. Les cellules dendritiques
Les cellules dendritiques sont les acteurs principaux de l’élaboration de la réponse immunitaire, puisqu’elles établissent la communication entre différentes cellules de l’immunité innée et acquise. Elles ont pour rôle de reconnaître et capturer différents antigènes et pathogènes pour ensuite les présenter aux lymphocytes T. De plus, les cellules dendritiques permettent de contrôler la spécificité et la magnitude de la réponse immune résultant de la présentation d’antigènes. Ces cellules proviennent de cellules précurseurs CD34 positives se situant dans la moelle osseuse (114-116). Les cellules dendritiques matures expriment en grande quantité le complexe majeur d’histocompatibilité II (CMH-II ou Human Leukocyte Antigen – DR isotype ou HLA-DR) et n’expriment pas les marqueurs de la lignée lymphoïdes, comme le CD3, le CD19 et le CD20 (117).
3.1. Altération des cellules dendritiques par le VIH-1
Comme les lymphocytes, les cellules dendritiques sont aussi affectées par l’infection par le VIH-1. Leur fréquence dans le sang diminue très tôt durant l’infection et corrèle inversement avec la charge virale (118, 119). Cette diminution est irréversible et serait liée à la sécrétion aiguë et chronique d’interféron de type 1 causée par l’infection, puisque l’IFNα altère la différenciation des cellules dendritiques (120, 121). Dans les organes lymphoïdes secondaires, le nombre de cellules dendritiques serait en revanche augmenté (122, 123). Le VIH-1 peut induire leur maturation partielle et augmenter leur expression du CCR7, menant à leur migration jusqu’aux organes lymphoïdes secondaires (124). De plus, les fonctions des cellules dendritiques sont altérées par l’augmentation de la quantité de cytokines inflammatoires et de microparticules apoptotiques provenant de la mort cellulaire des lymphocytes par le virus (125). En fait, la sécrétion d’IL-12, de TNFα et d’IL-6 par la
10
stimulation des récepteurs de type toll (toll-like receptors ou TLR) 2, TLR 3, TLR 4 et TLR 7/8 est inhibée par ces facteurs. Ainsi, les cellules dendritiques ne parviennent plus à activer les lymphocytes T CD4 Th1 (126, 127). De plus, la présence de particules microbiennes dans le sang contribuerait à rendre les cellules dendritiques tolérantes (127). La présentation des antigènes du VIH-1 est altérée par l’affinité des protéines de surface du virus avec certains récepteurs des cellules dendritiques et aussi par le récepteur impliqué en particulier (128-130). Chez les patients contrôleurs de VIH-1, les cellules dendritiques sont plus efficaces pour reconnaître le VIH-1, supportent moins la réplication virale et permettent une meilleure réponse anti-VIH par les lymphocytes T (131, 132).
Les cellules dendritiques participent à la phase précoce de l’infection et font le pont entre la réponse immunitaire anti-VIH innée et adaptative. Chez les patients contrôleurs de VIH-1, elles ont de meilleures fonctions anti-VIH (21, 133-135). Il existe cependant plusieurs familles de cellules dendritiques qui ont chacun des rôles qui leur sont propres. Afin de mieux comprendre le rôle des différentes cellules dendritiques dans l’infection par le VIH-1, il est nécessaire de bien nuancer les différences entre chacune de leurs classes.
3.2. Classes de cellules dendritiques
3.2.1.
Cellules dendritiques myéloïdes
Il existe de nombreux groupes de cellules dendritiques différents, dépendamment de leur origine et de leur expression de récepteurs de surface (Figure 4). Il y a d’abord les cellules dendritiques myéloïdes périphériques, qui sont séparés en deux sous-groupes selon leur expression du récepteur CD1c (aussi appelé blood dendritic
cell antigen 1 ou BDCA -1) et du CD141 (aussi appelé blood dendritic cell antigen 3 ou BDCA-3) (117, 136). Les
cellules dendritiques myéloïdes CD141/BDCA-3 positives se trouvent dans les ganglions lymphatiques, dans les amygdales, la rate ainsi que dans la moelle osseuse, en plus de pouvoir également se trouver dans des tissus non lymphoïdes comme la peau, les poumons et le foie (137-141). Ces cellules dendritiques CD141/BDCA-3 positives permettent la présentation croisée des antigènes aux lymphocytes T CD8 en attrapant des cellules mortes ou nécrotiques via leur récepteur C-type lectin domain family 9 member A (CLEC9A) et des virus avec les TLR 3 et 8 (137, 139, 141-144). Les cellules dendritiques myéloïdes CD1c/BDCA-1 positives, elles, expriment le CD11c, le CD11b, le CD13, le CD33 ainsi que le CD45RO (136, 138, 145). On les retrouve dans le derme, dans le sang ainsi que dans des tissus lymphoïdes (rate et ganglions lymphatiques) et non lymphoïdes (peau, foie, poumon et intestins) (114, 146). Les cellules dendritiques CD1c/BDCA-1 positives expriment plusieurs récepteurs d’antigènes comme les TLR 1 à 10, dectin-1 et dectin-2, qui leur permettent la présentation croisée des antigènes aux lymphocytes T CD8 (137, 142, 144, 147-150). Il existe un troisième
11
groupe de cellules dendritiques myéloïdes, qui représente un intermédiaire entre un phénotype de cellules dendritiques et de monocytes/macrophages. Ces cellules dendritiques expriment le CD14, un marqueur de monocytes, ainsi que le CMH-II et le CD11c, ces deux derniers en grande quantité. Elles expriment aussi d’autres marqueurs de monocytes, comme le CD163, le CD11b, le CX3C chemokine receptor 1 (CX3CR1), le facteur IIIa (FXIIIa) ainsi que le DC-SIGN et n’expriment pas le CCR7 (149, 151-153). Les cellules dendritiques CD14 positives expriment les TLR 1 à 9 et se retrouvent dans les tissus lymphoïdes et non lymphoïdes (154, 155). Elles ne peuvent pas stimuler de lymphocytes T CD4 efficacement et sont considérées comme moins matures que les cellules dendritiques CD1c positives (114, 152, 156, 157).
3.2.2.
Autres cellules dendritiques
Il existe aussi une lignée de cellules dendritiques présente dans l’épiderme que l’on appelle les cellules de Langherans, qui contiennent des granules de Birbeck (158) et qui expriment le CD36, le récepteur d’immunoglobuline E à haute affinité (high-affinity immunoglobuline E receptor ou FcεR1), le CD1a, la langerine et les molécules d’adhésion epithelial cell adhesion molecule (ou EpCAM) et cadhérine E. On retrouve aussi les cellules dendritiques plasmacytoïdes, qui expriment les CD123, CD303 (ou BDCA-2) et CD304 (114, 159). Les cellules dendritiques plasmacytoïdes constituent environ 50% des cellules dendritiques du sang, ce qui représente moins de 0,1% des leucocytes du sang (136). Elles se retrouvent dans les ganglions lymphatiques et les amygdales et ont des fonctions pro-inflammatoires par leur production d’IFNα (116, 160-163). Ces cellules dendritiques expriment fortement les TLR 7 et 9 (respectivement jusqu’à 800 et 350 fois plus que l’ubiquitine selon la figure 2 de l’article de Kadowaki, 2001 (164)) et peuvent ainsi reconnaître l’ARN simple et double brin viral ou endogène (114, 164, 165). Finalement, il existe aussi des cellules dendritiques du derme, qui expriment le CD1a, le CD14 et le CD1c (166, 167).
12
Figure 4 : Phénotypes, expression de récepteurs de pathogènes et localisation des différents groupes de cellules dendritiques chez l’humain.
pDC = cellules dendritiques plasmacytoïdes; LC = cellules de Langerhans; PRR = pattern-recognition receptors; ND = non déterminé. Figure tirée de la figure 6 de la revue Merad, 2013 (116)
3.3. Interaction des cellules dendritiques avec le VIH-1
Ce sont les cellules dendritiques de la muqueuse (CD1a et CD14 positives) qui jouent un rôle dans la pathogenèse de l’infection au VIH-1. Puisqu’elles se situent à la muqueuse vaginale et rectale, elles sont les premières cibles du virus lors de l’infection (163, 168-173). Les virions peuvent se lier à des récepteurs à la surface des cellules dendritiques, comme des lectines de type C, puis peuvent être internalisés par endocytose dans la cellule dendritique (163, 172-175). Les virions internalisés dans les cellules dendritiques peuvent être dégradés, mais l’internalisation du virus par certaines lectines de type C peut mener à une dégradation incomplète (176-180). En fait, la liaison de la protéine virale gp120 à certaines lectines de type C n’apporterait pas un changement de conformation suffisant du récepteur pour permettre une infection efficace de la cellule dendritique (172). Les virus sont alors retenus dans des compartiments cellulaires de la cellule dendritique, dans lesquels ils peuvent rester protégés de la dégradation, tout en demeurant infectieux. La cellule dendritique migre alors jusqu’à un organe lymphoïde secondaire. Le compartiment cellulaire contenant les virions peut se fusionner à la surface des cellules dendritiques et permettre ainsi le transfert de virions intacts (via une synapse virologique) des cellules dendritiques à d’autres cellules comme les lymphocytes T et les lymphocytes B, menant à la propagation de l’infection (181, 182). On appelle ce processus l’infection en trans. En fait, on sépare en
13
deux types d’infection le transfert de virions des cellules dendritiques aux lymphocytes T : L’infection en trans et l’infection en cis (176). Durant la phase trans (Figure 5), les virions sont transférés à d’autres cellules immunes
via une synapse virologique par proximité entre les deux cellules (168, 177, 182-184).
Figure 5 : Rôle des cellules dendritiques dans la transmission du VIH-1.
Étapes (1 = entrée du virus, 2 = transfert du virus) du transfert des virions du VIH-1 des cellules dendritiques de la muqueuse aux lymphocytes des ganglions lymphatiques. Figure inspirée de l’article Hubert, 2015 (185).
L’infection en cis, elle, implique la réplication productive du virus dans la cellule dendritique. Par conséquent, elle consiste en l’infection de la cellule par l’intégration des gènes viraux dans le génome de la cellule dendritique. La progéniture virale peut par la suite infecter de nouvelles cellules et propager l’infection (176, 178, 186). Puisque les cellules dendritiques sont très peu infectées de façon productive par le VIH-1, la propagation de l’infection par les cellules dendritiques se fait en majorité par infection en trans. En fait, les cellules dendritiques possèdent certains facteurs de restriction empêchant leur infection productive par le VIH-1 (163, 187).
3.4. Facteurs de restriction des cellules dendritiques à l’infection par le VIH-1
Les cellules dendritiques possèdent plusieurs facteurs de restriction de l’infection par le VIH-1 (illustrés à la Figure 3). Il y a entre autres l’enzyme apoplipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G orLectine Exosome VIH-1 Cellule dendritique CD4 CD8 Lymphocyte T CD4 Lymphocyte T CD4 Th17 Lymphocyte T CD8 Cellule apoptotique 1. Entrée du virus 2. Transfert du virus Muqueuse Ganglion lymphatique
14
3F (APOBEC3G/3F), le facteur de restriction bone marrow stromal cell antigen-2 (tetherin/CD317) et l’enzyme sterile alpha motif and HD domain 1 (SAMHD1) (188, 189). Le facteur de restriction ayant le plus de potentiel
est SAMHD1, qui permet de bloquer les étapes suivant l’entrée du virus dans la cellule dendritique (190, 191). SAMHD1 agit comme une triphosphohydrolase et permet de réduire le réservoir de désoxyribonucléotides triphosphate (ou dNTP) intracellulaire. Cette réduction inhibe la transcription inverse et la synthèse d’ADN complémentaire, ce qui bloque la réplication du VIH-1 (192, 193). Les cellules dendritiques myéloïdes et les cellules dendritiques plasmacytoïdes expriment de fortement SAMHD1 (194). En plus des trois facteurs nommés précédemment, la production de cytokines menant à la maturation des cellules dendritiques (comme les interférons de type I, comprenant l’IFNα produite par les cellules dendritiques plasmacytoïdes) bloque aussi l’infection productive des cellules dendritiques par le VIH-1 (195, 196).
De nombreux mécanismes de modulation de la réponse immunitaire, impliquant les cellules dendritiques, jouent un rôle dans la propagation précoce de l’infection au VIH-1. De plus, la réponse immunitaire chez les patients contrôleurs du VIH-1 est plus efficace et leurs cellules dendritiques ont de meilleures capacités anti-VIH. Ces faits signifient qu’il est possible d’agir sur la réponse immunitaire des patients afin de moduler leur réponse anti-VIH-1. Ainsi, plusieurs équipes de recherche ont tenté de développer des thérapies immunomodulatrices contre l’infection au VIH-1. Ces thérapies permettraient au système immunitaire d’être plus efficace contre le VIH-1, contrairement à la trithérapie actuelle qui cible majoritairement des protéines virales participant à la réplication du virus.
4. Thérapies en développement contre le VIH-1
Il existe plusieurs stratégies de développement d’une cure pour l’infection au VIH-1. Par exemple, on pourrait remplacer partiellement ou complètement certains éléments du système immunitaire des patients par modification génétique afin de les rendre résistants à l’infection (197-199). On parle aussi de la méthode shock
and kill, décrite comme une réactivation des virions en latence du réservoir viral permettant l’élimination de
toutes les cellules infectées par, par exemple, les lymphocytes T cytotoxiques (200-203). Cependant, ces stratégies possèdent des failles pouvant altérer leur efficacité. Une stratégie de cure fonctionnelle, ciblant des récepteurs immunomodulateurs, propose d’utiliser à notre avantage les capacités immunomodulatrices intrinsèques de ces récepteurs afin de moduler les interactions entre le VIH-1 et son hôte et la transduction de signaux cellulaires impliqués dans l’infection (204-207).
Une telle stratégie est déjà utilisée en clinique comme traitement contre plusieurs cancers ciblant le récepteur inhibiteur PD-1 et son ligand PD-L1. En effet, l’immunothérapie du cancer cible des protéines que l’on appelle
15
des points de contrôles immunitaires, ou checkpoints immunitaires. Ces checkpoints sont des récepteurs qui freinent les fonctions immunitaires et permettent aux cellules cancéreuses d’échapper à la réponse immunitaire et de proliférer (208). Le récepteur PD-1 fait partie de ces checkpoints. La liaison de PD-1 (exprimé sur des monocytes et des lymphocytes T et B activés) et de son ligand PD-L1, (présent à la surface des cellules cancéreuses) a pour effet l’immunosuppression des cellules immunitaires activées (208-211). L’inhibition de PD-L1 ou de PD-1 empêche leur liaison l’un avec l’autre et permet de maintenir les cellules immunitaires activées. Ces cellules activées peuvent par la suite éliminer les cellules cancéreuses d’elles-mêmes. Les cellules cancéreuses de la vessie, de la peau, des reins, des poumons, de la tête et du cou, des ovaires et du sang peuvent exprimer PD-L1 (212, 213). De nombreux inhibiteurs de PD-L1 et de PD-1 sont présentement utilisés dans des études cliniques. D’autres inhibiteurs ont été approuvés et font maintenant partie des traitements utilisés contre le cancer (Tecentriq® ou atezolizumab contre le cancer de la vessie, Nivolumab pour les
mélanomes non résécables, etc.) (214-216).
Cette stratégie peut s’appliquer à d’autres maladies caractérisées par une dérégulation de la réponse immunitaire, ce qui en fait un bon candidat pour le traitement de l’infection par le VIH-1. En fait, puisque l’expression de PD-1 est augmentée sur certains lymphocytes durant l’infection par le VIH-1, l’utilisation d’un anti-PD-1 a aussi été étudiée dans le contexte de l’infection par le VIH-1 (217-219). Une étude (Mylvaganam, 2018) menée sur des macaques rhésus a déterminé que l’utilisation d’anti-PD-1 combinée avec la thérapie antirétrovirale améliore les fonctions antivirales des lymphocytes T et diminue le réservoir viral, permettant un meilleur contrôle de l’infection. De plus, cette thérapie combinée aurait pour effet de restaurer la population de lymphocytes T CD4 Th17 de la muqueuse rectale et de diminuer l’inflammation des muqueuses (217). Il a aussi été montré in vitro que l’utilisation d’anti-PD-1 sur une population de lymphocytes T CD8 améliore la capacité de ces lymphocytes à survivre et proliférer et augmente la production de cytokines et de molécules cytotoxiques en réponse à la reconnaissance d’antigènes (81).
Les résultats des thérapies anti-PD-1 sont prometteurs et montrent qu’il est possible de réguler la réponse immunitaire en notre faveur contre certaines maladies. Mis à part le PD-1, d’autres protéines ayant un rôle de régulation de la réponse immunitaire pourraient aussi être de bonnes cibles thérapeutiques contre l’infection par le VIH-1 et aussi contre bien d’autres maladies caractérisées par un dérèglement de la réponse immunitaire. Il est donc nécessaire de déterminer quelles sont ces protéines potentielles qui permettraient, dans le contexte de l’infection par le VIH-1, de rehausser la réponse immunitaire anti-VIH-1 afin de contrôler la charge virale.
16
5. Les lectines et les lectines de type C
Les lectines sont des protéines à reconnaissance de motifs (aussi appelées PRR pour pattern recognition
receptors) capables de reconnaître et de se lier à une grande variété de glycanes (220-223). Certaines lectines,
que l’on dit être de type C, sont dépendantes du calcium et possèdent un domaine de reconnaissance des hydrates de carbone (carbohydrate recognition domain, CRD) (224-226). Elles permettent l’adhésion cellulaire, le changement de concentration de glycoprotéines dans le sérum et aussi la réponse immunitaire innée contre de potentiels pathogènes (227-229). Il existe 17 sous-familles de lectines de type C qui sont identifiées de I à XVII. Dans ce mémoire, il n’en est mentionné qu’une seule : la famille des récepteurs de type II (aussi nommés
asialoglycoprotein receptors) (223, 230-232). Les récepteurs asialoglycoprotein receptor (ASGR), macrophage galactose‐binding lectin (MGL), DC-SIGN, CD23, langerine et dendritic cell immunoreceptor (DCIR) font entre
autres partie de cette famille (232). Les récepteurs de type II se lient au mannose, à l’antigène de Lewis Lex, au fucose et à d’autres ligands exogènes. Ils permettent la capture d’antigènes, l’adhésion cellulaire et la costimulation des lymphocytes T (225, 227).
Certaines lectines de type C permettent le transfert de virions des cellules dendritiques aux lymphocytes T durant la phase précoce de l’infection au VIH-1. Par exemple, il a été montré que le macrophage mannose
receptor (ou MMR), faisant partie d’une autre famille de lectines de type C (récepteurs de type VI ou multi‐CTLD endocytic receptors), permet la liaison et la transmission du VIH-1 par les macrophages (233-235). Le DC-SIGN,
un récepteur de type II pouvant reconnaître les glycanes riches en mannose à la surface de la particule virale du VIH-1, permet l’infection en trans des lymphocytes T CD4 et des lymphocytes B par le VIH-1 via les cellules dendritiques (85, 180, 183, 205, 206, 236). Puis, une autre lectine de type C faisant partie du groupe des récepteurs de type II, le DCIR, permet aussi la transmission des particules virales du VIH-1 par les cellules dendritiques lors de l’infection (172).
6. Le DCIR
Le DCIR est une lectine de type C de la famille des récepteurs de type II (226). La liste des autres noms du DCIR est présentée au Tableau 1.
17
Autres noms du DCIR Abréviations
C-type lectin domain family 4 member A CLEC4A
Lectin-like Immunoreceptor LLIR
Cluster of differentiation 367 CD367
C-type lectin superfamily 6 CLECSF6
Tableau 1 :Différents noms du DCIR chez l’humain et leur abréviation.
Cette lectine a été d’abord clonée par Bates et al en 1999 (237) et est décrite comme un récepteur présentateur d’antigènes (238). Elle est majoritairement exprimée dans les cellules dendritiques du tissu interstitiel et du derme (CD14+/CD1a-) en comparaison avec les cellules dendritiques épithéliales et épidermiques (CD1a+/CD14-) (225, 239). Le DCIR est exprimé dans différents tissus et organes, notamment dans le tractus intestinal, la rate, la moelle osseuse, les ganglions lymphatiques, la vessie, la vésicule biliaire et les poumons (239). Ce récepteur est exprimé chez les cellules dendritiques immatures (226, 240), les monocytes, les macrophages, les lymphocytes B (225, 226), les basophiles (241, 242) ainsi que les neutrophiles (224, 243). De plus, l’expression du DCIR peut être induite chez des lymphocytes T CD4, ainsi que des lymphocytes T CD8 et des cellules NK (244, 245). En effet, l’expression du DCIR est augmentée à la surface des lymphocytes T CD4, CD8 et NK dans les articulations des patients atteints de polyarthrite rhumatoïde (244) et aussi dans les lymphocytes T CD4 apoptotiques et infectés par le VIH-1 (245). À l’inverse, l’expression du DCIR à la surface des neutrophiles et des cellules dendritiques est diminuée dans des conditions pro-inflammatoires, notamment en présence de TNFα, d’IL-1 alpha et de LPS (Figure 6) (243-245).
Figure 6 : Expression du DCIR dans différents types cellulaires et différentes conditions.
Le DCIR est faiblement exprimé dans des cellules dendritiques et des neutrophiles activées par du LPS ou du TNFα, tandis qu’il est fortement exprimé dans des lymphocytes T CD4, CD8 et NK de patients atteints de polyarthrite rhumatoïde, apoptotiques ou infectés par le VIH-1. LT : Lymphocytes T, DC : Cellules dendritiques.