THESE
Pour l’obtention du Grade de
Docteur de l’Université de Poitiers
Faculté des sciences fondamentales et appliquées(Diplôme national – arrêté du 7 août 2006)
Ecole doctorale : Ingénierie Chimique Biologique et Géologique Secteur de Recherche : Aspects moléculaires et cellulaires de la biologie
Présentée par :
Lucie Chevrier
________________________________________________________________
Modulation de l’expression de MYCN et de SNAP-25 au cours de la différenciationinduite par le neuropeptide VIP dans des lignées de neuroblastome humain.
__________________________________________________________________________________ Directeur de thèse : Pr Jean-Marc Muller
Co-directeur de thèse : Dr Corinne Chadéneau Soutenue le 30 mai 2008
Devant la commission d’examen JURY
Vincent LELIEVRE, Professeur des Universités, Université Louis Pasteur, Strasbourg Rapporteur Marie-Hélène RATINAUD, Professeur des Universités, Faculté de Médecine, Limoges Rapporteur Alain MOREL, Professeur des Universités, CRLCC Paul Papin, Angers Examinateur Jean-Marc MULLER, Professeur des Universités, Université de Poitiers Examinateur Corinne CHADENEAU, Maître de conférences, Université de Poitiers Examinateur
REMERCIEMENTS
Je remercie le Professeur Jean-Marc Muller pour m’avoir accueillie au sein de son équipe et pour son aide pour la rédaction de ce mémoire.
Je tiens à remercier tout particulièrement le Docteur Corinne Chadéneau pour m’avoir encadrée pendant ce stage et pour m’avoir fait partager ses connaissances et sa rigueur. Merci pour votre disponibilité et votre soutien.
Je prie les personnes qui m’ont fait l’honneur d’accepter de juger ce travail de croire en l’expression de ma reconnaissance respectueuse.
Je remercie également toutes les personnes du laboratoire pour l’aide et le soutien qu’ils ont pu m’apporter au cours de ce stage.
J’adresse ma plus vive reconnaissance à la Ligue Contre le Cancer, ainsi qu’au Lions Club de Melle pour leur soutien financier, leur gentillesse et l’intérêt qu’ils ont porté à ce travail.
Merci également à tous mes proches qui m’ont soutenue et aidée de près ou de loin pendant ces trois ans, et même beaucoup plus pour certains…
TABLE DES MATIÈRES
liste des abréviations ... 4
INTRODUCTION ... 6
1
èrepartie : Les neuroblastomes ... 7
I - Données cliniques ... 7
1) Définition et épidémiologie ... 7
2) Origine ... 8
3) Pathologie ... 9
a) Les neuroblastomes localisés ... 9
b) Les neuroblastomes disséminés ... 10
c) Les neuroblastomes de stade 4S... 11
4) Diagnostic ... 11
II - Données biologiques... 13
1) Contenu en ADN... 13
2) Amplification de l’oncogène MYCN... 14
3) Gain du chromosome 17 ... 15
4) Perte du bras court du chromosome 1... 16
5) Perte du bras long du chromosome 11... 17
6) Altération de l’expression des gènes des récepteurs des neurotrophines ... 18
7) Voie de l’apoptose ... 19
a) La caspase 8 ... 20
b) Les protéines inhibitrices de l’apoptose (IAP)... 21
c) Les protéines de la famille BCL2... 22
8) La protéine p53 ... 22
III - Classification... 24
IV - Traitements et taux de survie ... 29
V - Nouvelles stratégies thérapeutiques ... 30
1) Immunothérapie anti-tumorale... 30
2) Angiogénèse... 31
3) Thérapie de différenciation, exemple de l’acide rétinoïque... 32
a) Biosynthèse de l’acide rétinoïque ... 33
b) Récepteurs de l’acide rétinoïque... 34
c) Effets de l’acide rétinoïque dans les cellules de neuroblastome ... 36
in vitro ... 36
2
èmepartie : Le système VIP - récepteurs ... 41
I - Les neuropeptides de la famille du VIP... 41
1) Généralités ... 41
2) Biosynthèse des neuropeptides ... 41
a) Biosynthèse du VIP... 41
b) Biosynthèse du PACAP ... 43
c) Dégradation des neuropeptides ... 45
II - Les récepteurs du VIP et du PACAP ... 46
1) Structure des récepteurs ... 46
2) Nomenclature des récepteurs du VIP et du PACAP... 47
3) PAC1... 48
4) VPAC1 et VPAC2 ... 49
5) Distribution des récepteurs PAC1, VPAC1 et VPAC2 ... 51
6) Principales voies de signalisation ... 51
7) Récepteurs du PHI insensibles au GTP ... 53
III - Fonctions des neuropeptides VIP et PACAP ... 54
1) Prinicpales fonctions des neuropeptides ... 54
2) Contrôle de la prolifération et/ou de la différenciation... 55
a) Rôle dans le développement de l’embryon ... 55
b) Rôle dans le développement et le fonctionnement du système nerveux... 56
Neuropeptides et neurones sympathiques ... 57
Neuropeptides et cellules chromaffines ... 58
c) Rôle dans la tumorigénèse ... 59
IV - Système VIP-récepteur et pathologies ... 59
1) Pathologies non cancéreuses... 59
a) Pathologies liées à une hyperproduction de VIP... 59
b) Pathologies liées à une déficience en VIP ou en PACAP... 60
c) Autres pathologies... 62
2) Pathologies cancéreuses... 62
3) Neuropeptides et thérapie ... 64
a) Vectorisation et développement d’analogues stables... 64
b) Neuroprotection et les maladies neurodégénératives... 66
c) Maladies inflammatoires et auto-immunes ... 67
d) Thérapie anti-cancéreuse ... 68
e) Essais cliniques chez l’Homme... 69
V - Le système VIP-récepteurs dans les neuroblastomes ... 70
1) Découverte du système VIP-récepteurs dans les cellules de neuroblastome... 70
2) Régulation de l’expression du système VIP-récepteurs dans les cellules de neuroblastome. ... 71
3) Signalisation intracellulaire associée au système VIP-récepteur dans les cellules de
neuroblastome ... 74
3
èmepartie : Présentation du sujet de recherche et des objectifs de la thèse78
I - Objectif 1 : Modulation de l’expression de MYCN par le VIP ... 781) Importance de l’oncogène MYCN dans les neuroblastomes ... 78
2) Généralités ... 79
3) Régulation de l’expression de MYCN au cours du développement ... 80
4) Amplification de l’oncogène MYCN... 81
5) Rôles de MYCN dans les neuroblastomes... 82
a) Dans le développement des neuroblastomes... 82
b) Dans le contrôle du cycle cellulaire, de la prolifération et de la différenciation . 83 c) Dans la progression des neuroblastomes... 85
6) Thérapies ciblant l’expression de MYCN... 86
II - Objectif 2 : implication de SNAP-25 dans la différenciation neuronale... 87
1) Généralités ... 89
a) Identification de la protéine SNAP-25... 89
b) Complexes SNARE ... 90
2) Expression et rôles de SNAP-25 au cours du développement neuronal... 92
a) Expression de SNAP-25... 92
b) Rôles de SNAP-25 dans la neuritogénèse et la synaptogénèse... 93
c) Apport des modèles animaux transgéniques ... 93
PRESENTATION DES TRAVAUX ... 96
Présentation de l’article n°1 ... 97
Présentation de l’article n°2 ... 131
DISCUSSION GENERALE ET PERSPECTIVES ... 168
I - Discussion générale... 169
II - Perspectives ... 171
ANNEXE ... 178
LISTE DES ABREVIATIONS
Les termes suivis d’un astérisque sont anglais.
4-HPR : N-(4-hydroxyphényl) rétinamide 9cis-RA : acide 9-cis rétinoïque
13cis-RA : acide 13-cis rétinoïque AC : adénylyl cyclase
AMPc : adénosine 3’5’-monophosphate cyclique at-RA : acide tout-trans rétinoïqe
BDNF : brain derived neurotrophic factor* BHE : barrière hémato-encéphalique CaM : calmoduline
CNTF : ciliary neurotrophic factor* DM : double minute chromosome* DR : domain repeat*
E : jour de vie embryonnaire EC : boucle extracellulaire
Erk : extracellular-regulated kinases* GTP : guanosine triphosphate
HSR : homogeneously staining region* IAP : inhibitor of apoptosis proteins* IC : boucle intracellulaire
INPC : International Neuroblastoma Pathology Committee* INRG : International Neuroblastoma Risk Group*
INSS : International Neuroblastoma Staging System* IP3 : inositol triphosphate
kDa : kilodalton
LIF : leukemia inhibitory factor* MAPK : mitogen-activated protein kinase* MIBG : métaiodobenzyl-guanidine
MKI : mitosis-karyorrhexis index*
MYCN : v-myc avian myelocytomatosis viral-related oncogene,
neuroblastoma-derived*
P : jour post-natal
PAC1 : récepteur spécifique du PACAP
PACAP : pituitary adenylate cyclase-activating polypeptide* PHI/M : peptide having carboxyterminal isoleucine/methionone* PI3K : phospho-inositide 3 kinase
PKA : protéine kinase dépendante de l’AMPc PKC : proteine kinase dépendante du Ca2+ PLC : phospholipase C
PRP : PACAP-related peptide* RAR : retinoic acid receptor* RNAi : interférence de l’ARN RXR : retinoid X receptor* SCLC : small cell lung cancer* shh : sonic hedgehog
SNAP-25 : synaptosomal-associated protein of 25 kDa*
SNARE : soluble N-ethylmaleimide-sensitive factor attachment protein receptor* TGF-ß : transforming growth factor β*
TM : domaine transmembranaire TNF: tumor necrosis factor*
TPA : 12-O-tetra-décanoylphorbol 13-acétate Trk : tyrosine kinase receptor*
TH : tyrosine hydroxylase
VEGF : vascular endothelial growth factor* VIH : virus de l’immunodéficience humaine VIP : vasoactive intestinal peptide*
1ère
partie : Les neuroblastomes
I - Données cliniques1) Définition et épidémiologie
Les neuroblastomes représentent la plus fréquente des tumeurs extracrâniennes de l’enfance. Leur prévalence est de 1 pour 7000 naissances et ils sont responsables d’environ 15 % de la mortalité par cancer chez l’enfant (pour revues, Raguénez et al., 2001 ; Maris et al., 2007). Ces tumeurs touchent essentiellement les jeunes enfants. L’âge moyen au moment du diagnostic est de 18 mois, 40 % des cas sont diagnostiqués avant l’âge d’1 an, 75 % avant 4 ans et 97 % avant 10 ans (pour revue, Brodeur, 2003). Le taux de survie à 5 ans est de 45 % (pour revue, Grosfeld, 2000). Les neuroblastomes se développent à partir des cellules de la crête neurale qui sont, entre autre, les précurseurs du système nerveux sympathique. Les tumeurs primaires peuvent donc être localisées tout au long des structures du système nerveux sympathique (pour revue, Edsjö et al., 2007).
L’étiologie des neuroblastomes n’est pas connue. Il semble que des facteurs environnementaux ne soient pas impliqués. Cependant, une prédisposition génétique existe dans 1 à 1,5 % des cas. Ces neuroblastomes familiaux ont une transmission autosomique dominante avec une pénétrance incomplète (pour revue, Tonini et al., 2003). Le gène paired-like homeobox 2B (PHOX2B) pourrait être un gène de prédisposition aux neuroblastomes. En effet, une mutation de ce gène dans la lignée germinale a été identifiée dans deux familles présentant des neuroblastomes et des pathologies souvent associées à ces tumeurs, comme par exemple la maladie de Hirschsprung (pathologie des chaînes ganglionnaires intestinales dérivant de la crête neurale la plus commune chez les enfants) (Bourdeaut et al., 2005).
2) Origine
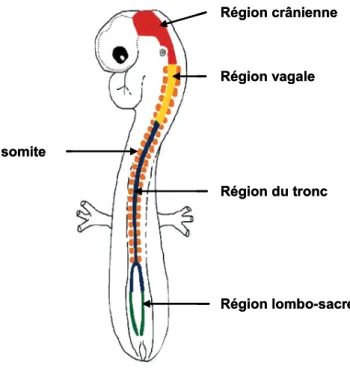
La crête neurale est une structure embryonnaire constituée de cellules progénitrices multipotentes qui présentent une forte activité de prolifération et de migration. La crête neurale est divisée en quatre parties (Figure 1) en suivant l’axe antéro-postérieur :
- la région crânienne, dont les cellules sont à l’origine des ganglions sensoriels et du cartilage de la face,
- la région vagale et la région lombo-sacrée, qui donnent naissance au système nerveux entérique,
- la région du tronc, dont dérivent les mélanocytes, les cellules de Schwann, la médullo-surrénale et les chaînes ganglionnaires sympathiques.
Région crânienne Région vagale Région du tronc Région lombo-sacrée somite Région crânienne Région vagale Région du tronc Région lombo-sacrée somite
Figure 1 : Structure de la crête neurale (d’après Dyer, 2004).
La crête neurale est divisée en quatre régions : la région crânienne en rouge, la région vagale en jaune, la région du tronc en bleu et la région lombo-sacrée en vert.
Les tumeurs primaires des neuroblastomes apparaissent préférentiellement au niveau des chaînes ganglionnaires et de la médullo-surrénale, ce qui suggère que les cellules de neuroblastomes dérivent des cellules du tronc de la crête neurale (pour revue, Dyer, 2004).
3) Pathologie
Les neuroblastomes présentent une grande hétérogénéité clinique qui dépend de la localisation de la tumeur primaire ainsi que de la présence ou non de métastases. La tumeur primaire peut se situer tout le long du système nerveux sympathique, mais dans 65 à 70 % des cas, elle se trouve dans l’abdomen, le plus souvent au niveau de la glande surrénale. La tumeur primaire peut également se situer au niveau du thorax (17 à 30 % des cas), du cou ou du pelvis (5 à 10 % des cas). Les nourrissons présentent le plus souvent des tumeurs au niveau du thorax et du cou. Les métastases touchent essentiellement la moelle osseuse et les os (environ 50 % des cas), les ganglions lymphatiques (28 % des cas), le foie (22 %) et la peau (12 %). Les métastases au niveau des poumons et du cerveau sont rares au moment du diagnostic mais peuvent apparaître lors des récidives (pour revue, Maris et al., 2007 ; D’Andon et al., 2004 ; www.fnclcc.fr). Au moment du diagnostic, le neuroblastome est soit localisé, soit métastatique.
a) Les neuroblastomes localisés
Lors du diagnostic, près de 40 % des patients présentent un neuroblastome localisé. Dans ces neuroblastomes, la tumeur est souvent asymptomatique. De ce fait, la maladie est souvent diagnostiquée par hasard. Les tumeurs situées au niveau de l’abdomen provoquent des douleurs abdominales et un inconfort. En cas d’augmentation brutale de la masse tumorale, la douleur devient plus forte et des saignements peuvent avoir lieu dans la tumeur ou à proximité. Les tumeurs situées dans le pelvis induisent des troubles de la miction et des problèmes intestinaux. Certaines tumeurs dites en sablier pénètrent le canal contenant la moelle épinière et provoquent des symptômes liés à la compression des racines nerveuses et de la moelle (troubles de la marche, de la miction et de la défécation). La neurochirurgie doit
alors être associée à la chimiothérapie afin d’éviter que ces symptômes ne deviennent définitifs (pour revue, Maris et al., 2007).
Deux symptômes paranéoplasiques sont associés aux neuroblastomes localisés. La sécrétion de VIP (vasoactive intestinal peptide) provoque des diarrhées hydriques profuses et une hypokaliémie. Ces troubles, qui touchent 7 à 9 % des patients, disparaissent généralement après ablation de la tumeur. Le syndrome opsoclonus-myoclonus, caractérisé par des mouvements rapides des yeux et une ataxie, est également associé aux formes localisées. Les enfants présentant un neuroblastome localisé et le syndrome opsoclonus-myoclonus (2 à 4 % des cas) ont un devenir favorable. Cependant, 70 à 80 % de ces patients présentent des déficits neurologiques à long terme (retards cognitif et moteur, troubles du langage et du comportement). Il semble que ces déficits puissent être atténués grâce à la chimiothérapie ou à l’administration par voie intraveineuse de glucocorticoïdes, d’hormone adrénocorticotrope (ACTH) ou d’immunoglobulines IgG (pour revue, Matthay et al., 2005).
b) Les neuroblastomes disséminés
Près de 60 % des patients présentent des métastases au moment du diagnostic. Ces patients ont souvent une fatigue générale. Les métastases touchant la moelle osseuse et les os peuvent provoquer des douleurs osseuses, une boiterie, une anémie ou des hémorragies. Des hématomes de l’orbite et un proptosis sont fréquents dans ces neuroblastomes métastatiques et sont dus à l’infiltration de la tumeur dans la cavité orbitale. Dans certains cas, les patients souffrent d’hypertension, qui peut être due à la compression d’une artère rénale (pour revue, Maris et al., 2007).
c) Les neuroblastomes de stade 4S
Il existe une troisième grande catégorie de neuroblastome appelé stade 4S (S = spécial) (D’Angio et al., 1971) , qui représente environ 5 % des cas. Ces neuroblastomes sont caractérisés par une petite tumeur primaire localisée et des métastases au niveau du foie. La peau et la moelle osseuse peuvent également être touchées. Le syndrome de Pepper est une forme particulière de neuroblastome de stade 4S caractérisée par des métastases hépatiques très volumineuses, pouvant provoquer des difficultés respiratoires. Dans environ 30 % des cas, les neuroblastomes de stade 4S présentent la particularité de pouvoir régresser spontanément probablement par un phénomène d’apoptose. De ce fait, le taux de survie des patients est de 85 à 92 %. Cependant, certains de ces neuroblastomes présentent des caractéristiques biologiques défavorables et une progression tumorale rapide. Dans les cas de récidives, ils se présentent alors comme des neuroblastomes disséminés (pour revue, Kerdudo et al., 2004).
4) Diagnostic
Le diagnostic est réalisé grâce à des marqueurs biologiques comme l’acide homovanilique (HVA) et l’acide vanillylmendélique (VMA). En effet, l’augmentation dans les urines de ces substances dérivées des catécholamines est observée dans 90 à 95 % des cas. De plus, la quantité de ces molécules augmente avec la masse tumorale, ce qui permet de suivre l’évolution de la tumeur. La lactodeshydrogénase (LDH) est un autre marqueur biologique important puisque son taux augmente dans le sang dans 75 % des neuroblastomes (d’Andon et al., 2004).
Le diagnostic est affiné grâce à des techniques d’imagerie. L’échographie permet de localiser les tumeurs situées au niveau de l’abdomen, du thorax ou du pelvis, alors que l’imagerie par résonance magnétique met en outre en évidence les compressions éventuelles de la moelle
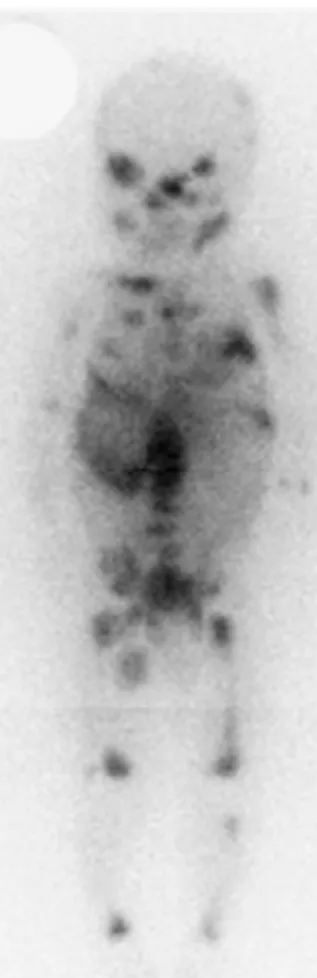
épinière. Ces deux techniques sont également utiles pour révéler les extensions locales ou régionales des tumeurs. Les tumeurs osseuses sont visualisées grâce à la scintigraphie à la métaiodobenzyl-guanidine (MIBG) (Figure 2) ou au 99Tc-diphosphonate. Les biopsies des tumeurs permettent de réaliser des analyses histologiques et biologiques visant à caractériser la présence de certaines anomalies génétiques caractéristiques des neuroblastomes (pour revue, Ishola et Chung, 2007).
Figure 2 : Marquage de métastases par scintigraphie à la métaiodobenzyl-guanidine (MIBG) (d’après Howman-Giles et al., 2007).
II - Données biologiques
Les neuroblastomes présentent de nombreuses anomalies génétiques ou des dérèglements de l’expression de certaines protéines qui peuvent constituer des facteurs importants dans l’établissement du pronostic de la maladie (Tableau I).
Facteur pronostique Pronostic Index ADN = 1 - Index ADN > 1 + Amplification de MYCN - Gain du chromosome 17 + Gain du 17q - Délétion du 1p - Délétion du 11q - Expression de TrkAII + Expression de TrkB -
Tableau I : Résumé des facteurs ayant une signification dans le pronostic des neuroblastomes.
- : facteur associé à un mauvais pronostic, + : facteur associé à un pronostic favorable.
1) Contenu en ADN
Les neuroblastomes présentent des anomalies au niveau de leur contenu en ADN. Grâce aux analyses cytogénétiques, il est possible de distinguer 4 niveaux de ploïdie :
- Les tumeurs presque diploïdes et les tumeurs presque tétraploïdes, sont trouvées chez les enfants de plus d’un an, et associées à un pronostic défavorable. Ces tumeurs sont caractérisées par des réarrangements chromosomiques tels que des amplifications ou des délétions, provoquant une grande instabilité génomique et par conséquent une grande agressivité.
- Les tumeurs presque triploïdes et les tumeurs presque pentaploïdes sont associées à un devenir favorable. Elles sont caractérisées par des gains ou des pertes de chromosomes entiers, sans réarrangements structuraux. Cela crée une instabilité mitotique avec anomalies de la ségrégation chromosomique engendrant une faible agressivité tumorale.
Cependant, le contenu en ADN ne constitue un facteur pronostique que chez les enfants de moins de 1 an (pour revues, Brodeur, 2003 ; Westermann et Schwab, 2002).
2) Amplification de l’oncogène MYCN
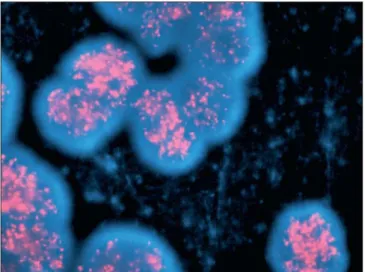
En 1983, Schwab et al. ont mis en évidence l’amplification du gène MYCN (v-myc avian myelocytomatosis viral-related oncogene, neuroblastoma-derived) sous forme de chromosomes minuscules doubles (DM pour double minute chromosome en anglais) ou des régions chromosomiques présentant un marquage homogène (HSR pour homogeneously staining region en anglais) dans des lignées cellulaires dérivant de neuroblastomes humains et dans une tumeur primaire (Figure 3). Par la suite, Seeger et al (1985) ont démontré que l’amplification de MYCN était associée aux neuroblastomes ayant une progression rapide et un mauvais pronostic.
Figure 3 : Amplification de l’oncogène MYCN dans des noyaux de cellules de neuroblastome mise en évidence par la technique de FISH (fluorescent in situ hybridation) (d’après Maris et al., 2007).
La présence de multiples copies du gène MYCN est détectée grâce à une sonde spécifique marquée en rouge.
Le gène MYCN, localisé sur le bras court du chromosome 2, code deux phosphoprotéines localisées dans le noyau, où elles exercent leur fonction de facteur transcriptionnel quand elles sont complexées à la protéine MAX (MYC-associated factor X). La protéine MYCN est
impliquée dans la prolifération, la croissance cellulaire et la synthèse protéique (Ramsay et al., 1986 ; pour revue, van Noesel et Versteeg, 2004).
L’amplification de l’oncogène MYCN est l’anomalie génétique la mieux caractérisée dans les neuroblastomes. Elle apparaît dans 22 % des neuroblastomes et constitue un facteur pronostic essentiel (pour revues, van Noesel et Versteeg, 2004 ; Plantaz, 2001). Le gène MYCN est également amplifié dans d’autres tumeurs comme des rhabdomyosarcomes, des médulloblastomes, des rétinoblastomes, des glioblastomes, des astrocytomes et des cancers du poumon à petites cellules (pour revue Strieder et Lutz, 2002).
Aujourd’hui, seule l’amplification du gène MYCN constitue un facteur pronostique, la signification du taux d’expression de l’ARNm MYCN ou des protéines MYCN étant controversée : selon les études, le taux d’expression de MYCN peut être corrélé ou non au stade de la maladie (Cohn et al., 2000 ; Tang et al., 2006).
Le rôle de MYCN dans les neuroblastomes sera développé dans la troisième partie de cette introduction (p 78).
3) Gain du chromosome 17
Le gain du chromosome 17 est l’anomalie génétique la plus fréquente dans les neuroblastomes, puisqu’elle concerne 50 % des cas. Cette anomalie, quand elle touche la totalité du chromosome 17, est associée à un bon pronostic, alors que le gain du bras long de ce chromosome est associé aux stades avancés (Vandersompele et al., 2001). Cette anomalie est généralement due à des translocations déséquilibrées entre les chromosomes 1 et 17, qui conduisent à la perte de l’extrémité distale du chromosome 1 et à la trisomie partielle de l’extrémité distale du chromosome 17. Des translocations entre les chromosomes 17 et 11 peuvent également avoir lieu. Les points de cassure sur le bras long du chromosome 17 sont hétérogènes. La région 17q22-qter a été identifiée comme la plus petite région commune de
gain de matériel chromosomique. Cette région porte probablement un ou plusieurs gènes contribuant à la tumorigénèse en cas de surexpression (pour revue, Maris, 2005).
Plusieurs gènes concernés par le gain du chromosome 17 sont actuellement étudiés. C’est le cas du gène NM23-H1 localisé en 17q22. Ce gène code pour une kinase de nucléotides diphosphates. NM23-H1 a tout d’abord été identifié comme un gène suppresseur de métastases dans les cancers du sein et les mélanomes (pour revue, Lombardi, 2006). Mais dans d’autres cancers tels que les lymphomes non Hodgkinien et les neuroblastomes, NM23-H1 est considéré comme un oncogène. Dans les neuroblastomes, l’expression élevée de nm23-H1 dans le sérum est associée à un mauvais pronostic et une grande agressivité (Hailat et al., 1991). Il a également été montré que le gène NM23-H1 est une cible transcriptionnelle de l’oncogène MYCN (Godfried et al., 2002). Un autre gène semble être également impliqué dans le développement des neuroblastomes. Il s’agit du gène de la survivine, localisé en 17q25, (Ambrosini et al., 1998). La survivine appartient à la famille des inhibiteurs de l’apoptose (IAP pour inhibitor of apoptosis protein). Dans les neuroblastomes, l’augmentation de l’expression de la survivine est associée aux tumeurs ayant un pronostic défavorable (Islam et al., 2000).
4) Perte du bras court du chromosome 1
La délétion du bras court du chromosome 1 concerne 30 à 35 % des neuroblastomes. Cette anomalie est souvent associée au gain du bras long du chromosome 17. La région minimale commune de délétion a été identifiée en 1p36.31 (White et al., 2005). La taille de la délétion varie en fonction de l’amplification de MYCN. En effet, elle est grande (1p35-1pter) dans les neuroblastomes présentant plusieurs copies de cet oncogène et petite (1p36.3) dans les tumeurs n’ayant qu’une seule copie du gène MYCN. La plupart des neuroblastomes présentant l’amplification de MYCN présente également la délétion du chromosome 1p, alors que tous
les neuroblastomes ayant la délétion en 1p ne présentent pas l’amplification de MYCN. Cela suggère que la plus large délétion du chromosome 1 pourrait précéder l’amplification de l’oncogène MYCN. Cette anomalie, bien que souvent associée à l’amplification de MYCN, constitue cependant un indicateur indépendant de devenir défavorable (pour revues, Brodeur, 2003 ; van Noesel et Versteeg, 2004). La perte du bras court du chromosome 1 semble induire la perte d’expression de gènes suppresseurs de tumeur. En effet, l’introduction du segment chromosomique 1p dans la lignée de neuroblastome NGP diminue la malignité de ces cellules et induit leur différenciation (Bader et al., 1991). L’identification du ou des gènes suppresseurs de tumeur localisés en 1p36 a donné lieu à de nombreuses études. Le gène p73, lors de son identification, a été considéré comme étant le gène suppresseur de tumeur potentiel dans les neuroblastomes (Kaghad et al., ,1997) mais cela n’a pas été confirmé par la suite. A ce jour, aucun gène suppresseur de tumeur n’a été clairement identifié sur ce fragment chromosomique, mais le gène CHD5 dont le produit est impliqué dans le remodelage de la chromatine, apparaît être un excellent candidat (Okawa et al., 2008).
5) Perte du bras long du chromosome 11
La délétion du bras long du chromosome 11 est observée dans environ un tiers des neuroblastomes. Cette anomalie est associée à un mauvais pronostic mais n’est pas liée à l’amplification de MYCN. Les altérations du bras long du chromosome 11 peuvent se manifester sous différentes formes : translocations impliquant les régions 11q21 et 11q22, délétion de la région 11q23, inversion entre les segments 11q21 et 11q23 ou perte allélique (pour revues, van Noesel et Versteeg, 2004 ; Maris, 2005). La délétion de ce fragment chromosomique est souvent associée à la perte du bras court du chromosome 3 (Vandesompele et al., 1998 ; Breen et al., 2000; Spitz et al., 2003).
6) Altération de l’expression des gènes des récepteurs des neurotrophines
Les neurotrophines sont des molécules impliquées dans la croissance, le développement, la survie et les mécanismes de la réparation du système nerveux. Elles agissent via les récepteurs Trk (tyrosine kinase receptor). Il existe trois types de Trk :
- TrkA qui lie préférentiellement de NGF (nerve growth factor),
- TrkB dont les ligands sont le BDNF (brain-derived neurotrophic factor) et NT-4/5 (neurotrophin-4/5),
- TrkC, spécifique de NT-3 (neurotrophin-3).
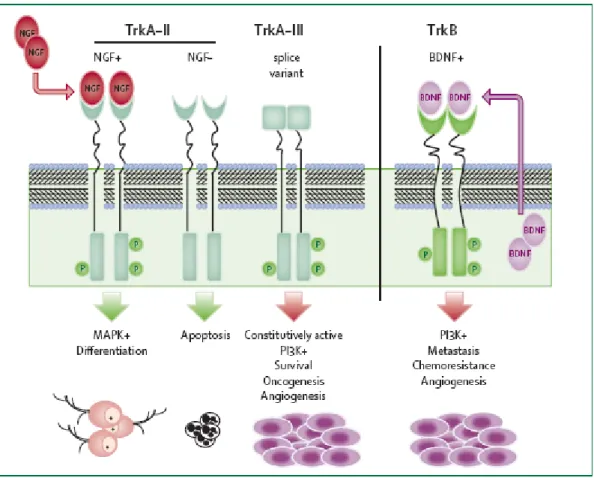
Un quatrième type de récepteur des neurotrophines a été identifié. Il s’agit du récepteur p75NTR qui est polyvalent mais présente une moindre affinité. In vivo, le système neurotrophine-Trk induit la survie ou la différenciation neuronale (Figure 4) (pour revue, Nakagawara, 2001). Le récepteur TrkAII est surexprimé dans les neuroblastomes ayant un pronostic favorable, notamment dans les cas de régression spontanée. Dans ces tumeurs, la surexpression de TrkAII et la production (même faible) du NGF par les cellules de Schwann environnantes pourraient permettre de stimuler la différenciation, alors qu’en absence de ligand, l’apoptose serait induite dans les cellules tumorales (Nakagawara et al., 1992 ; 1993 ; Kogner et al., 1993). Une autre isoforme du récepteur TrkA, nommée TrkAIII et résultant de l’épissage alternatif du gène TrkA, a également été identifiée dans les neuroblastomes, en particulier dans ceux de stades avancés. L’expression de cette isoforme protège les cellules de neuroblastome de la mort induite par la doxorubicine et favorise l’angiogénèse in vivo (Tacconelli et al., 2004). Dans les neuroblastomes agressifs, TrkB est fortement exprimé. Dans les formes de tumeurs exprimant TrkB et ses ligands, une stimulation autocrine/paracrine semble favoriser l’invasion et donc les métastases (Matsumoto et al., 1995). En ce qui concerne le récepteur TrkC, il est préférentiellement exprimé dans les neuroblastomes favorables mais son implication reste à déterminer (Yamashiro et al., 1996).
Figure 4 : Représentation schématique de la signalisation Trk dans les neuroblastomes (d’après Maris et al., 2007).
Le récepteur TrkAII, quant il est exprimé dans les neuroblastomes, peut induire la différenciation des cellules en présence de son ligand, le NGF, ou l’apoptose en absence de NGF. Lorsque le récepteur TrkB et son ligand, le BDNF, sont présents, ils favorisent l’invasion des cellules tumorales.
7) Voie de l’apoptose
L’apoptose est un mécanisme de mort cellulaire physiologique qui permet de contrôler le nombre de cellules de l’organisme au cours du développement par exemple. L’apoptose peut être déclenchée par des agents cytotoxiques ou les rayons γ. Ainsi, les substances utilisées en chimiothérapie agissent en provoquant l’apoptose des cellules cancéreuses (pour revue, Fulda et Debatin, 2003). L’apoptose peut être médiée par deux voies : la voie des récepteurs de mort membranaires (voie extrinsèque) ou la voie mitochondriale (voie intrinsèque). Ces deux voies
aboutissent à l’activation en cascade des caspases, des protéases à cystéines qui vont conduire au clivage de diverses protéines et à la mort de la cellule (pour revue, Couzinet et al., 2002). Certains neuroblastomes sont résistants aux drogues de chimiothérapie à cause d’altérations de certains effecteurs de l’apoptose, tels que la caspase 8, les protéines inhibitrices de l’apoptose (IAP), les protéines de la famille BCL2.
a) La caspase 8
La caspase 8 est impliquée dans l’apoptose médiée par les récepteurs du TNF (tumor necrosis factor) (pour revue, Couzinet et al., 2002). Le gène CASP8 codant cette enzyme est localisé en 2q33, une région associée à une perte d’hétérozygotie dans certains neuroblastomes (Teitz et al., 2000). Il a également été démontré que ce gène est méthylé dans environ 60 % des neuroblastomes. Cette modification épigénétique semble être statistiquement corrélée à l’amplification de l’oncogène MYCN (Teitz et al., 2000 ; Banelli et al., 2005 ; Yang et al., 2007). Malgré le fait que la méthylation inhibe généralement la transcription, l’expression de la caspase 8 est indépendante de l’état de méthylation du gène (Banelli et al., 2000). La majorité des neuroblastomes n’expriment pas la caspase 8. Cependant, quand elle est exprimée, son taux d’expression n’est pas corrélé à l’amplification de MYCN (Fulda et al., 2006). Il apparaît donc que, bien que la méthylation du gène CASP8 soit liée à l’amplification de MYCN, l’expression de la caspase 8 ne soit pas corrélée à ces deux événements.
En plus de son rôle dans l’apoptose médiée par les récepteurs du TNF, la caspase 8 est également impliquée dans la mort cellulaire dépendante des intégrines (integrin-mediated death ou IMD). Ce phénomène est connu pour empêcher la dissémination métastatique de cellules de neuroblastome. En effet, dans deux modèles animaux de dissémination métastatique (embryon de poulet et souris nude), l’injection de cellules de neuroblastome exprimant la caspase 8 n’induit pas la formation de métastases, contrairement à l’injection de
cellules n’exprimant pas cette caspase. Lorsque les cellules tumorales quittent le site de la tumeur primaire, elles se retrouvent dans un environnement différent dans lequel les intégrines ne sont plus au contact de leur ligand. Les intégrines non liées vont alors activer la caspase 8 et provoquer la mort des cellules. Ces travaux mettent en évidence le rôle de suppresseur de tumeur que peut avoir la caspase 8 dans les neuroblastomes (Stupack et al., 2006).
b) Les protéines inhibitrices de l’apoptose (IAP)
Les IAP sont des protéines qui inhibent l’apoptose en se liant à certaines caspases. Les IAP peuvent être neutralisées par les protéines Smac/Diablo ou Omi libérées par les mitochondries. Ces protéines sont fréquemment surexprimées dans plusieurs types de cancer. C’est le cas par exemple de la XIAP (X-linked inhibitor of apoptosis protein) surexprimée dans les gliomes ou cIAP1 dans les cancers ovariens (pour revue, Goldsmith et Hogarty, 2005). Dans les neuroblastomes, la IAP la plus étudiée est la survivine dont la surexpression est corrélée aux stades avancés de neuroblastomes avec un mauvais pronostic (Islam et al., 2000). Le gène BIRC5 qui code la survivine, est localisé en 17q25 (Ambrosini et al., 1998), région fréquemment associée à un gain de chromosome dans les neuroblastomes. Cependant, la capacité de la survivine à inhiber les caspases n’est pas claire. En effet, la survivine ne possède pas la séquence d’acides aminés essentielle à la liaison aux caspases (Verdecia et al., 2000). La survivine pourrait agir indirectement. En effet, dans des carcinomes hépatocellulaire, la survivine n’est capable de lier la pro-caspase 9 qu’en présence de la protéine HBXIP (hepatitis B X-interacting protein) (Marusawa et al., 2003).
c) Les protéines de la famille BCL2
Ces protéines sont importantes dans la voie mitochondriale de l’apoptose. En effet, elles inhibent ou permettent la libération du cytochrome c par la mitochondrie. Cette famille regroupe des protéines anti-apoptotiques comme BCL2 ou BCL-XL, et des protéines pro-apoptotiques comme BAX ou BID (pour revue, Fulda et Debatin, 2003). La protéine anti-apoptotique BCL2 est surexprimée dans de nombreux neuroblastomes, mais des travaux actuellement contradictoires ne permettent pas de lier cette surexpression aux caractéristiques cliniques des neuroblastomes (Castle et al., 1993 ; Ikeda et al., 1995 ; Mejia et al., 1998 ; Abel et al., 2005).
8) La protéine p53
La protéine p53 est un facteur de transcription qui stimule l’expression de gènes comme p21WAF1 et BAX, induisant respectivement l’arrêt du cycle et l’apoptose à la suite d’un stress. Dans de nombreux cancers, la protéine p53 est inactivée par mutation du gène (pour revue, Tweddle et al., 2003). Dans les neuroblastomes, les mutations de p53 sont très rares puisqu’elles affectent environ 2% des cas (Imamura et al., 1993 ; Hosoi et al., 1994) et sont généralement observées dans les neuroblastomes récurrents. Ces mutations semblent apparaître à la suite de la chimiothérapie et pourraient être responsables de la chimiorésistance de certains neuroblastomes (Keshelava et al., 2000 ; 2001 ; Tweddle et al., 2001). Ces données suggèrent un rôle de p53 dans l’apoptose induite par les drogues utilisées en chimiothérapie.
Bien que les mutations du gène p53 soient rares dans les neuroblastomes, des anomalies de la voie p53 ont été observées dans ces cancers. Les premières études concernant p53 dans les neuroblastomes ont mis en évidence un fort taux d’expression de l’ARN p53 ainsi que de la protéine, dont la demi-vie est augmentée par rapport à des cellules de la glande surrénale
(Sidell et Koeffler, 1988 ; Davidoff et al., 1992). Il a ensuite été démontré que la protéine p53 est anormalement localisée dans le cytoplasme dans des neuroblastomes indifférenciés et des lignées cellulaires (Moll et al., 1995 ; 1996 ; Goldman et al., 1996). Dans le cytoplasme, la protéine p53 est visible sous la forme d’agrégats protéiques dans lesquels l’extrémité C-terminale est masquée (Ostermeyer et al., 1996). La protéine Parc (p53-associated, Parkin-like cytoplasmic protein) a été identifiée comme responsable de la séquestration cytoplasmique de p53. La liaison de Parc et p53 implique le domaine N-terminal de Parc et le domaine C-terminal de p53 (Nikolaev et al., 2003). Il semble également que la séquestration cytoplasmique de p53 puisse être due à son hyperubiquitinylation (Becker et al., 2007). La séquestration cytoplasmique de p53 a laissé penser que, par ce biais, cette protéine était inactive dans les neuroblastomes. Cette hypothèse a d’abord été confirmée par une étude démontrant que dans des cellules de neuroblastomes, des dommages de l’ADN n’induisent pas d’arrêt du cycle en phase G1 car p53 n’est pas transloquée dans le noyau (Moll et al., 1996). Cependant, la localisation cytoplasmique et la non fonctionnalité de la protéine p53 sont remises en cause par plusieurs études, la plus récente datant de 2007. En effet, dans des tumeurs et des lignées de neuroblastome, p53 est détectée majoritairement au niveau nucléaire quel que soit l’état de différenciation des tumeurs et des cellules (Chen et al., 2007). Ces résultats contradictoires avec les travaux précédents pourraient être dus à l’utilisation d’un anticorps anti-p53 donnant un marquage cytoplasmique non spécifique (Wolff et al., 2001 ; Chen et al., 2007).
La voie p53 peut être inhibée par MDM2, le principal inhibiteur de la protéine p53. En effet, la protéine MDM2 peut être surexprimée dans les neuroblastomes d’une part à la suite de l’amplification du gène MDM2 et d’autre part à la suite de l’amplification de MYCN, ce facteur transcriptionnel régulant positivement l’expression de MDM2 (Corvi et al., 1995 ; pour revue, Slack et Shohet , 2005).
III - Classification
Du fait de leur grande hétérogénéité clinique, il a été important de bien caractériser et classer les différentes formes de neuroblastome afin d’en améliorer le diagnostic et par conséquent la thérapie. En 1984, Shimada et al., ont proposé une classification des neuroblastomes (et des tumeurs neuroblastiques en général) basée sur des critères histologiques. Cette étude a servi de base à la classification de l’ « International Neuroblastoma Pathology Commitee » (INPC). A cette même époque, un effort a été mené afin de développer des systèmes de classification des neuroblastomes plus complets, prenant en compte divers critères cliniques et biologiques. C’est ainsi qu’en 1995, la classification de l’ « International Neuroblastoma Staging System » (INSS) a été validée. Cette classification prend en compte la dissémination du neuroblastome. Ainsi, on distingue les formes localisées de stades 1, 2 et 3, des formes métastasiques de stade 4 (Tableau II). Parallèlement, Brodeur (1994) a proposé de classer les neuroblastomes en fonction des anomalies génétiques (amplification de MYCN, contenu en ADN, délétions chromosomiques, expression de TrkA). La dernière classification mise au point par l’ « International Neuroblastoma Risk Group » (INRG) prend en compte des critères de chacune des classifications précédentes et permet d’évaluer les neuroblastomes en fonction du risque de récurrence de la maladie et de déterminer le type de traitement à appliquer (Tableau III) (pour revue, Castleberry, 1997).
L’âge au moment du diagnostic est un critère important : le devenir d’un neuroblastome, quel que soit son stade, est meilleur chez un enfant de moins d’un an. Cependant, cet âge limite traditionnellement fixé à un an est actuellement discuté au vu de nouvelles études qui montrent qu’un âge limite situé entre 15 et 18 mois serait plus adapté (London et al., 2005 ; Schmidt et al., 2005 ; George et al., 2005).
Stade Description Stade 1 Tumeur localisée avec une exérèse macroscopiquement complète, avec ou
sans résidu microscopique ; les ganglions homolatéraux sont histologiquement sains (les ganglions adhérents à la tumeur et enlevés avec elle peuvent être envahis).
Stade 2A Tumeur localisée avec une exérèse macroscopiquement incomplète ; les ganglions homolatéraux non adhérents sont histologiquement sains (les ganglions adhérents à la tumeur et enlevés avec elle peuvent être envahis). Stade 2B Tumeur localisée avec ou sans exérèse macroscopique complète avec
ganglions homolatéraux non adhérents à la tumeur envahis histologiquement. Les adénopathies controlatérales doivent être histologiquement saines.
Stade 3 Tumeur latéralisée inextirpable, infiltrant la ligne médiane (= définie par la colonne vertébrale), avec ou sans envahissement ganglionnaire régional, ou tumeur latéralisée avec envahissement ganglionnaire controlatéral, ou tumeur médiane avec extension bilatérale par elle-même ou par envahissement ganglionnaire.
Stade 4 Toute tumeur primitive avec dissémination à distance dans les ganglions, l’os, la moelle osseuse, le foie et/ou d’autres organes (sauf si cela correspond à la définition des stades 4S).
Stade 4S Tumeur primitive localisée (comme définie pour les stades 1, 2A ou 2B), avec dissémination hépatique, cutanée et/ou au niveau de la moelle osseuse (restreint aux enfants de moins d’un an). Dans le stade 4S, l’envahissement médullaire doit être minime, c’est-à-dire moins de 10 % de cellules tumorales parmi les cellules nucléées sur la biopsie ostéo-médullaire ou le myélogramme. Un envahissement plus important doit faire classer le patient en stade 4. De plus, la scintigraphie à la MIBG ne doit pas montrer d’hyperfixation squelettique.
Tableau II : Classification des neuroblastomes selon l’ « International Neuroblastoma Staging System »
Cette classification permet de distinguer les tumeurs localisées de stade 1, 2 et 3, des tumeurs métastatiques de stade 4.
Dans la classification de l’INRG, le statut de MYCN est déterminé par le nombre de copies du gène : à partir de dix copies par cellule, la tumeur est considérée comme amplifiée pour l’oncogène MYCN, ce qui est le plus souvent associé à un mauvais pronostic.
Stade 1 Age MYCN 2 Histologie 3 Index
d’ADN récurrence Risque de
1 - - - - Faible
< 1an - - - Faible
Non amp - Non app Faible
Amp Fav Non app Faible
2A/2B
> 1 an
Amp Défav Non app Fort
Non amp - - Intermédiaire < 1an
Amp - - Fort Non amp Fav Non app Intermédiaire
Non amp Défav Non app Fort 3
> 1an
Amp Tout Non app Fort
Non amp - - Intermédiaire < 1 an
Amp - - Fort 4
> 1 an - - Non app Fort
Non amp Fav > 1 Faible Non amp - = 1 Intermédiaire Non amp Défav - Intermédiaire
4S < 1an
Amp - - Fort
Tableau III : Classification des neuroblastomes selon l’ « International Neuroblastoma Risk Group »
1 : d’après la classification de l’ « International Neuroblastoma Staging System » 2 : statut de MYCN : amplifié (amp) ou non amplifié (non amp)
3 : d’après la classification de l’ « International Neuroblastoma Pathology Commitee » : histologie favorable (fav) ou défavorable (défav).
- : quel que soit l’âge, le statut de MYCN, l’histologie ou l’index ADN. Non app : non applicable
L’histologie est définie selon les critères de la classification de Shimada qui prend en compte :
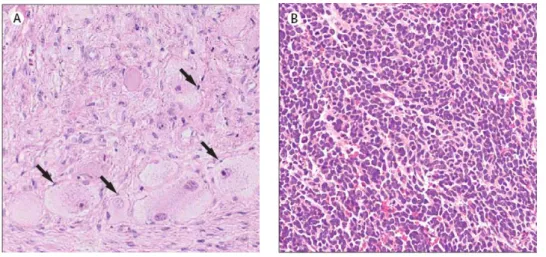
- le niveau du développement stromal : le stroma est un tissu conjonctif qui accompagne les cellules tumorales. Dans le cas des tumeurs neuroblastiques, le stroma contient plus ou moins de cellules de Schwann. On distingue alors les tumeurs à stroma riche (ganglioneuromes et ganglioblastomes) ou pauvre (neuroblastomes) en cellules de Schwann (Figure 5).
Figure 5 : Histologie de coupes de tumeurs neuroblastiques (tiré de Maris et al., 2007).
A : tumeur neuroblastique à stroma riche en cellules de Schwann (mises en évidence par les flèches). B : neuroblastome, caractérisé par des petites cellules rondes.
- le degré de maturation neuroblastique : tous les stades de la différenciation ganglionnaire sympathique sont représentés dans les tumeurs neuroblastiques, qui sont divisées en trois classes : indifférenciée, peu différenciée ou différenciée.
- l’index de mitose-caryorrhexis (MKI pour mitosis-karyorrhexis index), qui correspond au nombre de cellules tumorales en mitose ou en caryorrhexis sur 5000 cellules tumorales. On distingue alors les tumeurs de faible MKI (index < 2 %, ce qui équivaut à moins de 100 cellules en mitose et en caryorrhexis sur 5000 cellules), les tumeurs à MKI intermédiaire (index entre 2 et 4), et les tumeurs à MKI fort (index > 4 %).
A ces critères histologiques s’ajoute l’âge au moment du diagnostic.
Ainsi, la classification de l’INPC, reprise dans la classification de l’INRG, permet de définir trois types de tumeurs et de les classer en tumeur à histologie favorable ou non favorable :
- les ganglioneuromes sont des tumeurs au stroma riche en cellules de Schwann, dépourvues de neuroblastes mais composées de quelques cellules ganglionnaires sympathiques matures,
- les ganglioneuroblastomes présentent également un stroma riche en cellules de Schwann et des neuroblastes peu ou pas différenciés,
- les neuroblastomes sont des tumeurs au stroma pauvre en cellules de Schwann. Le degré de maturation neuroblastique est variable, tout comme le MKI. De ce fait, les neuroblastomes peuvent présenter une histologie favorable ou non (Tableau IV), alors que ganglioneuromes et ganglioneuroblastomes présentent une histologie favorable.
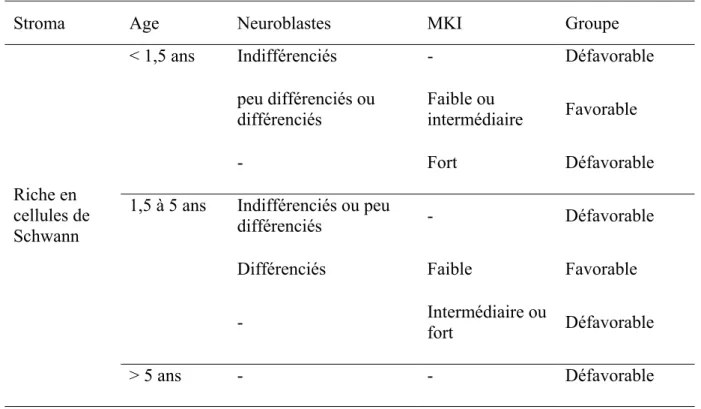
Stroma Age Neuroblastes MKI Groupe
Indifférenciés - Défavorable peu différenciés ou différenciés Faible ou intermédiaire Favorable < 1,5 ans - Fort Défavorable Indifférenciés ou peu différenciés - Défavorable
Différenciés Faible Favorable 1,5 à 5 ans - Intermédiaire ou fort Défavorable Riche en cellules de Schwann > 5 ans - - Défavorable
Tableau IV : Evaluation pronostique des neuroblastomes selon l’ « International Neuroblastoma Pathology Commitee » (tiré de Burgues et al., 2006)
- : quel que soit le stade de différenciation des neuroblastes ou le MKI
La relation entre le développement du stroma et l’état de différenciation des neuroblastes s’explique par le fait que les neuroblastes sécrètent des facteurs mitotiques et chimiotactiques qui permettent de recruter les cellules de Schwann présentes dans les tissus environnants non néoplasiques. Une fois dans le tissu tumoral, les cellules de Schwann sécrètent des facteurs anti-prolifératifs et différenciateurs nécessaires à la différenciation des neuroblastes (pour revue, Shimada et al., 1999).
IV - Traitements et taux de survie
Les neuroblastomes localisés de stade 1 ou 2 et considérés comme ayant un risque faible sont traités par exérèse de la tumeur. La chimiothérapie peut être appliquée lorsque la tumeur induit une compression de la moelle épinière ou dans les neuroblastomes de stade 4S pour lesquels les enfants présentent des problèmes respiratoires dus à une masse hépatique trop volumineuse. Ces neuroblastomes présentent un taux de survie d’environ 95 % à 5 ans. Les neuroblastomes localisés mais un peu plus invasifs (stade 3) sont tout d’abord traités par chimiothérapie pour ensuite permettre l’ablation complète de la tumeur. Le taux de survie à 5 ans de ces patients présentant un neuroblastome de risque intermédiaire est de 75 %. Les neuroblastomes présentant un risque fort, c’est-à-dire les formes métastasiques ayant des caractéristiques défavorables comme l’amplification de l’oncogène MYCN, subissent un protocole lourd, comportant successivement : une chimiothérapie d’induction intense, une chirurgie de la tumeur associée à une radiothérapie, puis une chimiothérapie myéloablative associée à une greffe de cellules souches hématopoïétiques. Enfin, un traitement de six mois par l’acide rétinoïque est administré. Malgré ce traitement très lourd, le taux de survie de ces patients n’est que de 30 % à 5 ans (pour revues, Tonini et Pistoia, 2006 ; Maris et al., 2007). La majorité des neuroblastomes est chimiosensible. En effet, les chimiothérapies d’induction provoquent en général une réponse partielle ou complète. Cependant, il existe des neuroblastomes résistants à la chimiothérapie. Ce phénomène peut être dû à la surexpression des gènes MRP1 (multidrug resistance-associated protein 1) ou MDR1 (multidrug resistance 1), codant des transporteurs de type ABC (ATP-binding cassette) qui permettent l’efflux des drogues et par conséquent diminuent le temps d’exposition des cellules à ces drogues (pour revues, Raguenez et al., 2001 ; Goldsmith et Hogarty, 2005). Afin d’améliorer la thérapie des neuroblastomes, il est donc important de développer de nouvelles stratégies reposant sur des
mécanismes différents de ceux des substances chimiothérapeutiques actuelles afin d’éviter l’apparition de résistances aux drogues, et de limiter le développement de tumeurs résiduelles.
V - Nouvelles stratégies thérapeutiques
Les nouvelles stratégies actuellement développées sont l’immunothérapie anti-tumorale, la thérapie anti-angiogénique et l’utilisation d’agents différenciateurs.
1) Immunothérapie anti-tumorale
Le principe de l’immunothérapie anti-tumorale repose sur le fait que les cellules tumorales expriment des antigènes qui leur sont spécifiques. Ceci est particulièrement vrai dans les neuroblastomes, qui, du fait de leur origine embryonnaire, expriment des antigènes peu ou pas exprimés par le tissu sain chez l’enfant. Parmi ces antigènes, le ganglioside GD2, fortement exprimé à la surface des cellules de neuroblastome (Wu et al., 1986), est devenu une cible privilégiée de l’immunothérapie. Il a en effet été montré que le traitement de cellules de neuroblastome par un anticorps anti-GD2 induit la lyse des cellules tumorales (Barker et al., 1991). Différents anticorps monoclonaux anti-GD2 ont été développés et utilisés lors d’essais cliniques de phase I ou II. Cette stratégie donne des résultats encourageants pour la thérapie des formes graves de neuroblastomes (Yu et al., 1998 ; Cheung et al., 1998, pour revue, Rousseau et al., 2006). De plus, les effets de ces anticorps peuvent être améliorés par l’administration conjointe du facteur de croissance des granulocytes (GM-CSF pour granulocyte-macrophage colony stimulating factor) (Barker et Reisfeld, 1993 ; Kushner et al., 2001), ou d’interleukine 2 (Frost et al., 1997).
Une autre forme d’immunothérapie anti-tumorale consiste à diriger le système immunitaire du patient contre des antigènes spécifiquement exprimés à la surface des cellules tumorales, via le système d’immunohistocompatibilité. Ainsi, en induisant in vitro la prolifération de
lymphocytes sensibilisés par un antigène donné puis en réinjectant ces cellules au patient, il est possible de tuer spécifiquement les cellules tumorales présentant l’antigène. Cependant, l’utilisation de cette technique est limitée dans les neuroblastomes par le fait que ces tumeurs expriment faiblement les protéines HLA (human leucocyte antigen). L’administration d’interféron-γ ou d’IL2 (interleukine 2) est alors nécessaire afin de stimuler l’expression des marqueurs HLA à la surface des cellules tumorales (Lampson et al., 1983 ; Corrias et al., 2001). Une autre approche permettant de contourner ce problème repose sur l’utilisation de cellules dendritiques à la place des lymphocytes. En effet, en chargeant les cellules dendritiques par un lysat de cellules de neuroblastome, il est possible de sensibiliser les cellules dendritiques à l’ensemble des antigènes tumoraux (Valteau-Couanet et al., 2002). Une étude clinique de phase I utilisant des cellules dendritiques pour générer des cellules T cytotoxiques montre qu’elle n’induit pas de toxicité (Geiger et al., 2001). Plusieurs stratégies sont testées afin de sensibiliser les cellules dendritiques. Par exemple, Morandi et al., (2006) ont démontré que des cellules dendritiques chargées par les ARNm de cellules de neuroblastome induisent la génération ex vivo de cellules T cytotoxiques capables de tuer spécifiquement les cellules tumorales. La sensibilisation des cellules dendritiques par des cellules de neuroblastome nécrosées est également efficace (Shilyansky et al., 2007).
2) Angiogénèse
L’angiogénèse permet la formation de nouveaux vaisseaux à partir de vaisseaux pré-existants. Elle nécessite la libération par les cellules tumorales de facteurs de croissance qui vont stimuler la prolifération de cellules endothéliales. De nombreuses études montrent que les cellules de neuroblastome libèrent des facteurs pro-angiogéniques et sont capables d’induire l’angiogénèse in vivo. De plus, la vascularisation est associée au mauvais pronostic et l’expression de certains facteurs angiogéniques tels que le VEGF (vascular endothelial growth
factor) ou le PDGF-A (platelet-derived growth factor-A) est augmentée dans les neuroblastomes de stades avancés (pour revue, Ribatti et al., 2002). L’angiogénèse constitue donc une cible thérapeutique intéressante pour les neuroblastomes, d’autant plus qu’elle nécessite l’intervention des cellules endothéliales qui ne sont pas touchées par l’apparition de phénomène de résistance aux drogues chimiothérapeutiques, contrairement à certaines cellules tumorales. Différentes approches ciblant la neutralisation de facteurs pro-angiogéniques, l’inhibition de la prolifération des cellules endothéliales ou la destruction des vaisseaux nouvellement formés donnent des résultats encourageants dans les neuroblastomes (pour revue, Tonini et Pistoia, 2006). Récemment, il a été démontré que l’administration de caplostatine, qui bloque l’angiogénèse en inhibant la perméabilité vasculaire, à des souris transgéniques ayant développés des neuroblastomes palpables induit une diminution importante de la masse tumorale, sans induire de toxicité (Chesler et al., 2007).
3) Thérapie de différenciation, exemple de l’acide rétinoïque
Les neuroblastomes sont les tumeurs présentant le plus grand nombre de ces de régression spontanée. Certains neuroblastomes, même s’ils présentent des métastases, sont capables de se différencier spontanément en une tumeur bénigne, le ganglioneurome. L’observation de ce phénomène de différenciation a poussé les chercheurs à étudier ce mécanisme in vitro, grâce à l’utilisation d’agents différenciateurs. Parmi ces molécules, on trouve les esters de phorbol, le NGF ou l’acide rétinoïque. Le cas de ce dernier sera développé ici pour deux raisons : il est déjà utilisé dans la thérapie de certains neuroblastomes et il a été utilisé dans les travaux réalisés au cours de la présente thèse.
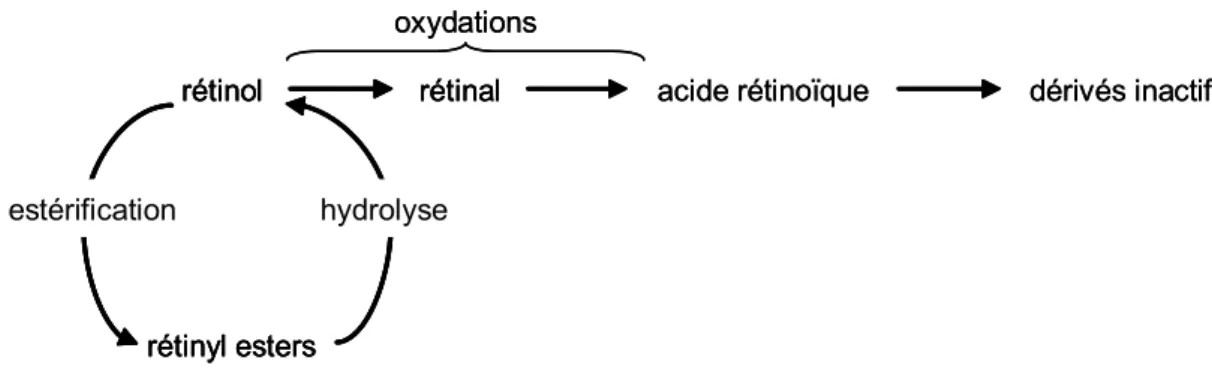
a) Biosynthèse de l’acide rétinoïque
L’acide rétinoïque est un dérivé du rétinol (également appelé vitamine A), une molécule hydrophobe inactive provenant de l’alimentation. Le rétinol est stocké dans le foie sous forme de rétinyl esters, qui peuvent ensuite être hydrolysés pour redonner le rétinol. Il est ensuite transporté aux cellules cibles grâce aux CRBP (Cellular Retinol-Binding Proteins). Dans le cytoplasme des cellules cibles, le rétinol est métabolisé en rétinal puis en acide rétinoïque grâce à deux réactions d’oxydation successives. L’acide rétinoïque est ensuite converti en dérivés inactifs (Figure 6), ou est pris en charge par les protéines CRABP (Cellular Retinoic Acid-Binding Proteins), dont le rôle précis n’est pas encore bien connu. Cependant, il semble que ces protéines puissent permettre de réguler la concentration cytoplasmique et nucléaire en acide rétinoïque, ou transporter l’acide rétinoïque dans le noyau (pour revue, Napoli, 1999).
acide rétinoïque dérivés inactifs oxydations
rétinol rétinal
rétinyl esters
hydrolyse estérification
acide rétinoïque dérivés inactifs oxydations rétinol rétinal rétinyl esters hydrolyse estérification rétinol rétinal rétinyl esters hydrolyse estérification
Figure 6 : Biosynthèse de l’acide rétinoïque à partir du rétinol.
Le rétinol, stocké dans le foie sous forme de rétinyl esters, est métabolisé en acide rétinoïque par des oxydations successives.
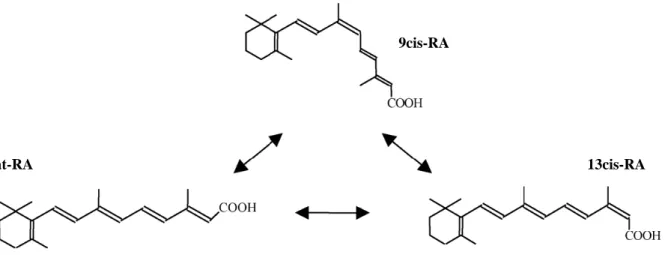
Il existe trois stéréo-isomères naturels de l’acide rétinoïque : le tout-trans-acide rétinoïque (at-RA pour all-trans retinoic acid en anglais), le 9-cis-acide rétinoïque (9cis-(at-RA) et le 13-cis-acide rétinoïque (13cis-RA) (Figure 7). D’un point de vue thermodynamique, le 9cis-RA est l’isoforme la plus instable, alors que l’at-RA, qui est le plus stable, représente la forme majoritaire de l’acide rétinoïque à l’équilibre (pour revue, Armstrong et al., 2005).
Figure 7 : Structure des trois isomères naturels de l’acide rétinoïque (d’après Armstrong et al., 2005).
b) Récepteurs de l’acide rétinoïque
Les effets de l’acide rétinoïque sont médiés par deux familles de récepteurs nucléaires : les RAR (Retinoic Acid Receptor) et les RXR (Retinoid X Receptor). Chaque famille est constituée de trois membres (α, β, γ), codés par des gènes différents. Les RAR présentent une haute affinité pour les isomères at-RA et 9cis-RA, alors que les RXR lient spécifiquement le 9cis-RA. Le 13cis-RA ne lie pas ces deux types de récepteurs pour lesquels il a une affinité cent fois plus faible que l’at-RA et le 9cis-RA, mais il agit via son isomérisation en at-RA (pour revue, Parisotto et al., 2006).
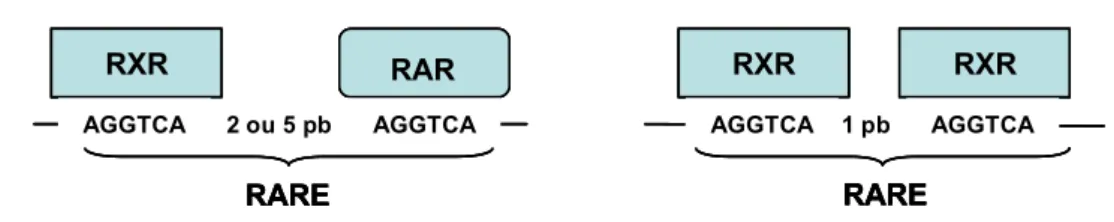
Ces récepteurs agissent comme des facteurs transcriptionnels, sous forme de dimères RAR-RXR ou RAR-RXR-RAR-RXR. Les trois isoformes de RAR peuvent s’associer sans distinction aux trois types de RXR, ce qui donne un grand nombre de combinaisons et par conséquent une régulation fine de l’expression des gènes cibles de l’acide rétinoïque. Sur les gènes cibles, les récepteurs se lient à un élément de réponse à l’acide rétinoïque, appelé RARE pour « retinoic acid response element », dont la séquence est AGGTCA. Les hétérodimères RAR-RXR lient deux de ces motifs répétés séparés de deux ou cinq paires de bases (DR2 (domain repeat 2) ou DR5), alors que les homodimères RXR-RXR lient les motifs séparés par une paire de bases (DR1) (Figure 8).
9cis-RA
2 ou 5 pb AGGTCA RXR AGGTCA RAR RARE 1 pb AGGTCA RXR RARE AGGTCA RXR 2 ou 5 pb AGGTCA RXR AGGTCA RAR RARE 2 ou 5 pb AGGTCA RXR AGGTCA RXR AGGTCA RAR AGGTCA RAR RARE 1 pb AGGTCA RXR RARE AGGTCA RXR 1 pb AGGTCA RXR AGGTCA RXR RARE AGGTCA RXR AGGTCA RXR
Figure 8 : Liaison des dimères de récepteurs de l’acide rétinoïque sur les RARE
Les dimères de récepteurs RXR et/ou RAR se lient sur les séquences AGGTCA, qui peuvent être séparé par 1, 2 ou 5 pb.
En absence de ligand, les dimères interagissent avec des protéines co-répresseurs qui vont permettre le recrutement de protéines à activité histone désacétylase, empêchant ainsi la transcription. La liaison de l’acide rétinoïque sur son récepteur provoque un changement conformationnel de ce dernier : le co-répresseur est alors libéré du complexe et remplacé par un co-activateur. Cette protéine recrute une protéine à activité histone acétyltransférase, ce qui permet la transcription (Figure 9) (pour revue, Reynolds et al., 2003).
AGGTCA RXR AGGTCA RAR Co-rép HD – AGGTCA RXR AGGTCA RAR RA RA Co-act HA + a : en absence de RA b : en présence de RA AGGTCA RXR AGGTCA RAR Co-rép HD – AGGTCA RXR AGGTCA RAR RA RA Co-act HA + a : en absence de RA b : en présence de RA AGGTCA RXR AGGTCA RAR Co-rép HD – AGGTCA RXR AGGTCA RAR AGGTCA RXR AGGTCA RAR AGGTCA RXR AGGTCA RXR AGGTCA RAR Co-rép HD – AGGTCA RXR AGGTCA RAR RA RA Co-act HA + AGGTCA RXR AGGTCA RAR RA RA Co-act HA + a : en absence de RA b : en présence de RA
Figure 9 : Activation des récepteurs de l’acide rétinoïque.
RA : acide rétinoïque, Co-rép : co-répresseur, HD : protéine à activité histone désacétylase, Co-act : co-activateur, HA : protéine à activité histone acétyltransférase, – : inhibition de la transcription, + : activation de la transcription.
En absence de ligand, les récepteurs de l’acide rétinoïque sont associés à des co-répresseurs et des protéines à activité histone désacétylase, empêchant ainsi la transcription des gènes. La fixation de l’acide rétinoïque sur ses récepteurs induit un changement de conformation de ces derniers, qui vont alors recruter des co-activateurs et des protéines à activité histone acétyltransférase, permettant ainsi la transcription.
Alors que les récepteurs RXRα, ß et γ semblent avoir des fonctions différentes au cours du développement (pour revue, Germain et al., 2006), le rôle de ces trois récepteurs dans la différenciation des neuroblastomes induite par l’acide rétinoïque n’est pas encore connu. En ce qui concerne les RAR, davantage de données sont disponibles. RARα, ß et γ sont exprimés dans les neuroblastomes, aussi bien dans les tumeurs que dans les lignées (Li et al., 1994) et leur expression est stimulée par l’acide rétinoïque dans les cellules de neuroblastomes (Chu et al., 2003). Chaque isoforme possède une fonction spécifique au cours de la différenciation de cellules de neuroblastome induite par l’acide rétinoïque. En effet, RARα jouerait un rôle important dans l’augmentation de l’expression de RARβ qui, lui, serait impliqué dans l’inhibition de la croissance cellulaire induite par l’acide rétinoïque (Lovat et al., 1997 ; Cheung et al., 1996). Le récepteur RARγ, quant à lui, serait plutôt associé au processus apoptotique (Meister et al., 1998).
c) Effets de l’acide rétinoïque dans les cellules de neuroblastome in vitro
In vitro, l’acide rétinoïque induit la différenciation de cellules de neuroblastome. Cette différenciation se caractérise au niveau morphologique par la formation de pseudoganglions et une neuritogénèse pouvant aller jusqu’à la mise en place d’un réseau neuritique (Siddel, 1982 ; Pence et Shorter, 1990 ; Lovat et al., 1997). Au niveau moléculaire, la différenciation est caractérisée par l’augmentation de l’expression de la protéine GAP43 (Growth Associated Protein 43), de marqueurs neuronaux (neurofilaments de haut poids moléculaire, tyrosine hydroxylase TH), l’augmentation de l’activité de l’énolase spécifique des neurones (NSE pour Neuron Specific Enolase ) (Morton et Buss, 1992 ; Chu et al., 2003 ; Pahlman et al., 1984). L’acide rétinoïque induit également la diminution de l’expression de l’oncogène MYCN (Thiele et al., 1985 ; 1988). Cependant, les trois isomères de l’acide rétinoïque n’ont pas tous
la même efficacité. En effet, le 13cis-RA est plus efficace que l’at-RA en ce qui concerne l’induction de la différenciation morphologique (neuritogénèse et/ou pseudoganglions), l’inhibition de la croissance et la diminution de l’expression de MYCN dans plusieurs lignées de neuroblastome (Reynolds et al., 1994). Dans la lignée SH-SY5Y, le 9cis-RA induit une neuritogénèse plus importante que l’at-RA (Lovat et al., 1997). La différenciation de cellules de neuroblastome par l’acide rétinoïque fait intervenir différentes voies de signalisation. En effet, l’activité du complexe transcriptionnel AP-1 (Activator Protein 1) est augmentée par l’acide rétinoïque dans les cellules de neuroblastome N1E-115 (de Groot et Kruijer, 1991). De plus, dans la lignée de neuroblastome humain SH-SY5Y, les kinases MAPK (mitogen-activated protein kinase) et JNK sont nécessaires à la croissance neuritique induite par l’acide rétinoïque (Sing et al., 2003 ; Yu et al., 2003), alors que la voie PI3K (phosphoinositil 3 kinase)-Akt est requise pour la diminution de l’expression des protéines Id (inhibitor of differentiation), qui séquestrent des facteurs de transcription pro-neuraux (Lopez-Carballo et al., 2002). Il a également été démontré que le traitement par l’acide rétinoïque régule le cycle cellulaire via une augmentation du taux de la protéine p27 dans les cellules SMS-KCNR et LA-N-5 (Matsuo et Thiele, 1998 ; Borriello et al., 2000).
in vivo
Les résultats prometteurs des études in vitro ont conduit à tester l’acide rétinoïque in vivo. Des essais cliniques concernant le 13cis-RA avaient déjà été menés dans des pathologies comme les leucémies, des syndromes myélodysplasiques (hémopathies clonales caractérisées par un trouble de la maturation des précurseurs myéloïdes), les lymphomes cutanés à cellules T et les carcinomes squameux de la peau de stade avancé. Lors des premiers essais cliniques sur les neuroblastomes, le 13cis-RA fut administré en continu à une dose de 100 mg/m2 de surface corporelle/jour, correspondant à la dose maximale tolérée chez les adultes
(Villablanca et al., 1995 ; Khan et al., 1996 ; Kohler et al., 2000). Cependant, ce dosage ne permettait pas d’atteindre une concentration plasmatique équivalente à la concentration nécessaire pour induire la différenciation in vitro (Reynolds et al., 1994). L’administration de 13cis-RA à haute dose (160 mg/m2/jour) de manière discontinue a alors été testée, cette dose permettant d’atteindre une concentration plasmatique supérieure à celle utilisée pour les études in vitro (Villablanca et al., 1995 ; Khan et al., 1996 ). Une étude menée sur des patients atteints de neuroblastomes à risque de récurrence élevé a montré que l’administration de 13cis-RA à haute dose de manière discontinue présente un intérêt clinique puisque cela permet d’augmenter la survie de 20 % par rapport au groupe témoin (Matthay et al., 1999). Le 13cis-RA présente une meilleure efficacité clinique que l’at-RA grâce à ses propriétés pharmacocinétiques spécifiques (Tableau V). En effet, la dose maximale tolérée de 13cis-RA est supérieure à celle de l’at-RA (160 mg/m2/ jour pour le 13cis-RA contre 90 mg/m2/jour pour l’at-RA), ce qui permet d’atteindre des concentrations plasmatiques supérieures aux concentrations induisant un effet in vitro. De plus, l’at-RA présente une demi-vie courte (inférieure à 45 minutes) car il est rapidement métabolisé en dérivés inactifs, ce qui n’est pas le cas du 13cis-RA, dont la demi-vie est par conséquent significativement plus longue (5 heures). De ce fait, l’exposition de l’organisme au 13cis-RA est plus durable (pour revue, Reynolds et al., 2003).
Isomère Dose maximale tolérée (mg/m2/jour) Durée de vie
at-RA 90 < 45 mn
13cis-RA 160 5 h
Tableau V : Comparaison des propriétés pharmacocinétiques de deux isomères de l’acide rétinoïque, l’at-RA et le 13cis-RA.
Les essais cliniques menés sur l’acide rétinoïque ont conduit à une modification des protocoles thérapeutiques : dans les cas de neuroblastomes métastatiques, la chimiothérapie