\
,
REAGT1QNS D'ORGANpSILANES AVEC DES COMPLEXES
ORGANOM~TALLIQUESDE L'URANIUM, DU
THORI~M,DU ZIRCONIUM Er DE L'HAFNIUM .
..
par
_, J.,e a n - Pie r r e Bar r y
.
, )~..-Thèse ttrésentée à la FacuJ--t-é des EtuCles'-Supér leures et CIe
Reeher~he en vue CIe l'obtention du grade de Maitre ès SCiences (M. Sc.) Département de Chimie Unlvers-Ité MeGIll Montréa l,Québec Canada JUillet 1967
-\
..
\
"
,JEAN-~IE~RE
BARRY
Réa ct Ion dl or ganos lIanes avec des 'Comp 1 exes
"
,
•
d eU, Til, Z r etH f .
..
.'
c
..
!
\
RESt/HE·
\
-Les complexes CPz*HHe2 (où Cp*
=
n5-C5(CH3}5et M
=
U.,!h) f,~j.,ren"t préparés et ut t 1 isés pour _pol Y mé ris
e
r I e ph éri
y 1 s fla ne •. 0 ans 1 e cas' duC Pz" UMe 2 'JU
il
,,po'lymère contenant 10
..à
12 atomes de Slllcl~m a, pu' êtret
IsOlé· et Jdentlflé. Le.cpz*ThMe2
nia
donné Que dul ' dlp'hénYI-1,'Z d,lsllane. O',ver.s essais ont été tentés afin
"
-
~d'obtenir des 1 ~ens thor lum sl..Ilclum. Les résu,ltats obtenus
ont été 'discutés en f.on<ptlon de la stabilité de ces liens.
Des réactions avec le Phénylsllane et les
comPlexes'cp'z'zr--/1ez (Cpi
=
115_C5H4CH3)'
~CPz*ZrHe2'
(C'pZZ.rNZ)'ZNz
et
CP2H'f~ez {Cp=
n5-C5H5} ont auss i êt'é" Q
\
effectuées et lea résultats son~ Interprétés'à la lumière
des Observatlon~ déJà rapportees pour des SY~témes
's
emb 1 ab 1 es. ..."[a polymérlsatlo/':l du phénylsllane_p.ar' le
\
Cpz'ZrHez a aussI été tentée en présence de dlffér~nts
ale è n e 5 et a don né dan s I e 'c a
5
d lÎ c y CIO h ex é n e uni' ply nié r epar t 1 e 1 1 eme nt h Y. d r e 5 i l.Y 1 é . {'
-.' ;
l
, , "•
• 1 ABSTRACT -", .\"
The complexes cP2*MMe2 (where cp*
=
,,5_C5'(CH3)s-. and M
=
~, T~)
were pr'epared' and found tocatalytlc~lly Iflduce.the polymerlzatJ
lon of phenylsllane, •
. When CP2I1U~2 wa~ used on pRenylsllane, a silicon contalnlng
- pOlymèr ( 10
:J
12 s,'t,lcon atoms) was.'
1 5 0,1 a t e d, and
~
•
cha r .. a é ter' 1 z e d . ln a simllar reaction, the complex CP2*ThMe2
gave only 1,2-dJphenyld'Sllane. These results are d/Iscu$sed w.-'th reS!;lect to bond stablllty.
Reactions of pl}enylsilane ln the presence of C P 2 1 Z r M e 2 , ( C P 1 =',,5 - C 5 H 4 C H 3), C p 2 Il Z r M e 2 ',[ (C P 2 Z r N 2 ) 2 N 2 )
and C~P2-HfMe2 (Cp
=
"S-C5H5) were attempted and the. '
,
...
results are compared wlth those of a similar sys'tem.
i n the pre 5 e n c e 0 f var i 0 usa l-k-e n e s wa 5 a 1. s o t rie d. A hydrosilylated pOlymer was Isolated ln the case of
',cye 1 ohexéne.
, , '
..
'f " " " t ,
\
-
\ :'(
W-)
"
.
dirécteur ,(je thhe,
.
~
:
'\0. l' 1 REMERCIEMENTSl
'v ,~I
Il , ! 1 à remercier' l,e pou r lIa 1 d e\~
e t ,Dr John Harrod, mon
généreusement fourni 'tout au 1 0
if
1 de me n t r a va, 1 d e,
recherche. Me~ remerciements vont aussI au Dr Françoise
\
Sa'urlOI pour ses conseils
sur_Îa~RMN,
au Dr Udaltlll pouravoir enregistré les chro~atogrammes par perméatlon de gel
~~nsl qu·au Dr Clare Aitken pour ses enc~uragements
chale~reux et nos nombreuses djscusslons.
1
Je veux aussI' remercier toute IléQu'lpe -.du l'ab
..
435, plus partJcullèremen1; FrançOIs Gauvin par QUI tout est
arr, vé al ns 1 Que Jacques Demers et S,tep'tlen Mof'l1' 1 s pour 1 eurs
conseils, leur amItié et leur aIde lors de la prépa~atj'on de
, ce ma nus cr i't
.-/ Des remercjem~nts vont aussi au CRNSG et au fobds
f'CAR pour le support t'inancier qU·lls m·ont fourni.
Un merCI tout sp6cial i Louise pou~ son temps,
.-son amour et &a force qUI m10nt soutenu tout ~u long de ~es
"
études. Enfin, merci à
Ami~l, Méra~,
Sarrel et Viréo pourleur visite hedomadaire.
)
O
'! ,.
Q" " , ' 0 ,;~
-
' l ' -, 1 " \'1
AB LE "0 E S HAT 1 ER E S .f..
.
Rés umé •..••...•. :. ; • •• , i-t • • , • • . . . t , • • • • • • • , • • , • • • •·
..
'-' , \ Abstract .••..•.•.• -••. r, •...
. .
.
. . .
• • • • • • • • , f , , " 1 L R eme r C i eme nt s . ~. , ... , ~ .. , . , , . , ,. . , , ... , . , ... , .. · . , . . . , ' .. ' i v Table'des matières •..•• • , • , • • • • • • • • , . . . , • , • r , .~.· .. v
l ntr.odu~t l,on généra 1 rtés ... l ' . , • • • • • • • • ', • • • 1 ~.. , • •• 1 o r 9 a no pol Y s i '1 0 x a n es. . • . . . , • , • • • • , • • • • • , , t • , , .. 2o r
9a no
P
0, 1 Y Sila
n es. . • . . . . ". . . . . ~ . '. . ... . . . . . . . . '\. -utilisation Industr lelle des or.ganopolys Ilanes ... 9/
préparat.'on catalytlQu~ des organopolysilanes •... 12
Ch~pltr,e 1. Actinides et organosilanes.
1 ntrodu-ct Ion • . . . . • • . . . : PartIe expér Imenta l.e'
/
1 n, s t ru me n ta t Ion'.
, ma n 1 p
u
1 a:t Ion s. • •. . . .
.~...
~. ... .
~..
.
.
. .
Rés u 1 t a t 5 'e t dis 'c u s 5 ion ,
-,
,
Synthèse des catalY5eur~ du thorium
.-
-•. .
• •. 15 " . -. . . . • 22 • • • t • • ~ • • • 23 et de" l 1 Ur:.
a n i u m '"' •• f 'v' • .', , . . . . , • • • • ' , ' • • •• • •• I, , : • • • '32 _!_réactlon ,de l'~ranlum avec le phénylsïlane ••.•.•• 35
..
r
~a c
t Ion dut h 0r
1 Um
a
ve
cIe p h é<cl Y 1 sila n:e . .. • '... ..
4 7';]
.
Cl'lap'd.re I l . Le "-l.ien 5(1 ic!um acti'nide.
,
,,
.
Intro<tu'ctlon .•..••••••. : •. · . • . . . . • . 57
Par,t 1 e--expér imeAta Je
ma n
l 'Pu lat
Ion s • .' ~ , . . . • . . . t • • 63,j'
,""
, "
c
.) .~c
" " • • • • , , If 66 <.réaction du avec les complexes
101'_" ~ dut h 0 r 1 ume t d e lUranlum" . . . ;,",.,: . . . ,72 Chapitre III. Réaction de 'l'hafnium. et du, zirconium
/
~vec les organos lIanes ..
In~ro"duction,,,.,, ..•• ,,,,,,,,,,,,,, I l . t • • " " " " " . , "
.
, " , , • • 76 Partie expérimentile , , ma n , p u 1 a t ion s , , " " , . , . , " • , , . , " " . t , , · .82 Rês{ltats et discussion / -.
~ .. - 1t-
. " ,
'
(
Y..--_~ e ne r a i es" t , , , , " t,. , " " , • • , , , " • , , " , , .. " l f , . . . " , " • 89( C\
•
-réa~_tion
du phénylsi lanS" avec les complexesdu zirconium .... " • • , " , , • t , " , , , t ' , • -. · .89
réaction du phéhylsilane av~c les comple~es
ae
llhafn-,um . . . , ... , •.•• , .•..•.
~_.,.,.97polymérisation du phênylsllane en présence d,Jun ,
.. , • , t " " , , \ , , .. ,,98
••••.•• •• \ ••• 105 complexe ~u Zlrconl~m et d'alcênes •
, C !=l n c 1 U 5 Ion" " . . . • "-t .. t .. • , ""~' ... " , • " • , • , , • , , " , .. , , "
.
' B i b 1 ; 09
ra
phi eF' , : • " " , " , , , , • , , .t" " , , , • I t " , • , , " .,...
\~.1?8
.
'"
\
\
\ 1 ,\
, , (. " ,1 NTRODUCT ION
Génér,a lités:
~
-Le term~ polym~re est en~o~e 'fortement assocl~
'"
danS-I~esprit d~,plusleurs au carbone. Cela tient à ce
que'-t-dans la nature, les polym~res du carbone sont de lOin le5
.
~plus répa'ndus et les plus varIés. Il est vrai Q.ue certains
autres éléments ont aussI la possibll"tté de former des
.
"
chaînes pOlymér IQues dont l'e squelette est const 1 tué d'un
seul élément (par exemp)e le soufre QUI eXiste à 1'6tat
.
~ \natu-rel sous forme de Sa) mais aucun autre élément ne donne
.
,
.une complexIté_et une"> var iété comparables à celle du
~~ar
bone. Ce qui peut surprendre toutefoIs lorsqu'on\ o
-'
consulte un tableau périodique, c'est la singularité de ce phénomène. En effet, le silicium, Qui est dans la même
famille que 1er
carbone, ne se rétrouve pas' naturellement
SOUS forme polymériQue. Oe'svrcroît, aucun être vivant ne
----...~ _ _ M ... _ ' " \ .
) , u
t.
1 1 Ise d' une f·a 9 0 n 9 é n é r a J. '1~
é e .1
/
L'abond'ance du SiliCium sur terre semble pourtant
----~-\ /
Jouer en sa faveur pUIsqu'il se retrouve au second rang des
O
· ,
f,;r " , j
.< ,
composantes de l'écoroce terrestre
aïors
que Je carbonef-igure a.u quatorzIème rang étant près de mIlle fOIS mOins
,
(
(
(
éJéments sont, sur le pLan chimique, a~se2 éloignés l'U,n de
1 ' a,u t r e. Lep rem 1 e r Indice de cette disparité du
.,d'
comportement chimique nous est donné par l'énergie de
diSSociation des 1 iens C-C comparée à celle de-s liens SI-SI
(2). Dans le premier cas, ,on trouve une valeur tYPique de
347 KJmOI-1 alors Que pour le Silicium cette valeur est de
.
2 2 2 K J mo 1 - 1. Cet t e d 1 f f é
r
e nc e
des t aIl
1 l i t é peu t ë t r e{
,expliquée en par~le par un mOins Don recouvrement des
..
o
orbltales.!1ü 'à la tai-Ile supérieure de l'atome de Silicium
.
c.ompar'ée à·celle du carbone (1,11A pour le Silicium et.O,77A
pour le carbOne).
de stab 1
ll~~
desPlus remarquable encore est la tllfférence
liens C-O et SI-O, où. les valeurs sont
respec~lvement de 360 KJmol-1 et de 452 KJmol-1 • Cette
1 mp 0 r tan te d 1 f f é r en c e qUI d émo n t r e '1 a gr and e a x 0 phil 1 G 1 té s:J u
5~1 iClum, situe assez bien les difficultés rencontrées lors
\ de la prépat'atlon des organopoJysllanes en présence
d'oxygène ou d'eau, ce qu 1 aura It été le cas pour un
or 9 a n I.S me
v
1 van t' sur ter r e. Lat enda
ne e qu'a 1 e sil 1 c 1 u m à-liens 81-0 qUI sont Inertes (e.g. sable, Quartz)
o
à e II e se.u 1 e J a Quas i absence de cet élément dans
1 e ma n d ev,
van
t .
Organopo 1 ys Il oxanes:
I l ne faudrait pa~ ~roire pour autant QU'aucun
poJymè~e contenant des atomes de slliclum'n'a ét-é préparé.
-Ainsi
en
1872, Landenburg (-3) déSlgn.era d'u nom de "silIcone" \ \un composé obtenu par l'action de 1.'eau sur des
d 1 ch. 1 0 r 0 d , a 1 k y.}' s ,la n es. La" c é ton e" QU'" 1 cr O l t a v 0 , r
\
dé'couve~e n'est pas un monomère mais plutôt un polymère
contenant une chaine répét1tlve de liens Si-O-SI et des
groupements organ IQues attachés aux atomes de sil, c J um. Le
• nom sllr-c-orre-est maintenant utilisé dans le domaine
•
Industriel pour désigner des p'olymères du s,loxane de po,ds , 0
molacula,re élevé. Le 3 cher cheu r s l' on
t
r emp 1 a c ë pa r I eterme organopolys,loxane.
\
L'utll isa'tion' courante <les organopolys.ï loxanes1
•
n'a . vrp
i me n t dé bu.f
é que 10 r s ~ u' i l' fut po S S J b 1 e deI esobtenir de façonorental;lle: En effet, la synt/1èse des !
"si 1 icones" repose aujourd'hui encore principalement sur
l',hYdrol yse des organochloros 1 lan~s:
(
n
,lRR'SiCl + n H 0 2 2 t+.
- ___ ) l
o-L
l
+ 20 Hill1
t,
In
schéma \,. \Vers 1940, Rochow (4) m,s au point une synthèse
•
dIrecte de ces organochlorosllanes en faIsant passer du
qhtoruré de méthyle sur du &Itlclum m~talllQue chauffé (ca.
250°C) en présence d'un catalyseur métall'Que
(
, Q..
\
(~
(
..
.Chabitueliement du ~Iyre'),
IJ
obtint ainsi en majorité ledim~thyldichlorosilane avec un bon rendement:
Cu
- - - " " - - >
(C H31
2 SiC 1 2+
6
H3
SIC 1 3 + (C H 3)--s SIC 1 \2500~ produit produits mIneurs majeur
schéma 2
.
Les 'çlrganopolysllOXanes çevenant ainsI plus
facile à obteJllr, on Vit apparaître d~ nouveaux polymères
,possédant des proprIétés Intéressantes et uniques Qui ont
été raPidement mises à profit par l'Industrie, Citons par
exemple l'excellente résistan'ce thermique ainsI
Qu'électrique QUI leur a valu une place de choIX aussI bien
d'a n sIe SIS 0 1 a n t S é 1 e' c tri que S que dan s i e s I l qUI des 'd e
réfr Igératlon (5).
J
Organopolysilanes: (6,7)
\
L e fa 1 t que 1 e s pol Y SI,) 0 x a n e 5 a t t end ire n t
Jusqu~én 1940 pour attelnareçvralment leur rythme de
C r Q 1 S t ère e n r e che r che e t dan S I ; u t i lis a t ion i n dus t r ~ 1 e ') 1 e
~
eut sur pre n d ré.
'~I
est e n cor e plu s
éton n a n
td\ con
5t~},e
rIe
1 ent chem 1 nement des or 9a nopo) ys 1) anes tcha i ne~ 81 -8 l')" La
.
D
o
\
..;n.
.'
\en
19~4
alorsqu~it
f i t réagir du diChlorodiPh!nylsilaneavec du sodium métallique. Cette découverte ne SUscita, pas
un très grand Intérét dans la communauté sCientifique.
pUisqu'II aura fallu attendre jusqu'en 1949 avant de vOir .)
resurgir les ofgaliopolYSllanes. Cette fOIS ce fut à partir
du travail de Burkhard (9) qUI, en utl1 isant une synthèse
semblable à cell,e de KipPlng, obtint des PolyméthYlslla~es
(schéma 3). Par malheur le
polym~re
obtenu est un -eomposéblanc-insoluble dans les solvants usuels et qUI se décompose
. en chauffant à 2000C. Burkhard avanca toutefo 1 S 1
a
formu 1 e( He 2 SI) n' Cet t e des cri pt l-crn peu en cou r age an t e b 1 en Que
préCise des propriétés de ce nouveau polymère expliQue
-probablement l'Clbsence de recherche subs,équente dans ce
domaine pendant 20 ans.
Me
1
-SI1
CI n Me SiCI + 2n Na2
2
+ 2n NaCI Me n schéma 3Il faudra la découverte d'une application des
organopolysllanes pour vOir une prolifération des
,
publications. C'est~ la préparation de fibres de carbure de
.
\
Silicium par yaJlma et Hayashl (10,1',12,13) Qui en relança' - '
l'Intérêt. Ces deux chercheurs ont chaùffé- du
,
polyméthylsilane obtenu ,par la métho,ge de BurKhard à 4S0oC
, ,
(
o
f
(
"
ce Qui leur a donné un polycarbos.ilane (-SiH(CH3)-CH2-)n (où
n~1500). Ce polymère p'eut être mo~lé en {j-bres QUi J par la
su 1 te , son t 0 x y dé e s pa r t 1 e 1 1 eme.r t à l'a 1 r ou . ré t i cu 1 é e s a fin
de garder leur "forme intacte. Les 'polycarbos i lanes sont
ensUite soumis
..
à une autre étape de chauffage à .1250,OC-sousatmosPl1ere Ine.r...te pour donner le carbure de sil icium (SiC)
ayant
des
propriétés physIques très intéressantes;He
Me1
1
SI-SIJe
Me'1
n H1
SI-CH~e
2
n H H 450°C1
1
) SI-CH - S I-.-CH1
21
Me He 0 ~ ---) SIC + CH + H 2 4 schéma 4 2 n\
4' /_____ Malgré les nombreux travaux QUI ont été pUbliés sUite
li' à cette d~couverte, on 'remarque que la synthèse de
~ 0
\
ces organopoJ ys i Janes a très peu Changé. La réac~ion
- <> -" "
ba s e de
des or 9 a n 0 chI or 0 s j 1 an es a v e c l es métaux a 1 c·a 1
.
i n 5 . ( l i " Na, K '.. ~ ~ /
et des mé 1 anges de ceux-c 1) d~meur e encor e. 1 a seu Je méthode
connue de les obtenir.
.
.
,
U
,..·
..
(
;. r
/'
n
-L~s premler~ travaux de s~nth~se des compos~s
~ ,
contenan't des at'omes de sil iClum rel i~s entre eux fu-rent
concentrés principalement sur I~obtentlon de cycles ou de
'"
cha i ne 5 de pet 1 te ta 1 Ile (1 4 1 15 , 15 a). - Pa p' ex emp 1 e Bu r K ha r d
dans les travaux précités obtint, en plus des potymères, un ,
.
composé QU~II IdentifIa comme étant le
r
•
p e r mé t h Y 1 he
x
a c y cIo s i l an e . De nos jours, ce composé estobtenu avec un excellent rendement en utilisant ùn alliage de sO,dium et de potassium et en laissant réagir le
dlmethyldlchlqrosllane à '46°C (t6) . ..JI a aussI été possible
d 1 1 5 ole r e
t
de car a c t é r, Ise r' des c y cIe 5 con t e n a n t 4, 5 , 7 e tméme jusqu'à 35 atomes de Silicium (17,16). Bien que ces
rés u 1 ta t 5 soi en t 1 fi té r e's 5 an
t
5 , I l e 5 t r ema r Qua tJ 1 e den 0 ter1 e peu de var 1 a n c e r e n con t rée -d ans ces S y n thé 5 es.
En ajoutant un trlchloroalKylsllane au systëme ,
déJà,connu, West
t l
l i
(19) obtrnrent en p.etlte Quant"të de~composés ayant un squelette de silicium polycycliQue"
•
.~esicli + HeZSICIZ +Na
(1---)
1l'.
'...:--.'11
1;· 1
.
~.
" -/ '.
.
pro d U 1 t .ma jeu r
schéma 5
Les systèmes' obtenus sQnt toutefoIs rélatlvement Simples
\, -..:.. I I ' "
..
c
,
'\
et aucune spéc i f ici té n" a pu à ce jour \ favor i ser un composé en pa..r tic u 1 j e r •
/
\
Un des dé·veloppements..: majeurs obtenus dans ,lé
_ champ des organopolysiJanes fu.t, sans nul doute, l'obtention
~': de polymères solubles. Comme ~I ,a été mentionné
précédemment, 1 l'''i
n
sol u b i l i t é ,d e s po " -1 Y m ë re
s p -r-
é par é s1 n t t 1 a 1 eme nt a v ait b eau cou pre f roi d i l ' a r de u r des
cherctfeurs. W-est, en 1978, (20,21) découvr i t Que des
polymères solubles pouvaient être préparés sImplement en
ajoutant un mélange de dlméthyldjchlo~osllane et de
phénylméthyldichlorosllane à du sodium: \ Ph
Me
1 .
L·
m PhMeSICI + n Me SiCI +Na
>
SI 2 2. 2-a
1
r
He Me m'n
Y' schéma 6 lce copolymëre est rela'tivement soluble dans les
solvants organIques et fond au lIeu de se décomposer!
1 0 r s q u,l i 1 e ~-
t
cha u f f é . I l s emb t e que 1.J ad dIt Ion degroupements phényles en alternance le long ~e la chaine du
poly(rlère ré~uise substantiellement la 'crlstallinité d{! ce.
\
,
(
o
)
-\
o
der nie r et 1 u i con f ë r e c'e s no u v el} es pro p r i été s. -A v e c
l/avènement d'un polymère soJuble et fusible, un obstacle ma jeu r à 1 1 U t i 1 i s a t ion i n dus t r i e Ile des 0 r 9 an 0 pol ys i 1 an e s ~
venait de tomber. De plus ce nouveau polymère se
t r ans for
~e
é
n c ar~b
ure dê
s~
ici u m' san 5 a v 0 i r à pas 5 e r par 1 e pol)'.carbosilane. Ainst- on peut par simple chauffage, opérer<:p cette- trans:tion (22).UtilisatIon industr,elle des organopolysilanes:
c-L'avenlr industriel des organopolysllanes semble
très prometteur P~ISQU/On ... leur a déjà trouvé trois,
appl icstions dIfférentes SOIent: les fibres de carbure de
•
silicium (1,1,12), les photorésïstances négatives et
Positives (23,24;25) et comme Initiateur de polymér.lsat.on, (26) •
.
,Comme i l a dé,jà été montr~ plus haut. les
organopOlysllanes peuvent être transformés en carbure de
sil~iclum par SImple pyrolyse sous des condItIons·
,
1 ...
appropriées. De plus, avec l'avènement des'organoPolYSilanes P
, '
~
sol u b 1 es, i 1 est ais é de mo dei e r ces der nie r 5 d 1 r e ct erne nt
selon les formes déSirées sans avoIr à passer par "'a
p
r
e,m i ère é ta p e •c'e
5 t - à - d ire 1 a t ra n s f 0" ma t ion, des.c
t
c:
,permis d'augmenter ~e~slblement les rendements de carbure de
5'Î 1 i
c,
um...
.,.... les fibres de carbure de silIcIum sont parmi lessubstances les plus résistantes connues à la traction. On
comprend aisément pourquoi elles sont si intéres.santes.
la seconde appl,cation est toute aussi
importante. Oès le dé~ut de la rechèrche sur .les
,
.
,_organopolysllanes, leur comportement face à la tumière
ultraviolette 8\attlrè l'attention. Les organopolysilanes
.1 • j
absorbent très fortement la lum,ère ultraviolette dans la
,
'régr-on al laot de ~oo à 350 nm:' Plus la chaine de SIl icium
est longue, plus l'absorption se retrouve à plus faIble
..,
. 'énergie et plus le coeffiCient d'extinction molaIre est
élevé. Ce comportement a été attribué à la capacité des
atomes de Silicium Cie délocaliser ,les électrons cr (21'.26).
En oiÙre .• les recherches dans .ce· domaine ont montré Que
l'irradiation ultraVIolette provoque. soit la
sC)~on'de
la-. "
:~~~ li ...
chaine de ·sillcium donnant des un'Ités de polymères plus
,
petites, soit ~ne réticulation du polymère-en permettànt aux
substituants organiques de r~aglr directement avec le'
sil iCI u m, for ma n t a j n s j des bJ" a n che s 5 e C' 0 n da 1 r es ( 29) •
/
ce
double:ëffet de la lumrère ultraViolette(absorption et photo-dégradation) a amené une autre application des organopolysi lanes: celle des
photorésistances. Ces polymères s'y prStent p~rfaitement
o
O·
.'4tbi
'--puiSQU',ils peuvent être utilisés aussi b~ tant que
photorésistance pOSitiv~ que négative. Pour obtenir une
~
photorésistance négative, il suffit d'exposer une partle'du
polymère à la ,Iumlér.e ultrav/_olette .. Cette surfac"'expoSée
se réticUle: changeant ainsi ses propriétés par rapport à la
partie non-expOSée. Il est alors possible d'attaquer
sp-éciflquement la surface sous le polyalkylsilane non \
ma d 1 fié. Par con t r e pOl! r ce qui est deI' U.t i 1 i sa t ion Ct u
pol ymé r e comme ph 0 t 0 r
é'S
1 st an cep 0 S 1 t ive, c,l e'$ t 5 a pro p r i é t.éde photodégradation QUI est mise à profit. On utilise un'
polymère Qui, cette fOIS, se décompose sous l'effet de la
luml~re uitravlolette PQur donner des unités plus peiltes
1 ais s a ~ t 1 a par t i e non - exp 0 5 ë e 1 nt a ete. 0 n peu t par ex e m p 1 ~e ,
à.. l'a i de d' un I.a s e r , découper a
v
e c une t r ès gr and e p.é C 1 s 1 onun patron Quelcqnque sur la surface se trouvant sous leI
polymère expOSé. De plus, puisQu'en se décomposant le
.
polymèr~ voit sa 'capacité ~'abàorptlon des radlat/ons
réd u 1 te (I e co e f fiC, 1 en t d' ex tin c t ~ 0 n mo 1 air e d / min u a nt) 1
pluS rIen n'empêche l'Interaction de la lumIère avec la
couche se trouvant en dessotis. La partie non-'exposée à la
,
lumIère Quant à elle, peut-être oxydée ou encore pyrolysée
et former ainsi une membrane protectrice.
La dernière ,utilisation QUi a été suggérée
récemment consIste à se servir du pOlymère comme
photoinitlateur de polymérisation. West ~!l ont découvert
JO
que photoinitiateurs' de la polymérisation du
styrèn~
ét
du 'méthylmêtacrylate simplement en leur aJoutant un peu de'"
poly~ëre et en ex~osant le tout à la lumière. Ce résultat
,', ~
est particulièrement surprenant sion conSidère qUé rien
nia
été fait afin d/élimlner les,traces d'air QU"i se trouvaient dans le système. Pour expl iQuer ces rë!ultats, West a
"
!}'uggéré la formation, sous l'action de la lumière, de
sil è n e s qui s e rai en t die x celle n t-s cap te urs d ' 0 x y 9 è n e e t QUI pourraient en même temps déclencher la POlyméris,tlon des autres composés. CecI expl iQuerait, toujours selon West" le
- falbl~ rendement photonique de l~organopolysl~ane en tant
-~Que photoinltlateur pUisque l'espèce initiatrice (le
"Silène") est SI réactive qu_e très peu survivent assez
longtemps pour provoquer une polymér isatlon.
Prépartion catalytique des organopolysilanes:
"
J
,,-Les organopolysilanes obtenus jusqu/à présent ont tous été synthétisés de la même manière, c'estàd'ire par la -réa ct ion de mé tau x a t cal 1 n s a v e c d 1 f f é r en t's
di~hlo~odlalkylsilanes. Ce mode de préparatiorr étant non
ca ta 1 y t i que, 1 1 fa u t de u x mo 1 e s de m é t a 1 par mo 1 e d e
chloroalkylsllane consommé., On fait face à des problèmes .. importants
+
s 1 on dès Ire faire de plus grandes quantités dec:
polymère. De plus, 1 es condlt,lons de synthèse sont assez drastiques ce qui 1 imi te souvent 1a
nature des su bs 1 tuantso
,'1.
. l
Qu"on 'veut avoir-sur le s"i 1 icium. Avant ce travai l,
seulement deux',groupes av'alent rapporté leurs résultats sur
la polymérisation ca~alytiQue des organosilanes i l'aide de •
./
compQsés org~nométaU iques. Ojim~'tl!.l,(30) en 1973 ont
décrIt la réaction du tr
is(triphenylphosphlne)chloro-rhodium(l) avec dIfférents organos; lanes. 1 (1 s rappor.tel)t
des échang~s parmi les groupement~ organiques des
-'"
organosllanes et dans certaIns cas une dlmérisatlon ou même
.
.
une trq"érisatlon de ces produIts. BIen qU'II soit questlon~
dans leur artÎcle de ,"polymère de poids molécul,8lre plus
élevé" aucune caractérisation de ce produIt .n~est rapportè~
.
et,
â
notre connaissance. aucun autre résultat n~a été_"'t~
publ lé 'depuLs 'lors sur ce sujet .
Le second groupe à aVOH pUQllé ses travaux sur
ç_e s u jet
-e
ste e 1 u ide Ha r r 0 d t l.u
(3 1 • ·,3 2 1 3 3 , 34). Cesderniers ont obtenu une polymér Isatlon de certaIns
mono-alkylsiJanes ~t de mono-aryls,lanes à "aide de
.
d 1 f f é r e n t seo mp osé ~ dut 1 tan e e t du z ire 0 n J u m . 8 i en q u-e 1 e
,
mécanisme de réactIon reste encore,dlfflcile à déduire en
dépit de -l'Isolation de certains complexes Intermédial,res
tout à fait unIques en leur ,genre, Harrod propo-se, pour
expliquer la 'formatIon de polymère,. I" insertion répétit'ive
de Silène au lien métal SilIcium. Notons QU'Il s'agit du
premier exemple bien déta,ll~ pUbl,é dans la 1i/ttérature
pour la_polymérisation catalytique des organosllanes à
);
ol"alde Oe métaux -de transition.' L'originallt~ et l'intér!t
'
.
-(
J
•(
./
.de cette synthèse reposent dans le fa i t Q,u' ici s~u 1 emen,t une
faible quantité·de métal est requise pour polymériser les
organos i l anes. Le pOids moléculaire du polymère obtenu
n'est toutefois pas très élevé (typiquement ceux-ci sont
, comp1Osés de 15 à 20 atomes de si 1 iCiUm)-mals 'la vOie ouverte
par cette _synthèse est d'un très grand intérêt.
Pour faire suite aux travaux ci-haut ~mentLon'nés,
no usa von sen t r e p r, 1 S no t r ~ r e che r che dan sIe d a ma 1 ne' dei a
pol y mê ris a t Ion Gat a 1 y t i
Q
u'~'
des -0 r g an 0 sil an es. No usa von sdonc tenté des expérfences similaires â celle fa~tes par
Harrod et al mais sur des systèmes organométalliques
- , -
.
l
différ~nts et nous nous sommes intéress'és à divers a~pect
,
"
des interactions des compos.és du silicium et des mèt.a.ux de
transition, domaine peu.explor.é jusqu'à présent. ,
.\
.' )\
"..
' " /; , .,
\
CHAPITRE l'
.
-ACTINIDES
ETORGANOSILANES
INTRODUCTION
\
, " - -, ' /La chimie organométall IQùe des métaux de
.
,.
·tr ans
1t ion au
5en 5
0 Ùel 1_ e est c omp ris e den
0s
JO Ùr
5est' urie
chi mie
S
5e z r é ce nt e .
Ce n" est v
jr, tue 1 1 eme nt qu' a v e c 1 a
première
ynthèse du' ferrocène
(35)que cette brancJle de la
chi mie s' st v rai me nt dé v e-l
0p p é e . Lac h
1m \ e des a c tIn i d
~ 5et
des lanthanides
Q~ant àelfe ne fut
s~rJeusement explor~eQue b eau cou p plu s ta rd.
C'e
5t fin ale men t e n 1 9 66 a
Ile c ..
~a
f
synthèse de l'uranocène (biscylooctatëtraènyle uranlum(IV)
·(36) Qu'on,vlt naitre l'intérêt pour cette famille
d'éléments.
Les premiers complexes des organoact,n,des
\ ' Comrne n c
ër en t
à êt reis olé s e t 1 eu r
5car a c t
éris
ti Que
5pro pre
5étud ; ées.
La prIncipale raison de leur absence pour une
SI, 1
Ion gue p
ériO de dei a sc
én e deI a ch, mIe
0r 9 an
0mé ta 1 1
1que e 5
tla réactIvIté extrême dont Ils font preuve face
àl'oxygène.
Ce prOblème n'a pu être résolu que par le développement'
, récent des méthodes anaéroblques et anhydres. AUJourd'huI
y
.
.-encore la recherche en organométallique des actInides
demeure une chasse gardée d'une pOIgnée de
chercheur~.(
()
Led eux f ème é Il é n eme n t ; mp 0 r tan t pou r J a chi mIe
des actinIdes et QUI allait attIrer l'attention des chercheurs, fut sans doute l'isolatIon de composés
contenants' des 11 ens act j n 1 de car bone. En effet, pendant
longtemps on a cru Que de tels lIens ne seraIent JamaIs Isolés ce qUI entraina un certaIn désintéressement initIaI pUisQu'une grande partIe de l'originalité de la chimIe organométallIque repose sur le falto Que de tels liens
ouvrent la porte à une sérIe de nouvelles réactions et de
nouveaux comportements. Cette croyance au sujet de
l'instabIlité des liens act/n/de carbone reposaIt
prIncipalement su~ l'échec rencontré lors du proJet
-Manhatt.an (37) Où on ava It tenté sans succès de préparer,
~
des composés, du genre UR~ QUI, le souhaitaIt-on, auraIent
permIs en étant volatIls de séparer 'les isotopes de l ' ur él n i um par S 1 mp J e dis
t
1 lIa t i, 0 n.\
C'est en 1?69 que différents groupes Indépendants
(38.39,40) décou~rlrent ciu'en chOisissant avec soin
l'enVIronnement autour du méta)., Il était possIble d'obtenIr des Ile n s mé t a.J car bon e d' une st ab 1 l i t é r ema r Qua b 1 e . 1 1 fu t . alors possible de synthétIser toute une gamme de nouveaux
composés du type CP3HR (où Cp
=
cyclopentadlènyle, M=
U
ouTh et R
=
alKyle ou aryle). Ces composés se montrèrentd'une grande stabilité thermique (38,41,42) ce qUI
contredisait les appréhensions des premiers chercheurs.
De
fait, I l deVint rapIdement éVIdent que cette stabIlIté
"
t _
D
aIl aIt nUI r eau x po s s t b 1 1 ~I tés c a ta 1 y t i Que s .p 0 te n t i e Ile s des
jJ
~
actinides P~lsQu/el-le s/~ccompagnalt d'une faible
réactivité, MarKs et son éqUipe mOdIfIèrent alors les
groupements autour du métal. Comme on pouvait S't attendre,
-les complexes dIts homQleptiQues (c'est-à-dire pe contenant Que
9
es 1 j en s mé ta 1 car b Q n e d u t V P e (1) .5 e mo n t r é r en t t r èsréactffs
(43,44)
mais en ~ëme temps laissaient peu d'espOirQuanOt à leur utd Isatlon et leur
J
isolation, conclUSIons QUI ,
avaient déJà été émises lors des recherches Initiales. Les
complexes' ayant une formule ~u type CP2MR2 furent étudiés
{45,46)
mai~ on se rendIt compte que de telS. composés ne,
pou val en t ê t r e i sol é s pu 1_ S qu' Ils s e t ra n s for ma i en t
raptdement en epMR~'ns ~où S
=
solvant comme le THF) et, ,
ep~MR. Par contre, en changeant l'encombrement stér,IQue
autour du métal, soit en utilIsant le lIgand
pentaméthyl-cycopentadlènyle (eplt) , i l fut Posslbre d'Isoler des
complexes ayant deux l-iens métal carbone
(47,48).
Cettedè-couverte fut pour beaucoup dans ,l'_avancement de la chimie
des organoactlnides car les composés contenant deux
groupements eplt et deux groupements alKyles furent largement utilisés par lâsulte dans de nombreuses études, leur grande polyvalence vient princIpalement du faIt Que de tels
1
compOSés se trouvent à mi-chemin entre les complexes du type
pour ce QUI est de la réactIvIté tandIs Que
" ,
leur stabilIté est ,assez_.p~oche de celle du CP3MR (4-9)
La, chimIe de ces \compoSés allait rf/server
.
,.", "
---l
,-' , " ~ 1 l "(
'--
,,,,
, '.
.
, \ , 1 \ëer~aines
surprtBeS, prin.cipalement düe)5 au faIt~
les
-actinides ont une très grande affinité pour l/oxygène. Ain SI, Mar k s déc 0 U v r i t Que 10 r s..Q U e 1 e d 1 mé t h Y 1 e
,
bls(pentaméthylcylopentadlènyle)uraniu~(IV) était mIs en
/
.
présence de monoxyde de carbone, il formait 1
e
comp'l exe1 (50) , . , ;
-\,
Me /Me\
C=C
/
"-
\
/0
0 ,"
IlcPZ*u
/
UCPZ
*
\
0 0 1"f"c /
JI,
1
\
Me He 1,
-,
1.C e mo
le,
der é a c t Ion fa c eau C 0 est t_ n hab 1 tue 1pUIsque ordInaIrement on observe un lien métal carbone (51),
rI eXiste pourtant d/autres métaux QU. donnent le même effet ,
.
notamment 1 e
21
rcon 1 um (52) e"t 1 e titane(53).
Marks et son.
~éqUipe ont réussI à isoler~ d'autres 'comPlexes qUI démontrent
- - Q
Que 1 a p r
lm
i ère é t a pee 0 n sis tee n un - 1 , end u t y P e a c y ) e 1 0 Ù'"
le carbone et I/ox\'f'9éne prennent part au lien avec le méta,l
- r •
(54,55,56).
r Dans de tels complexes le carbone ~~ comporte opratiquement comme un carbène (~).
,,/
'~
-'"
.
,,~
v:
-o
Marks a aussI d~montr~ Que certains complexes JU
thorlUm.et de "ur'anll.lm .!tva',ent la capacité d'ac't,ver les
liens carbone hydrogêne (sChéma 7) (57.58,59). Ce type de .. d'activation avait déjà été remarqué pour 'e~ complexes
}
o
contenant des métaux de 1 a fam,l 1 e des 1 anthan, des (1 e lutécium par exemple) (60).
\
schéma 7
,Du pOint (Je v.ue catalyse et ny("og~n,t,on
•
catalytIque, les complexes
d,'lkJlee
~esont
montr~&trte
performants.
Harks a d!couvert entre autre Que
le/
,
,
"
diméthylbis(pentaméthylcyclopentadienyle)UraniUm(IV) était
un des meille'urs ca/talyseurs de la polymérisation de
/
l'éthylène (61).
SUI~e aux résultats obtenus par les c~ercheurs
dans le domaine des ,Qrganoa~tlnides, i l nous sembla très
éVident Que de tels systèmes seraient trés intéressants i
,
étud.er pour la polymérisation des organosllanes. En effet,
b 1 e n Que l ' u r a n 1 ume t 1 eth o,r i u m n e soi e n t pas dan S 1 a m ê m e
famille Que le tItane et le zirconium, leur chimIe est t"rês
souvent vo',slne. Par exemple, comme,pour les éléments du
groupe 4, Je niveau d'oxydation IV "est Je plus stable même
f
SI l'uranium et le titane peuvent facilement changer de
niveau. De plus, l'uranium et le thorium possèdent
habituellement quatre ligands tout
comme
le titane et le~
Z· 1 r con 1 u m . F 1 na 1 eme nt, a 1 n s i QU' 0 n l'a vu plu 5 ha ut, 1 a
réactiVité des actinides face au
CO
entre autre est partagée"
POU r I e 5 de u X gr 0 u p e s de mé tau x. Ble n 5 û r, 1 1 Y a au S 5 1 des
dIfférences majeures,
comme
par exemple le faIt Que les,
actinIdes ne SUivent pas ,la régie des 18 électrons et QU'Ils {
{
possèdent des orbitales f qUI peuvent être partIellement
r emp 1 Les.
/
~us
avons essayé d'obtenir des réactIons~s}mllalres à celles observées pour les métaux du groupe 4
aveè 'es or ganos i l anes. Pour cela, il a fallu tout d'abord
s cie n t i f 1 Que e
t
1 e s mo d i fie r a fin dei e 5 a da pte r A nosressources, PUIS
nous
avons
exploré les
réactions
de
ceso • •
C omp 1 ex e & a v e c i e sor 9 a nos l I a ne s. L ~ s
rés u 1
t 8 ts
0b
t
e nus
furent très encourageants bien Qu'en -même tempspartIculIèrement compliqués.
Contra Irement aux métaux
du,
gr 0 u ~ e 4. 1 1 fut i mp 0 s 5 1 b 1 e d' 1
sol
e r et d' 1 den t i f 1 e rclairement
des
Intermédiairesde
réaction./ 'v o " , / /
"
(
(
CHAPITRE
PARTIE EXPERIMENTALE
\ J nstr umentat 1 on:Les 5pectr~5- Infra-rouge des pol ymères d'es
organos lIanes furent obtenus en plaçanot une m,nce couche de,
l',ééhantillion étudié dlr~ctement sur une plaque de chlorure
de potAiss'lum. Tous les spectres furent enregistrés sur un
appareIl du type PèrKin-Elmer 297 entre
.
40.0.0. et 60.0.. '
cm- 1 . Laréférence choIsie fut la bande d'absorptIon située à 160.1
cm- 1 d'un film de poly,styréne. La réSOlution 'est de 4 cm- 1.
Les spec~res de résonnance magnétIque nucléaIre
ont été,obtenus sur, le modèle XL-200 de Var lan à .transformée
de' Fou r rIe r. Les d é p-l ace men t
s
chi m 1 Que 5 (± 0 , 0 5 p pm) u t I I i s é splus lOin sont donnés par rapport au tétraméthylsilane (0.,00
.
ppm) , bien QU'en aucun cas le tétraméthYlsi Jane ne' fut
ajouté à l'échantillon. La référence fut ob~nue à "aIde
\
~u PIC résiduel da au solvant.
--Les mesures cl'e,pOlds moléculaire (PH) furent
obt~nues soit i l'aide d'uh osmom~tre i pression de vapeur (OPV) (Corona-Vle'scan mod~Tle 232A) en utt 11 sant comme solvant
le toluène maintenu i une température de 500C (i 70
Daltons), SOit par la chromatographie de perméatlon de gel (CPG) (Var l'an modèle 5000) muni -d'une colonne 103 "
Ultrastyragel de Waters ~~soclates et éluée avec le
,
tétrahydrofuranne. Les standards utilISés consIstaient
en
dupolystyrène (fournIs par Toyo Soda manufactur 1119 Co Ltd). La
préCISion de cette méthode est évaluée à ±40 Daltons.
Man i
P
u 1 a t, Ion s :'Sauf dans' des cas particuliers QUI seront décr Its
,
lès manipulatIons-ont t~utes été effectués
sous
.
\atmosphère inerte par la teChnIque de Schlenk (62, ( 3 ) .
"'argon et Ilazote (LInde) utIlIsés comme gazs Inertes, ont
été désoxygénés en les faisant réagir avec le catalyseur
,
R~-11 (Chemalo~n PUIS séchés sur du tamis moléculaIre .H
pour éliminer toute tra'C~ d'eau.
L'e tétrahydrofuranne (THf'), 1'6ther dl~ttlyltQue
(EtZO)t le dlméthoxy6thane
(ch,O,
le tolu~ll'e (TOI) • le1
pentane (Pent) et l'hexane (He)() furent' 5~Ch6$ sur un
..
(
(
c
mélange de sodium et de benzophénone, puis distillés sous
a tmo s p h ère 1 ne rte ( 64). A van t d' ê t r eut 1 1. i 's é s , l e s
différents solvants furent complètement dégazés par de
nombreux cycles de gel-dégel. Les
s~lvants
deutérésutil~SéS
pour obtenir les spectres de résonnance magnétique nucléaire
( R H N), 1 e ben z è*n e - d 6 , l e toI u è n e - d 6 et 1 e c y cIo h e x an e - d 1
f
(Herk, Sharpe and Pome) furent traités de la même manière. Ils ont de plus été transférés par disti llatlon sous
pression réduite Immédiatement avant leur usage.
Les pro~ults_suivants: le tétrachlorure de
thorium (Alfa), le chlorure d'lsopropylmagnéslum(ll) (A 1 d r 1 ch), 1 e br 0 mo - 2 but è ne - 2 (A 1 d rie h ) - , l e 1 j th 1 U m
(A 1 d r 1 ch), 1
e
t r i chIo r 0 p hé n y 1 sil a n e ( Pet r arc h), l ' h Y d r .u r ed'aluminium et de lithium (Aldrich), ·'''acétate 'd'éthyle ('1Ildrlch), Je méthyllithium en solution dans l'éther
,
éthyll<;lue (A!drICh), le trloxYde d'uranlum(IV) (Alfa) et
"hexachloFopropène (Aldrich) furent obtenus com~ia'ement
"
et UtIliSés tel Que reçus. Le pentaméthyl-1,2,3,4,5
Cyl 0 Den ~ ad 1 è n e - 1 ,3 (C P ~ H) f. ut pré par é selon une mé t h 0 de
publ,'ée (65) et fut obtenu avec un rendement de 50 ~ 60Z,
puis conservé à ... 10oC sous atmosphère inêr;.e-et à l'abri ,de
la lumière. Le ph_ényls I,'.,ane fut préparé teton la méthode
décrite par Flnholt ~
Al
(66) et conservé sous atmosphèreinert~. Le tétrac~lorure d'uranium fut préparé selon une
mé th 0 de con nue à par tir dut rio X Y de d' ur an i u m ( 1 V) ( 67) et
con 5 e.r v élu i au s S I S 0 usa tmo 5 p h ère 1 ne rte. Le
)
o
o
-("S-C5(CH3)S)2UCI2 (CP2*UCI2) et le " \
(fI~-C5·(CH3)S)2ThCI~
(CP2ltThClë)f~rent pr6P~r6S
selon la méthode décrite par Harks~ ~ (~e)
etfure~t
obtenus avec\ ~,
des rendements respect 1 fa de 76 et 80r.. F' 1 na l'ement 1
e
(l1S-CS(CH3)S)2U(CH3)2 (CP2"UHe2) et le
{fl5_C5(CH3)S)2Th(CH3)2 (CP2*ThMeZ) furent obtenus grace à 1
a
mé th 0 d e déc r i t e par Mar ~_ 5 ( 46) et fur en t 1 sol ~ 5 a v e cdes rendements respectifs de 65 et
72%.
Prëparat',on du Cs (CH3)SM9C\' THF' (Cp"MgC-l' THF)
"
-/
La préparation de ce composé' estobasée sur celle Clécr Ite par
Marks (48) sauf pour Quelques dIfférences dans l'extraction et I~obtentlon du produit final. 127 ml d'une solution 2H de chlorure d',sopropylmagnéslum (254 mmol) dans l'ét.her
,
éthyl'Que sont placés dans un ballon d'une capa·clH Cle 350
ml.
l'éthe" éthylique est ensuite évaporé au maximum 50USpressIon rédUite à l'aide d'un ba,n d'eau tl~de. Le préCIpité blanc gélatineux QUI est obtenu est mis en suspension dans 125 mL ge toluène séché préalablement et désoxygéné. Sous atmo'sPhère Inerte, une trappe r e f r o i d i e ' ---760C
I est alors placée entre le ballon et le sysUme de
d,'Strlbutlon de gaz. l'addlt.ion d'un seul coup de 40 mL d.e
C_P"H (255,4 mmol) DI=-odult une lég~re évolution de gal
(prC'pane) QUI dey.en,t plus éVidente lorsque le 5Y5t~me est le ballon est a 1 or,,! chauffé à '10o C. ce QUI augmente
, 1
\
.
(
(
d"éther éthyl ique). la SiOlution prend alors une coloration brunâtre. Véther éthylique est recueilli dans la trappe
tandis Que le propane est ëllmlné. Ce
~ëlange
est laissé à '-réagir pour environ 2 à 3 heures (Jusqu'à la fin de l"ëvolution de gaz) puis Il est retiré de la source de
ohaleur et laissé revenir à température de la p.éce. Après
ce temps, '40 ml de THF sec et désoxygéné y sont ajoutés d'un ....
seul cou'p, dégageant un peu de chaleur qu) peut être
\ é'
suffisante pour dissoudre le produit temporairement. Un préCIpIté se forme rapIdement et i l est ensuite laissé à
/
cr, 5 t'a 1 l , 5 e r à une t e m p é rat ure de 5 0 C p 6 u r l a nui t. L e
solIde est f i ltré sous atmosphère Inerte et lavé avec du
tolUène frOid JusqU"à l''obtent,on d"un f i l t r a t ,nco/ore.
Ce
f i Itrat peut être légèrement ,réévaporé sous press ion
r édu 1 te, et r efro 1 dl pour obten i r une seconde
cr Istal Iisation. Rendement: 80 à 85Y.
Réa~tlon du CP2*UMe2 avec le phénylsi lane
A ) R a p p 0 r t mo 1 air e d e ' : 2
, 1
Une 'réaction typique est décrite ICI.
On
ajoute 5 mL detoluène sec à un ba lIon contenant déJà 0,5 9 de CP2*UMe2
",
(0 ,,9 mmo'). 8 i en, qu' Ç> n u t I I i 5 e un toI u è n e t r ès 5 e c • 0 n no te
toujours une légère décomposition du complexe
organométalli'que. 0,22 ml de phényl.sllane. préalablemen~
,-
'1.;"ii f
...
dé 9 a z é son t, a JOu tés i Jas 0 1 u t j,o n l i ' a ide d ' une 5 e r i n g.u e .
On n'observe pas ~e changement de la couleur de la solution
nI d'évolutIon éVidente de gaz. Certaines de ces réactions
1
ont été effectuées en' chauffant le mélange réactionnel ,
JUSQu'à 600
e
sans Qu'~n puisse noter de changement1 mp 0 r tan t . A P r ès a VOl r i a 1 s s é réa 9 1 r , pen dan t une P é rio de,
allant de Quelques heures à plusieurs Jours, le solvant est
D
évaporé à sec pour obtenir une hUIle brune sensIble à l'aIr
1
et soluble dans la plupart des solvants usuels. De nombreux
(
e s ~ ais de CI"l st ail 1 sa t Ion 0 n t été te nt é s i d e ~ t emp é rat ure 5 (
,allant Jusqu'à -7aoc et ont permis de recueillir' unlQuemen~
de petItes Quant 1 tés du p'r 0 du 1 t de départ.
.r
'\1)
Bl,
Rapport mo 1 a 1'1" e de 1 : 1 5
-Pour ces réactIons, le protocole e)(p~rlmental utllls~ est
semblable à celuI décrIt CI-haut sauf pour ce qUI a trait j
1 a Qua nt 1 té ut 1 Ils é e. 0, 6 __ 9 de Cp 2. Il U M
è
2 ('. 1 ~mo 1) son tdiSSOUS dans 5 ml de toluène. A l'aide d'une seringue, on
Intr,odult 2. ml de phénylsllane (16,0 mmol). la réactIon est
agitée à la température de la pièce pour une pér Iode lJIlant
CIe {) à 24 heures. Après ce temps, le solvant est
"comp 1 ètement évapor é et on Obt 1 ent une hu, 1 e tH' une,
'habItuellement plus vIsQueuse Que celle obtenue:avec un
rapP1rt molaire de 1:2. Cette hUIle est alors IntroClU.lte
dans une colonne i Chromatographie pr~altblement rempl
le
de(
(
(
Toute§ ces
manipul~t;onssont effectuées sous atmosphère
1
n e r(t e . A u con tac t dei' a d sor ban t , l ' hui 1 e bru n
e
r ê agi t
violemment
et
pren~une couleur noire. La colonne est éluée
avec
~OOml de toluène pour Oonner une
sOlutlO~Incolore Qui
,
est évaporée sous pressIon rédUIte laIssant le polymêre du
ph.nYlsl lane sous forme d'une Pite blanche QUI peut atre
lavée avec
30 mld'hexane.
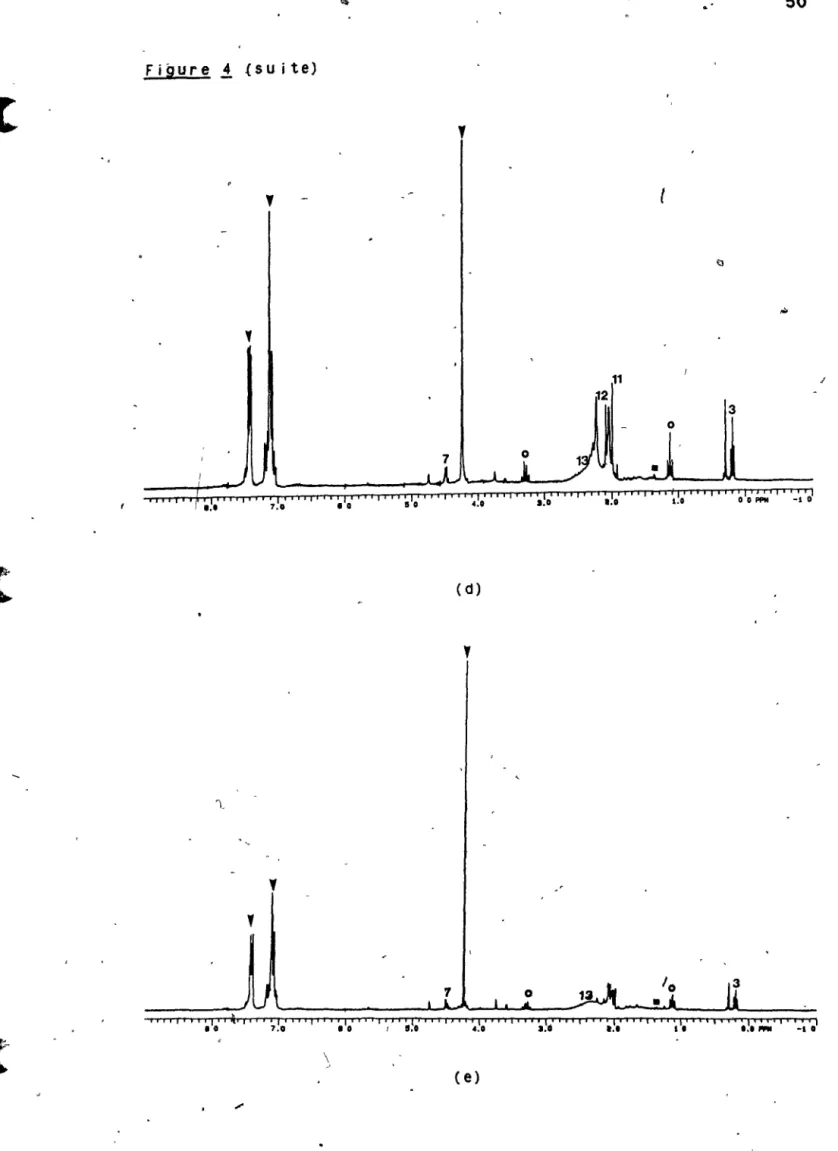
Caractéristiques du
polymèr~: RHN (e606):m.
large,
d 7,90-6,62 (5H, Ph)m.
large,
d 0,25-4,10 (1H, SI-H) IR v(Si-H) 2080cm-
1 ,"q"
1100cm-
1 Jd(SI-Hl,
915cm-
f ,-\PH
(CPG) :entre
600et
800 0,
la même r ê a c t ion fut au s s
1te nt é e en ut
1Ils a nt l' éther
éthylique comme solvant. Oans
un
bal Ion contenant
1 9 ~eCPZ*UMez
(1,8mmol),
10ml d'EtZO et 3 mL de phénylsllane
..
,.
( 2 4 mmo 1) son t a JOu t
ës t
0u r
àt O,U r. A p r 'è s 20 min u tes,
0n
remarque Que la sOlu,;on Dasse du 'rouge au vert ivei une
..
\
importante évolution de gaz.-Cette
éV~lution'cesse
a"près une
•
heu r e ma i s peu t
ét r e réa c t
r
v é e a v e-e
t'
a JOu
t <Id"a 1
iQUo t s
Sil
PP 1 éme nt aIr es de ph é n
y1
5
i 1 an e , b
ien QU'a 1
0r s {a réa c t Ion
soit mOIns violente. le solvant est alors évaporé
àsec mais
)
o
\
• 1
1-0 r 5 Que pla c é e SOU S V f de, 1 a sol ut f 0 n v f r e ra p i d eme n tau
r- ()
u gel nit i al. L'a d dit 1 0~
su p P 1 éme n' ta 1 r~
de ph é n y 1 sil an e
-( N 1 mL) ne pro v 0 Que a u c u n cha n g e m-e nt. Lep 0 1 Y m ère 0 b t e n u
peut Stre isol~ de la manl!r! d~crlte-ci-h~ut et pré5ert~
des caractéristiques en tout pOint semblables à celles du
polymère préc~dent.
Réaction du CP2*ThMez avec le phénylsilane.
A ) R a p p 0 r t mo 1 air e" de 1; 2
o·,
5 9 de' C p 2 If Th Me
2 (0, 9 mmo 1) son t pla c é 5 dan sun bal Ion 5 e c_, et dégazé. 5 ml de toluène sont ensuite aJ~uté5 ainSI
Q\
0,24 mlde Phényls,lane
(2-mmoq.'
Avec le temps, on noteune légère coloration Jaune de
18
solution mais Il n'ya
au
c une
ë volu
tjonc
de
g'a.r~·o
t 8 b 1e.
A pr ès
une p é rIO de d ete m D Sallant de 2 â 48 heures, la solutIon est ~vapor~~ â sec pour
donner une pâte Jaune. Ce composé est très senslbl-e à l'aIr
J
et à l'humIdité et fut Impossible à caractér.ser,malgré de
nombreux efforts pour le recristallIser. Cette réaction fut
o
répétée à dIverses températures allant de 40 à 90
Oc
sansr J
rien 0 b'ten 1 r de plu 5 •
, /
.
B) Ra p po r
t
mo 1 air e dei: 1 2\
Ai" 1 9 d e C p 2
*
Th M e 2 ( 2 1 1 mmo 1 ) sont aJoutés dans l'ordre 5, \
'.
ml des 0 1 van t ( toI u é ne, ben 2
ë
ne, C y c 1 0 h e x a ne 0 u he x an e) et 3ml de p l'l ê n y 1 sil an e '( 24 ,1 mmo 1 ). las 0 j ut ion pre n d a 1 0 r sun e
coloration jaunâtre et demeure ainSI pour le res'te des
ma n î pu 1 a t ion S" A pré 5 2, à 3 J o-u
r
s , l a sol ut Ion estêva
po réè
à5 e cet 1 a p â t e j a
u
n â t r e' est c h r 0 ma t 0 9 ra
phi é e 5 ur
gel de f l o r l s i l avec 300 ml de toluène. "-prés év.ap-ora1ion del#éluant, on o~ti~nt typiquement 0,6 9 de sol Ide planc
con t e na n t p r i ne 1 pal e me nt. 1 e d i P h é n y 1 - 1 ,2 dis i 1 a ne.
RMN (COCI3):
m
d 7,50 {4H o-P.!!.Jm
<5 7,12 (SHm
et p-Ph) S <5 4, 52 (4 H S, -H2) l 'If~ v(Sî-H) 2150 -cm- 1 , <:S(SI-H) 910 cm:' 1 ture ( 1 5 a)' : r , , RMN (C60S) : s <5 4,39'"
/ ~.
(pa s. de valeurs rapportées pour. _1 es groupemen,ts phényleS)
C
\"
SM:
M+
214"
•
\
1
0
-
. .
La mame réaction fut tèntée en utilisant l'éther éthylique ,
comme solvant. 0,9 g de CP2*ThMe2 (1,7 mmol) sont dissous
.
dans 15 ml d'Et20. 1 ,S mL de phé'nylSI lane j(12 mmol) y sont
'"
aJout~s pUIS le m~lang~ es~ laissé â agiter
W
températuri,de1 apI è ce. A pré S· une heu r e der é
a
c t 1 on , l a COU 1 e u r deia
~ < / , '
solution passe du Jaune cl~ir.â l'orangé puiS au rouge sang.
On-peut noter alors la prés~nce diun préCIpité blanc QUI est
recueilli et Identlf'lé par'RHN'. la solution rouge perd
rapidement sa couleur Initiale et le préCIpité blanc se fart
\ '0
~IU5 Important. Lorsqu'on évapore la solution Immédiatement
\
a p r ès l ' a p par 1 t ion deI a col o'r a t , 0
n
r 0 u g e ,- 0 n 0 b t 1 en t une'/
pate orangée qUI est décolorée.par le toluène ét l'hexane
se,c et qu 1 donne un' spectre de RHN inextricable et
non-reproductible d'une fOIS à l'autre. Cette hUile rouge
réa g 1 t' au s 5 1 a v e cie pll é n y
l
5 lIa n e .et
JJ nec h r 0 ma t 0 g r a phi e du\
rés U 1 ta t de cet i. e réa c
t
Ion '~o nt
r e l a pré 5 e n cep r 1 n c 1 pal e men t du d 1 ph é n y 1 - 1 • 2 d ~ 5 1 ta' ne.Caractérisation du composé Isolé:
J'; RMN (C6 0 6):
m.
1 6 2,23 (CS ( CH 3) 5) >t~ 5 • • 6 1 9 t 1 (Th- H).
-? /c.
\ ( ! • o bHAP brRE•
RESULTATS ET DISCUSSION
/1S'_y n t h è s e des c a t a 1
y
s e urs du t h 0 r 1 ume t d e l ' u r a n 1 u m :Le
choIx des organoactlnide~comme catalyseurs potentiels de la polymérisation des organosllanes peut être expliqué sion tient compte de la simila-rlté QUI eXiste~
entre cette famIlle et celle de,s co'mplexes du groupe 4. De
plus" comme les premIers chercheurs l'ont rapidement
démontré, "la Chlmle,çJes actinIdes est à la fOIS très varlèe
.
et très/étendue tout en SUlvantr-assèz bIen- les vOies déjà
ü
établies. Finalement, la recherche dans ce doma Ine est sufflsament avancée pour ne pas à avoir à tâtonner sur la
1.'
préparatIon d'un catalyseur de départ:
,
,
1.
Afin de calquer le plus possible les réactl~ns de
°pO-lymérl~atlon.des organosilanes déJà obtenues par Harrod
et'-'ll
avec les composés du t'Itane et du zirconium (32,33,,
34,66),'11 fut décidé d~ préparer les complexes du type
"
·ii
CP2H(IV)Me2 .. (H
=
Th et U). Dan s I e . cas~'p e~,
5 a c tin 1 cre s,se u 1 ~J~s composés ayant la t'ormule Cp
=
pentaméthyl'" cyclopentadlènyle (Cp*).sont connus.--...
la préparation -de _ces 'complexes a d6ji 6t6
effectuée par Marks et son équ Ipe (-47). CeuX-Ct ont obte~u
tout d'abord le CPZ·MCIZ (où M
=
U et Th) par réaction avecle dér Ivé du type Gr Ignard du 'Igand
pentaméthyl-o "
cyclOpentadlènyle (schéma 6). PUIS, par Simple méttlylatlon 1
à l'aide du méthyliithlum, rIs obtinrent les complexes du
toluéne
Z CpIlMgCi'THF + MCl4 ---~> CPZIlMC IZ t 2 MgC 12 t- 2 THr~,
"aoc
y
46 hrs schéma 8c
I l reste toutefoIs Que la Dr~paratlon de ce~
prodUits de départ pose en elle m~me un défI asse! 1 mt> 0 r tan t. L a p r 1 n C 1 pal e cau 5 e de cet t e d, f fIc U 1 t é est
sûr.'!ment la senslbl Ilt~ extr!me dont t'ont preuve les
o\ganoactlnldes face j
l'ox~g!!ne.
La préparation dU.,
(
(
demande un certain doigté. Les synthèses de Marks et son équipe décrites cI-haut ont été suivies dans leurs grandes 1 1 g ne 5 ma i 5 ce r ta in e s é ta p e son t été ,.. ace 0 U r c 1. es p ~ r
ce
\
q U ' e Ile sn' 8 p P 0 r ta i e n t pas a s s e z d ' ,8 van ta 9 e s pou r I e s
pro b 1 ème s QU' e Ile s g é n é rai en t. Par e)( emp 1 e. 1 a
recristall,sat.on â -78 oC des composés 'd,chloro et diméthyle
..
aet I,n ides a été suppr .mée parce Que, plus souvent
QU"autrement, la manipulation de ces solutions froides
entraînait une Impor~ante eondensati~n d'eau provenenant du
milieu amblant et provoquait la décomposition parfol-s totale de l'échant,ll,on.
Il fut BUSS i remarqué Que la synthèse du CPZ*UCIZ
donnait de bien meilleurs résultats lorsqu'on ut,llsalt comme produit de départ le tétrachlorouranlum(IV)
fraîchement préparé à partir du trloxyde d'u,ranlum(VI)
plutôt QU'un produit commercial. CecI s"expllQue
probablement par la plus grande pureté du produit fraiS qUI
est,
~oute
f,n"
prat'!., exempt d'oxyde d'uran,um mIXte
P~oduit'par/la lente mars constante dégradatIon du
t~trachlorouranlum(IV} au contact de l ' a i r .
Let h 0 r 1 u m qua n t à 1 u i 1 cau s e b eau COU P mo 1 n s de
-probléme du poin~ de vue de synthèse. CecI est dO â ce Que
les complexes du thor ium sont légèrement plus résistants à
-" 0
o ,
\ '
solubilité dans les dIfférents solvants organiques rend leur
isolatIon et leur recr IstalllS3tlon plus faCII~.
FI
na 1
e~ent/l
a
seule caractérisatIon QUI futobtGl;1ue de ces complexes fut les spectres de résonnancJ!
i#
ma 9 n é t 1 Que n u clé
a
l'r
e " L a p r 1 nc
1 pal e rai son de cee h 0 1 X,«III
repose sur le faIt QU'II s'agIt de lOin de la méthode la
plu saI sée POU
r
dé tee ter 1 e 5 1 mpu
r:
e té 5e,
taus
s '.
c e QUI1
n'est pas négl,geable, un spectre de .RMN est beaucoup plus
fa cri e à 0 Q t e-n 1 r QU' uns p e ct r e ,In f ra
r
0 u g e (s 0 usa tmo 5 P hère
Inerte). De plus, °tous les composés de départ sont olen
connus et parfaItement caractérisés et donc aucun
(J
rens--.gnement Inédit ,n'aura ,t pu être obtenu par les
méthodes d'analyses conventlonelles (48)
...
Ré
le
t , 0 n de l ' u r an; U m~
I I
ph é ny
1 sil a ne:J
a) Ra p po r t mo 1 air e t : 2
/
,
Dan 6 l ' e Il POl r d ' 1 sol
et des
C 0 mp 1ex
e Il Clu
m~ me t y p e~ue ceux déjà obtenus avec les ana logues du groupe 4
tout
Justesuffisant pour
~ormerun c6mplexe contenant des
Ile n
![Fig ure ].: S p e c t r e i n f r a r 0 u 9 e du pol Y mè r e ex t rai t deI a réaction du CP2*UMeZ avec le phénylsllane](https://thumb-eu.123doks.com/thumbv2/123doknet/7670703.240061/50.918.101.843.65.1136/fig-ure-pol-mè-rai-réaction-umez-phénylsllane.webp)