The Structural Stability of P2-Layered Na-Based Electrodes during Anionic Redox
Texte intégral
Figure

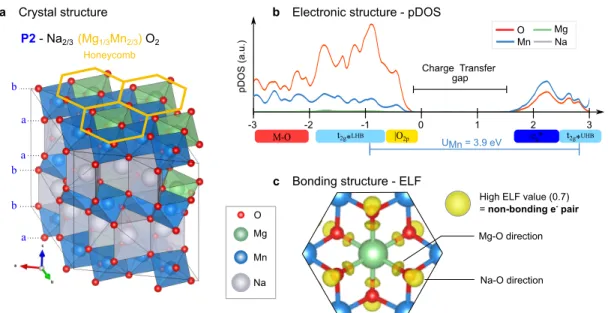
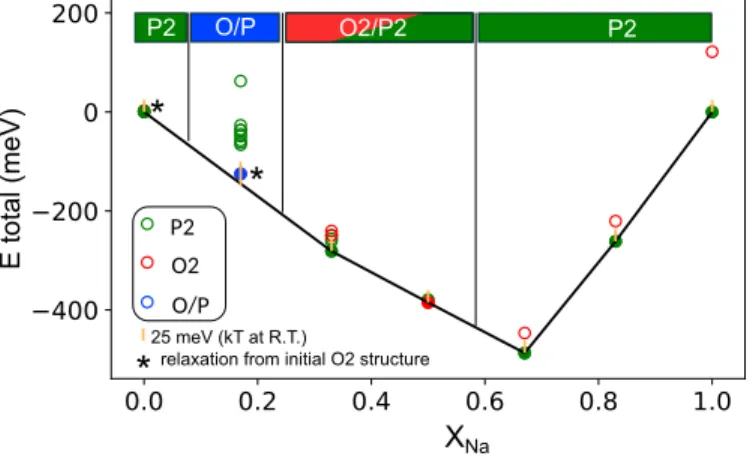
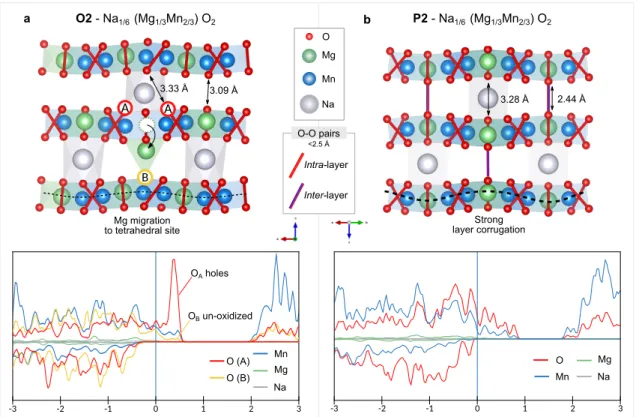
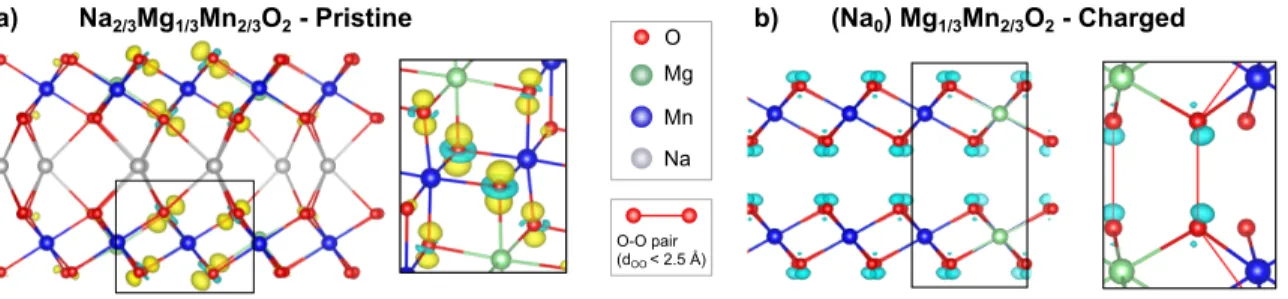



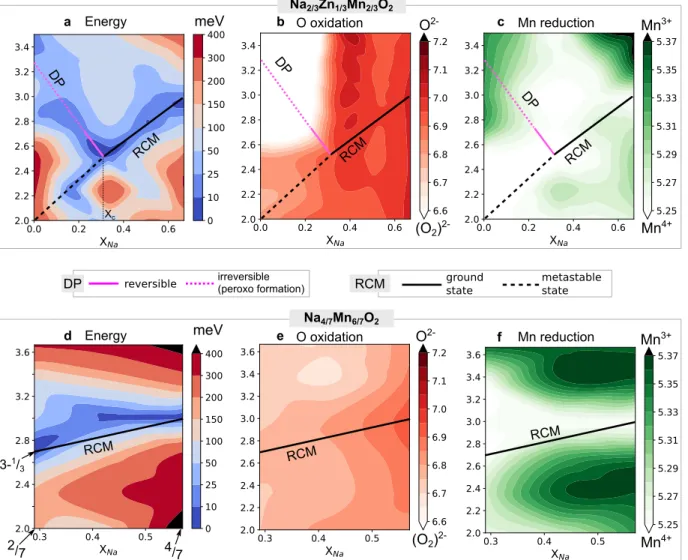
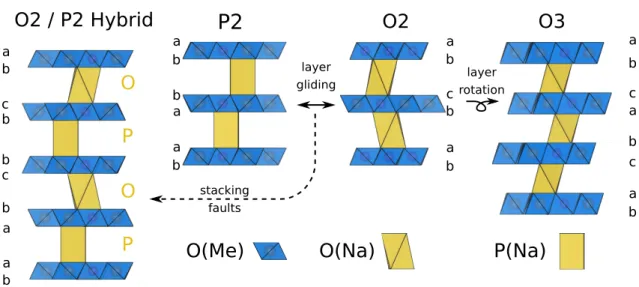
Documents relatifs
In sum, this book is an excellent introduction to the field of mathematical cognition that provides a succinct and clear overview of the most important concepts and findings.
This thesis demonstrates how Multi-Attribute Utility Theory (MAUT) can be extended to the group level to forecast future market shares by applying a distribution to
As a result, historical production quantity and inventory level is evaluated against the order replenishment lead-time and lead-time variability to measure the current
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Hermine Huot, Marie Buyse, Laurine Campanella, Ying-Yan Chen, Mei-Na Guo, Ya-Ying Li, Chang Liu, Wen-Shen Liu, Ye Liu, Claire Mariatte, et al. To cite
This first chapter described the principal aspects of the development of Li-ion batteries, from the beginning of intercalation chemistry in layered materials to
Finalement, une étude a été initiée en collaboration avec le SDI (Service de l’informatique cantonal) dans le but de trouver un logi- ciel permettant une gestion informatisée de
By applying state-changing techniques as a medium for material ac- tivation, we provide a framework for a two-part assembly process, starting from the manufacturing
