EXPLORATION DE LA CARTOGRAPHIE PAR
ANALYSE D'ASSOCIATION (« ASSOCIATION
MAPPING ») CHEZ L'ORGE (HORDEUM VULGARE L.)
Mémoire présenté
à la Faculté des études supérieures et postdoctorales de l'Université Laval dans le cadre du programme de maîtrise en biologie végétale
pour l'obtention du grade de maître es sciences (M.Sc.)
DEPARTEMENT DE PHYTOLOGIE
FACULTÉ DES SCIENCES DE L'AGRICULTURE ET DE L'ALIMENTATION UNIVERSITÉ LAVAL
QUÉBEC
2012
Mémoire cie fin d'études
Résumé
La cartographie par analyse d'association (« Association mapping »- AM) est une nouvelle approche prometteuse visant à identifier les déterminants génétiques (connus sous le nom de QTL) qui sous-tendent les caractères complexes chez les plantes cultivées. Malheureusement, les meilleures approches analytiques à employer pour limiter le nombre de faux positifs dans de telles analyses demeurent à déterminer pour chaque espèce. Le but de ce travail était d'explorer la cartographie AM chez une collection d'orges canadiennes. Diverses approches analytiques ont été comparées pour leur capacité à détecter des QTL simulés ou réels. La population utilisée comprenait 166 cultivars d'orge qui avaient préalablement été génotypes avec 866 marqueurs DArT (Zhang et al., 2009). Ces lignées ont également été génotypées pour sept locus SSR et ces données ont permis de simuler des QTL. La taille des plantes a été mesurée sur deux sites et deux ans pour fournir des données phénotypiques nécessaires à l'analyse des QTL contrôlant ce caractère. Des diverses approches analytiques, celle s'appuyant uniquement sur les relations de parenté entre les lignées (« kinship ») s'est avérée la meilleure. Cette approche a été employée pour identifier des QTL simulés (basés sur les SSR) ou réels. Les résultats nous ont montré que cette méthode était performante pour des caractères ayant une héritabilité modérée à élevée. De plus, les QTL que nous avons identifiés pour la taille des plantes étaient plus reproductibles que ce qui a été rapporté précédemment dans la littérature. Ces résultats suggèrent que la cartographie AM peut être appliquée avec succès dans un programme d'amélioration génétique, du moins pour certains caractères d'intérêt.
Abstract
Association mapping (AM) is a novel and promising approach to identify QTLs for complex traits in crop plants. Unfortunately, the best analytical approaches to be used in order to limit the number of false positives in such analyses remain to be determined for each species. The aim of this study was to explore AM in a collection of Canadian barley cultivars. Various analytical approaches were compared for their ability to detect simulated or real QTLs. The population we used was composed of 166 barley cultivars that had been previously genotyped with 866 DArT markers (Zhang et al, 2009) These cultivars were also genotyped at seven SSR loci, and these data were used to simulate QTL. Plant height was measured at two sites and two years to provide phenotypic data necessary for the analysis of QTLs controlling this trait. Of the tested analytical approaches, the model using genetic relationship among the lines ('kinship') as sole covariate proved to be the simplest and best approach. This model was used to identify simulated QTLs (based on SSR markers) and real QTLs (for plant height). The results indicated that AM could be used successfully to analyze traits with moderate to high heritability. Moreover, the QTLs identified for plant height were more reproducible than those have been previously reported in the literature. These results suggest that AM mapping can be applied successfully in a breeding program, at least for some traits of interest.
Mémoire de fin d'études
Avant-Propos
Je remercie bien évidement mon directeur de recherche, Dr François Belzile qui m'a accordé toute sa confiance en me proposant ce projet de recherche et pour son encadrement durant ces années d'étude ainsi que sa grande disponibilité tout au long du projet. Je le remercie en particulier pour son soutien financier qui m'a donné la deuxième opportunité à travailler encore dans son laboratoire et aussi qui m'a permis de mener à terme ce projet pour l'obtention d'un diplôme de maîtrise.
Un grand merci à Martine Jean, la «Madame wiki-dictionnaire» du labo, pour tous ses coups de main, tous ses conseils, de travaux au labo à l'analyse statistique et ainsi que sa disponibilité d'écoute en tout temps pour résoudre des problème qui m'ont permis aussi de repartir de l'avant lors de mes instants de difficulté.
Je remercie également Suzanne Marchand pour m'avoir initié à la culture de l'orge et à la prise de données sur la date d'épiaison. Merci tout particulier pour son sympathique cadeau d'origine québécois à chaque fois que j'effectuais un retour à mon pays.
J'aimerais exprimer toutes ma reconnaissance à Martin Lacroix et son équipe de stagiaires qui m'ont donné beaucoup de coups de mains dans le cadre du travail au champ pour la prise de données.
J'adresse un grand merci à tous mes amis au Québec et d'ailleurs, pour tous ces bons moments de franches rigolades avant, pendant et surtout après le boulot au cours de ces trois ans et demi passés au laboratoire : Thanh Tung Huynh, Mébakek Lamara, la famille d'Elmer Iquira, Maxime Bastien et Patricio Estives.
Je tiens à remercier tout spécialement au Ministère de l'Éducation et de la Formation du Viet Nam, pour son soutien financier pour mes trois années de maîtrise, de même que la Coop fédérée pour son soutien financier pour le projet de recherche.
J'exprime enfin, un merci tout particulier à mes parents pour leur soutien constant et leur générosité sans limite et mes plus tendres remerciements à ma petite famille, ma femme Anh Phuong et mes deux sages enfants, Anh Khoi et Julie. Anh Khoi, mon grand garçon, et Julie, ma plus belle fille, leurs présences m'avaient donné beaucoup de joies et d'énergie pour repartir lorsque le plus dur moment est survenu. Anh Phuong, ma belle femme traditionnelle, pour sa patience, sa compréhension et pour son support de mes deux enfants dans les moments difficiles.
Table des Matières
Résumé i Abstract ii Avant-Propos iii
Liste des tableaux f vi
Liste des figures vii
Annexe viii PARTIE I: INTRODUCTION GÉNÉRALE ix
1 INTRODUCTION GÉNÉRALE 1 PARTIE H: REVUE DE LITTÉRATURE.. 4 2.1 REVUE DE LITTÉRATURE 5 2.1.1 Cartographie conventionnelle 5 2.1.2 Cartographie par analyse d'association 6
2.1.3 Aspects techniques de la cartographie par analyse d'association 9
2.1.3.1 Taille de la population 9 2.1.3.2 Types et nombre de marqueurs 10
2.1.4 Analyses statistiques 14 2.1.5 Travaux antérieurs réalisés par le laboratoire 17
2.2 PROJET DE RECHERCHE 21 2.2.1 Hypothèse globale de la recherche 21
2.2.2 Objectifs de recherche 21 PARTIE III: MANUSCRIT 22
Résumé 24 Abstract 25 3.1 INTRODUCTION 26
3.2 MATERIALS AND METHODS 27
3.2.2 Genotypic data 28 3.2.3 QTL mapping on simulated and real data 28
3.2.3.1 Synthetic phénotypes based on SSR genotypes 28
3.1.1.1 AM with artificial phénotype 30 3.1.1.2 AM of plant height QTLs 30
3.2 RESULTS 31 3.2.1 Mapping synthetic QTLs 32
3.2.2 Mapping QTLs for plant height 35
3.3 DISCUSSION 37 3.3.1 Performance of different models used in AM 37
3.3.2 Performance of whole genome scans on simulated and simple phénotypes of
varying level of complexity 38 3.3.3 Whole genome scans for plant height 39
Mémoire de fin d'études
Reference 42 PARTIE IV : CONCLUSION GÉNÉRALE 46
3.4 CONCLUSION GÉNÉRALE 47
Bibliographie 51 Annexe 57
Liste des tableaux
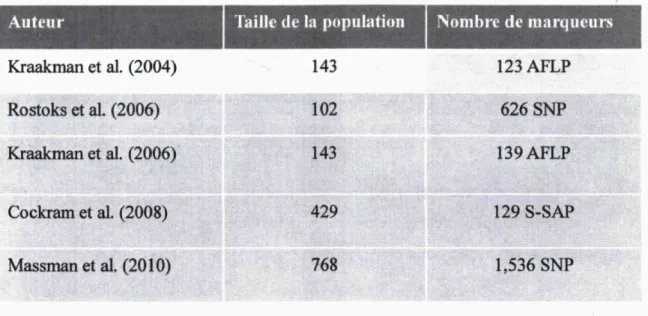
Tableau 1. Tailles des populations et nombre de marqueurs employés dans le cadre
de travaux de cartographie AM chez l'orge 9 Table 2. Results of simulated QTLs 33 Table 3. Plant height (PH) measured onl66 barley lines at two sites over two years. 35
Table 4. QTL for plant height 36 Table 5. Comparison of P-value 38
Mémoire de fin d'études
Liste des figures
Figure 1. Relation entre la puissance de détection et le « false discovery rate » (FDR)
en fonction de la taille de la population 10 Figure 2. Exemple de 'représentations génomiques' générées par le principe de la
technologie DArT. 12 Figure 3. La détection des marqueurs DArT (Wenzl et al., 2004) 13
Figure 4. Structure de la population au sein des orges canadiennes 18 Figure 5 Synthetic phénotypes based on two SSR genotypes 29 Figure 6. Comparison of different models for association mapping 32 Figure 7. Genome scans showing -log (p) for marker-trait associations 34 Figure 8. Genome scan showing -log (p) for marker associations with plant height. 36
Annexe
Mémoire de fin d'études
PARTIE I: INTRODUCTION GENERALE
1 INTRODUCTION GÉNÉRALE
L'orge commune (Hordeum vulgare L.) est une plante herbacée annuelle de la famille des Poacées domestiquée depuis plus de 10 000 ans (Crawford, 2003; Daniel and Maria, 2000; Pourkheirandish and Komatsuda, 2007). Il s'agit d'une espèce diploïde qui compte sept chromosomes (1H à 7H). Il existe des cartes génétiques denses de l'espèce dont une des plus récentes totalise plus de 3000 marqueurs distribués sur un peu plus de 1000 cM (Marcel et al., 2007; Mufioz-Amatriain et al., 2011; Thomas et al., 1995). La taille du génome a été évaluée à 5 Gb (Bennett et al., 1976) et des efforts sont en cours présentement pour sequencer complètement le génome de l'orge (Schulte et al, 2009).
L'orge est cultivée dans une centaine de pays et sa production se classait au quatrième rang mondial des cultures céréalières en 2010 (http://faostat.fao.org/). Le Canada est le plus grand producteur d'orge avec une production totale de 7,6 millions de tonnes et une superficie de 2,79 millions d'hectares en 2012 (Agriculture et Agroalimentaire Canada 2012). Les zones principales de culture se situent dans l'Ouest du pays (principalement le Manitoba et la Saskatchewan), mais l'orge représente tout de même une culture d'importance au Québec.
L'orge sert à diverses utilisations finales. L'orge brassicole est utilisée pour fabriquer des boissons comme la bière et le whisky, et sert comme édulcorant dans une variété d'aliments. L'orge sert à la fabrication de nouilles, de céréales pour petit déjeuner, et de préparations instantanées pour bébés. On l'utilise également comme composante importante de la moulée utilisée dans diverses productions animales (porcs, bovins et volailles). Au Québec, c'est son utilisation en alimentation animale, plutôt que brassicole ou pour l'alimentation humaine, qui motive le plus le choix de cette production.
En tant que culture importante, l'orge fait l'objet de divers efforts d'amélioration génétique afin de développer des cultivars qui répondent bien aux attentes des producteurs et des utilisateurs. Outre les efforts visant à augmenter le rendement, de grands efforts sont déployés pour améliorer la résistance génétique aux maladies. Dans ce contexte, la cartographie et la caractérisation moléculaire des locus fonctionnels sous-jacents à la
Mémoire de fin d'études
résistance aux maladies et aux caractères agronomiques permettraient l'utilisation de la sélection assistée par marqueurs dans les programmes d'amélioration variétale.
Malheureusement, la plupart des caractères agronomiques importants tels que le rendement, la tolérance aux stress abiotiques et la résistance à plusieurs maladies (incluant la fusariose), sont des caractères polygéniques complexes contrôlés par des locus de caractères quantitatifs (« Quantitative Trait Loci », QTL). Or, bien que plusieurs recherches aient focalisé leurs efforts pour cartographier de tels QTL, il est encore très difficile d'exploiter leurs résultats dans des programmes d'amélioration variétale et ce pour plusieurs raisons telles que l'utilisation de sources de résistance à faible potentiel agronomique comme parents des populations de cartographie, la faible résolution et reproductibilité des QTL identifiés, ainsi que l'utilisation de techniques de génotypage mal adaptées pour la cartographie à haute résolution.
Les marqueurs DArT (« Diversity Array Technology » permettent d'effectuer un génotypage à moyen débit chez l'orge (Wenzl et al., 2004). À l'aide de cette plateforme de génotypage, près de 1000 marqueurs DArT ont été utilisés pour étudier la diversité génétique et la structure de la population au sein d'une collection d'orges canadiennes (Zhang et al., 2009). Une telle évaluation systématique de la diversité des ressources génétiques de l'orge est une condition préalable à l'efficacité de son exploitation dans les programmes d'amélioration ainsi que pour le développement de stratégies pour une conservation optimale de la diversité génétique. De plus, la disponibilité d'un grand nombre de marqueurs tels que les SNP (Waugh et al., 2009; Yu et al., 2009; Zhu et al., 2008; Zhu and Yu, 2009) et les marqueurs DArT (Wenzl et al, 2004; Wenzl et al., 2006; Zhang et al, 2009) ouvre la porte à l'utilisation d'une nouvelle approche pour la cartographie à haute résolution des QTL, soit l'analyse d'association (« Association mapping », AM). L'analyse d'association est une nouvelle approche prometteuse pour identifier des QTL pour les caractères complexes chez des cultures comme l'orge (Oraguzie et al, 2007). Les premières études d'AM chez l'orge ont suggéré qu'elle pourrait être une approche valable pour identifier des caractères complexes tels que la résistance aux maladies, le rendement, et l'adaptation au stress (Caldwell et al., 2006; Choo et al., 2004b; Cockram et al., 2008; Kraakman et al., 2004; Rostocks et al., 2006).
Mon mémoire de maîtrise s'appuie sur ces avancées pour explorer la faisabilité de l'analyse d'association chez l'orge. Dans le cadre de ce mémoire, nous voulons évaluer l'impact de certains paramètres clés de cette approche de même que diverses approches analytiques sur des caractères progressivement plus complexes. Dans une seconde phase, nous souhaitons tirer profit de ces nouvelles connaissances pour tenter de réaliser la cartographie de caractères d'intérêt agronomique, comme la taille des plantes.
Mémoire de fin d'études
PARTIE II: REVUE DE LITTERATURE
2.1 REVUE DE LITTÉRATURE
La détermination de la base génétique des caractères d'importance économique est un objectif majeur de l'amélioration des plantes (Collard et al., 2005; Yu et al., 2006). L'étude génétique de ces caractères rencontre souvent des contraintes. En effet, des caractères quantitatifs complexes sont responsables de la majorité de la diversité génétique fonctionnelle d'intérêt dans les programmes de sélection (Collard et al., 2005; Kraakman et al., 2006; Kraakman et al., 2004). Ces caractères complexes sont contrôlés par des gènes multiples localisés en différents endroits dans le génome et dont l'expression est influencée par l'environnement. Leur analyse est donc largement accomplie en utilisant la cartographie QTL. Deux approches sont utilisées en cartographie QTL, soit la cartographie conventionnelle et la cartographie par analyse d'association (Collard et al, 2005).
2.1.1 Cartographie conventionnelle
La cartographie QTL conventionnelle s'appuie sur la force des associations entre les marqueurs génétiques dans une population en ségrégation développée en croisant deux parents ayant des valeurs phénotypiques contrastées pour un caractère d'intérêt (Doerge, 2002; Zhu et al., 2008). La cartographie conventionnelle est donc réalisée avec des populations F2, rétrocroisées, haploïdes doublées (HD), de lignées recombinantes autofécondées (« Recombinant Inbred Lines », RIL) et de lignées de substitution recombinantes (« Recombinant Substitution Lines », RSL) (Borém et al., 1999; Doerge, 2002; Flint-Garcia et al., 2003; Mackay and Powell, 2006; Thomas et al., 1995). La cartographie conventionnelle utilise des marqueurs polymorphes entre les génotypes parentaux pour identifier des marqueurs associés aux locus responsables de la variation de l'expression phénotypique du caractère évalué dans des environnements multiples (Collard et al., 2005).
Les deux principales approches de cartographie conventionnelle sont l'approche ponctuelle et la cartographie d'intervalle (Collard et al., 2005). L'approche ponctuelle mesure l'impact du QTL à l'endroit où se trouve chaque marqueur. Cette approche ne permet cependant pas de distinguer entre le cas d'un marqueur proche d'un QTL à faible impact et celui d'un
Mémoire de fin d'études
marqueur distant d'un QTL à fort impact. Par contre, la cartographie d'intervalle mesure
l'impact du QTL dans l'intervalle entre deux marqueurs. Pour ce faire, elle calcule le score
LOD (« Log of the odds »), c'est-à-dire le log du ratio de vraisemblance entre l'hypothèse
(HO) voulant qu'il n'y ait pas de QTL dans l'intervalle et l'hypothèse alternative (Hl)
voulant qu'il y en ait un, et estime un intervalle de confiance à l'aide d'une distribution de
probabilité (« P-value »).
Une des principales limites de la cartographie conventionnelle est qu'il n'y a que les QTL qui sont en ségrégation au sein d'un seul croisement qui puissent être localisés. De plus, la position des QTL est souvent d'un faible niveau de résolution à cause du nombre limité de recombinaisons pouvant être utilisés pour tester la liaison génétique entre les marqueurs et les caractères d'intérêt. Cette approche nécessite donc le phénotypage et génotypage d'un grand nombre de lignées pour accroître la résolution de la cartographie (Zhu et al., 2008).
2.1.2 Cartographie par analyse d'association
La cartographie QTL par analyse d'association constitue une stratégie alternative ou complémentaire à la cartographie QTL conventionnelle. Cette approche a tout d'abord été appliquée dans l'étude des maladies humaines et son utilisation a débuté récemment chez les plantes (Mackay and Powell, 2006). Elle offre trois avantages par rapport à la cartographie conventionnelle : i) augmenter la résolution de la cartographie, ii) réduire le temps de recherche, et iii) augmenter le nombre d'allé les utilisés (Flint-Garcia et al., 2003; Waugh et al., 2009; Yu and Buckler, 2006; Yu et al., 2008).
En effet, la cartographie par analyse d'association repose sur la caractérisation de lignées distinctes non-issues d'un croisement. Elle permet donc d'utiliser des collections déjà existantes de lignées caractérisés pour de nombreux phénotypes différents, et aussi de tirer profit des données historiques de phénotypes mesurés pendant le développement de nouvelles variétés (Waugh et al., 2009; Zhu et al., 2008). Elle permet ainsi d'exploiter une plus grande diversité allélique et bénéficie d'une plus grande résolution en raison de recombinaisons historiques plus nombreuses (Zhu et al., 2008).
La cartographie par analyse d'association se divise généralement en deux grandes approches: la recherche d'associations par balayage du génome entier (« Genome Wide Association », GWA) et l'approche par gènes candidats (Wilson et al., 2004; Zhu et al., 2008). L'approche par gènes candidats cherche à valider s'il existe réellement une association entre un caractère d'intérêt et certains gènes ou régions génomiques qu'on soupçonne de jouer un rôle dans l'expression de ce phénotype. Par contre, l'approche GWA examine tous les chromosomes pour identifier des régions reliées au caractère d'intérêt. Elle nécessite l'emploi d'un très grand nombre de marqueurs et la probabilité de succès dépend de l'étendue du déséquilibre de liaison.
La cartographie par analyse d'association s'appuie sur le déséquilibre de liaison (« Linkage disequilibrium », LD) pour identifier des associations entre les marqueurs et le caractère d'intérêt. Le LD correspond au degré d'association non aléatoire entre les alleles à différents locus. Plusieurs mesures peuvent être utilisées pour décrire le LD telles que le coefficient normalisé de déséquilibre (D') ou le carré de la corrélation allélique entre deux locus (r2) (Flint-Garcia et al., 2003; Gaut and Long, 2003; Lalouel, 1977; Slatkin, 2008;
Zhao et al, 2007a). Cependant, r2 serait la plus pertinente des mesures du LD pour
l'identification de marqueurs fortement associés à la variation d'un caractère phénotypique chez les plantes (Zhu et al. 2008). Des valeurs de r2 de 0,1 ou 0,2 sont souvent utilisées
comme seuil au-dessus duquel on conclut à la présence de LD (Cockram et al., 2008; Kraakman et al., 2004; Ramsay et al., 2000; Zhang et al., 2009).
L'étendue du LD génomique détermine la résolution et la densité de marqueurs nécessaires pour la cartographie par analyse d'association. Si le LD s'étend sur une courte distance, la résolution de la cartographie sera élevée, mais un très grand nombre de marqueurs sera requis (Gaut and Long, 2003; Lalouel, 1977; Zhu et al., 2008). Par contre, si le LD s'étend sur une longue distance, alors la résolution de la cartographie sera faible, mais un nombre plus restreint de marqueurs sera suffisant (Bradbury et al., 2007; Flint-Garcia et al., 2003). Par conséquent, une compréhension approfondie de l'étendue du LD génomique est une des clés de la conception et la réalisation de la cartographie par analyse d'associations.
Mémoire de fin d'études
Une première étude effectuée chez l'orge avait proposé que le LD s'étendait sur plus de 10 cM (Kraakman et al., 2004). Cependant, une seconde étude a semblé indiquer que le LD serait plutôt de 3.9 cM (Rostocks et al., 2006). En fait, les travaux de Cockram et al. (2008) ont révélé que le LD peut varier de 0,7 et 6 cM selon les régions du génome et les collections d'orge examinées. Enfin, selon Zhang et al. (2009), le LD au sein des lignées d'orge canadiennes se situerait entre 2 et 3 cM.
Cependant, l'étendue du LD est affectée par de nombreux facteurs tels que la fréquence allélique et le taux de recombinaison, la taille et la structure de la population, la mutation, le mode de reproduction et le métissage (Flint-Garcia et al., 2003). En effet, le mode de reproduction d'une espèce détermine en partie l'étendue du LD au sein d'une population diversifiée (Flint-Garcia et al. 2003). En règle générale, le LD s'étend sur une distance beaucoup plus grande chez les espèces autofécondées comme l'orge et le blé, que chez les espèces à pollinisation croisée, comme le maïs. De même, la présence de structure (subdivision) à l'intérieur de la population peut accroître le LD et ainsi conduire à la détection de fausses associations entre le génotype et le caractère d'intérêt. Des méthodes appropriées doivent donc être mises en œuvre pour tenir compte de telles structures de la population et augmenter la résolution lors de cartographie avec une population structurée (Falush et al, 2007; Flint-Garcia et al., 2003; Yu et al., 2006; Zhao et al., 2007b). Grâce à elles, des études récentes exploitant la cartographie par analyse d'association sur des populations structurées ont ainsi pu être réalisées malgré l'extension du LD sur plusieurs cM (Agrama and Eizenga, 2007; Cockram et al., 2008).
Récemment, la cartographie par analyse d'association a été utilisée pour étudier la génétique de certains caractères complexes chez des plantes tels que le riz (Iwata et al, 2007), le maïs (Yu and Buckler, 2006; Yu et al., 2008), Arabidopsis (Agrama and Eizenga, 2007; Zhao et al., 2007b) et l'orge (Choo et al., 2004b; Cockram et al., 2008; Haseneyer et al., 2009; Inostroza et al, 2009; Kraakman et al., 2006; Massman et al., 2010; Rostocks et al., 2006; Slatkin, 2008). Les études les plus récentes semblent indiquer qu'elle puisse être une approche valable pour identifier les QTL pour des caractères complexes tels que le rendement et l'adaptation aux stress et la résistance aux maladies (Caldwell et al., 2006; Kraakman et al., 2004; Rostocks et al., 2006; Wang et al., 2010; Waugh et al., 2009).
2.1.3 Aspects techniques de la cartographie par analyse d'association 2.1.3.1 Taille de la population
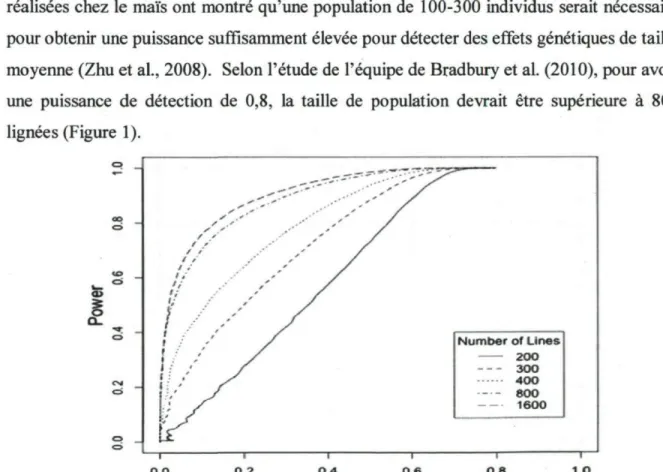
Comme en cartographie QTL conventionnelle, il serait théoriquement souhaitable de travailler avec de « grandes » populations pour maximiser la capacité de détecter des QTL dont l'effet est modeste. Les ressources, tant pour le phénotypage que le génotypage, n'étant pas infinies, on peut se demander quelle est la taille minimale permettant d'identifier des QTL utiles en sélection? Les travaux en cartographie par analyse d'association étant encore relativement peu nombreux, nous n'avons pas encore réponse à cette question. Jusqu'à présent, la taille des populations utilisées pour la cartographie par analyse d'associations est demeurée relativement faible. En effet, parmi 26 études recensées par Zhu et al. (2008), plus des deux tiers (19 sur 26) avaient été réalisées sur moins de 200 individus et la moitié (13 sur 26) sur moins de 100 individus. Chez l'orge, les quelques études publiées à ce jour comptent entre 100 et 800 lignées (Tableau 1). Des simulations réalisées chez le maïs ont montré qu'une population de 100-300 individus serait nécessaire pour obtenir une puissance suffisamment élevée pour détecter des effets génétiques de taille moyenne (Zhu et al., 2008). Selon l'étude de l'équipe de Bradbury et al. (2010), pour avoir une puissance de détection de 0,8, la taille de population devrait être supérieure à 800 lignées (Figure 1). a> % CL. C M . _» —-, _ " . " ^ * ^ . - ■ r\\\* — _ i ^ -— "T-* ■ "* " " *» *r *• ." " x f .* * s **■ . " * y * ." .' ' r * .' *" _.' / ' ' .-'* ** / * S •* * / - , ' ' /
, ' y /
! ■•' ■-' - ' / >• • / n ,' / 1; ' s _■ ' ' / ' ■ • • ' ' / Number of U n e s f / / 1 • ' / 200 1 •• ' / 300 t: é y 4 0 0 ':/ / 800 1 1 1 16O0 1 1 1 I I 0.0 0.2 0.4 0.6 0.8 L O F D RFigure l. Relation entre la puissance de détection et le « false discovery rate » (FDR) en fonction de la taille de la population employée (200, 300, 400, 800, and 1600) dans le cadre
Mémoire de fin d'études
Tableau 1. Tailles des populations et nombre de marqueurs employés dans le cadre de travaux de cartographie AM chez l'orge.
Kraakman et al . (2004) 143 123 AFLP Rostoks et al. (2006) 102 626 SNP Kraakman et al . (2006) 143 139 AFLP Cockram et al. Massman et al. (2008) (2010) 429 768 129 S-SAP 1,536 SNP
2.1.3.2 Types et nombre de marqueurs
Pour la cartographie par analyse d'association, un grand nombre (des centaines, voire des milliers) de marqueurs, sélectivement neutres et couvrant le génome entier, est nécessaire pour caractériser la composition génétique globale des individus. Le choix d'une technologie de génotypage dépend à la fois du nombre de marqueurs à tester et du nombre d'individus à génotyper (Kwok, 2000; Syvanen, 2005). Traditionnellement, les marqueurs SSR ont le plus souvent été choisis pour les études de cartographie QTL conventionnelle puisqu'ils sont des marqueurs multialléliques hautement polymorphes. Ceux-ci sont également très informatifs pour décrire la structure de la population au sein d'une collection de lignées non apparentées. Leur nombre relativement limité (quelques centaines) et le fait qu'ils ne se prêtent pas à des analyses hautement parallèles (« High throughput ») les rendent moins utiles cependant pour les analyses d'associations où, typiquement, le nombre de marqueurs souhaité est plus grand que dans une analyse QTL conventionnelle. Heureusement, depuis quelques années, deux nouvelles technologies permettant de caractériser un grand nombre de marqueurs en parallèle ont été mises au point chez l'orge. Il s'agit d'une part des puces de diversité ou « Diversity Array Technology » (DArT) (Wenzl et al. 2004) et des polymorphismes mononucléotidiques ou SNP (pour « Single Nucleotide Polymorphism ») (Waugh et al., 2009).
2.1.3.3 Les marqueurs SSR
Les marqueurs SSR (« Simple sequence repeats ») sont basés sur l'amplification PCR de microsatellites qui sont de courtes séquences d'ADN répétées en tandem. Bien que la fréquence relative des SSR chez les plantes soit considérée comme plus faible que chez les humains, des études effectuées chez l'orge suggèrent qu'il y a en moyenne un SSR à chaque 220 kb (Liu et al., 1996; Ramsay et al., 2000).
Les marqueurs SSR présentent plusieurs caractéristiques attendues d'un bon marqueur. C'est pourquoi ils sont fréquemment utilisés pour l'estimation de la structure de la population (Falush et al., 2007) et des relations de parenté entre les lignées (Zhu et al. 2008). De plus, la plupart des études de cartographie chez l'orge ont été effectuées avec ce type de marqueur (Choo et al., 2004b; Liu et al., 1996; Ramsay et al., 2000; Struss and Plieske, 1998; Varshney et al., 2007; Von Zitzewitz et al., 2005).
2.1.3.4 Les marqueurs DArT
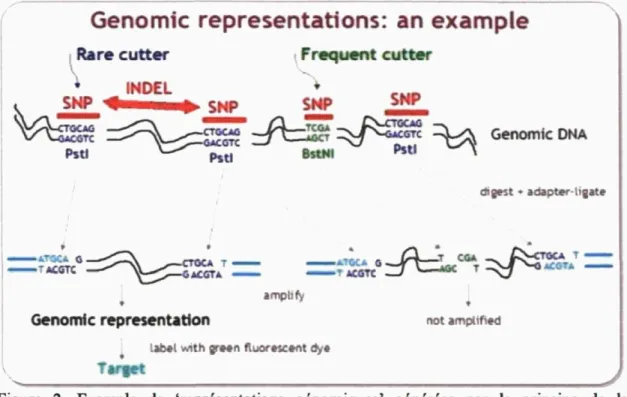
La technologie des puces de diversité (DArT) est une technologie basée sur l'hybridation de plusieurs fragments amplifiés par PCR sur des puces portant des milliers d'amplicons connus pour être polymorphes au sein d'une espèce. La technologie DArT repose sur la diminution de la complexité des échantillons d'ADN par l'obtention d'une 'représentation génomique' (Figure 2) de ces échantillons à l'aide d'une méthode combinant la digestion par des enzymes de restriction et la ligature d'adaptateurs permettant par la suite une amplification sélective de ces fragments (Wenzl et al., 2004).
:
Genomic representations: an example
Ra re cutter Frequent cutter
INDEL * SNP SNP Genomic DNA
A
P»tt > — ^ p ,t i BstHI " « ^ *gest • adapter-tigate -G_CCT_ —— TACfiTC — / *■"" nuT- ^ w amplifyGenomic representation not amplified
label with green fluorescent dye
* " * * _ _ _ .__> Figure 2. Exemple de 'représentations génomiques' générées par le principe de la
technologie DArT (Wenzl et al., 2004).
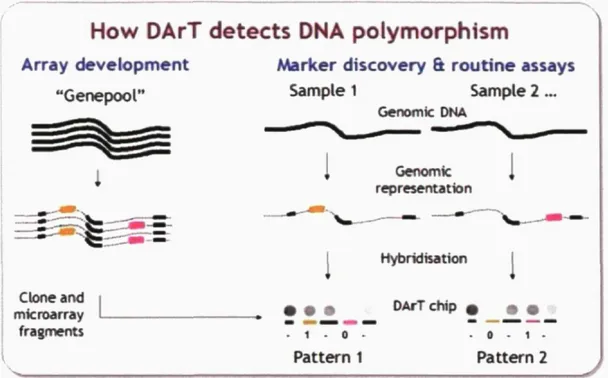
Cette représentation génomique contient deux types de fragments (Figure 3) : i) les fragments constants trouvés dans toutes les représentations préparées à partir des échantillons d'ADN, et ii) les fragments variables (polymorphes) appelés marqueurs DArT, que l'on retrouve dans certaines, mais pas toutes les représentations. Les fragments variables sont informatifs puisqu'ils reflètent des variations de séquence déterminant la fraction de l'ADN original qui sera compris dans la représentation Leur présence vs absence dans une 'représentation' génomique est examinée par hybridation de la représentation sur une puce d'ADN comportant un répertoire de marqueurs DArT présents chez cette espèce.
How DArT detects DNA polymorphism
Array development Marker discovery & routine assays
'Genepool' Sample 1 Sample 2
Genomic DNA Genomic J representation Hybridisation Clone and microarray fragments DArT chip 1 o -Pattern 1 o i -Pattern 2
Figure 3. La détection des marqueurs DArT (Wenzl et aL, 2004)
La technologie DArT permet maintenant d'obtenir rapidement et à un coût fort raisonnable (< 5000 $CDN) le génotypage de phis 2000 marqueurs sur un ensemble de 96 lignées d'orge par analyse (Wenzl et al., 2004). Afin d'intégrer l'information générée par la technologie DArT avec les données précédemment produites par les autres technologies de génotypage, une carte-consensus a été construite en combinant l'ensemble de données de dix populations dont la plupart avaient été génotypées simultanément par des marqueurs DArT, SSR, RFLP et/ou STS, SNP, OPA TDM, PERO, (Szucs et aL, 2009; Wenzl et aL, 2006).
2.1.3.5 Les marqueurs SNP
Les marqueurs SNP, comme leur nom le suggère, sont des différences d'une seule base dans la séquence d'ADN. Le polymorphisme de ce type est très abondant chez les plantes et des travaux récents chez l'orge suggèrent que de tels polymorphismes sont détectés à tous les 240 pb en moyenne (Duran et aL, 2009). Au-delà de leur grand nombre, c'est aussi l'existence de plateformes de génotypage SNP à haut débit qui justifie l'intérêt grandissant pour ces marqueurs. En ce moment, chez l'orge, la technologie Golden Gate d'Ilhimina 13
permet d'analyser jusqu'à deux jeux distincts de 1 536 marqueurs SNP sur 96 lignées à la fois (Waugh et aL, 2009)..
2.1.3.6 Le nombre de marqueurs à analyser
Le nombre de marqueurs employés dans les travaux réalisés à ce jour en cartographie par analyse d'association varie grandement. Parmi les 26 études relevées par Zhu et al. (2008), ce nombre variait entre 21 et près de 9000. Il faut noter cependant que dans le cas d'une analyse d'association à l'aide de gènes candidats, le nombre de marqueurs requis peut être relativement faible. Néanmoins, dans les cas où on souhaite pratiquer un balayage génomique (GWA), il serait évidemment souhaitable d'avoir un grand nombre de marqueurs également espacés. Bien que des analyses d'association aient été réalisées avec un peu plus d'une centaine de marqueurs chez l'orge (Kraakman et al. 2006; Cockram et al. 2008), Rostoks et al. (2006) ont conclu qu'il faudrait environ 1000 marqueurs ayant une bonne distribution pour assurer la couverture entière du génome chez l'orge. Pareillement, Zhang et al. (2009), ont estimé qu'en présence d'un LD s'étendant sur 2,6 cM en moyenne, près de 900 marqueurs DArT offraient une couverture assez complète (85 %) du génome.
2.1.4 Analyses statistiques
La cartographie QTL conventionnelle est bien développée et validée chez les espèces végétales (Doerge, 2002; Wei and Zeng, 2008; Zeng, 2005). En revanche, jusqu'à tout récemment peu d'efforts avaient été déployés pour développer la cartographie par analyse d'association chez les populations végétales. Dans une situation idéale, des analyses statistiques relativement simples seraient suffisantes pour déterminer si un marqueur est associé à un caractère ou phénotype. Par exemple, dans le cas d'un marqueur biallélique, un test de t permettrait de conclure si les individus qui possèdent un allele présentent un phénotype différent des individus qui possèdent l'autre allele. Malheureusement, l'existence d'une structure ou de relations familiales au sein de la population de cartographie peut générer de fausses associations marqueur-caractère, et des approches statistiques doivent être employées pour compenser pour ces facteurs. En effet, l'origine géographique, l'adaptation locale, l'histoire et le mode de reproduction font en sorte qu'on
retrouve habituellement à la fois une structure de la population et des relations de parenté au sein des collections de lignées pouvant être utilisées pour la cartographie par analyse d'association (Yu and Buckler, 2006). Or ces deux types de relations engendrent des faux positifs qui ont longtemps été considérés comme rendant impossible l'analyse d'association chez les plantes (Yu et al., 2008; Yu et al., 2006; Zhao et al, 2007b; Zhu et al., 2008). Heureusement plusieurs méthodes statistiques ont été proposées pour tenir compte des structures de population et des relations de parenté tels que le contrôle génomique (Devlin and Roeder, 1999), l'association structurée (Falush et al., 2007; Falush et al., 2003; Pritchard et al., 2000a), l'analyse en composantes principales (Price et al. 2006) et le modèle mixte unifié (MLM) (Yu et al. 2006). Ces approches utilisent l'information fournie par le génotypage de marqueurs répartis sur l'ensemble du génome pour décrire et tenir compte de la structure de la population ou des relations de parenté lors des analyses d'associations. Le modèle mixte unifié est typiquement utilisé pour tenir compte de la structure de la population et des relations de parenté car ils sont des meilleurs pour réduire l'effet de faux positifs (Waugh et al., 2009; Yu et al., 2009; Zhu et al., 2008). Des travaux chez Arabidopsis (Aranzana et al., 2005), le riz (Iwata et al., 2007) et aussi chez l'orge (Haseneyer et al., 2009; Massman et al., 2010) ont montré que le modèle mixte unifié était le mieux à même de réduire le nombre de faux positifs obtenus dans de telles analyses. Dans la plus récente étude chez l'orge, le modèle utilisant comme seule covariable les relations de parenté entre les lignées s'est avéré le meilleur (Bradbury et al., 2011).
Typiquement, dans un modèle mixte, il faut fournir au logiciel employé essentiellement quatre informations : le génotype de chaque individu, son phénotype, une description de la structure de la population sous forme de matrice (Q ou P, voir plus bas) et une description des relations entre les lignées (matrice K). Le logiciel TASSEL (Bradbury et al., 2007) est fréquemment employé pour réaliser de telles analyses et il permet, entre autres choses, de réaliser des analyses d'associations au moyen du modèle mixte de Yu et al. (2006 et 2009). Dans ce qui suit, nous nous pencherons brièvement sur les façons d'obtenir les matrices qui sont nécessaires et d'évaluer la vraisemblance des résultats d'une analyse d'association.
Mémoire de fin d'études
Le logiciel STRUCTURE est généralement utilisé pour produire la matrice Q (Pritchard et al., 2000a), laquelle correspond en fait à une matrice n x p, où n est le nombre de lignées et p est le nombre de sous-populations qui décrit le mieux l'organisation de la diversité génétique au sein de la population. L'analyse d'un ensemble de marqueurs aléatoires permet d'attribuer les individus à des sous-populations discrètes et d'estimer une matrice Q représentant la structure de la population. Cette matrice décrivant le degré d'appartenance des individus à chaque sous-population est ensuite utilisée lors du calcul des statistiques d'associations pour éviter des fausses associations causées par la structure de la population (Aldrich et al., 2008; Falush et al., 2003; Pritchard and Rosenberg, 1999; Pritchard et al, 2000a).
L'analyse en composantes principales (« Principal Component Analysis », PCA) est depuis longtemps utilisée dans les études de la diversité génétique et a récemment été proposée comme une façon rapide et efficace d'estimer la structure de la population pour les analyses d'associations (Price et al, 2006). Dans un tel contexte, le poids de chaque individu sur chaque composante principale est utilisé pour décrire l'appartenance des individus à des sous-populations. Ces données forment ce qui est appelé la matrice P. Le logiciel EINGENSTRAT permet d'estimer les composantes principales et de rectifier les statistiques utilisées pour l'analyse d'association. L'utilisation d'une matrice P en remplacement de la matrice Q dans une analyse basée sur le modèle mixte unifié a récemment permis d'obtenir des résultats prometteurs (Bradbury et al., 2011; Waugh et al., 2009; Weber et al., 2008; Zhao et al, 2007a).
2.1.4.2 Analyse des relations de parenté entre les lignées
Le logiciel SPAGeDi (Hardy and Vekemans, 2002) est souvent employé pour décrire les relations génétiques (« kinship ») entre les lignées et ainsi obtenir une matrice K. Le logiciel TASSEL intègre également un autre algorithme permettant de calculer une matrice K et nous sommes intéressés à comparer comment ces deux approches permettront de réduire le nombre de faux positifs.
2.1.4.3 Comment mesurer la vraisemblance d'une analyse d'association?
Un des grands défis de l'analyse d'association est de déterminer si les associations marqueur-caractère sont véridiques ou représentent plutôt des faux positifs. Divers outils ou approches sont utiles pour sonder cette question. Dans un premier temps, il est possible de réaliser des simulations. À titre d'exemple, Zhao et al. (2007) ont attribué des valeurs phénotypiques arbitraires à différents alleles de marqueurs SNP et ont ensuite déterminé laquelle des approches analytiques testées donnait la meilleure réponse, c'est-à-dire celle qui reflétait le mieux la situation qui avait été programmée. Sur des données expérimentales réelles, on peut également examiner la distribution cumulative des valeurs p. En théorie, cette fonction devrait être une droite entre le point 0 (aucune association n'a une valeur p=0) et 1 (toutes les associations ont une valeur p <1). Lorsqu'une analyse d'association ne tient pas suffisamment compte de la structure de la population ou des relations entre les lignées, il en résulte un excédent de faibles valeurs p (voir des exemples dans Zhao et al. 2007) qui sont potentiellement des faux positifs. Finalement, il peut être utile de comparer les QTL identifiés via une analyse d'association et ceux obtenus suite à une cartographie QTL conventionnelle. En cas de coïncidence, cela constitue un très bon argument en faveur de l'existence d'un QTL à cet endroit.
2.1.5 Travaux antérieurs réalisés par le laboratoire
Comme nous l'avons mentionné précédemment, la faible résolution de la cartographie QTL conventionnelle limite l'utilité de marqueurs liés aux QTL pour la sélection assistée de marqueurs et pour l'identification des gènes candidats pour ces QTL. Cette méthode nécessiterait une grande capacité de phénotypage et de génotypage pour générer les dizaines de milliers de données nécessaires pour compenser l'information limitée par le faible nombre de recombinaisons disponibles pour tester la liaison génétique entre les marqueurs et le caractère et accroître la résolution. C'est pour ces raisons que notre laboratoire est intéressé à explorer l'avenue de l'analyse d'association chez l'orge pour développer des marqueurs qui nous seront utiles dans le cadre de nos efforts de sélection.-Comme assise préalable à un tel travail, une étude a été réalisée afin d'explorer l'utilisation des marqueurs DArT chez une collection de 169 orges canadiennes représentatives de ce qui y est cultivé et employé dans les programmes de sélection (Zhang et al. 2009). Comme nous l'avons mentionné précédemment, cette étude a permis d'identifier 863 marqueurs
Mémoire de fin d'études
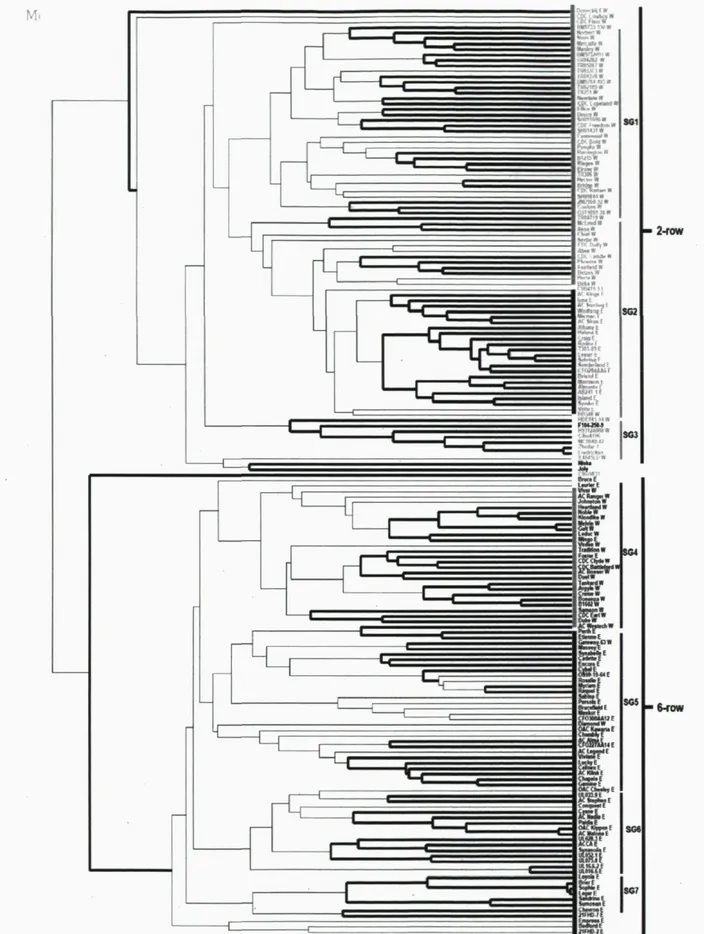
DArT polymorphes, de mesurer l'étendue moyenne du LD (2,6 cM) et de décrire la structure de la population au sein de cette collection (Figure 3). Une structure très marquée de la population a été notée en fonction du nombre de rangs de grains au sein de l'épi. À de très rares exceptions, les lignées à deux rangs formaient un groupe et les lignées à six rangs un second groupe bien démarqué du premier. Un groupement des lignées en fonction de leur origine géographique (est ou ouest du pays) était également évident et permettaient de définir des sous-groupes (SGI à 7).
- 2-fOW
- 6-row
Figure 4. Structure de la population au sein des orges canadiennes. Les 169 lignées forment deux groupes en fonction du nombre de rangs de grains (« 2-row » et « 6-row »). À un niveau plus fin, des sous-groupes (SGI à SG7) comprennent des lignées qui partagent une origine géographique commune. Tiré de Zhang et al, 2009.
Mémoire de fin d'études
Toujours dans le cadre de travaux antérieurs réalisés au sein du groupe et avec l'aide de plusieurs collaborateurs, des données phénotypiques ont été amassées pour cette collection de lignées. Une moitié des lignées a été caractérisée pour sa résistance à la fusariose (évaluée sur la base de la teneur en DON des grains) sur un total de quatre sites, sur deux ans à raison de trois répétitions par site. Pour l'autre moitié des lignées, nous possédons d'importantes données agronomiques historiques (rendement, poids spécifique, hauteur, date d'épiaison) de même que des données plus récentes découlant d'essais réalisés en 2007 et 2008 (huit sites, deux ans, deux répétitions par site). Par contre, aucun phénotype n'a encore été analysé sur l'ensemble des lignées dans le cadre d'essais orthogonaux, une situation que nous souhaitions corriger dans ce travail. Armé de ces données, nous avons réalisé des analyses d'association sur le caractère agronomique tel que la taille des plantes. Compte tenu de la structure très marquée de la population, nous estimons qu'il nous faudra être très prudents et méthodiques car, tel que mentionné précédemment, une structure de population marquée présente un risque accru d'identifier de fausses associations entre des marqueurs et le caractère à l'étude. C'est pourquoi le présent projet accorde une très grande importance à l'exploration de plusieurs paramètres ainsi que diverses approches analytiques afin de réaliser la cartographie de caractères complexes. De plus, aucune étude de ce type n'a encore été réalisée sur des orges canadiennes, le matériel qui est le plus employé dans nos travaux d'amélioration génétique.
2.2 PROJET DE RECHERCHE
2.2.1 Hypothèse globale de la recherche
La cartographie par analyse d'association (AM) avec les marqueurs DArT permet d'identifier des QTL pour des caractères polygéniques chez l'orge.
2.2.2 Objectifs de recherche
Le présent projet de recherche vise deux objectifs principaux correspondant aux deux volets suivants :
Volet 1 : Déterminer l'impact de différentes méthodes d'évaluation de la structure de la population et des relations de parenté entre les lignées sur la cartographie de locus génétiques simples à complexes.
Volet 2 : Réaliser une analyse d'association sur la taille des plantes et comparer les QTL ainsi identifiés aux QTL obtenus en cartographie conventionnelle.
Mémoire de fin d'études
PARTIE III: MANUSCRIT
Comparative analysis of different mixed linear models
for performing association mapping in Canadian barley
(Hordeum vulgare L.).
MANUSCRIT
C. Nguyen, M. Jean and F. Belzile.
Département de phytologie et Institut de Biologie Integrative et des Systèmes (IBIS), Université Laval, Québec (Qc) G1V 0A6.
Ce manuscrit a été rédigé dans le cadre d'un projet de maîtrise en biologie végétale. Il sera soumis sous peu à une revue avec comité de lecture.
Mémoire de fin d'études
Résumé
La cartographie par analyse d'association (AM) est une nouvelle approche prometteuse visant à identifier les déterminants génétiques (connus sous le nom de QTL) qui sous-tendent les caractères complexes chez les plantes cultivées. Malheureusement, les meilleures approches analytiques à employer pour limiter le nombre de faux positifs dans de telles analyses demeurent à déterminer pour chaque espèce. Le but de ce travail était d'explorer la cartographie AM chez une collection d'orges canadiennes. Diverses approches analytiques ont été comparées pour leur capacité à détecter des QTL simulés ou réels. La population utilisée comprenait 166 cultivars d'orge qui avaient préalablement été génotypes avec 866 marqueurs DArT (Zhang et al., 2009). Ces lignées ont également été génotypées pour sept locus SSR et ces données ont permis de simuler des QTL. La taille des plantes a été mesurée sur deux sites et deux ans pour fournir des données phénotypiques nécessaires à l'analyse des QTL contrôlant ce caractère. Des diverses approches analytiques, celle s'appuyant uniquement sur les relations de parenté entre les lignées (« kinship ») s'est avérée la meilleure. Cette approche a été employée pour identifier des QTL simulés (basés sur les SSR) ou réels. Les résultats nous ont montré que cette méthode était performante pour des caractères ayant une héritabilité modérée à élevée. De plus, les QTL que nous avons identifiés pour la taille des plantes étaient plus reproductibles que ce qui a été rapporté précédemment dans la littérature. Ces résultats suggèrent que la cartographie AM peut être appliquée avec succès dans un programme d'amélioration génétique, du moins pour certains caractères d'intérêt.
Abstract
Association mapping (AM) is a novel and promising approach to identify QTLs for complex traits in crop plants. Unfortunately, the best analytical approaches to be used in order to limit the number of false positives in such analyses remain to be determined for each species. The aim of this study was to explore AM in a collection of Canadian barley cultivars. Various analytical approaches were compared for their ability to detect simulated or real QTLs. The population we used was composed of 166 barley cultivars that had been previously genotyped with 866 DArT markers (Zhang et al., 2009) These cultivars were also genotyped at seven SSR loci, and these data were used to simulate QTL. Plant height was measured at two sites and two years to provide phenotypic data necessary for the analysis of QTLs controlling this trait. Of the tested analytical approaches, the model using genetic relationship among the lines ('kinship') as sole covariate proved to be the simplest and best approach. This model was used to identify simulated QTLs (based on SSR markers) and real QTLs (for plant height). The results indicated that AM could be used successfully to analyze traits with moderate to high heritability. Moreover, the QTLs identified for plant height were more reproducible than those previously reported in the literature. These results suggest that AM mapping can be applied successfully in a breeding program, at least for some traits of interest.
Mémoire de fin d'études
3.1 INTRODUCTION
Association mapping (AM) is a novel and promising approach to identify QTLs for complex traits in crop species such as barley (Oraguzie et al., 2007). This approach was first applied in the study of human diseases and it has recently been explored more widely in plants by several research groups around the world (Mackay and Powell, 2006). Using existing collections of germplasm already characterized for many different phénotypes with data typically produced during the development of new varieties, association mapping offers three advantages over conventional QTL mapping (Zhu et al., 2008): 1) an increase in the resolution of the map position of QTLs, 2) a reduction in the time required to identify QTLs, and 3) an increase in the number of loci and alleles that can be identified in a single study. The first such studies in barley suggested it might be a valid approach to identify QTLs for complex traits such as yield, disease resistance and adaptation to stress (Aghnoum et al, 2009; Caldwell et al., 2006; Kraakman et al., 2004; Massman et al., 2010; Rostocks et al, 2006) and yield, plant height (Inostroza et al., 2009).
Much remains to be learned, however, as to the required population size, marker density and the best analytical methods. In association mapping, a mixed linear model (MLM) is typically used to account for population structure and the relationship among the lines (kinship) in order to reduce the number of false positive associations between markers and traits (Waugh et al, 2009; Yu et al., 2009; Zhu et al., 2008). Population structure and relatedness among lines can lead to confounding effects and the identification of false associations simply because of the very large number of tests of association typically performed. Therefore, there have been several studies conducted to explore statistical methods to account for population structure and kinship (Devlin and Roeder, 1999; Falush et al., 2007; Falush et al, 2003; Pritchard et al., 2000a; Yu et al., 2006; Zhu et al, 2008; Zhu and Yu, 2009). These approaches use information provided by genotyping markers distributed throughout the genome to describe and account for population structure or kinship during the analysis. Work in Arabidopsis, a diploid and self-pollinated species like barley, showed that the full model MLM (combining kinship and structured association) was best able to reduce the number of false positives obtained in such analyses (Aranzana
et al., 2005; Zhao et al., 2007a). In recent work conducted on a collection of American barley fines, however, Bradbury et al. (2010) concluded that a simple MLM approach relying on kinship alone worked very well.
One of the challenges in comparing the efficiency of various approaches is that we need to know the "correct" answer in order to identify the analytical approach that yields the best approximation. Simulations using molecular markers as traits have long been used to compare different approaches or models in association mapping (Falush et al., 2007; Falush et al., 2003; Price et al., 2006; Pritchard et al., 2000a; Yu et al., 2006; Yu et al., 2009). In most such work done to date, biallelic markers (such as SNPs) have been used to genotype a collection of lines and a small subset of these markers are then used singly or in combination as "synthetic" QTLs. One possible pitfall in this approach is that it presumes that there can be only two alleles at any given QTL.
In a real-life situation, one could imagine that there are more than two alleles for a QTL within an association mapping population. Here, we use SSR data and real phenotypic data on plant height to: (i) test the efficacy of different methods for describing population structure and kinship; (ii) explore the capacity of a mixed linear model (MLM) approach to correctly map synthetic QTL of known location using simulated QTLs and real QTL (barley height).
3.2 MATERIALS AND METHODS
3.2.1 Plant material and phenotypic characterization
The population used in this work consisted of 166 barley cultivars/lines previously examined for linkage disequilibrium (LD) and population structure (Zhang et al., 2009). This collection is split almost equally between six-row (84) and two-row (82) lines and covers the main geographic areas where barley is grown in both eastern and western Canada (see table SI). As a test trait, plant height (PH) was measured in two orthogonal trials in 2009 and 2010 on two sites, St-Hyacinthe, Quebec (SH) and St-Augustin-de-Desmaures, Quebec (SA). The experimental design was a randomized complete block with two replications where each line was sown as a single 2m-row. Phenotypic summary
Mémoire de fin d'études
statistics were generated using SAS (SAS Institute 2002) including means, standard deviations and coefficients of determination (R2). The observed standard deviations were
used to calculate the heritability in simulation studies. Additional data on a subset of this population (64 lines, indicated in table SI) were obtained from previous work in which plant height was measured on 8 sites over two years (Kong et al., 1995).
3.2.2 Genotypic data
In addition to the 866 polymorphic DArT markers reported by Zhang et al. (2009), seven SSR loci were genotyped on this collection and used as traits in the simulation work described below. Young leaves (3-4 days) of each line were used for DNA extraction using the protocol suggested by Triticarte (http://www.triticarte.com.au). PCR for the SSR analysis was performed in a 10-ul reaction mixture containing: 1 uL of DNA, 0.5 pL (2.5 pM) of each SSR primer, 1 pL of dNTPs (2 mM), 0.2 pL (2 pM) of universal Ml 3 primer labeled with IDR700 and IRD800, 0.1 pL of Taq DNA polymerase and 0.15 pL Taq buffer supplied with the enzyme. DNA amplification was performed in a T Gradient thermocycler (Biometra, Gôttingen, Germany) in the following conditions: 1 cycle of 94°C for 3 min, 58°C for 1 min and 72 °C for 1 min; 30 cycles of 94°C for 45 sec, 55°C for 45 sec and 72°C for 45 sec; 1 cycle of 72°C for 5 min.
Amplified products were revealed on a 6.5% polyacrylamide gel and visualized on an automatic DNA analyzer (LI-COR Global Edition IR2 DNA Analyzer) set to 1,500 V
(temperature - 50°C) during lh45 to 2h30 (according to the size of PCR products). The genotype data analyzed by SAGA program was used to convert in the phénotype data for TASSEL analysis.
3.2.3 QTL mapping on simulated and real data
3.2.3.1 Synthetic phénotypes based on SSR genotypes
In the simplest test case (monogenic, biallelic), the two alleles of a biallelic SSR marker (WMC1E8, HVM67) were set to alter plant height by +/- 8 cm (Fig. 5). To introduce variable levels of heritability (h2=\, 0.75, and 0.5), the synthetic phénotype at each level of
heritability was created by adding the plant height standard deviation of each line with 1, 28
1.33 and 2 times respectively with the plant height after adjustment. In a second case (monogenic, multiallelic), phenotypic values were similarly generated for the alleles of multiallelic SSRs (Bmag0273a, Bmag0749, Bmag0718, HVM40 and Bmag0877) at the three heritability levels. Finally, in the last case (bigenic, multiallelic). the genotypes at both SSR loci were used to generate the synthetic phénotypes.
Figure 5. Synthetic phénotypes based on two SSR genotypes. A) The two alleles of a biallelic SSR marker (SSR1) were set to alter plant height by +/- 10 cm and +/- 5 cm for SSR2. B) The simulated height would be various depending on the synthetic phénotypes, varying from 85 to 115 cm. C) The QTL obtained were represented by the DArT markers near the SSR phénotype simulation.
3.1.1.1 AM with artificial phénotype
Association mapping using a mixed linear model approach as implemented in TASSEL was performed on the various synthetic phénotypes. Two analytical approaches were used to describe the population structure and one to estimate the relative kinship between the lines. Population structure was described with either a Q matrix generated by the software STRUCTURE v2.2 (Pritchard et al., 2000a) using 866 DArT markers and k = 2 (k value chosen based on results of Zhang et al. (2009) or using a P matrix generated by PCA analysis as implemented in the TASSEL 2.2. software (Bradbury et al. 2007). The marker-based relative kinship matrix was calculated on the basis of 866 DArT markers used to generate a K matrix with the appropriate module of the TASSEL software.
3.1.1.2 AM of plant height QTLs
QTL associated with PH were identified by a mixed linear model (MLM) approach as implemented in TASSEL with 866 DArT markers using only a kinship (K) matrix (Yu et al. 2006) generated within TASSEL for each data set. We used the parameters with the convergence criterion set at 1.0 x 10~10 and the maximum number of iterations set to 200.
To correct for multiple testing, we used the false discovery rate (FDR) method (Storey and Tibshirani, 2003) to choose the P-values based on the ^-values for each mapping analysis at FDR 10%.
3.2 RESULTS
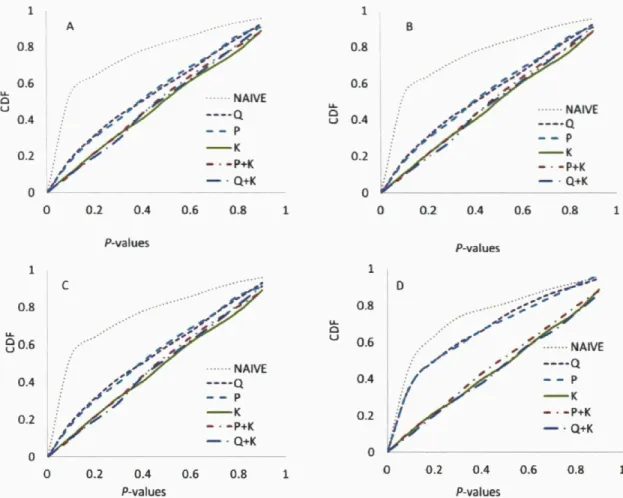
In a first step, we wanted to determine the best analytical approach to reduce the effects of population structure and kinship on our barley population. To this end, we compared the distribution of observed P-values for associations between the DArT markers and various simulated phénotypes as well as one real phénotype (plant height). We used two matrices (Q or P) to account for population structure and one matrix (K) to estimate relatedness among lines. These matrices were used singly or in combination to test a total of six different models. Figure 5 shows the cumulative distribution of P-values from analysis by AM scans across all genomic loci for all our simulated and real phénotypes. The colored lines correspond to several tests of association with/without correction for population structure and kinship. If these are well accounted for by the model, the cumulative distribution function (CDF) should be linear. For all of the phénotypes examined, in the absence of any correction (naive model), there was a large excess of associations with small P-values, suggesting a large number of false positives. When used alone, the P and Q matrices markedly reduced the excess of associations showing a low P-value, but did not result in a linear function. The models including the K matrix, alone or in combination with the Q and P matrices, proved the most effective in producing a linear relationship between the observed and predicted P-values. As the K matrix alone proved effective, it was used in all ensuing work.
1 0.8 0.6 0.4 0.2 0 A 4 r NAIVE Q - - p K
W
— Q-HC P+K 0.2 0.4 0.6 0.8 1 u °2 °4 °-6 P-values P-valuesFigure 6. Comparison of different models for association mapping. The cumulative distribution of P-values (CDF) under different simulated or true phénotypes using a single biallelic SSR (A), a single multiallelic SSR (6 alleles) (B), two SSR loci totaling 8 alleles (C) and plant height (D). The colored lines correspond to tests of association with or without (naive model) correction for population structure and kinship.
3.2.1 Mapping synthetic QTLs
To examine how well our association mapping approach and dataset could allow us to identify the loci underlying traits controlled by one or more loci with one or more alleles, using the K-only model, we first performed a set of simulations using the genotype at seven SSR loci as proxies for phénotype (see Materials and Methods) with three levels of heritability. Table 2 shows the results of these tests. At perfect heritability (h2 =1), DArT
markers closest to the SSR loci underlying the simulated phenotypic data were identified correctly in all seven cases where a single bi- or multiallelic SSR was used. Similar success was encountered for cases where 2 SSR loci were used to produce the simulated phénotype. 32
At h2 = 0.75, five of the seven single SSRs underlying the phénotype were identified
successfully. For simulations based on 2 SSR loci, when two biallelic SSR were used, both were correctly identified. When a biallelic and a multiallelic SSR were used, only the biallelic locus was identified as a QTL. At h2 = 0.5, only the biallelic SSRs were
successfully identified when used singly. In simulations with 2 SSR loci, the same partial success was obtained, as with h2 = 0.75.
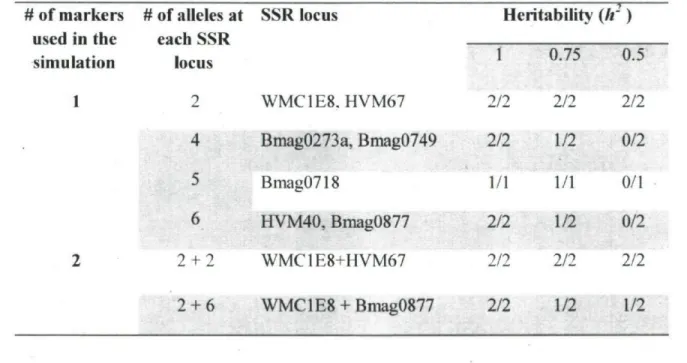
Table 2. Results of simulations using SSR loci as the genetic basis for phenotypic data. Biallelic and multiallelic SSR loci were used singly or jointly to simulate various levels of complexity at the loci underlying a trait. The success rate at three heritability levels is shown, with success meaning that the DArT marker(s) closest to the known position of the tested SSR(s) was significantly associated with the simulated trait.
# of markers # of alleles at SSR locus used in the each SSR
simulation locus 1 2 WMC1E8. HVM67 4 Bmag0273a, Bmag0749 5 Bmaa0718 Heritability (h ' ) 0.75 0.5 2/2 2/2 2/2 2/2 1/2 0/2 1/1 1/1 0/1 2/2 1/2 0/2 2/2 2/2 2/2 6 HVM40, Bmag0877 2 + 2 WMC1E8+HVM67 2 + 6 WMC1E8 + Bmag0877 2/2 1/2 1/2
The results of whole genome scans conducted on such simulated data are shown in Figure 7. In the case illustrated, the phenotypic data were simulated using two SSR loci with two (WMC1E8) alleles and six (Bmag0877) alleles at the three heritability levels (Fig. 7a, b and c). As shown in Fig. 7a, at h2 = 1, only the DArT markers closest to the two SSR loci used
in the simulation showed a significant association. At lower heritability levels (Fig. 7b and 7c), the DArT marker closest to WMC1E8 maintained a highly significant association, but the markers closest to the second SSR (Bmag0877) were no longer significantly associated with the simulated trait. False positive associations also started to appear, such as a DArT
l.OE-07 WMC1E8 (2)
i
l.OE-05 l.OE-Ol l.OE-07 l.OE-05 10E-03 10E-01 Bmag0877 (6) / WMC1E8(2) BJji
Bmag0877 (6) /(LiiiiUiiiA
1H 2H 3H 4H 5H 6H 7H h2 = 0.75 l.OE-07 l.OE-05 l.OE-03 l.OE-Ol WMC3JE8 (2) Bmag0877 (6)_k uikli _i_ u_i
1H 2H 3H 4H 5H 6H 7H
rV = 0.5
Figure 7. Genome scans along the seven chromosomes of barley showing -log (p) for marker-trait associations where the trait was simulated using data from two SSR loci totaling eight alleles and three heritability levels. A) h2 = 1, B) h2 = 0.75 and C) h2 = 0.5.
3.2.2 Mapping QTLs for plant height
We continued our evaluation of the association mapping approach by performing an analysis on a real trait, plant height. There were significant (P < 0.0001) differences among lines at the two locations in each of the two trial years (Table 3) with height varying from as little as 61 cm to as much as 117 cm. Within the trials, the data were highly correlated (R > 0.91) between the two replicates. Finally, plant height was lower and less variable among the two-row subset than among the six-row subset.
Table 3. Plant height measured on 166 barley lines at two sites over two years. For each test site and year, the mean height (± standard deviation) is indicated for the entire set of lines as well as for the subsets of six-row and two-row lines. The range of measured heights and the correlation between values observed in the two replicates at each site are also shown.
Site Year Mean height±s.d. (< mi) Range
(cm) R2 Whole population (166 lines) 6-row (84 lines) 2-row (82 lines) Range (cm) R2 St-Augustin 2009 87+10 88+10 85±09 62-117 0.95 2010 81±10 86±09 75±08 61-107 0.92 St-Hyacinthe 2009 90±07 92±06 88±07 63-113 0.94 2010 88±08 89±07 86±08 64-117 0.91
Using the same approach as described previously, we identified 4 chromosomal regions that were significantly associated with this trait. As illustrated in Figure 8 and summarized in Table 4, two QTLs (on 2H and 6H) were identified in three of the four environments (site-years), while two other QTLs (on 3H and 7H) were identified in two of the four environments. As variation for plant height was not equal among two-row and six-row lines (Table 3), we performed a similar analysis separately on these two subsets of lines. As shown in Table 4, among the six-row subset, 3 of the 4 QTLs for plant height detected among the entire population (2H, 3H and 7H) were also detected in at least one environment. Among the subset of two-row lines, none of the markers showed a significant association with plant height.
St-Augustin 2009
yiÉ_lLi_Mkl__iJJ'"*'il__
St-Hyacinthe2010
u
ijUJLiiLiiik
Figure 8. Genome scan showing -log (p ) for marker associations with plant height.
Table 4. QTLs for plant height detected in either the whole set of 166 lines or in a subset of six-row lines. Populatio n Chr • Positio n(cM) Marker P-value Populatio n Chr • Positio n(cM) Marker SA 2009 SA 2010 SH2009 SH 2010 2R+6R 2H 71.01 bPb-1836 3.0E-06 1.4E-04 0.0014 9.5E-06 2R+6R 3H 77.84 bPb-0079 0.001 1.1E-05 0.0069 2.2E-04 2R+6R 6H 73.44 bPb-8347 0.001 0.010 6.4E-05 1.5E-04 2R+6R 7H 106.93 bPb-4924 0.002 6.7E-04 0.007 6.1E-04 6R 2H 71.01 bPb-1836 4.3E-06 4.9E-04 0.002 0.044 6R 3H 77.84 bPb-0079 0.003 0.0012 0.006 9.5E-04 6R 6H 73.44 bPb-8347 ns ns ns ns 6R 7H 106.93 bPb-4924 0.002 0.0054 0.004 9.1E-04 36
3.3 DISCUSSION
3.3.1 Performance of différent models used in AM
Given the very strong population structure among this collection of Canadian barley varieties (Zhang et al., 2009), there was a potentially high risk of identifying false associations between markers and traits if an AM analysis was performed with this population. The first objective of this study was therefore to determine the best analytical approach to reduce the effects of population structure and kinship for AM analysis. Simulations based on SSR data and real phenotypic data on plant height were used to investigate the ability of two matrices (Q or P) to account for population structure and one matrix (K) to account for relatedness among lines.
Our results indicate that all models including a K matrix performed the best in accounting for population structure and kinship. Therefore, using a K-only model appears to represent an efficient and simple approach for AM in this collection. This is in contrast to what has been reported by some earlier studies in barley (Cockram et al., 2008; Massman et al., 2010; Ramsay et al., 2000; Rostocks et al., 2006) as well as other species such as rice (Agrama and Eizenga, 2007; Iwata et al., 2007), Arabidopsis (Zhao et al., 2007b) and maize (Yu et al., 2006) where it was found that the combined use of a kinship (K) and population structure matrix (Q or P) performed the best. Our results were in agreement, however, with the recent work of Bradbury et al. (2011) in a collection of North American barley where the authors concluded that the use of K-only model was better than the other MLM models. We hypothesized that in Canadian and North American barley populations, the kinship matrix probably more correctly modeled the true relationship than in the earlier studies in barley (Cockram et al., 2008; Massman et al., 2010; Ramsay et al, 2000; Rostocks et al., 2006) as well as other species such as rice (Agrama and Eizenga, 2007; Iwata et al., 2007), Arabidopsis (Zhao et al., 2007b) and maize (Yu et al., 2006). Indeed even the strong population structure found among our collection of Canadian barley varieties divided the population in only two main groups, corresponding to two-row and six-row barleys. Since these groups also correspond to mostly independent breeding pools, this may explain why the kinship matrix alone is sufficient to account for population structure.