T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Physiopathologie cellulaire, moléculaire et intégrée
JURY Dr Jean-François TANTI Pr Samy HADJADJ Dr Daniel METZGER Pr Philippe VALET Pr Remy BURCELIN Pr Pierre GOURDY
Ecole doctorale : Biologie, Santé et Biotechnologie Unité de recherche : INSERM U858
Directeur(s) de Thèse : Pr Pierre GOURDY, Pr Remy BURCELIN Rapporteurs : Dr Jean-François TANTI, Pr Samy HADJADJ
Présentée et soutenue par Elodie RIANT Le 10 Novembre 2009
Titre : EFFETS PROTECTEURS DES OESTROGENES SUR L’INSULINO-RESISTANCE ET LE
A Mikaël, C’est toi ma petite étoile…
Je tiens tout d’abord à remercier Messieurs les Professeurs Rémy Burcelin, Pierre Gourdy et Jean-François Arnal de m’avoir accueilli au sein de leurs équipes et de m’avoir permis de réaliser cette thèse.
Monsieur le Professeur Philippe Valet, merci de m’avoir fait l’honneur de présider la soutenance de cette thèse. Et Messieurs le Professeur Samy Hadjadj, Docteur Jean-François Tanti et Docteur Daniel Metzger, je suis très honorée de votre investissement dans la lecture et correction de cette thèse. Je vous prie de trouver ici le témoignage de ma reconnaissance et de mon profond respect.
Rémy et Pierre, je vous remercie pour la confiance que vous m’avez accordée et le soutien que vous m’avez apporté. Nos réunions du mercredi très enrichissantes m’ont beaucoup aidée.
Pierre, merci beaucoup pour ton soutien surtout ces derniers mois d’écriture pas toujours faciles. Je ne te remercierai jamais assez pour ton temps, ta patience et ton aide lors de mes moments de doute et de pleures. Merci encore.
Je voudrais bien sûr remercier l’ensemble des membres des DEUX !!! équipes qui ont rendu ces années très agréables et enrichissantes. Ça n’a pas toujours été facile d’être ainsi coupé en deux mais ce qui est sûr, c’est que j’ai eu la chance de rencontrer encore plus d’amis formidables.
Haude, tu es comme une petite sœur, adorable, toujours de bonne humeur et pleine d’entrain. Tu as été d’une très grande aide et je t’avoue que ce début d’année a été difficile sans toi… De toute façon, nous sommes quasi voisines et la maison est toujours ouverte…
Coralie, un grand merci à toi de m’avoir soutenue et encouragée…et trouvé les mots pour me réconforter.
Toutoune, nos discussions sont très agréables, nous avons beaucoup de points communs, je sais que tu comprends souvent ce que je ressens. Ah ! Au fait Jiss, merci de supporter toutes ces conversations de filles !!!!
Julie, il faudra que tu me donnes le secret de ta pêche et de ta bonne humeur.
Alexia, Amandine, vous m’avez manqué lors de cette journée, mais heureusement qu’il y a msn pour papoter et avoir vos idées culinaires qui font un tabac lors des apéros !!!
Aurélie et Elo, un immense merci pour cette journée Pretty Women qui m’a vraiment remonté le moral. Vous êtes des amies formidables !!
Anne, Sandra, courage les filles…
Je remerciement également Henrik, Françoise, Fred, Victo, Myriam, Matteo, Chantal, Sandrine, pour leurs conseils et leur gentillesse..
Et un grand merci à Dudu, Aurélie, Coco, Christian pour leur bonne humeur du midi qui change les idées le temps d’une petite pause !!.
Un grand merci à Lucie, une amie toujours présente malgré la distance. Fabien, Marielle, Emilie, Julien, merci d’être présent pour moi ainsi que mes beaux-parents sans oublier Tatie Mimi.
En dernier lieu, je remercie mes parents et ma famille de leur soutien inconditionnel et de leur amour qui m’ont permis d’aller jusqu’au bout.
SOMMAIRE
TABLE DES FIGURES 5
PRINCIPALES ABREVIATIONS 7
SITUATION DU SUJET 9
INTRODUCTION 11
Partie I : Oestrogènes et récepteurs des oestrogènes I. Les oestrogènes 13
II. Les récepteurs des oestrogènes 14
II.1. Le récepteur des oestrogènes α 14
II.1.1. Organisation du gène 14
II.1.2. Structure protéique 15
II.1.3. Modification post-traductionnelles 16
II.2. Le récepteur des oestrogènes β 16
II.2.1. Gène et structure protéique 16
II.2.2. Modifications post-traductionnelles 18
III. Les mécanismes d’action des récepteurs des oestrogènes 18
III.1. Le mécanisme classique : activité transcriptionnelle ERE dépendante 18
III.2. Activité transcriptionnelle ERE indépendante 19
III.3. L’activité initiée à la membrane 19
III.4. L’activité transcriptionnelle ligand-indépendante 21
Partie II : Oestrogènes et métabolisme glucidique I. Etudes épidémiologiques 23
II. Effets du traitement hormonal de la ménopause 24
III. Effets des xénoestrogènes 26
IV. Altération de la voie des oestrogènes : impact sur l’homéostasie glucidique 28
IV.1. Chez l’Homme 28
IV.1.1 Mutation de l’aromatase 28
IV.1.2. Mutation du ERα 29
IV.1.3. Polymorphisme du gène du ERα 29
Partie III : Physiologie de l’homéostasie glucidique et physiopathologie du diabète de type 2.
I. Physiologie de l’homéostasie glucidique 31
I.1. L’insuline et son récepteur 31
I.1.1. L’insuline 31
I.1.2. Le récepteur de l’insuline 33
I.1.3. Les voies de signalisation intracellulaire 34
I.2. L’Insuline et l’homéostasie glucidique 36
I.2.1. Action hépatique 36
I.2.2. Actions périphériques 37
I.2.2.1. Le muscle squelettique 37
I.2.2.2. Le tissu adipeux 38
II. Physiopathologie du diabète de type 2 38
II.1. Altérations de la sécrétion d’insuline 39
II.2. L’Insulinorésistance 40
II.2.1. Quelles conséquences pour les tissus cibles ? 41
II.2.1.1. Le foie 41
II.2.1.2. Le tissu adipeux 41
II.2.1.3. Le muscle squelettique 42
II.2.2. Quels sont les mécanismes impliqués ? 43
II.2.2.1. Rôle des acides gras libres 43
II.2.2.2. Rôle de l’inflammation 45
II.2.2.2.1. Les cellules de l’inflammation 47
II.2.2.2.2. Les effecteurs humoraux 49
II.2.2.3. La fonction endothéliale 52
III. Influence des oestrogènes sur la physiopathologie du diabète de type 2 54
III.1. Oestrogènes et insulino-résistance 54
III.1.1. Au niveau du foie 54
III.1.2. Au niveau du muscle squelettique 55
III.1.3. Au niveau du tissu adipeux 56
III.1.4. Implication de l’endothélium 57
III.2. Oestrogènes et insulino-sécrétion 58
III.2.1. Sécrétion de l’insuline 58
III.2.2. Prévention de la perte de la masse cellulaire β pancréatique 59
III.3. Oestrogènes et système nerveux central 60
III.4. Oestrogènes et réponses inflammatoires 63
III.4.1. Oestrogènes et immunité innée 63
RESULTATS EXPERIMENTAUX 67
RESULTATS EXPERIMENTAUX 1 69 I. Objectifs
II. Résultats
II. Discussion et perspectives
Résultats complémentaires 1 89 I. Introduction et Objectifs
II. Modèle expérimental et Méthodologie III. Résultats IV. Discussion Résultats complémentaires 2 97 I. Objectifs et Méthodes II. Résultats RESULTATS EXPERIMENTAUX 2 103 I. Introduction
II. Matériels et Méthodes III. Résultats
IV. Discussion
CONCLUSION ET PERSPECTIVES 131
TABLE DES FIGURES
Figure 1: Formules développées des oestrogènes naturels
Figure 2: Représentation schématique de l’organisation génique et protéique du ERα Figure 3: Structure du gène ESR2 et de la protéine ERβ
Figure 4: Mécanisme d’action des récepteurs aux oestrogènes
Figure 5: Prévalence du diabète traité selon l’âge et le sexe en 2007 en France, d’après les
données du régime général de la sécurité sociale
Figure 6 : Représentation schématique de la synthèse de l’insuline
Figure 7 : Représentation simplifiée de la structure secondaire du récepteur de l’insuline Figure 8 : Les voies de signalisation de l’insuline
Figure 9: Action de l’insuline dans le foie en condition physiologique
Figure 10 : Translocation du transporteur de glucose GLUT4 à la membrane en réponse à
l’insuline.
Figure 11: Action physiologique de l’insuline dans le muscle et le tissu adipeux
Figure 12: Evolution schématique des mécanismes physiopathologiques et de l’hyperglycémie au cours du diabète de type 2
Figure 13: L’inflammation et la résistance à l’insuline : voies de signalisation impliquées Figure 14 : Implication des cytokines pro-inflammatoires dans la dérégulation de la voie de
signalisation de l’insuline.
Figure 15: Hyperinsulinisme, résistance à l’insuline et dysfonction endothéliale
Figure 16: Hypothèses schématiques pour expliquer l’effet des oestrogènes sur le
métabolisme glucidique
Table 1: Effet du traitement de la ménopause (THM) sur l’incidence du diabète dans les
principales études d’intervention. (d’après Bonds et al., 2006 ; Margolis et al., 2004 ; Kanaya et al., 2003)
Table 2: Mesure des paramètres biologiques de base et après 1 an de traitement par
oestrogènes équins seuls ou placebo chez des femmes non diabétiques participant à l’étude WHI
PRINCIPALES ABREVIATIONS
ACC Acetyl-CoA Carboxylase
AF-1 / AF-2 Activation function ½
AGL Acide Gras Libre
AMPK AMP-activated protein kinase
Ar Aromatase
DBD DNA Binding Domain
E2 Oestradiol
ER α-β Sous-type alpha et bêta du récepteur des oestrogènes
ERE Estrogen Responsive Element
FAS Fatty Acid Synthase
GLUT4 Glucose Transporter 4
HERS Heart and Estrogen/progestin Replacement Study
HOMA-IR Homeostasis Model Assessment of Insulin Resistance
IRS Insulin Receptor Substrate
KO Knock Out
LBD Ligand Binding Domain
LPL LipoProtéine Lipase
LPS LipoPolySaccharides
NO Monoxyde d’Azote
OVX Ovariectomie
RER réticulum endoplasmique rugueux
SERM Selective Estrogen Receptor Modulator
Stat3 Signal transducer and activator of transcription 3
TNFα Tumor Necrosis Factor alpha
TZD Thiazolidinediones
SITUATION DU SUJET
Les modifications progressives du mode de vie favorisant la sédentarité et les excès caloriques, notamment la consommation d’aliments riches en glucides et en graisses, se traduisent par une progression épidémique de l’obésité et du diabète de type 2, en particulier dans les pays occidentaux. Le fait inquiétant est que la prise de poids représente un facteur de risque important pour la survenue des complications métaboliques telles que le diabète de type 2, mais aussi les affections vasculaires. Le diabète de type 2 est une pathologie multifactorielle impliquant une prédisposition génétique et des désordres métaboliques acquis, qui conduisent à la détérioration progressive de la sécrétion et de l’action de l’insuline. Ces anomalies de sécrétion et d’action de l’insuline peuvent être induites ou accélérées par des facteurs environnementaux tels que le changement de qualité, les excès alimentaires (Western diet), et la sédentarité. Ainsi, les facteurs environnementaux liés à l’hygiène de vie sont actuellement considérés comme un déterminant majeur et sociétal de la genèse de la prise pondérale, de l’insulino-résistance et de l’hyperglycémie.
Le diabète de type 2, anciennement nommé diabète non insulino-dépendant (DNID), est le diabète le plus fréquent (environ 90% des diabètes connus). Les coûts qu’il engendre posent un problème de santé publique important et croissant. En effet, la prévalence du diabète de type 2 augmente parallèlement au vieillissement des populations, à l’urbanisation, à la sédentarisation et au développement de l’obésité. De plus, cette prévalence est généralement sous estimée car l’hyperglycémique peut évoluer de façon insidieuse et silencieuse pendant de nombreuses années avant que le diagnostic ne soit porté.
En France, une étude récente, réalisée à partir des données du régime général de la sécurité sociale en 2007, a évalué le taux de prévalence du diabète traité à 3,95%, correspondant à 2,5 millions de personnes. Cette étude met également en lumière un taux de prévalence élevé à 5,7% après 60 ans et un accroissement des disparités géographiques (Kusnik-Joinville, 2008).
Les données épidémiologiques internationales concernant la prévalence du diabète de type 2 montrent des disparités importantes entre les différents pays et ethnies étudiés. Par contre, elles témoignent uniformément d'une augmentation considérable de sa fréquence dans
les pays industrialisés ou en voie de développement. Les prévisions à l'échelon mondial estiment que le nombre de sujets diabétiques de type 2 passera de 171 millions en 2000 à 366 millions en 2030 (Wild et al. 2004).
Dans ce contexte préoccupant, la mise en place de mesures diagnostiques, préventives et thérapeutiques efficaces représente donc un enjeu incontournable en termes de santé publique. En complément des modifications de l’hygiène de vie, il paraît donc indispensable d’identifier de nouvelles cibles moléculaires capables d’inhiber ou de limiter la progression de l’obésité et du syndrome métabolique. Un faisceau d’arguments épidémiologiques, cliniques et expérimentaux, accumulés au cours des dernières décennies, plaide en faveur d’un effet favorable des œstrogènes sur le plan pondéral et métabolique. Il paraît donc intéressant de comprendre comment les œstrogènes et leur voie de signalisation influencent le métabolisme glucidique, et dans quelles proportions ils sont capables de prévenir l’apparition du diabète de type 2, en particulier dans le cadre d’un stress nutritionnel.
Au cours de la première partie du manuscrit, je rappellerai en premier lieu les caractéristiques des oestrogènes et de leurs récepteurs. Ensuite, je reviendrai sur les arguments cliniques et épidémiologiques liant les oestrogènes au métabolisme glucidique et au diabète de type 2. Après un rappel sur la physiologie du métabolisme glucidique et la physiopathologie du diabète de type 2, j’évoquerai enfin l’influence des œstrogènes sur les mécanismes conduisant au diabète de type 2.
La seconde partie sera consacrée à l’exposé de mes travaux de recherche, comprenant un article publié en 2009 dans la revue Endocrinology et des résultats complémentaires consacrés à l’influence des oestrogènes sur le métabolisme basal et le développement du tissu adipeux dans des conditions expérimentales de régime hyperlipidique.
Partie I : Oestrogènes et récepteurs des œstrogènes.
I. Les œstrogènes
Les œstrogènes, hormones sexuelles femelles primaires, sont un groupe de dérivés stéroïdiens en C18 du cholestérol. Chez la femme, les trois œstrogènes naturels sont : le 17β-œstradiol (E2) (majoritaire), l’oestriol (E3), présent uniquement durant la gestation, et l’oestrone (E1) (Figure 1).
17β-oestradiol (E2) Oestriol (E3) Oestrone (E1)
Figure 1: Formules développées des oestrogènes naturels.
Le 17-β œstradiol est l’œstrogène majoritaire, dont la biosynthèse provient du métabolisme du cholestérol au niveau des cellules endocrines des ovaires en période d’activité génitale. Chez les femmes ménopausées, la synthèse d’E2 s’interrompt au niveau des ovaires, mais persiste au niveau des tissus périphériques, du fait de la conversion des androgènes par l’aromatase. Ces hormones favorisent le développement des caractères sexuels secondaires, et sont également impliquées dans le contrôle du cycle menstruel. Chez l’homme, l’E2 est également produit grâce à la conversion des androgènes par l’aromatase périphérique (Simpson et al. 2002), mais les concentrations circulantes sont beaucoup plus faibles que chez la femme. L’oestriol est sécrétée lors de la gestation, et son origine est essentiellement placentaire. Enfin, l’oestrone n’est détectée que dans le liquor folliculi durant la phase lutéale du cycle.
Outre ces molécules naturelles synthétisées dans l’espèce humaine, il existe d’autres composés possédant des analogies structurales et fonctionnelles avec les oestrogènes naturels. Parmi eux, nous retrouvons les phyto-oestrogènes qui sont des composés d’origine végétale.
Ces composés proviennent soit directement de la plante d’origine, soit sont ingérés sous forme de précurseurs qui sont ensuite métabolisés in vivo par la flore intestinale tels que les isoflavones, les lignans (comme l’entérolactone), les flavonoïdes ou encore les stilbènes (comme le resveratrol). Ils sont considérés comme des modulateurs sélectifs des récepteurs des œstrogènes (SERM). Les SERM sont des composés de synthèse dont la caractéristique principale est de posséder des activités mixtes antagonistes et/ou agonistes vis-à-vis des récepteurs des oestrogènes (ER), selon le tissu cible.
II. Les récepteurs des oestrogènes
Les ER appartiennent à la superfamille des récepteurs nucléaires et à la classe des récepteurs des hormones stéroïdes. L’hypothèse de l’existence d’un ER, émise à partir de l’observation d’une fixation d’E2 tritié sur des cellules utérines de rattes (Jensen 1962), n’a été confirmée que bien plus tard par le séquençage du cDNA puis le clonage de premier récepteur des oestrogènes (Walter et al. 1985; Green et al. 1986).
II.1. Le récepteur des oestrogènes α
II.1.1. Organisation du gène
Le gène du récepteur des œstrogènes α (ERα), nommé ESR1, est localisé sur le chromosome 6 q25.1 chez l’homme (Gosden et al. 1986; Menasce et al. 1993). Le gène ESR1 est constitué de 8 exons codants séparés par 7 introns, qui s’étendent sur environ 140 kB. L’exon 1 code pour le domaine A/B. Les exons 2 et 3 et une partie de l’exon 4 codent pour le domaine C. Le domaine D est codé par l’exon 4. Le domaine E est codé par une partie de l’exon 4, les exons 5, 6, 7 et enfin une partie de l’exon 8. L’exon 8, quant à lui, code pour le domaine F (Greene et al. 1986). La transcription du gène ESR1 peut conduire à la synthèse d’ARNm de tailles variables, due à l’utilisation de promoteurs multiples dans la région 5’. Cette région 5’ est la seule qui ne semble pas conservée entre les espèces et serait à l’origine de l’expression de variants de façon tissu-spécifique (Grandien et al. 1995; Kos et al. 2000). Ceci conduit à l’expression d’ARNm de séquence variable au niveau de la région 5’ non traduite, dont la plupart traduisent l’expression d’une protéine de pleine taille de 66 kDa (Reid et al. 2002). La mise en évidence d’un épissage alternatif de l’exon 1 a permis de montrer
l’existence d’une protéine tronquée de 46 kDa, où le domaine A/B est manquant (Flouriot et al. 2000).
II.1.2. Structure protéique
La protéine ERα de pleine taille est donc constituée des domaines A/B, C, D, E, F (Figure
2). Le domaine NH2 terminal A/B a pour principale caractéristique de posséder une fonction
de transactivation AF-1 (Kumar et al. 1987), impliquée dans des interactions protéines-protéines (McInerney et al. 1996; Webb et al. 1998), et initie les actions récepteur ligand-indépendantes mais aussi ligand-dépendantes. La région C correspond au domaine de liaison à l’ADN, caractérisée par la présence d’une séquence particulière permettant la fixation spécifique à des séquences d’ADN nommées ERE (Estrogen Responsive Element). Cette région est aussi impliquée dans la dimérisation du récepteur (Ruff et al. 2000). Le domaine D semble posséder une séquence signal de localisation nucléaire et peut être le siège de modifications post-traductionnelles (Wang et al. 2001). La région E contient le domaine de fixation du ligand, une seconde fonction de transactivation AF-2, un domaine d’homo ou d’hétérodimérisation, et une partie du signal de localisation nucléaire (Nilsson et al. 2001). Enfin la région F aurait un rôle dans la spécificité de reconnaissance de la liaison du récepteur à un ligand agoniste ou antagoniste (Nichols et al. 1998).
Figure 2: Représentation schématique de l’organisation génique et protéique du ERα (adapté de (Billon-Gales et al. 2009)).
II.1.3. Modifications post-traductionnelles
Le ERα peut être le siège de nombreuses modifications post-traductionnelles qui sont étroitement liées à ces différents types d’action. L’acétylation des résidus Lys 266 et Lys 268 augmente la capacité du récepteur à se fixer à l’ADN via le domaine C (Wang et al. 2001). La méthylation au niveau de l’Arginine 260 du domaine C a pour effet de localiser le récepteur des oestrogènes au niveau cytoplasmique (Le Romancer et al. 2008). La nitrosylation par le monoxyde d’azote (NO) de résidus Cystéine dans le domaine C induit une baisse de la capacité du récepteur à se lier à l’ADN (Garban et al. 2005). La palmitoylation sur le résidu Cystéine 447, dans la région E est la modification post-traductionnelle indispensable à la localisation membranaire du ERα et à son interaction avec la Cavéoline-1 (Ascenzi et al. 2006). On dénombre aussi de multiples sites de phosphorylation de ce récepteur sur les résidus Sérine 104, 106, 118, 167 dans le domaine A/B. Ces phosphorylations résultent de l’activation de différentes kinases comme Akt (Martin et al. 2000). La phosphorylation de la Ser 236 dans la région C par la Protéine Kinase A régule la dimérisation du récepteur (Chen et al. 1999). Au niveau du domaine E, la phosphorylation de la Tyr 537 par c-Src module sa capacité de liaison à l’ADN et sa dimérisation (Arnold et al. 1997). Les phosphorylations de la fonction AF-1 participent à l’activité transcriptionnelle du récepteur par le recrutement de co-activateurs. Enfin, l’ubiquitination du récepteur sur des résidus Lysine de la région C, plus précisément au niveau du site de liaison à l’ADN, permet la dégradation du récepteur, en absence de ligand, vers le protéasome.
II.2. Le récepteur des oestrogènes β
En 1996, un deuxième récepteur des oestrogènes a été cloné à partir de la prostate de rat et nommé ERβ (Kuiper et al. 1996). Les gènes du ERβ chez l’homme et la souris ont été clonés par la suite (Mosselman et al. 1996; Tremblay et al. 1997).
II.2.1. Gène et structure protéique
Le gène humain du ERβ (ESR2) est localisé sur le chromosome 14 en position q23.2 et sa taille est d’environ 63,2 kb. La protéine ERβ est produite à partir de huit exons espacés par sept introns, les exons 0N 0K en position 5’ et l’exon ox en position 3’ n’étant pas traduits
(figure 3). Un épissage alternatif peut intervenir entre l’exon 7 et l’exon ox afin de produire une isoforme ERβ2 (Kuiper et al. 1996). La protéine ERβ humaine est constituée de 530 acides aminés (Ogawa et al. 1998), alors que celle de souris en comprend 539 (Leygue et al. 1998). ERβ, en tant que membre de la superfamille des récepteurs nucléaires, possède des caractéristiques structurales communes aux autres membres de cette famille et plus particulièrement à ERα, à savoir cinq domaines distincts A/B, C, D, E, F. En comparant la séquence des protéines ERα et ERβ de pleines tailles, on remarque une forte conservation des acides aminés dans le domaine C (95% d’homologie). En revanche, pour les autres domaines, ce niveau de conservation n’est pas retrouvé : A/B (20%), D (30%), E (55%) et F (20%). Malgré ces différences de séquences, ces domaines conservent des fonctions analogues à celles décrites précédemment pour ERα. Il faut noter que les domaines de liaison du ligand du ERα et ERβ ont des structures tri-dimentionnelles très proches, mais les acides aminés reliant le ligand et la poche diffèrent en deux positions (Brzozowski et al. 1997). Enfin, la taille de la poche de ERβ est significativement plus petite, ce qui pourrait avoir de forte implication pour l’affinité et la pharmacologie des ligands. De façon classique, ERβ possèdent deux domaines de transactivation AF-1 et AF-2, situés respectivement dans les régions A/B et E (Figure 3).
Figure 3: Structure du gène ESR2 et de la protéine ERβ (Zhao et al., 2008).
Ok ON 1 2 3 4 5 6 7 8 ox A/B C D E F ATG TAG ESR2 RE -Activation Trancriptionelle -Localisation nucléaire -Dimérisation -Fixation à l’ADN -Fixation du ligand AF-1 AF-2 Ok ON 1 2 3 4 5 6 7 8 ox A/B C D E F ATG TAG ESR2 RE -Activation Trancriptionelle -Localisation nucléaire -Dimérisation -Fixation à l’ADN -Fixation du ligand AF-1 AF-2
II.2.2. Modifications post-traductionnelles.
Le ERβ, sans doute en raison d’une découverte plus tardive, a jusqu’ici été moins étudié que le ERα. Toutefois, certaines modifications post-traductionnelles ont été mises en évidence récemment. ERβ peut-être soumis à la palmitoylation nécessaire à sa localisation au niveau de la membrane plasmique (Galluzzo et al. 2007). Le ERβ peut également être phosphorylé au niveau de la fonction AF-1 par les MAPK, ce qui provoque le recrutement de SRC-1 (Steroid Receptor Coactivator-1) et induit une activation transcriptionnelle (Tremblay et al. 1997).
III. Les mécanismes d’action des récepteurs des oestrogènes
Après leur diffusion à travers la membrane plasmique, les oestrogènes peuvent activer la transcription de leurs gènes cibles par diverses voies de signalisation, génomiques ou bien extra-génomiques (Figure 4).
III.1. Le mécanisme classique
: activité transcriptionnelle ERE
dépendante
Les ER sont localisés dans le cytoplasme ou dans le noyau des cellules sous forme de monomères associés à des protéines chaperonnes HSP (Heat Shock Proteins), principalement HSP70 et HSP90 ou bien sous la forme de dimères libres. L’entrée du ligand par diffusion à travers la membrane plasmique et sa liaison au récepteur induit la dissociation des HSP du complexe. La conséquence est le repliement de ER et en particulier de l’hélice H12 du LDB menant à la dimérisation du récepteur de façon stable et enfin à sa translocation nucléaire (Shiau et al. 1998). Dans le noyau, il se fixe sur des séquences ERE (Estrogen Responsive Element) situées en amont des gènes cibles via le domaine de liaison à l’ADN. Cette fixation entraîne le recrutement des coactivateurs essentiels à l’activité transcriptionnelle, grâce aux fonctions de transactivation AF-1 et AF-2. Selon le gène et le type cellulaire concernés, ces fonctions de transactivation agissent en coopération ou de façon indépendante, et leurs activités sont modulées (Berry et al. 1990; Tzukerman et al. 1994).
III.2. Activité transcriptionnelle ERE-indépendante
Les récepteurs des oestrogènes sont capables de moduler l’expression de gènes qui ne possèdent pas de séquence ERE dans leur région promotrice, lors d’une activation du récepteur par son ligand. Une fois la translocation nucléaire des ER effectuée, ils sont capables d’interagir directement avec des facteurs de transcription actifs, qui sont fixés à la région promotrice des gènes cibles. Par exemple, ERα est capable d’interagir avec le facteur de transcription NF-κB (Nuclear Factor κB), et plus particulièrement avec la sous unité p65 (Galien et al. 1997). Dans ce cas, cette interaction préviendrait la liaison de NF-κB au niveau des séquences promotrices du gène de l’interleukines-6, inhibant ainsi sa transcription.
Par ce mécanisme, le ERα peut également jouer un rôle d’amplification de l’activité transcriptionnelle. Par exemple, il est capable d’interagir avec le facteur de transcription Sp1 qui favorise la fixation à l’ADN et le recrutement de co-activateurs. Cet effet a été observé dans le cas du gène codant pour le récepteur aux LDL (Low Density Lipoprotein) dont l’activation transcriptionnelle dépend de Sp1 (Li et al. 2001). Enfin, le ERα est capable de combiner des effets d’activation et de répression transcriptionnelle par l’interaction avec un même facteur de transcription, tel AP-1. En effet, il favorise la transcription du gène de l’IGF1 (Umayahara et al. 1997) et réprime celle du gène codant pour le TNFα (An et al. 1999), la transcription des ces deux gènes étant régulée par AP-1. Cependant, ces différents effets de l’E2 ont été décrits uniquement in vitro, et leur pertinence reste à démontrer in vivo.
III.3. L’activité initiée à la membrane
La première observation d’effets rapides des oestrogènes, montrant que l’ajout d’E2 sur des cellules de l’endomètre était suivi d’un influx de calcium intracellulaire immédiat, a été réalisée en 1967 (Szego et al. 1967). Ce type d’action des oestrogènes a également été révélé au niveau de cellules de l’hypophyse, induisant un influx de calcium intracellulaire dans la minute suivant l’exposition à l’E2 (Morley et al. 1992). Ces observations ne sont pas compatibles avec les mécanismes décrits précédemment, car l’activité transcriptionnelle des ER ne peut donner un effet biologique aussi rapidement. Par la suite, l’observation d’une sécrétion rapide de prolactine par les cellules tumorales de l’hypophyse suivant une stimulation par l’E2 a constitué une première piste révélant l’existence d’un ER membranaire, depuis longtemps suspectée. En effet, le marquage par un anticorps anti-ERα était positif au
niveau des membranes de ces cellules, qui n’étaient pourtant pas perméabilisées (Pappas et al. 1995).
Depuis, ces effets ont été également étudiés dans différents types cellulaires afin de caractériser les mécanismes moléculaires mis en jeu. Ainsi, l’E2 active rapidement la voie cAMP/PKA dans les neurones de l’hippocampe et dans les cellules musculaires lisses vasculaires par exemple (Farhat et al. 1996). Ces actions rapides de l’E2 sont aussi capables d’induire l’activation de la phospholipase C et de la PKCα, de produire un influx de calcium dans diverses populations cellulaires (Picotto et al. 1996; Perret et al. 2001). Les MAPK (Mitogen-activated protein kinase) sont quant à elles également activées par l’E2 dans certains types cellulaires, tout comme les ERKs (extracellular signal-regulated kinases) et la p38 (Chambliss et al. 2005). De plus, l’E2 peut diminuer l’activité de la MAPK phosphatase 1 (Filardo et al. 2002) permettant l’amplification des effets sur les ERK (Razandi et al. 2004). Enfin, il est montré que l’E2 active la voie des Pi3K/Akt (Levin 2005). La plupart des études font intervenir le ERα dans les effets rapides de l’E2. Ce récepteur serait par conséquent l’acteur principal de ces phénomènes.
Enfin, plus récemment, ERα a été montré comme étant capable de recruter des membres de la famille des petites protéines G et ainsi d’activer des voies de signalisation impliquant la protéine kinase A (PKA) ou la protéine kinase C (PKC), ou encore d’agir sur des canaux calciques intracellulaires (Zhang et al. 2006). Un récepteur membranaire des oestrogènes, appelé GPR30, a également été récemment identifié (Filardo et al. 2002;Revankar et al. 2005). Ce récepteur appartiendrait à la famille des récepteurs couplés aux protéines G et agirait au niveau de la membrane plasmique (Funakoshi et al. 2006). GPR30, provoquerait alors la modulation de cascades de signalisation comme celle de la PKA (Filardo et al. 2002). Il serait impliqué dans certains effets non génomiques, notamment au niveau du système nerveux central (Brailoiu et al. 2007; Jacobi et al. 2007). L’étude de certaines souris déficientes en GPR30 à mis en évidence une tendance l’hyperglycémie et à l’intolérance au glucose chez ces animaux (Martensson et al. 2009). Toutefois, une revue récente décortiquant chacun des modèles transgéniques disponibles met en lumière de sérieuses discordances et suggère que la liaison de l’oestradiol à GPR30 n’est pas assez forte pour considérer cette molécule comme un troisième récepteur aux oestrogènes. Les phénotypes de ces différents modèles murins sont manisfestement influencés par des
paramètres qui ne sont pas toujours exclusivement liés à l'inactivation de GPR30 (Langer et
al., 2009 sous presse).
III.4. L’activité transcriptionnelle ligand-indépendante
L’activation de certaines voies de signalisation peut moduler l’activité du ERα, en absence de ligand, grâce à des phosphorylations. Par exemple, les facteurs de croissance sont considérés comme des activateurs de ce récepteur. En effet, l’Epithélial Growth Factor (EGF) est capable de mimer les effets biologiques de l’E2 sur le système reproducteur (Nelson et al. 1991). L’effet de l’EGF est fortement inhibé en présence d’un antagoniste pur du ERα (Curtis et al. 1996). De plus, des travaux ont montré que l’activation du récepteur à l’EGF (EGF-R) induit la phosphorylation de la Ser 118 localisée au niveau de la fonction AF-1 de ERα par les MAPK ERK1 et ERK2 dans les cellules cancéreuses (Bunone et al. 1996). Bien qu’étant relayée par la fonction AF-1 dans les cellules vasculaires, endothéliales et musculaires lisses, cette activation du ERα par l’EGF n’implique ni la voie des MAPK, ni la Ser 118 (Karas et al. 1998). Par ailleurs, d’autres facteurs de croissance tels que l’insuline (Patrone et al. 1998), l’Insulin Growth Factor 1 (IGF-1) (Ma et al. 1994) peuvent activer le ERα. En effet, l’activation du ERα par l’insuline dans la lignée neuronale SK-N-BE, est initiée par l’activation de la fonction AF-2, et non de la fonction AF-1 (Patrone et al. 1998), considérée comme le site préférentiel de phosphorylation des kinases induites par les facteurs de croissance.
Figure 4 : Mécanisme d’action des récepteurs aux oestrogènes.
1- Mécanisme d’action classique (direct) : après activation par le ligand, les dimères de récepteurs se lient à l’ADN au niveau de séquences spécifiques (ERE).
2- Mécanisme d’action ERE indépendant (indirect) : les dimères de récepteurs se lient à l’ADN via des intéractions protéiques.
3- Mécanisme d’action ligand-indépendant : des facteurs de croissance activent des kinases qui phosphorylent les récepteurs et se fixent à l’ADN au niveau des séquences spécifiques ERE.
4- Mécanisme d’action non-génomique : les récepteurs localisés à la membrane activent des kinases, conduisant à des modifications rapides de protéines cytoplasmiques ou bien à des régulations transcriptionnelles.
Partie II : Oestrogènes et métabolisme glucidique
Un faisceau d’arguments épidémiologiques, cliniques et expérimentaux, accumulés au cours des dernières décennies, plaide en faveur d’un effet bénéfique des oestrogènes sur le métabolisme glucidique, et plus précisément vis-à-vis du risque de survenue du diabète de type 2. Cependant, les mécanismes responsables de cet effet métabolique bénéfique restent encore flous.
I. Données épidémiologiques
La prévalence du diabète de type 2 et du syndrome métabolique, comme la survenue d’évènements cardio-vasculaires, est moindre chez la femme que chez l’homme dans les tranches d’âge moyen (35-64 ans) (Balkau et al. 2007). Ainsi, selon l’étude de population MONICA, incluant des sujets choisis au hasard sur la listes électorales dans trois régions françaises, la prévalence ajustée du diabète de type 2 dans la population française d’âge moyen en 1997, était de 5,1% chez les femmes et de 7,3% chez les hommes et celle de l’hyperglycémie à jeun respectivement atteignait 5,2% et 11,8% (Gourdy et al. 2001). Toujours en France, mais en 2007, selon une étude réalisée à partir des données du régime général d’assurance maladie, les hommes présentaient, à partir de 40 ans, un risque plus élevé (+ 44,5%) d’être diabétique que les femmes. Ce sur-risque masculin était maximal (+ 59,3%) pour la classe d’âge 60-64 ans (Kusnik-Joinville et al. 2008) (Figure 5).
Parmi les raisons évoquées pour expliquer ces discordances entre les hommes et les femmes, l’influence des hormones stéroïdes sexuelles sur la répartition des masses adipeuses, et plus généralement sur la sensibilité à l’insuline, pourrait jouer un rôle déterminant (Gale et al. 2001). Dans ce contexte, il a été montré que les femmes en période d’activité génitale présentaient une meilleure sensibilité à l’insuline (environ 41%) que les hommes d’âge comparable et en bonne santé (Nuutila et al. 1995; Mittendorfer 2005). De plus, au moment de la ménopause, les femmes connaissent fréquemment une prise de poids et une modification de la répartition de la masse adipeuse, conduisant à l’accumulation de tissu adipeux au niveau abdominal et péri-viscéral. Comme nous le verrons ensuite, les conséquences fonctionnelles de cette répartition « androïde » de la masse grasse s’avèrent délétères en termes de sensibilité à l’insuline et de tolérance au glucose.
Figure 5: Prévalence du diabète traité selon l’âge et le sexe en 2007 en France, d’après les données du régime général de la sécurité sociale (Kusnik-Joinville, 2008).
II. Effets du traitement hormonal de la ménopause
Plusieurs travaux cliniques, principalement menés chez des femmes ménopausées non diabétiques, ont suggéré que l’administration chronique d’un traitement hormonal de la ménopause exerce des effets favorables sur l'homéostasie glucidique et la composition corporelle, et améliore la sensibilité à l’insuline, sur la base des valeurs d’insulinémie à jeun (Espeland et al. 1998). Cependant, d'autres essais ont conclu à l’absence de bénéfice, ou même à une altération de la tolérance au glucose (Vehkavaara et al. 2000). Toutefois, l’interprétation de ces travaux doit rester prudente compte tenu de disparités méthodologiques importantes, tant sur le profil des femmes (en particulier leur âge et l’ancienneté de la ménopause) que sur la taille des effectifs, le type et la durée du traitement hormonal, et les critères de jugement biologiques utilisés. En revanche, les résultats de deux grandes études d’intervention américaines de grande envergure, randomisées et réalisées en double aveugle, permettent d’affirmer l’effet favorable du traitement hormonal de la ménopause vis-à-vis de la survenue d’un diabète. Ainsi, dans les études WHI (Women’s Health Initiative) et HERS
(Heart and Estrogen/progestin Replacement Study), l’administration d’oestrogènes équins couplés à de l’acétate de médroxyprogestérone, a permis une réduction de 21% à 35% de l’incidence du diabète chez les femmes sous traitement par rapport à celles recevant un placebo (Table 1) (Bonds et al. 2006; Margolis et al. 2004; Kanaya et al. 2003; Salpeter et al. 2006). Chez les femmes hystérectomisées participant à l’étude WHI, l’administration d’oestrogènes équins seuls s’est quant à elle soldée par une réduction non significative de 12% de l’incidence du diabète (Table 1). Cependant, dans un sous-groupe de patientes, des mesures biologiques standardisées ont mis en évidence une réduction significative des valeurs glycémiques et insulinémiques à jeun, et donc une amélioration significative de l’index HOMA-IR (Table 2).
Placebo THM HR 95% CI P HERS (E+P) 9,5 % (n=1030) 6,2 % (n=999) 0,65 (0,48-0,89) 0,006
WHI (E+P) 4,2 % (n=7627) 3,5 % (n=8014) 0,79 (0,67-0,93) 0,004
WHI (E) 9,3 % (n=4906) 8,3 % (n=4806)) 0,88 (0,77-1,01) 0,072
Table 1: Effet du traitement de la ménopause (THM) sur l’incidence du diabète (E+P : œstrogène + medroxyprogestérone ; E : œstrogène seul, n : effectifs) dans les principales études d’intervention. (d’après Bonds et al., 2006 ; Margolis et al., 2004 ; Kanaya et al., 2003)
Oestrogènes seuls Placebo Différence p
Moyenne DS Moyenne DS Moyenne ES Glucose (mmol/l) Niveau Basal 5,42 0,97 5,42 0,82 0,00 0,07 1,00 Evolution à 1 an -0,22 0,74 0,01 0,56 -0,24 0,06 <0,01 Insuline (mU/l) Niveau Basal 11,98 7,75 12,46 7,05 -0,47 0,54 0,38 Evolution à 1 an -1,64 4,91 0,36 4,04 -2,00 0,48 <0,01 HOMA-IR Niveau Basal 2,94 2,12 3,11 2,15 -0,17 0,15 0,26 Evolution à 1 an -0,52 1,50 0,12 1,29 -0,63 0,14 <0,01
Table 2: Mesure des paramètres biologiques de base et après 1 an de traitement par oestrogènes équins seuls ou placebo chez des femmes non diabétiques participant à
Dans la population française, alors que les schémas thérapeutiques prescris diffèrent le plus souvent de ceux utilisés dans les études américaines, les données issues de la cohorte E3N (1993) ont également récemment suggéré que la prise d’un traitement hormonal de la ménopause, notamment les combinaison oestro-progestatives par voie orale, est associée à un risque plus faible de développer un diabète, par comparaison aux femmes ménopausées non traitées (de Lauzon-Guillain et al. 2009).
III. Effets des xénoestrogènes
Les xénoestrogènes, perturbateurs endocriniens, sont des substances environnementales, naturelles (phytoestrogènes) ou synthétisées par l'homme, qui exercent une activité estrogénique d’intensité variable.
Au cours des dernières années, différents travaux ont également mis en évidence l’effet bénéfique de certains phytoestrogènes alimentaires vis-à-vis du risque d’obésité et de diabète. En effet, des données épidémiologiques et des études d'intervention nutritionnelle chez l'homme, comme dans des modèles animaux, suggèrent que les phytoestrogènes pourraient prévenir les symptômes de la ménopause et l’incidence de certaines maladies cardio-vasculaires, ainsi que les anomalies lipidiques ou encore l'ostéoporose. Plusieurs études ont ainsi montré que les protéines contenues dans le soja réduisent le taux de cholestérol plasmatique total et le LDL cholestérol (Beynen et al. 1990; Prasad 1999). Il apparaît également que la consommation de suppléments ou d'aliments riches en phytoestrogènes aurait un effet bénéfique sur le diabète et l'obésité (Bhathena et al. 2002). L’étude clinique randomisée, en double aveugle, rapportée par l’équipe de Jayagopal en 2002, confirme cette observation en montrant que l’addition de phytoestrogènes de soja à l’alimentation modifie favorablement la sensibilité à l'insuline, le contrôle de la glycémie, et le taux de lipoprotéines chez des femmes ménopausées présentant un diabète de type 2, améliorant ainsi leur profil de risque cardiovasculaire (Jayagopal et al. 2002).
L’étude de modèles murins aboutit à la même constatation : un régime alimentaire riche en soja réduit la prise de poids et l’accumulation de masse adipeuse chez des souris adultes (Cederroth et al. 2007). Ce phénotype est associé à une augmentation de l'oxydation des lipides, en raison de l’utilisation préférentielle des lipides comme source énergétique, et
une augmentation de l'activité locomotrice. La modulation du bilan énergétique est associée à un effet des phyto-oestrogènes sur l'expression hypothalamique de neuropeptides comme AgRP (Agouti-related protein) (Cederroth et al. 2007). Dans les tissus périphériques de souris nourries avec des phytoestrogènes, en particulier dans le tissu adipeux blanc, la phosphorylation de l'AMP-activated protein kinase (AMPK) et celle de l'acétyl-CoA carboxylase sont augmentées, et l'expression de gènes impliqués dans l'oxydation des acides gras et dans la biogenèse mitochondriale est augmentée. Ces souris sont également sujettes à une réduction de l’insulinémie et du contenu pancréatique en insuline. Tout ceci est associé à un effet bénéfique sur la sensibilité à l'insuline, due à l'augmentation de la capture du glucose par les muscles squelettiques par des mécanismes indépendants de la signalisation insulinique (Cederroth et al. 2008).
D’autre part, depuis plusieurs décennies, des observations épidémiologiques animales et humaines ont suggéré que des substances environnementales possédant une activité estrogénique peuvent participer à la physiopathologie de plusieurs affections touchant, dans les deux sexes, le développement, le fonctionnement ou l’oncogénèse de l’appareil de reproduction (Toppari et al. 1996; Golden et al. 1998). Les xénoestrogènes recouvrent un très large spectre de diverses structures moléculaires avec au premier plan les molécules organochlorées. Les substances les plus connues et étudiées sont actuellement le bisphénol A (BPA) et les phtalates esters comme le benzylbutyl-phtalate (BBP), qui sont largement utilisés dans la manufacture des plastiques. La plupart des études sur les effets du BPA sur la santé se sont concentrées sur une bonne documentation de l’activité oestrogénique au niveau de l’appareil reproducteur (Takeuchi et al. 2004), mais des rapports ont mis en évidence d’autres effets délétères (Okada et al. 2008), incluant des atteintes hépatiques (Elsby et al. 2001; Tyl et al. 2002), des perturbations de la fonction des cellules bêta pancréatiques (Ropero et al. 2008), et des effets favorisant l’obésité (Newbold et al. 2008). Une étude analysant les concentrations de BPA et l’état de santé de la population générale adulte aux Etats-Unis a mis en évidence une corrélation entre les concentrations urinaires de Bisphénol A et le diabète (Lang et al. 2008). Dans des modèles murins, le BPA génère une signalisation de l’insuline excessive induisant une résistance à l’insuline dans le foie et le muscle ainsi qu’un épuisement des cellules β. Par conséquent, ce type de perturbateur endocrinien pourrait contribuer au développement du diabète de type 2 (Hugo et al. 2008; Nadal et al. 2009).
IV. Altération de la voie des oestrogènes
: impact sur
l’homéostasie glucidique
Chez l’Homme, l’absence d’oestrogènes ou de leurs récepteurs exerce un effet délétère sur le métabolisme des glucides et des lipides. Deux types de mutations majeures ont été identifiés comme possible cause de la déficience congénitale de la voie des oestrogènes chez l'homme:
(a) une mutation du gène codant pour l'aromatase (Morishima et al. 1995; Carani et al. 1997), enzyme responsable de l'aromatisation du cycle A des androgènes, permettant la synthèse des oestrogènes.
(b) une mutation du gène du récepteur α aux oestrogènes (Smith et al. 1994).
Par ailleurs, différentes études de cohortes incluant des sujets d’origines ethniques différentes, mettent en évidence une association entre le polymorphisme du gène ESR1 du récepteur des oestrogènes et le diabète de type 2 (Speer et al. 2001).
IV.1. Chez l’Homme
IV.1.1. Mutation de l’aromatase
Plusieurs cas de mutation du gène de l’aromatase ont pu être identifiés chez des hommes et des femmes. En 1993, l’équipe de Simpson présente le cas d’une déficience congénitale de l’aromatase chez une femme adulte, due à deux mutations de l’exon 10 du gène de l’aromatase. En 1995, Morishima rapporte le cas d’une nouvelle mutation de l’exon 9 dans le gène de l’aromatase, et d’autres cas cliniques suivront (Mullis et al. 1997; Carani et al. 1997). Cette déficience en oestrogènes conduit à l’apparition précoce d’un surpoids « androïde », d’une insulino-résistance et d’une dyslipidémie (Faustini-Fustini et al. 1999). L’implication des oestrogènes dans l’homéostasie du glucose et de l’insuline a été confirmée notamment par la restauration de la sensibilité à l’insuline, mesurée par test de tolérance à l’insuline, lors de l’administration chronique d’oestradiol chez un sujet atteint de déficience congénitale de l’aromatase (Rochira et al. 2007).
IV.1.2. Mutation du ERα
Contrairement aux cas de mutation du gène de l’aromatase, un seul cas de mutation du ERα a été recensé. Or en 1994, l’équipe de Smith rapporte le cas d’un homme de 28 ans dont la résistance aux œstrogènes est causée par une mutation du gène du récepteur α aux œstrogènes (Smith et al. 1994). En plus des anomalies osseuses, de contrôle de la fonction gonadique et d’infertilité, cette mutation entraîne également une augmentation du taux de glycémie à jeun, une intolérance au glucose et une insulino-résistance. Cette résistance aux oestrogènes induit également une modification du profil lipidique caractérisé par de faibles taux sérique de cholestérol total, de VLDL, de HDL et de LDL, avec cependant un taux normal de triglycérides. L’absence de ce récepteur conduit également à une obésité de type androïde.
La voie des oestrogènes semble donc fortement impliquée dans l’homéostasie du métabolisme, notamment glucidique, et son altération semble favoriser l’évolution vers un diabète de type 2.
IV.1.3. Polymorphisme du gène du ERα
Des études génétiques menées dans différentes cohortes mettent en évidence l’influence du gène ESR1, codant le récepteur α des oestrogènes, sur l’homéostasie du glucose. Une étude récente montre que certains polymorphismes de l’intron 2 du gène ESR1 sont corrélés avec la survenue du diabète de type 2 dans une large cohorte d’américains d’origine européenne (Gallagher et al. 2007). D’autres polymorphismes, dans l’intron 1 et 4 ont également été associés avec le diabète de type 2 dans des cohortes d’Afro-Américains (Keene et al. 2008), de Français (Dahlman et al. 2008), ou encore de Chinois (Huang et al. 2006).
IV.2. Dans les modèles animaux
Modèle classique de carence oestrogénique, l’ovariectomie bilatérale de souris femelles se traduit par une accélération de la prise de poids et une augmentation des taux sanguins de glucose, témoignant d’une évolution progressive vers l’intolérance au glucose.
L’administration d’oestrogènes exogènes normalise ces anomalies, ce qui implique que la voie des oestrogènes joue un rôle important dans le métabolisme du glucose (Heine et al. 2000).
Le développement de modèles murins transgéniques, déficients en récepteur des oestrogènes α (ERα KO) ou en aromatase (Ar KO), a permis de confirmer le rôle clef des oestrogènes pour la régulation de l'homéostasie glucidique et énergétique.
Le rôle crucial des oestrogènes dans l’homéostasie du glucose a été ainsi rapporté dans un modèle de souris déficientes en aromatase (ArKO), enzyme nécessaire à la conversion des androgènes en oestrogènes (Fisher et al. 1998). Le phénotype de ces souris ArKO males et femelles est caractérisé par une réduction de l'oxydation du glucose, une augmentation des taux plasmatiques d'insuline et une augmentation de la masse de tissu adipeux (Jones et al. 2000). A long terme, ces anomalies conduisent au développement d’un diabète de type 2. Une étude a montré que chez les souris mâles ArKO présentant une intolérance au glucose et une résistance à l'insuline, un traitement par 17β-oestradiol reversait ce phénotype (Misso et al. 2003).
Dans le modèle murin de déficience en récepteurs α des œstrogènes (ERαKO) (Heine et al. 2000), mais pas dans celui de souris ERβKO, le poids corporel, la masse de tissu adipeux et le taux de cholestérol sont augmentés. Les souris mâles et femelles ERαKO développent une intolérance au glucose, suggérant que l’effet régulateur des oestrogènes sur le métabolisme glucidique est médié par le récepteur ERα (Cooke et al. 2001).
Partie III : Physiologie de l’homéostasie glucidique et
physiopathologie du diabète de type 2.
I. Physiologie de l’homéostasie glucidique
Le maintien d’une glycémie normale, i.e. l’homéostasie glucidique, dépend de mécanismes endocriniens, nerveux, et métaboliques qui contrôlent l’entrée et la sortie du glucose dans l’organisme (Broberger 2005). En phase absorptive, le glucose alimentaire absorbé au niveau digestif est plus important que la production hépatique de glucose, l’hyperglycémie est ensuite détectée au niveau de la veine hépatoportale (Burcelin et al. 2000). Le glucose est alors utilisé en quelques dizaines de minutes par les tissus de l’organisme, notamment le foie, les muscles et le tissu adipeux. La glycémie est ainsi maintenue à sa valeur initiale. A jeun, le maintien d’une glycémie normale, permettant une disponibilité suffisante pour les tissus périphériques, est principalement assuré par le foie. Dans ces conditions, les muscles et tissus adipeux n’utilisent que peu de glucose. Les tissus splanchniques sont alors responsables de la majeure partie du métabolisme énergétique. Les mécanismes endocriniens permettant de maintenir l’homéostasie glucidique à jeun sont essentiellement dépendants du glucagon, des catécholamines et des glucocorticoïdes. Par contre, à l’état nourri, l’insuline est la principale hormone impliquée. Son action anabolisante est indispensable pour le développement des tissus, la croissance, et l'homéostasie glucidique.
I.1. L’insuline et son récepteur
I.1.1. L’insuline
La molécule d’insuline est un polypeptide d’un poids moléculaire de 6 kDa. C’est un hétérodimère constitué de deux chaînes polypeptidiques, la chaîne A et la chaîne B, reliées entre elles par deux ponts disulfures. Dans la plupart des espèces, comprenant l’Homme, la chaîne A comporte 21 acides aminés, avec un pont disulfure intracaténaire reliant les acides aminés 6 et 11, et la chaîne B en comporte 30.
Le gène de l’insuline humaine est situé sur le bras court du chromosome 11. Il contrôle la synthèse d’un précurseur de 11,5 kDa, la pré-pro-insuline. Les 25 premiers acides aminés
de la molécule représentent une séquence riche en résidus hydrophobes et permettent la pénétration du peptide au cours de la synthèse dans la lumière du réticulum endoplasmique rugueux (RER). Dès que cette séquence a traversé la membrane du RER, elle est éliminée par des peptidases spécifiques. Reste donc, dans les citernes du RER, le peptide tronqué : la pro-insuline, de 86 acides aminés, contenant l’insuline (51 acides aminés) et un autre segment, le peptide-C, reliant la fin de la chaîne A au début de la chaîne B (Docherty et al. 1994). Dans le réticulum, les molécules de pro-insuline s’associent en hexamères, en présence de zinc (Figure 6).
Il existe une maturation post-traductionnelle de la proinsuline qui implique le fonctionnement coordonné du RER et de l’appareil de Golgi. Elle aboutit au stockage de l’insuline dans des vésicules sécrétoires lisses, matures, stockées dans le cytosol puis libérées par exocytose au niveau de la membrane plasmique. Une cellule bêta humaine contient en moyenne 10 000 vésicules de sécrétion.
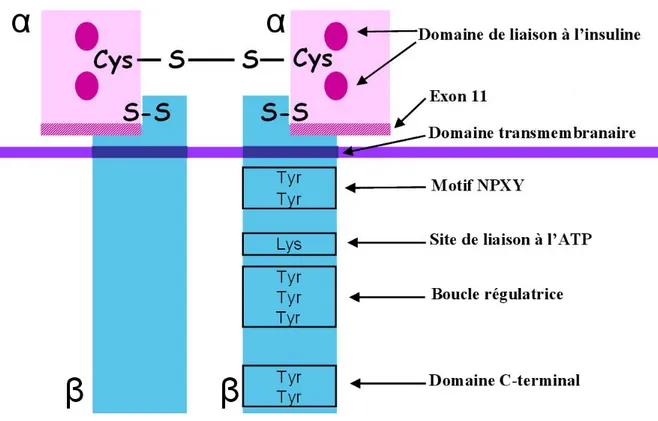
I.1.2. Le récepteur de l’insuline
Pour expliquer les effets métaboliques de l’insuline, l’existence d’un récepteur spécifique localisé dans la membrane plasmique a été postulée il y a près d’un demi-siècle, entre autres par Rachmiel Levine, un pionnier de la diabétologie qui montra, dès 1949, que l’insuline stimule l’entrée du glucose dans les cellules (De Meyts 2004). L’ADN complémentaire du récepteur insulinique est cloné en 1985 par les groupes d’Axel Ullrich à Genentech (Ullrich et al. 1985) et Bill Rutter à Chiron (Ebina et al. 1985). La séquence du gène, déterminée peu après, a fourni des données essentielles sur la structure et l’organisation moléculaire du récepteur.
Le récepteur de l’insuline appartient à la superfamille des récepteurs à tyrosine kinase (RTKs). On dénombre une soixantaine de RTKs dans le génome humain, groupés en 19 familles selon l’architecture du domaine extracellulaire, lequel utilise une variété de modules protéiques pour créer des sites de liaisons spécifiques pour le ligand, hormone, cytokine ou facteur de croissance (De Meyts 2005).
Dans sa configuration naturelle à l’état non activé, le récepteur de l’insuline se présente sous la forme d’un hétérodimère constitué de deux unités β et de deux sous-unités α, liées de façon covalente par des ponts disulfures. L’holorécepteur a un poids moléculaire de 350 kDa. Chaque moitié du dimère comporte une sous-unité de 135 kDa et une sous-unité de 95 kDa. La sous-unité α est entièrement extracellulaire et contient le site de liaison à l’hormone. La sous-unité β est transmembranaire et porte l’activité tyrosine-kinase (Figure 7). La liaison de l’insuline à deux régions spécifiques de l’unité α produit un changement de conformation. Ce changement libère l’activité catalytique de récepteur, se traduisant par une autophosphorylation. Plusieurs sites tyrosine sur la sous-unité β sont phosphorylés ; la phosphorylation sur un résidu tyrosine en position 960 crée un motif de reconnaissance pour l’ancrage des effecteurs cellulaires protéiques IRS-1 et IRS-2 (Insulin receptor substrats) (Le Roith et al. 2001). Les IRS sont ancrés à la membrane plasmique à proximité du récepteur grâce à un domaine PH (pleckstrin homology) localisé dans leur région N-terminale. Leur interaction avec le récepteur de l’insuline activé induit la phosphorylation de plusieurs résidus tyrosyl de leur région C-terminale permettant la fixation de protéines possédant un domaine SH2 comme l’unité p85 du complexe p85/p110 de la
PI3-deux protéines sont les médiateurs des fonctions de l’insuline sur le métabolisme et la croissance cellulaire, respectivement.
Figure 7 : Représentation simplifiée de la structure secondaire du récepteur de l’insuline
I.1.3. Les voies de signalisation intracellulaire
L’insuline active de nombreuses voies de signalisation intracellulaire représentées schématiquement et de façon non exhaustive dans la Figure 8.
La transduction du signal initiée par la fixation de l’insuline mobilise plusieurs seconds messagers possédant des domaines SH2. La tyrosine kinase activée recrute une famille de protéines d’accostage telles que IRS 1-6, Shc, Gab1, dont les tyrosines, phosphorylées par le récepteur, lient une variété de molécules de signalisation. Deux voies principales de signalisation sont alors activées : la voie des PI-3 kinases et la voie des MAP kinases.
La première cascade de signalisation, impliquant la voie de la PI-3 kinase, concerne essentiellement les effets métaboliques de l’hormone. Après son activation par les IRS, la PI-3 kinase induit la production de PIP3 (phosphoatidylinositol 3, 4, 5 phosphate) qui se lie au
domaine PH de la PDK-1 (PI-3-K-dependent kinase). A son tour, celle-ci phosphoryle et active la sérine/thréonine kinase B ou Akt, ainsi que les isoformes atypiques ζ et λ de la Protéine Kinase C. Ces deux isoformes sont impliquées dans la translocation du transporteur de glucose GLUT4 exprimé dans les muscles squelettiques et le tissu adipeux (Okada et al. 1994). Akt phosphoryle également GSK-3 (glycogen synthase kinase-3), qui, en inhibant l’effet freinateur de cette enzyme sur la glycogène synthase, stimule directement la glycogénogénèse (Shepherd et al. 1995).
L’autre voie, associée aux effets de l’insuline sur la croissance, met en jeu le système MAP Kinase (mitogen-activated protein) via l’activation de Grb2 et aboutit au recrutement des ERK-1 et ERK-2 (extracellular signal regulated kinase), responsables de la phosphorylation de facteurs de transcription dont Elk-1.
Figure 8 : Les voies de signalisation de l’insuline.
La voie PI3-K regule le métabolisme du glucose dans le muscle squelettique, le tissu adipeux blanc et le foie ; elle contrôle la production de NO et la vasodilatation au niveau des vaisseaux. La voie MAPK contrôle la croissance et la prolifération cellulaire. IRS, Insulin Receptor Substrate ; NO, nitric oxide ; eNOS, endothelial Nitric Oxide Synthase, PDK, phosphoinositide-dependent protein kinase ; PKC, Protéine Kinase C. Figure adaptée de (Muniyappa et al. 2007).
I.2. L’insuline et l’homéostasie glucidique
A travers ses actions intégrées sur le métabolisme des hydrates de carbones et des lipides, l’insuline joue un rôle fondamental sur la régulation de l’homéostasie glucidique. Ses effets glucorégulateurs s’exercent essentiellement sur le foie, le muscle et le tissu adipeux.
I.2.1. Action hépatique
Le foie est un organe clef dans le contrôle de l’homéostasie des carbohydrates et des lipides (Postic et al. 2004). L’insuline inhibe la néoglucogénèse et la glycogénolyse et favorise le stockage de glucose sous forme de glycogène (Boden 2004) (Figure 9).
Figure 9: Action de l’insuline dans le foie en condition physiologique.
En ce qui concerne le métabolisme des lipides, une augmentation du flux d’acides gras libres est retrouvée dans de nombreux modèles de défauts d’action de l’insuline au niveau hépatique (Bergman et al. 2007). Dans des conditions physiologiques, l'insuline augmente l'expression des gènes d'un certain nombre d'enzymes de la voie de biosynthèse des triglycérides (Gonzalez-Baro et al. 2007). Au niveau hépatique, l’insuline inhibe la production de VLDL (Very Low Density Lipoprotein). L’effet inhibiteur de l’insuline sur la production hépatique de VLDL a clairement été mis en évidence in vivo. C’est ainsi que chez le sujet sain, il est observé sous l’effet de l’insuline une inhibition de la production de VLDL-triglycérides (–67%) et de la production d’apolipoprotéines B (apoB) des VLDL (–52%) (Lewis et al. 1993; Malmstrom et al. 1998). L’insuline apparaît réduire la production de
VLDL, nonseulement en diminuant le taux des acides gras libres dans la circulation (limitant ainsi les substrats nécessaires à la formation des VLDL), mais aussi par un effet inhibiteur direct dans l’hépatocyte (Malmstrom et al. 1998).
I.2.2. Actions périphériques
I.2.2.1. Le muscle squelettique
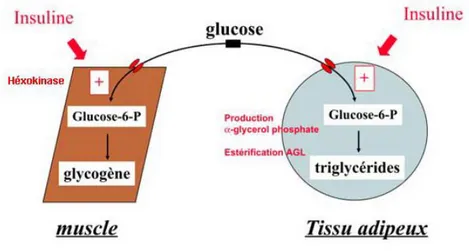
Le muscle squelettique est le principal site d'utilisation de glucose en réponse à l’action de l'insuline. Dans le muscle, l’insuline active la translocation à la membrane plasmique du transporteur de glucose GLUT4 (Figure 10) (Bjornholm et al. 2005). Des vésicules périnucléaires comportant les protéines transmembranaires GLUT4 sont recrutées à la membrane plasmique. Celles-ci migrent sur les faisceaux d’actine, fusionnent à la membrane à l’aide de protéines d’ancrage par des mécanismes consommateurs d’énergie, puis les transporteurs sont exposés à la membrane plasmique. Ces derniers peuvent alors transporter librement le glucose suivant un gradient de concentration allant du liquide interstitiel vers le cytoplasme. Le glucose est alors très rapidement pris en charge par l’héxokinase 2 qui génère le glucose 6-phosphate. Ce métabolite se trouve au carrefour de la synthèse du glycogène et de la voie de la glycolyse. L’insuline active les deux voies, et notamment les enzymes de la glycolyse telles que la phosphofructokinase, la pyruvate synthase et la pyruvate déshydrogénase.
Figure 10 : Translocation du transporteur de glucose GLUT4 à la membrane en réponse à l’insuline.
I.2.2.2. Le tissu adipeux
Une des fonctions du tissu adipeux est de stocker les acides gras et le glucose sous la forme de triglycérides. Dix pour cent du glucose sanguin sont captés par les adipocytes sous l’action de l’insuline. Cet effet biologique dépend cependant de la masse totale du tissu adipeux, ainsi que de la sensibilité à l’insuline des adipocytes. Ce mécanisme nécessite également la translocation du transporteur de glucose GLUT4 à la membrane plasmique des adipocytes (James et al. 1994; Smith 2002), ainsi que l’activation de la lipogénèse et l’inhibition de la lipolyse des triglycérides. La résultante est que l’insuline augmente la charge du tissu adipeux en triglycérides (Figure 11).
Figure 11: Action physiologique de l’insuline dans le muscle et le tissu adipeux.
II. Physiopathologie du diabète de type 2
Le diabète de type 2 est défini par une hyperglycémie chronique due à des mécanismes complexes et intriqués, responsables d’une insulinodéficience et d’une insulinorésistance. Ces anomalies entraînent une production excessive de glucose par le foie, ainsi qu’un défaut de son utilisation par les tissus périphériques, notamment les muscles et le tissu adipeux. L’hérédité, l’âge, la sédentarité ou encore l’obésité représentent des facteurs favorisant le développement du diabète de type 2.
II.1. Altération de la sécrétion d’insuline
L’installation progressive d’un diabète de type 2 implique une dérégulation de la sécrétion d’insuline dont les mécanismes moléculaires peuvent être multiples (Kasuga 2006). Dans la plupart des cas, la survenue de l’hyperglycémie est le signe d’une défaillance des cellules β pancréatiques, lorsque les îlots de Langerhans ne peuvent plus assurer une réponse compensatoire à la résistance à l'insuline (Figure 12). Puis, l’hyperglycémie chronique contribue également à amplifier le dysfonctionnement des cellules β et la perte de la masse des cellules β par apoptose : c’est l’effet glucotoxique (Prentki et al. 2006).
En 1986, l’équipe de Hales a authentifié un réel déficit en insuline mature en dosant la pro-insuline et l’insuline. En effet, dans le diabète de type 2, ce déficit insulinique a été longtemps masqué par un excès de pro-peptides circulants. Ces peptides immatures peuvent atteindre plus de 40% des formes circulantes chez les diabétiques alors qu’ils représentent seulement 5% en conditions physiologiques (Temple et al. 1990).
En 1995, l’application du HOMA chez des sujets inclus dans l’étude de l’UKPDS (United Kingdom Prospective Diabetes Study) a montré que le défaut de sécrétion insulinique jouait un rôle majeur dans leur élévation glycémique progressive (Matthews et al. 1998), mais cette sécrétion est déjà réduite de 50% au moment du diagnostique de diabète de type 2, et anormale à des stades plus précoces avec une baisse supplémentaire de 15% observée six ans plus tard (Tripathy et al. 2000). Les mécanismes proposés pour expliquer la réduction progressive de l’insulinosécrétion sont nombreux. Au delà des prédispositions génétiques et des phénomènes récemment identifiés de programmation fœtale, l’hypothèse la plus plausible à ce jour fait intervenir les concepts de glucotoxicité (Weir et al. 2007) et de lipotoxicité (Unger 1995) conduisant à l’apoptose des cellules β. L’exposition chronique de la cellule β à l’hyperglycémie et à des concentrations élevées de triglycérides et d’acides gras libres altère en effet de façon progressive et irréversible l’insulinosécrétion induite par le glucose (Prentki et al. 2006).