Quasi-One-Dimensional Metal-Insulator Transitions in Compound Semiconductor Surfaces
Texte intégral
Figure
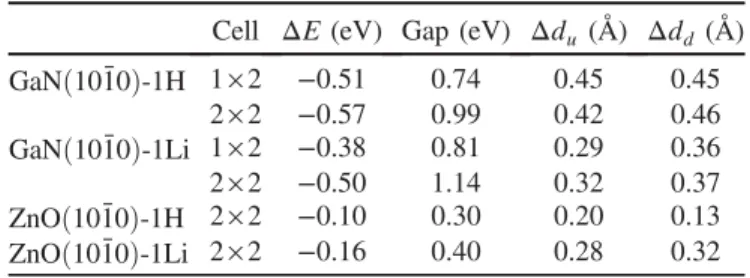
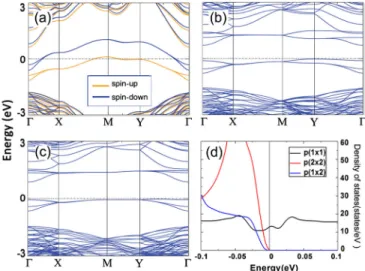
Documents relatifs
16 Secondly, a rigorous thermodynamic description of the interplay between adsorption and deformation including structural transitions is provided by the use of
It should be noted that if covalency had been treated more rigorously (not just by determining effective d-d integrals), then the pushing up of the a, band in V203
(the degeneration of the two e, band being maintained under the trigonal symmetry). - Density of states for corundum structure with one elec- tron per site in a doubly
qu'il y a diffusion de l'énergie parmi les ions Gd3+ et piégeage par des impuretés à haute température et par des ions G d 3 + perturbés à basse
Abstract.- The Landau theory of phase transition is extended to include the singular free-energy due to a metal-insulator (MI) transition and to discuss the condensation
As it is known [15, 16, 19] this phenomenology is exact in the low temperature limit of the continuum models and the results of Trullinger and Sasaki [21] and those of the present
- Some topics about metal-non- metal (M-NM) transitions induced by electron corre- lations in several disordered systems such as doped semiconductors, disordered
La prescription d’une antibiothérapie par fluoroquinolones durant les trois (infections respiratoires) à six mois (infections urinaires) précédant le traitement mis en place dans le
