Transition-Metal-Free Reduction of Carbon
Dioxide
Thèse
Marc-André Courtemanche
Doctorat en chimie
Philosophiae Doctor (Ph.D.)
Québec, Canada
© Marc-André Courtemanche 2015
iii
Résumé
Seulement neuf années se sont écoulées depuis la découverte que les ‘’Paires de Lewis Frustrées’’ (PLF) peuvent promouvoir le clivage de l’hydrogène, mais plus d’un millier d’articles scientifiques ont déjà été publiés sur le sujet. Au début des travaux décrits dans cette thèse, les catalyseurs pour la réduction du CO2 étaient excessivement rares et peu
efficaces. La présente thèse porte donc sur le développement de nouveaux systèmes sans métal de transition pour la réduction catalytique du CO2 en molécules riches en énergie et
plus précisément, en méthanol.
D’abord, la préparation d’un nouveau système basé sur les PLF et sa capacité à activer le CO2 de façon réversible est présenté. En présence de catécholborane, le CO2 est
catalytiquement réduit en méthoxyboranes, espèces facilement hydrolysables en méthanol. Surprenamment, un produit de décomposition est identifié comme étant responsable de l’activité catalytique.
En effet, l’espèce ambiphile 1-Bcat-2-PPh2-C6H4 constitue le premier exemple d’un
catalyseur sans métal de transition pour l’hydroboration du CO2. L’activité de ce catalyseur
excessivement simple surpasse celle des meilleurs systèmes basés sur des métaux.
Des études mécanistiques détaillées révèlent que l’activation simultanée du borane et du CO2
est d’une importance critique. Une investigation poussée révèle que la formation d’un adduit entre le catalyseur et le formaldéhyde résulte en un organocatalyseur d’autant plus actif. Il est aussi démontré que les phosphazènes, super bases organiques, sont des organocatalyseurs très actifs pour la transformation du CO2 en dérivés de formate ou de
méthanol. De façon intéressante, le DMF (N,N-diméthylformamide) peut promouvoir l’hydrosilylation réductive du CO2 en absence de catalyseur.
Une nouvelle stratégie d’hydrogénation a été développée en étudiant les aspects fondamentaux de l’hydrogénation par les PLFs, permettant ainsi la conception d’un système pour l’hydrogénation du CO2 en conditions ambiantes. Même si une voie de décomposition
inattendue rend le processus stoéchiométrique, une optimisation du catalyseur pourrait générer le premier catalyseur sans métal pour l’hydrogénation du CO2.
v
Abstract
Only nine years have passed since the seminal discovery that Frustrated Lewis Pairs (FLPs) could split dihydrogen and yet, more than a thousand research papers have already been published on the subject. As the work presented herein commenced, metal-free systems capable of catalytically transforming CO2 could be counted on a single hand while
transition-metal based systems were almost as scarce. As such, the present thesis deals with the development of novel transition-metal-free systems for the catalytic reduction of CO2 to
energy rich materials, most notably methanol.
Firstly, the preparation of a new FLP system bearing three pendant phosphine groups Al(C6H4(o-PPh2))3 and its ability to activate carbon dioxide in a reversible fashion are
presented. In the presence of catecholborane, CO2 is catalytically reduced to
methoxyboranes, species which are readily hydrolyzed to methanol. Interestingly, a decomposition product is shown to be responsible for the catalytic activity
Indeed, species 1-Bcat-2-PPh2-C6H4 is the first report of a catalyst for the metal-free
hydroboration of carbon dioxide. The activity of this excessively simple catalyst surpasses that of the best transition metal systems while using the cheap and high hydrogen content borane BH3.SMe2.
In-depth mechanistic studies reveals that simultaneous activation of both the borane and CO2
molecules is of critical importance. Further investigation reveals that the formation of an adduct between the catalyst and formaldehyde affords an even more potent organocatalyst. It is also shown that phosphazene superbases are very active organocatalysts for the transformation of CO2 to either formate or methanol derivatives. Unexpectedly,
N,N-dimethylformamide (DMF) can promote the reductive hydrosilylation of CO2 in the absence
of any catalyst.
Finally, the challenging task of developing a metal-free system for the hydrogenation of CO2
was undertaken. A novel strategy was developed by studying the fundamental aspects of FLP mediated hydrogenations, allowing us to achieve CO2 reduction under ambient conditions.
While an unexpected decomposition pathway hampered catalysis, optimisation of the catalyst design is expected to yield the first metal-free catalyst for the hydrogenation of CO2.
vii
Table of Contents
Résumé ... iii
Abstract ... v
Table of Contents ... vii
List of Tables ... xiii
List of Schemes ... xvii
Acknowledgements ... xxvii
Inserted research articles and author contributions: ... xxix
1 Introduction ... 1
1.1 A note on chronology: ... 1
1.2 A note on compound identification: ... 1
1.3 General introduction ... 2
1.4 Carbon dioxide and the greenhouse effect ... 3
1.5 Using CO2 as an energy vector: the methanol economy ... 5
1.6 Challenges in the transformation of carbon dioxide ... 7
1.7 The carbon dioxide molecule ... 8
1.8 Organometallic binding of carbon dioxide ... 10
1.9 Carbon dioxide for energy storage: general aspects ... 13
1.10 Metal enzymes for CO2 reduction ... 14
1.11 Transition metal catalyzed reduction of CO2 to energy rich materials ... 15
1.12 Photochemical reduction of carbon dioxide ... 17
1.13 Transition-metal free transformations of carbon dioxide ... 18
1.14 A short history of Frustrated Lewis Pair chemistry ... 20
1.15 Carbon dioxide activation and reduction using Frustrated Lewis Pairs ... 23
1.16 Original hypotheses and motivation ... 24
1.17 Non-frustrated aluminum based ambiphilic molecules for CO2 capture ... 27
1.18 Scope of thesis ... 30
2 Experimental methods ... 31
2.1 Inert atmosphere chemistry ... 31
2.1.1 Glovebox ... 31
2.1.2 Schlenk line ... 32
2.2 Working with gases. ... 33
2.3 Nuclear Magnetic Resonance (NMR) spectroscopy. ... 34
viii
2.5 DFT calculations ... 36
2.6 Gas chromatography ... 38
3 Aluminum based ambiphilic molecules fort the capture of carbon dioxide. ... 39
3.1 Advances in the transition-metal mediated reduction of CO2 ... 39
3.2 Advances in the binding of CO2 by FLPs ... 41
3.3 Fundamental aspects of CO2 chemistry: the general concept. ... 42
3.4 Overview of the project ... 44
3.5 Research article: A Tris(triphenylphosphine)aluminum Ambiphilic Precatalyst for the Reduction of Carbon Dioxide with Catecholborane ... 47
3.5.1 Résumé ... 47
3.5.2 Abstract ... 47
3.5.3 Introduction ... 48
3.5.4 Results and discussion ... 50
3.5.5 Conclusion ... 61
3.6 Experimental Section ... 62
3.6.1 General procedure ... 62
3.6.2 Synthesis of compounds ... 63
3.6.3 CO2 reduction catalytic tests ... 64
3.6.4 Crystallographic studies ... 65
3.6.5 Computational details ... 67
4 Metal-Free catalytic Reduction of CO2 to methanol ... 69
4.1 Research article: A Highly Active Phosphine-Borane Organocatalyst for the Reduction of CO2 to Methanol using Hydroboranes... 69
4.1.1 Résumé ... 69
4.1.2 Abstract ... 69
4.1.3 Introduction ... 70
4.1.4 Results and discussion ... 72
4.1.5 Conclusions ... 81
4.2 Experimental ... 82
4.2.1 General experimental ... 82
4.2.2 Synthesis of 1 ... 83
4.2.3 General procedure for catalytic reduction of carbon dioxide ... 84
4.2.4 General procedure for big scale catalytic reduction of carbon dioxide ... 85
4.2.5 Reactivity with methylformate ... 85
ix
4.2.7 Crystallographic information ... 86
4.2.8 Computational Details: ... 88
4.3 Conclusions and perspectives ... 89
5 Mechanistic investigations ... 91
5.1 Metal mediated catalytic reductions of CO2... 91
5.2 Metal-free activation of CO2 ... 93
5.3 Metal-free reduction of CO2 ... 94
5.4 Research article: Reducing CO2 to Methanol using Frustrated Lewis Pairs: On the Mechanism of Phosphine-Borane Mediated Hydroboration of CO2 ... 95
5.4.1 Résumé ... 95
5.4.2 Abstract ... 95
5.4.3 Introduction ... 96
5.4.4 Computational details ... 98
5.4.5 General remarks ... 99
5.4.6 First reduction step: CO2 to HCOOBcat ... 100
5.4.7 Second reduction step: from HCOOBcat to CH2O and derivatives ... 106
5.4.8 Third reduction step: reducing CH2O and derivatives to CH3OBcat ... 111
5.4.9 Discussion ... 115
5.4.10 Conclusion ... 117
5.5 Experimental Data:... 118
5.5.1 General experimental ... 118
5.5.2 Synthetic methodology ... 118
5.5.3 Additional computational information regarding the catalyzed reduction of CO2 to HCOOBcat ... 119
5.5.4 Additional computational information regarding the catalyzed reduction of HCOOBcat to CH2O and derivatives ... 120
5.5.5 Additional computational information regarding the catalyzed reduction of CH2O and derivatives to CH3OBcat ... 121
5.5.6 Additional computational information regarding the possible involvement of IM3C in catalysis ... 122
5.5.7 Additional computational information regarding the uncatalyzed reduction of CO2 to CH3OBcat ... 123
5.6 Conclusion and perspectives ... 126
6 Metal-free catalytic hydrosilylation of CO2 to methanol ... 131
6.1 Base catalyzed hydroboration of CO2 ... 131
x
6.3 Research article: Phosphazenes: Efficient Organocatalysts for the Catalytic
Hydrosilylation of Carbon Dioxide ... 136
6.3.1 Résumé ... 136
6.3.2 Abstract ... 136
6.3.3 Introduction ... 137
6.3.4 Results and discussion ... 139
6.3.5 Conclusions ... 145
6.4 Experimental ... 146
6.4.1 General experimental: ... 146
6.4.2 Initial test experiment: ... 147
6.4.3 Rearrangement of 1 to 4 ... 147
6.4.4 Rearrangement of 2 to 5 ... 148
6.4.5 Rearrangement of 3 to 6 ... 149
6.4.6 Catalytic reduction using 13CO2... 150
6.4.7 Following the reaction in DMF, with various loadings of silane: ... 150
6.4.8 Reaction under 5 atm. of CO2 ... 151
6.4.9 General procedures for catalytic tests: ... 152
6.5 Conclusions and perspectives ... 156
7 Metal-free hydrogenation of carbon dioxide ... 159
7.1 Introduction to FLP mediated hydrogenations ... 159
7.2 Understanding FLP hydrogenation... 160
7.3 Research article: Intramolecular B/N Frustrated Lewis Pairs and the Hydrogenation of Carbon Dioxide ... 169
7.3.1 Résumé ... 169
7.3.2 Abstract ... 169
7.3.3 Introduction ... 170
7.3.4 Results and discussion ... 171
7.3.5 Conclusions ... 178
7.4 Experimental ... 179
7.4.1 General experimental: ... 179
7.4.2 Synthesis of compounds 1, 2 and precursors ... 180
7.4.3 HD scrambling with 1 ... 183
7.4.4 Synthesis of 3 from 1 ... 183
7.4.5 Hydrogenation of CO2 - J Young NMR Tube Experiments ... 184
xi
7.4.7 Computational details: ... 188
7.4.8 DFT calculation for the protodeborylation of 1 and 2... 189
7.4.9 DFT study of the possible isomers of 3 ... 193
7.4.10 DFT study of the hydrogenation of CO2 ... 194
7.5 Conclusions and perspectives ... 198
8 Conclusion and perspectives ... 199
8.1 General conclusions ... 199
8.2 Ongoing and future work ... 202
8.2.1 Non-CO2 related chemistry ... 202
8.2.2 CO2 related chemistry ... 203
8.3 Philosophical discussion ... 205
8.3.1 Carbon dioxide ... 205
8.3.2 Frustrated Lewis Pairs ... 208
8.4 Personal bibliogaphy ... 210
xiii
List of Tables
Table 1-1: Summary of key isolated transition metal CO2 complexes. ... 12
Table 2-1: NMR nuclei of relevance to this project. ... 34
Table 4-1: Reduction of CO2 with various hydroboranes. ... 77
Table 4-2: Crystal data and structural refinements for compound 1. ... 87
Table 5-1: Crystal data and structural cefinements for compound 2. ... 125
Table 6-1: Catalytic hydrosilylation of CO2 by phosphazene bases. ... 144
Table 7-1: Hydrogenation of carbon dioxide by 1 and 2 ... 175
xv
List of Figures
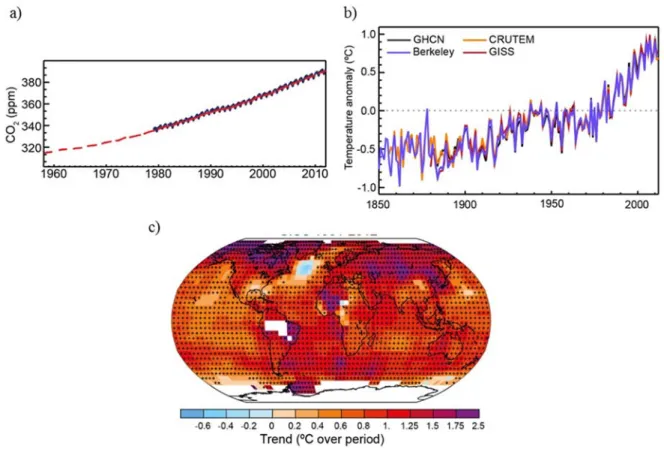
Figure 1-1: a) CO2 concentrations in the atmosphere b) Global temperature anomalies c) Trend of
global temperature variation. ... 3
Figure 1-2: Conceptual representation of the use of CO2 to produce energy vectors. ... 6
Figure 1-3: Representation of the ambiphilic character of the CO2 molecule. ... 8
Figure 1-4: Walsh diagram of the CO2 molecule ... 9
Figure 1-5: Transition state in the process of CO2 reduction by CO dehydrogenase.* ... 15
Figure 1-6: Proposed mechanistic pathways for the base-catalysed hydrosilylation of CO2. ... 19
Figure 1-7: Proposed mechanism for the FLP-mediated hydrosilylation of CO2. ... 23
Figure 1-8 Reported CO2-FLP adducts- October 2010. ... 25
Figure 1-9: Reported CO2-FLP adducts- June 2011. ... 26
Figure 2-1: A glovebox workstation. ... 31
Figure 2-2: a) A Schlenk line b) Schlenk flasks. ... 32
Figure 2-3: The cluster that was used for calculations: Colosse. ... 38
Figure 3-1: Catalysts for the reduction of CO2 a) Ni pincer catalyst b) Ir Pincer catalyst c) Pd or Pt pincer catalyst. ... 40
Figure 3-2: New reported FLP systems for CO2 binding. ... 42
Figure 3-3: Qualitative thermodynamic analysis of Lewis pair reactivity a) Classical Lewis pair reactivity b) Frustrated Lewis Pair reactivity. ... 43
Figure 3-4: Structures of the previously reported ambiphilic ligands. ... 48
Figure 3-5: ORTEP drawing of the first molecule of 1 in the asymmetric unit cell. ... 51
Figure 3-6: Optimized structure of 2 using DFT calculations. ... 53
Figure 3-7: Optimized structure of 2 using DFT calculations. ... 55
Figure 3-8: Turnover number (TON) for the formation of CH3OBcat. ... 57
Figure 3-9:ORTEP drawing of one independent molecule of 5. ... 60
Figure 4-1: ORTEP drawing of 1 in the asymmetric unit cell. ... 72
Figure 4-2: Turn-over numbers (TON) for the formation of CH3OBcat. ... 74
Figure 4-3: Turn-over numbers (TON) for the formation of (CH3OBO)3. ... 75
Figure 4-4: Enthalpy profile (in kcal mol-1) for the reduction of CO2 by 1 and catecholborane. ... 79
Figure 4-5: Generation of a formaldehyde adduct from 1. ... 80
Figure 4-6: Assignment of spectra for 1. ... 83
Figure 5-1: New metal based catalytic systems for the reduction of CO2... 92
xvi
Figure 5-3: Important intermediates and transition states for the catalyzed reduction of CO2 to
HCOOBcat. ... 105
Figure 5-4: Relative energies of transition states and intermediates for the reduction of HCOOBcat to CH2O or catBOCH2OBcat. ... 110
Figure 5-5: ORTEP drawing of 2. ... 111
Figure 5-6: Relative energies of transition states and intermediates for the reduction of CH2O to CH3OBcat. ... 114
Figure 5-7: New transition states for CO2 reduction involving a formaldehyde adduct as catalyst . ... 128
Figure 6-1: New catalysts for the hydroboration of CO2. ... 131
Figure 6-2: Assignment of spectra for 4. ... 147
Figure 6-3 Product ratio over time for Test A ... 150
Figure 6-4 Product ratio over time for Test B ... 151
Figure 7-1: Qualitative analysis of hydrogen splitting by FLP systems. ... 160
Figure 7-2: Schematic representation of an hydrogenation reaction mediated by the Noyori catalyst. ... 164
Figure 7-3: General strategy for the hydrogenation of CO2 by FLP systems. ... 165
Figure 7-4: A CO2 molecule with the partial charges. ... 165
Figure 7-5: Hydrogen transfer from ammonia-borane to CO2. ... 166
Figure 7-6: DFT study of the thermodynamics of H2 splitting and CO2 binding by a variety of aryl bridged FLP systems. ... 167
Figure 7-7: ORTEP depiction of 4. ... 176
Figure 7-8: Geometry of TS for reaction of C with CO2. ... 177
Figure 7-9: First protodeborylation step for 1. ... 189
Figure 7-10: Second protodeborylation step for 1. ... 190
Figure 7-11: First protodeborylation step for 2. ... 191
Figure 7-12: Second protodeborylation step for 2. ... 192
Figure 7-13: DFT optimized structures of the possible isomers for compound 3. ... 193
Figure 7-14: Hydrogenation of CO2 by 1. ... 194
Figure 7-15: Hydrogenation of CO2 by 1 (continued). ... 195
Figure 7-16: Hydrogenation of CO2 by 2. ... 196
Figure 7-17: Hydrogenation of CO2 by 2 (continued). ... 197
Figure 8-1: Timeline of the important systems for CO2 reduction to energy rich material. ... 201
xvii
List of Schemes
Scheme 1-1: Schematic representation of the amine scrubbing process. ... 4
Scheme 1-2: Generation of Aresta's complex. ... 10
Scheme 1-3: Aerobic combustion of methane. ... 13
Scheme 1-4: Stepwise CO2 hydrogenation. ... 14
Scheme 1-5: Cascade catalytic hydrogenation of CO2 to methanol. ... 16
Scheme 1-6: Catalytic hydroboration of CO2 by a Ni pincer complex. ... 16
Scheme 1-7: Dual interaction of reduced CO2 with an Ir pincer complex. ... 17
Scheme 1-8: Lewis acid catalyzed hydrosilylation of CO2. ... 18
Scheme 1-9: Formation of a classical Lewis adduct. ... 20
Scheme 1-10: Trapping of benzyne by a phosphine-borane Lewis pair. ... 21
Scheme 1-11: Trapping of hydrochloric acid using an amino-borane Lewis pair. ... 21
Scheme 1-12: Representation of Frustrated Lewis Pair reactivity and hydrogen splitting. ... 22
Scheme 1-13: Reversible metal-free activation of CO2 by FLPs. ... 22
Scheme 1-14: FLP mediated CO2 hydrogenation and formation of methanol. ... 23
Scheme 1-15: FLP mediated stoichiometric reduction of CO2 by ammonia borane. ... 24
Scheme 1-16: Preparation of ambiphilic compounds 1 and 2. ... 27
Scheme 1-17: Reactivity of ambiphilic molecules 1 and 2 with CO2. ... 28
Scheme 1-18: Formation of a new spirocylic CO2 activation product. ... 29
Scheme 3-1: CO2 hydrogenation by Ru catalyst. ... 39
Scheme 3-2: Hydroboration of CO2 by a Ruthenium polyhydride catalyst. ... 41
Scheme 3-3: Proposed dual activation strategy for the catalytic hydroboration of CO2 ... 45
Scheme 3-4: Decomposition of 1 into 2. ... 45
Scheme 3-5: Synthesis of Al(C6H4(o-PPh2))3 (1). ... 50
Scheme 3-6: Generation of 3 upon exposure of 1 to CO2. ... 54
Scheme 3-7: Proposed protection of the phosphine moieties of 1. ... 58
Scheme 3-8: Synthesis of species 5. ... 59
Scheme 4-1: Reduction of CO2 in presence of HBcat and catalyst 1. ... 73
Scheme 4-2: Synthesis of 1. ... 83
Scheme 4-3: Catalytic reduction of methylformate by 1 and HBcat. ... 85
Scheme 5-1: Catalytic hydroboration of CO2 after ring-expansion of an NHC-9-BBN adduct. ... 94
Scheme 5-2: Schematic representation of the stepwise hydroboration of CO2 to methoxyboranes using hydroboranes (H[B]). ... 99
xviii
Scheme 5-3: Reaction of 1 with CO2, generating IM0 illustrating the potential binding sites for HBcat. ... 100 Scheme 5-4: Pathway A: hydroboration reaction of CO2 through a classical 4-membered transition state. [B] = Bcat. ... 101 Scheme 5-5: Pathway B: hydroboration through coordination of HBcat to the catechol fragment followed by intramolecular hydride delivery. [B] = Bcat. ... 102 Scheme 5-6: Pathway C: hydroboration through simultaneous Lewis base activation of the borane and Lewis acid activation of CO2. [B] = Bcat. ... 103 Scheme 5-7: Pathway D: CO2 reduction through the generation of a boronium / hydridoborate ion pair. [B] = Bcat. ... 104 Scheme 5-8: Calculated catalyst-free mechanism for the hydroboration of HCOO[B]. ... 106 Scheme 5-9: Experimental verification for the hydroboration of formic acid by catecholborane. [B]=Bcat. ... 107 Scheme 5-10: Attempt to isolate HCOOBcat, leading to the exclusive formation of 2 (IM2C’). [B]= Bcat... 107 Scheme 5-11: Possible interactions and rearrangements of HCOOBcat with catalyst 1. [B]=Bcat. ... 108 Scheme 5-12: Suggested pathway for the catalyzed reduction of HCOO[B] involving the catalyst [B]=Bcat. ... 109 Scheme 5-13: Formation of IM2C’(2) through the rearrangement of catBOCH2OBcat to CH2O. [B]=Bcat. ... 112 Scheme 5-14: Experimental verification for the catalytic role of 1 in the hydroboration of 4-bromobenzaldehyde by catecholborane. [B]=Bcat ... 113 Scheme 5-15: Catalyzed reduction of formaldehyde to CH3OBcat, regenerating the catalyst. [B]=Bcat ... 113 Scheme 5-16: Proposed mechanistic pathway including important transition states for the reduction of CO2 to CH3OBcat by 1. [B]=Bcat. ... 116 Scheme 5-17: Calculated pathways for the reduction of CO2 to HCOOBcat. [B]=Bcat... 119 Scheme 5-18: Calculated pathways for the reduction of HCOOBcat to CH2O and derivatives. [B]=Bcat. ... 120 Scheme 5-19: Calculated pathways for the uncatalyzed reduction of CO2 to CH2O. [B]=Bcat. .... 121 Scheme 5-20: Calculated pathways for the catalyzed reduction of CO2 to HCOOBcat from IM3C. [B]=Bcat. ... 122
xix Scheme 5-21: Proposed mechanism for the catalyst free reduction of carbon dioxide to CH3OBcat.
[B]=Bcat. ... 123
Scheme 5-22: Labelling experiments with 13CH2O... 127
Scheme 6-1: CO2 capture by dimethylamine. ... 132
Scheme 6-2: Base catalzyed reduction of CO2 to methylamines. ... 133
Scheme 6-3: Protonation of a phosphazene superbase. ... 134
Scheme 6-4: Proposed strategy for the hydrosilylation of CO2 using a phosphazene catalyst. ... 135
Scheme 6-5: Rearrangement of phosphazenes in the presence of CO2 with proposed intermediate. ... 140
Scheme 6-6: Product ratio after the catalytic reduction under 5 atm of CO2 and after addition of an extra loading of silane in the absence of CO2. Yield based on Si-H. ... 142
Scheme 6-7: Preparation of 4 from 1. ... 147
Scheme 6-8: Preparation of 5 from 2. ... 148
Scheme 6-9: Assignment of signals for 5. ... 148
Scheme 6-10: Preparation of 6 from 3. ... 149
Scheme 6-11: Assignment of signals for 6. ... 149
Scheme 7-1: Hydrogen activation by a BCF/Et2O Lewis Pair. ... 162
Scheme 7-2: Catalytic hydrogenation by a BCF/ET2O Lewis Pair ... 162
Scheme 7-3: FLP mediated reduction of CO at a rhenium center using a strong phosphazene base in combination with a weak Lewis acid. ... 164
Scheme 7-4: Preparation of 1-2. ... 171
Scheme 7-5: DFT study of possible isomers of 3. ... 172
Scheme 7-6: DFT study of H2 activation and protodeborylation events. ... 173
Scheme 7-7: Synthesis of 3 from 1. ... 183
Scheme 7-9: Synthesis of 4 from 2 (bigger scale hydrogenation). ... 186
Scheme 7-10: Assignment of the NMR spectra for 4. ... 187
xxi
List of Abbreviations
{1H} = proton decoupled 13CO
2 = carbon 13 labeled carbon dioxide
6-31+G** = a basis set for DFT calculations 9-BBN = 9-borabicyclo[3.3.1]nonane
Å= angstrom
ADH = alcohol dehydrogenase AlF = Al(C6F5)3 B3PW91 = a DFT functional B97D = a DFT functional BCF = B(C6F5)3 BMe3 = trimethylborane BPh3 =triphenylborane
COSY = NMR experiment correlating 1H and 1H nucleus
d = doublet
DCM = dichloromethane dd = doublet of doublets
DFT = Density Functional Theory DMSO = dimethylsulfoxide Et2O = diethyl ether
FalDH = formaldehyde dehydrogenase
FateDH = formate dehydrogenase
FID = flame ionization detector FLP = Frustrated Lewis Pair
FTIR = Fourier transform infrared spectroscopy GC = gas chromatography
gHSQC = NMR experiment correlating 13C and 1H nucleus
xxii
HBPin = pinacolborane HMB = hexamethylbenzene
HOMO = highest occupied molecular orbital LUMO = lowest unoccupied molecular orbital Lutidine =2,6-dimethylpyridine m = multiplet Me = methyl MeOH = methanol Mes = 2,4,6,-trimethylphenyl Mes’ = 2,4,5,-trimethylphenyl Ms = mass spectroscopy Mt = mega tonne
MTO = methanol to olefin process
NADH = nicotinamide adenine dinucleotide NBO = natural bond order
NMR = nuclear magnetic resonance NOx = mono-nitrogen oxides
Ph = phenyl
PNN = (2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine) PPh3 = triphenylphosphine
ppm = parts per million r.t = room temperature s = singlet
SDD = a basis set for DFT calculations that was developed by Stuttgart and Dresden t = triplet
THF = tetrahydrofuran
TMP = 2,2,6,6-tetramethylpiperidine TOF = turnover frequency
xxiii TON = turnover number
TS = transition state
w-B97XD = a DFT functional δ = chemical shift
ΔH= enthalpy
ΔHform = enthalpy of formation
Π = pi orbitals σ = sigma orbitals
xxv
To my parents, Serge and Lise
‘‘The more original a discovery, the more obvious it seems afterwards’’ - Arthur Koestler
xxvii
Acknowledgements
First and foremost, I would like to thank my supervisor, Frédéric-Georges Fontaine for a wonderful time in the Fontaine group. Thank you for the continuous support, for believing in me more than I believed in myself, for always backing up my crazy ideas and suggesting even crazier ones. Thank you for pushing me a little bit further every single time, for sending me to countless conferences and for the helpful discussions about life, chemistry, scotch, chess, and of course Game of Thrones. To be sure, there is so much more to say, but let me put it that way: if it could be possible to start a second Ph.D. in your group, I would do it again without the slightest hesitation.
I feel like I have grown incredibly in the past five years, and that is mostly due to the amazing people I have been working with. Thank you Ambreen, Maria, Etienne, Fred, Marc and Nicolas for being so awesome and for the countless discussions, whether it was about politics, religion, the meaning of life or about how robots will take over the world, every moment spent with you guys is cherished and you will be dearly missed. Special thanks to the new guys: Étienne, Nicolas, Julien and Matthew for being awesome colleagues and for collaborating on many of the new projects, you guys have a great future ahead.
I don’t know how I could possibly use words to express my gratitude for meeting and working with my partner Marc-André Légaré. It was not always easy, but through the years, projects and experiences, we have done so much together and have become so close. I’m not sure how it’s going to be when it comes to the point where our roads have to split, but I’d rather not think about that and reflect on the awesome times drinking pivo, playing games, discovering chemistry together and all the rest…. Thank you so much for everything! Of course, none of this would have been possible alone and I would like to thank every other person who have contributed to the projects and/or worked in the Fontaine lab. Jérémie, Simon, Celia, Sékou, Guillaume and the new interns Lydia and Thomas, it was a pleasure to get to know you all.
xxviii
Special thanks also to Laurent Maron and Christos Kefalidis, not only for welcoming me in Toulouse and teaching me the dark arts of DFT, but also for the good times. Without you, my Ph.D would have never been the same. Special thanks also go to Douglas W. Stephan for being such an open minded, down to earth person. It is really a great honor and pleasure to have the opportunity to work with such an outstanding person and chemist. Similarly, huge thanks to Alex Pulis for the time invested in the hydrogenation project, it was a real pleasure collaborating with you and I sure hope we will have the opportunity to do so again in the future. Special thanks also to the team in Toulouse (Didier Bourissou, Ghenwa Bouhadir and Richard Declerq) for the collaborative effort on pinpointing the role of the formaldehyde adduct. I am also highly indebted to the research professionals in the department. Pierre Audet and Wenhua Bi for their continuous help with the NMR and X-ray experiments. I would also like to thank the members of my thesis committee, professors Douglas W. Stephan, Peter H. McBreen and Stephen Westcott for taking some of their precious time for reading my thesis and coming to my defense.
None of this would have been possible without the support of my family throughout the course of my undergraduate and graduate studies. In fact, without this support, I don’t think I would have even set foot in a university! Thank you so much, mom, dad and France for believing in me. I was doing this for you guys and I still am, but thanks to your continuous encouragement to push forward, I finally found what I was meant to do. Also thanks to all the Aubut and Courtemanche family, my aunts, uncles, and everyone else! I cannot continue without thanking my friends who made the last 7 years in Québec city seem like nothing at all. Thanks for the all the fun Antoine, Félix, Pierre, Oli, Vince, Marc-Alex, Frank, J-R, Thomas and all the others… Last but not least, the continuous support through the good and hard times, the encouragement as well as the understanding of my life partner Maude is what keeps me going every day. Warmest thanks to you my love.
xxix
Inserted research articles and author contributions:
The following section lists the published work that is included in every chapter as well as the contribution of authors for every publication. All of the publications that are included in this thesis have been published at the time this thesis is being deposited.
Chapter 2: Courtemanche, M.-A.; Larouche, J.; Légaré, M.-A.; Wenhua, B.; Maron, L.; Fontaine, F-G A Tris(triphenylphosphine)aluminum Ambiphilic Precatalyst for the
Reduction of Carbon Dioxide with Catecholborane. Organometallics, 2013, 32, 6804-6811. Invited contribution
Author contributions: Most of the experimental work was made by MAC but MAL and Jérémie Larouche (JL) helped with the synthesis of compounds. JL performed the V.T. experiment. The manuscript writing and editing was mostly done by MAC and FGF. The manuscript was reviewed and edited by MAL. All of the DFT calculations was done by MAC. Laurent Maron (LM) supervised the DFT work.
Chapter 3: Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F-G. A Highly Active Phosphine–Borane Organocatalyst for the Reduction of CO2 to Methanol Using
Hydroboranes J. Am. Chem. Soc. 2013, 135, 9326-9329.
Author contributions: The catalyst was discovered, isolated and crystallized by MAC. While
the catalytic activity was discovered and the first catalytic experiments were done by MAC, most of the experimental screening and the in-situ generation of the formaldehyde adduct were done by MAL. The catalytic tests with methyl formate and the DFT calculations were performed by MAC. The manuscript was written by MAC, FGF and MAL. LM supervised the DFT work.
Chapter 4: Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F-G. Reducing CO2
to Methanol using Frustrated Lewis Pairs: On the Mechanism of Phosphine-Borane Mediated Hydroboration of CO2. J. Am. Chem. Soc. 2014, 136, 10708-10717.
Author contributions: All of the DFT calculations as well as all the experimental work were
xxx
kinetics, data analysis and possible reaction pathways. The manuscript was written by MAC, with revisions and edits by FGF and LM. LM supervised the DFT work.
Chapter 5: Courtemanche, M.-A.; Légaré, M.-A.; Rochette, E.; Fontaine, F.-G. Phosphazenes: efficient organocatalysts for the catalytic hydrosilylation of carbon dioxide
Chem. Commun, 2015, 51, 6858-6861.
Author contributions: The general concept of the reaction was developed by MAC and MAL.
The isolation of the reaction products with CO2 was made by MAC. The catalytic screening
experiments were performed by MAC and Étienne Rochette (ER). The manuscript was written by MAC, then reviewed and edited by all the authors.
Chapter 6: Courtemanche, M.-A.; Pulis, A. P.; Rochette, R.; Légaré, M-A.; Stephan, D. W.; Fontaine, F.-G. Intramolecular B/N Frustrated Lewis Pairs and the Hydrogenation of
Carbon Dioxide. Chem Commun, 2015, 51, 9797-9800
Author contributions: The concept and original reactivity of CO2 hydrogenations were
developed by MAC. Alexander P. Pulis (APP) and MAC performed most of the experimental work, with occasional synthetic help from ER and MAL. The manuscript was written by MAC and heavily edited by Douglas W. Stephan (DWS) after discussions. All the authors reviewed and edited the manuscript prior to publication. Most of the DFT calculations were performed by ER under the supervision of MAC.
Note: the data collection and refinement as well as resolving of all the crystal structures were made by Wenhua Bi.
1
1 Introduction
1.1 A note on chronology
The field of carbon dioxide reduction that is being covered by this thesis has been evolving at an incredible pace since the beginning of my doctoral studies. As such, it would be difficult for the reader to properly situate the present work if all of the modern systems were presented in the introduction. For these reasons, many results by other research groups will be reported in the main chapters according to the chronological order in which the work was published. Therefore, the literature review in the introduction will be mostly limited to contributions dating before the first results contained in this thesis were published. Again, this is so that the reader may have a better sense of the mindset in the field at the time the work was taking place. Before each new chapter, a short introduction section describing the advances in the field will be included in order to get the reader up to speed on the developments. Occasionally, anachronisms relating to future systems may be included for a more in-depth analysis. In such cases, the timescale will be explicitly discussed as to avoid confusion.
1.2 A note on compound identification
In this thesis, every chapter will present a unique numbering of compounds as to avoid the necessity of going back and forth through chapters to be reminded of the structure of a certain compound. In the cases where compounds appear in more than one chapter, they will be explicitly presented once again and they will be assigned a new number for the chapter.
2
1.3 General introduction
The industrial revolution is one of the most important events in human history.1 The transition
to new industrial processes involving the use of steam or coal powered machines and large scale chemical processes allowed the rapid development of manufacturing industries. For instance, the large scale production of acids such as sulfuric acid or polyvalent bases like sodium carbonate proved critical in the early development of the textile, soap and paper industries.2 From that point, the fields of science and technology started evolving
dramatically. Continuous industrialization, development and evolution has consistently led to a significant increase of living standards and consequently, to an uninterrupted increase of global population. While the world population today stands around 7 billion, it is expected to increase to up to 11 billion by 2050, a 57% increase in less than 40 years. 3
Such a striking expansion inevitably leads to an augmented need for primary resources. In an increasingly technological society, energy has become one of the most important of such resources. In 2004, the worldwide energy consumption was about 15 terawatts (TW). In 2025, it is expected that this number will increase to 21 TW and even reach 30 TW in 2050, doubling the energy consumption in less than 50 years. The production of such an enormous amount of energy would require 30 000 nuclear power plants of 1 gigawatt (GW) annual output, meaning that one such power plant would need to be built every single day for the next 50 years in order to meet the energetic demand.4
Today, over 86% of the energy that is being used originates from some form of fossil fuel such as petroleum, oil, natural gas, coal and others.5 The limited amount of these resources
stands in contrast to the ever growing energy demand of society, up to a point where the earth’s natural reserves are threatened of depletion.6 The burning of tremendous amounts of
fossil fuels is not only a short term solution, but is also at the origin of many serious environmental problems. Thus, one of the most critical issues of the 21st century is to identify
how the energetic needs of our society can be satisfied without jeopardizing the future of our planet.
3
1.4 Carbon dioxide and the greenhouse effect
Burning hydrocarbons generates primarily water (H2O) and carbon dioxide (CO2), which are
released in earth’s atmosphere. While CO2 is essential to life, it can absorb thermal radiation
from the surface of the earth and re-radiate this energy to the lower atmosphere, resulting in a net increase of earth’s temperature, a process called the greenhouse effect.7 Emissions of
this greenhouse gas (GHG) have been increasing constantly since the industrial revolution and the concentration in the atmosphere has recently reached over 400 ppm for the first time in the history of mankind (Figure 1-1 a).8 Up until now, most of the negative effects from the
increase in CO2 concentration have been mitigated by natural CO2 sinks such as oceans, trees
and soil.9 Still, the increased amount of CO
2 in the atmosphere has already led to an overall
rise of temperature on a planetary scale (Figure 1-1 b,c).10
Figure 1-1: a) CO2 concentrations in the atmosphere b) Global temperature anomalies c)
4
The consequences of this global warming, which will worsen as the capacity of the natural sinks reach their limit, include the retreat of glaciers, the rise of sea levels, changes in precipitation patterns, ocean acidification and species extinction due to a quick shift of temperature regime.9 While they are difficult to predict, the ramifications of such
environmental disasters will certainly engender serious consequences on the lifestyle of mankind.
The sequestration and underground storage of carbon dioxide was proposed as a possible avenue to reduce the atmospheric concentration of this GHG. Unfortunately, such a solution remains temporary and presents serious security issues.11 Another solution that comes to
mind is the limitation of CO2 emissions. Fortunately, technologies toward the direct recovery
of carbon dioxide from large industrial emitters have made significant progress during the past few decades, making it possible to significantly reduce emissions. One of the most promising avenues for CO2 capture was first invented by R. R. Bottoms in 1930.12 While the
process was continuously optimized until today, the core concept relies on the same principle: CO2 is transformed into carbamates by reaction with an amine and is subsequently released
by heating (Scheme 1-1).13
Scheme 1-1: Schematic representation of the amine scrubbing process.
Unfortunately, except for the carbon tax concept, of which the development is stagnating, there is very little economic incentive for the industry to recover carbon dioxide emissions as this waste product does not have significant commercial value. However, the possibility of transforming it in value added products could provide an incentive for industrial companies to recover the greenhouse gas since the recovery process would become lucrative.14 In fact, a number of processes utilize CO
2 directly as a feedstock for the synthesis
of fine chemicals, but since these are mostly carried out on relatively small scales, they are of limited interest in the fight against global warming.15
5 The industrial processes that uses by far the most carbon dioxide are the synthesis of urea and propylene polycarbonate.16,17 In 2004, 72 megatonnes (Mt) of CO2 were used to produce
urea and as much as 100 Mt were used to produce propylene polycarbonate.18 Looking at the
bigger picture and comparing these numbers to the anthropogenic emissions of 25.6 gigatonnes (Gt) per year, we find that the combined processes utilize barely 0.7% of the excess CO2 that is produced by humans every year.
In a serious analysis of CO2 transformation products, the economical aspect must not be
neglected. In order for the market to stay balanced, the offer must not exceed demands, but in a scenario where gigatonnes of products must be used to have an actual impact on the atmospheric concentrations, finding a product in continuous demand on such a scale leaves very few candidates. Even though the transformation of CO2 into value added products such
as carbonates, esters, lactones, carbamates, pyrones, isocyanates and many more commodity chemicals provides a promising, green alternative to the actual processes, the scale at which these products are used is not sufficient to have a significant impact on the CO2 concentration
in the atmosphere.19 For this reason and to remain succinct, this thesis will mostly be directed
towards the transformation of CO2 to highly energetic material such as methane, methanol
and others.
1.5 Using CO
2as an energy vector: the methanol economy
Indeed, one of the only fields in which value added products derived from carbon dioxide could be used on a scale sufficient to have an impact on the atmospheric concentrations is the field of energy. In fact, while most of the energy sources used by mankind produce either heat or electricity, the latter cannot be efficiently stored for prolonged periods of time. Furthermore, the transportation of electricity over long distances is inefficient and wasteful.20
Consequently, there is much room for improvement in terms of energy storage, transportation and distribution. Hydrogen (H2) has been suggested as a clean, renewable and green
alternative energy medium.21 However, despite intense investments from governmental and
private instances as well as many efforts in terms of research and development, a number of underlying fundamental problems associated with the use of H2 remain unsolved. Hydrogen
is a highly volatile and explosive gas, raising safety concerns for its large scale transportation or storage. Furthermore, current automotive technologies are not adapted to use hydrogen as
6
a fuel and the required technology transfer to replace service-stations would engender astronomical costs.20
On the other hand, the use of CO2 to produce energy vectors, that is the storage of energy in
carbon dioxide through chemical transformations followed by the release of said energy by combustion, could provide an entirely carbon neutral strategy to the energy storage problem. Indeed, the simple concept of transforming the greenhouse gas into highly energetic hydrocarbons such as methane (CH4) or methanol (CH3OH) would provide a very practical
way to store energy (Figure 1-2).
Figure 1-2: Conceptual representation of the use of CO2 to produce energy vectors.
While the use of methane gas raises concerns similar to those that were described for hydrogen, methanol would be the ideal candidate due to its similarity with the hydrocarbons that are used on a massive scale every day. It is a relatively non-toxic liquid that can easily be stored and transported on a large scale.22 The combustion of methanol generates only
water and carbon dioxide, making it a very green alternative to hydrocarbons which generate polluting mono-nitrogen oxides (NOx), sulfur dioxide (SO2) and other polluting particles.
Furthermore, very little modifications would be required to incorporate the use of methanol into everyday life. A simple, cheap reconfiguration of combustion engines would allow modern cars to run exclusively on methanol fuel. An investment of only $50 000 would be required to transform a regular hydrocarbon dispensing based station into a fully functional methanol dispensing gas station, a sharp contrast with a transformation into a hydrogen based gas station necessitating an investment of $1 000 000.22
7 The use of methanol as a fuel is far from being a new concept. In fact, one of the world’s first cars, the Ford model T was powered by methanol fuel. With the technological advances of the modern world, methanol can now be used from public transportation buses to highly performant race cars.20
On the other hand, hydrocarbon based fossil fuels offer much more than energy. From plastics to synthetic materials, from polymers to pharmaceuticals, hydrocarbon derivatives are essential to everyday life. Fortunately, most of these can easily be derived and produced from methanol, making it possible to imagine an economic system based solely on the use of methanol as a raw material to replace fossil fuels, a concept developed and promoted notably by Nobel laureate George A. Olah and defined as ‘’The methanol economy’’.20,22–24 Through
the methanol to olefin process (MTO), methanol can be converted to ethylene or propylene, generating the required basic building blocks for most of the petrochemical based products.25
Furthermore, methanol can be used as synthetic material for the generation of commodity chemicals such as polymers, acetic acid, paints, dyes, adhesives and much more. Exploiting the concept of the methanol economy to its full potential would allow us to become entirely independent from fossil fuels and stop emitting carbon dioxide in the atmosphere, slowing down global warming significantly. All of this without affecting our capacity to produce hydrocarbon based commodity chemicals.
1.6 Challenges in the transformation of carbon dioxide
In order to become entirely independent from fossil fuels and generate a sustainable economy, the energy must come from a cheap, green and renewable source. Much research is ongoing towards the use natural sources of energy such as solar, hydroelectric, wind and geothermal processes.26 These technologies need to be developed to provide the world with
the required energy, but while this problem is of fundamental importance, it is beyond the scope of this thesis to discuss the details of such technologies.
Indeed, another underlying problem needs to be solved before clean energy can be implemented into the methanol economy. It is not a coincidence that such a tremendous amount of CO2 ends up in the atmosphere. Factually, carbon dioxide is a very stable molecule
(ΔHform=-392.5 kJ.mol-1). Therefore, its transformation often requires highly reactive
8
Such prerequisites are problematic as a lot of energy is being wasted. For instance, methanol is currently produced on a very large scale (100 Mt/year), mostly by steam reforming, a non-sustainable process that combines non-renewable methane from natural gas and water to generate methanol, hydrogen and water. The process involves a Cu/ZnO/Al2O catalyst and
operates at high temperature (250 °C) and high pressure (250 MPa).27 Other technologies
involving the use of hydrogen or geothermal heat are emerging, but they are still at the stage of early development and both still require high temperature and pressures to effect reduction to methanol. Consequently, new, more efficient processes for CO2 reduction must be
uncovered in order to facilitate the storage of energy into the small molecule. To develop such processes, the basic properties and reactivity patterns of CO2 must be understood. Only
then can new concepts and avenues towards its reduction can be revealed and it is around the context of opening such new avenues that this thesis is constructed.
1.7 The carbon dioxide molecule
Carbon dioxide is a thermodynamically stable molecule that belongs to the D∞h point group
and is linear in its ground state, making it non-polar. However, due to the possible resonance forms of carbon dioxide and the electronegativity of the oxygen atoms, the molecule possesses an ambiphilic character. Indeed, the oxygen atoms are slightly nucleophilic, while the carbon atom itself is electrophilic (Figure 1-3). Therefore, two main strategies can be employed in order to promote a reaction with the molecule. First, the oxygen atoms could bind to an electron deficient active center, or an electron rich atom could interact with the carbon atom.
9 The Walsh diagram of CO2, which plots the change of molecular orbital energy levels with
respect to the molecule’s geometry is depicted in Figure1-4. A close look at the diagram reveals that upon bending of the CO2 molecule, the energy variation of the π orbitals is much
more pronounced than that of the σ orbitals. In fact, the sigma (σ) orbitals show little to no destabilization, while the degeneracy of the pi (π) orbitals are split upon bending (2πu to 6a1).
For the highest occupied molecular orbital (HOMO), it is clear that the bending is not favorable since both components are energetically higher upon bending, meaning that if one binds the CO2 molecule through oxygen interaction with an electron deficient molecule, the
molecule will remain essentially linear.
10
In contrast, while one component of the lowest unoccupied molecular orbital LUMO remains largely unaffected, the out of plane component (corresponding to the anti-bonding orbital localized on the carbon atom) is drastically stabilized, to the point where it starts mixing with other bonding orbitals. Translating this observation in terms of reactivity reveals that introduction of electron density in this LUMO orbital will lead to bending of the CO2
molecule, drastically changing its fundamental properties. The ambiphilic nature of carbon dioxide and the consequences that stem from this unique characteristic will be underlined throughout the introduction since it constitutes the foundation of the research project motivating the work described herein.
1.8 Organometallic binding of carbon dioxide
While many believe that Aresta was the first to trap carbon dioxide using the now well-famed electron-rich Ni0 species depicted in Scheme 1-2,28 the coordination of carbon dioxide to
organometallic species was discovered almost a decade before this seminal discovery. In a 1972 review, Volpin already describes a number of transition metal CO2 adducts, albeit with
very limited knowledge of the involved coordination modes.29 One year before, the first
crystallographically characterized complex of CO2 was reported. Unfortunately, the
limitations of the method at the time did not allow the authors to unambiguously determine the coordination mode of the carbon dioxide moiety.30 In 1974, Floriani was already taking
advantage of the ambiphilic nature of CO2 and reported a cobalt salen complex that was
proposed to bind CO2 through nucleophilic interaction of the electron rich cobalt center to
the electrophilic carbon atom of CO2. The negative charge on the oxygen atoms was proposed
to be stabilized by the sodium counter-ion.31Nonetheless, Aresta’s complex was the first
completely structurally characterized metal complex of CO2 and constitutes a major
breakthrough in the field (Scheme 1-2).
11 In this complex, the nucleophilic and electrophilic properties of CO2 are both exploited. In
the years that followed, the rich reactivity of carbon dioxide with metal complexes was slowly uncovered. The key results as well as the coordination modes for a variety of transition metal centers are summarized in Table 1-1. 19
12
Table 1-1: Summary of key isolated transition metal CO2 complexes.
Coordination mode Structure Metalref C-O bond length (Å)
η1-O U32 1.122(4), 1.277(4)
η1-C Rh33, Ir34 1.20(2), 1.25(2)
η2-C-O Ni,35–37 Rh,38 Fe,39 Pd40 1.17, 1.22
μ2-η2 M1 O C O M2 Pt 41,42 Ir/Zr,43 Ir/Os,44 Rh,45 Ru46,47 1.229(12), 1.306(1) μ2-η3 Re/Zr,48 Ru/Zr,49
(Ru/Ti, Fe/Zr, Fe/Ti)50
1.285(5), 1.281(5) μ2-η3 Re/Sn,51 Fe/Sn52 1.269(1), 2.257(7) 1.252(3), 2.394(2) μ3-η3 Os,53,54 Re55 1.276(5), 1.322(5) 1.28, 1.25 μ3-η4 Co31,56,57 1.20(2), 1.24(2) μ4-η4 Ru58 1.283(2), 1.245(2) μ4-η5 Rh/Zn59 1.29(14), 1.322(1)
13 Looking at Table 1-1, it can be determined that by increasing the coordination number of CO2, the C-O bond order diminishes, resulting in longer C-O bond lengths. Even with such
a wide range of unique CO2 complexes, none exhibited catalytic activity in the reduction of
the small molecule. However, some metal complexes were developed for the catalytic transformation of CO2 into value added chemicals. For an in-depth discussion on the
formation of value added products such as carbonates, esters, lactones, carbamates, pyrones and isocyanates, the reader is directed to recent review articles.60,61 Indeed, as it was stated
before, the scope of this thesis is limited to the transformation of CO2 into energy rich
materials.
1.9 Carbon dioxide for energy storage: general aspects
Simply put, the combustion of hydrocarbons is an oxidation reaction. Let us take the combustion of methane as an example: in the presence of heat, the carbon atom is oxidized from its -4 state to +4 oxidation state, an exothermic reaction that generates energy in the form of heat (Scheme 1-3).
Scheme 1-3: Aerobic combustion of methane.
It is logical that in order to store energy in the CO2 molecule, the opposite reaction must take
place. In order to achieve this, some form of energy is required. Also, due to the high stability of the CO2 molecule, a catalyst is often required to accelerate the reaction. In Scheme 1-4,
the reduction of CO2 using hydrogen as primary energy source is depicted. By transferring
two, four, six or eight electrons to carbon dioxide, it would be possible to generate formic acid, formaldehyde, methanol or methane respectively. 62,63
14
Scheme 1-4: Stepwise CO2 hydrogenation.*
*Energies are reported in kJ mol-1. Thermodynamic values are given for aqueous solutions of H2 and CO2, while the products are in liquid phase.
This multi-step process is at the core of the challenge of CO2 reduction. In fact, a dichotomy
exists in this reaction: the inherent stability of the CO2 molecule mandates the use of highly
reactive catalysts, yet the catalyst must remain stable in the presence of all the fundamentally different reaction products. Another major challenge resides in the design of a catalyst that will selectively generate a single desirable reduction product.
1.10 Metal enzymes for CO
2reduction
As early as 1976, evidence for the possibility of a catalytic CO2 reduction process was put
forward. An enzyme, formate dehydrogenase (FateDH) was shown to catalytically reduce
carbon dioxide to formate by using a reduced form of nicotinamide adenine dinucleotide (NADH) as an electron source.64 In 1999, the ingenious use of a cascade of three different
enzymes, namely FateDH, formaldehyde dehydrogenase (FalDH) and alcohol dehydrogenase
(ADH) catalyzed the reduction of CO2 all the way to methanol.65 However, the use of the
NAD/NADH couple for reduction is not a viable option in terms of large scale transformation due to the high cost of this energy source.66 Nonetheless, an in-depth study of these biological
systems could lead to hints regarding the important parameters in CO2 reduction chemistry.
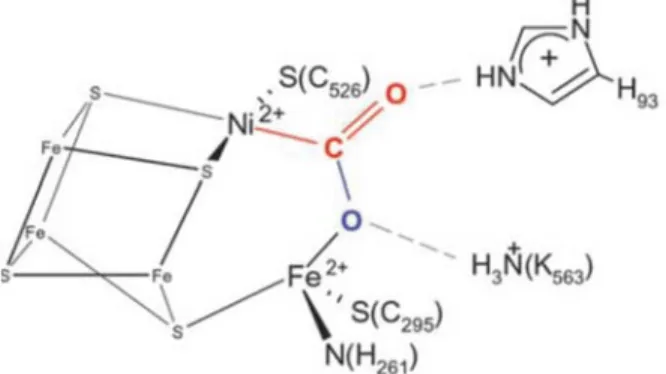
In 2007, a major breakthrough in enzyme mediated carbon dioxide reduction was made when an intermediate of CO2 reduction was crystallized, allowing the postulation of a plausible
15 In one of the key steps of the reaction, it was found that the ambiphilic activation of carbon dioxide, which can be defined as the simultaneous activation by an electron rich and an electron withdrawing fragment, was of crucial importance. In Figure 1-5, which represents a key transition state, it is possible to observe that the electrophilic iron center interacts with the oxygen atom of CO2 while the nucleophilic nickel center interacts with the carbon atom.
Figure 1-5: Transition state in the process of CO2 reduction by CO dehydrogenase.*
*Figure reproduced from (Jeoung, J.-H.; Dobbek, H. Science, 2007, 1461-1464). Reprinted with permission from AAAS.
1.11 Transition metal catalyzed reduction of CO
2to energy rich materials
Meanwhile, a number of heterogeneous catalytic systems capable of hydrogenating carbon dioxide to methanol have been reported. Yet, such catalysts require elevated operating pressure (40-50 atm) and temperatures (200-250°C). Not only does the high temperature regime consume important amounts of energy, but it also limits the theoretical yield of the reaction since the products are entropically disfavored.62
These limitations encouraged chemists to develop homogeneous catalysts capable of operating at lower temperatures and pressures, but the task proved challenging. Perhaps inspired by the enzymatic cascade stratagem, Sanford reported in 2011 that the use of three homogeneous catalysts (PMe3)4Ru(Cl)(OAc), Sc(OTf)3, and (PNN)Ru(CO)(H) PNN =
(2-(di-tert-butylphosphinomethyl)-6-(diethylaminomethyl)pyridine) operating in sequence could lead to the formation of methanol (Scheme 1-5).68 While interesting in concept, the
turnover number (TON) remained limited (TON = 21) and the reaction conditions were still far from ideal (135 °C, 10 atm H2, 30 atm CO2).
16 13CO 2 A,B,C 12CH 3OH 13CH 3OH + H2O Ru PMe3 Me3P PMe 3 Me3P Cl OAc Sc(OTf)3 Ru CO N N P(t-Bu)2 (A) (B) (C)
Scheme 1-5: Cascade catalytic hydrogenation of CO2 to methanol.
Utilizing hydrosilanes as a primary source of chemical energy and an iridium catalyst, Eisenberg managed to reduce CO2 to methoxysilanes, products that can be readily
hydrolyzed to methanol, albeit with limited efficiency.69 Then, it was shown that the use of
a zirconium catalyst in combination with the highly Lewis acidic B(C6F5)3 (BCF) promoted
the hydrosilylation of carbon dioxide all the way to methane, reaching a TON of 225.70 In
2010, Guan and collaborators broke the record in efficiency for the reduction of CO2 to
methanol. Using catecholborane (HBcat) as a reducing agent, they were able to show that a nickel pincer catalyst could reduce CO2 to methanol derivatives with an impressive turnover
frequency (TOF) reaching 495 h-1 (Scheme 1-6).71,72
17 While the reduction of CO2 to energy-rich materials was still only emerging, a number of
highly active systems had been reported for the hydrogenation of CO2 to formates.73–79 Even
though the detailed description of such systems is beyond the scope of this thesis, one particular system must be discussed in more detail.
In 2011, Hazari and Crabtree reported an iridium based pincer catalyst exhibiting impressive reactivity for the hydrogenation of CO2 to formic acid (Scheme 1-7).80 As was discussed
earlier, the generation of formic acid is thermodynamically disfavored and must be trapped with a stoichiometric amount of base. Nonetheless, the system reached turnover numbers up to 348 000 with a TOF of 18 000 h-1. Once again, drawbacks of the system include high
operating temperature 185 °C and high pressure (55 atm).
Scheme 1-7: Dual interaction of reduced CO2 with an Ir pincer complex.
However, the key observation in this system was the particular mode of action of the catalyst where CO2 interacts through hydrogen bonding with the N-H moiety of the catalyst. The
authors suggest that this weak interaction is key in promoting efficient catalysis, once again underlining the importance of synergistic interactions with CO2.
1.12 Photochemical reduction of carbon dioxide
The ability of certain metal centers or semiconductors to absorb sunlight has led to development of catalysts for the photochemical conversion of CO2 to energy enriched
reduction products using water as the primary electron source. Much advance has been made in the last two decades and the subject has been recently reviewed.81,82 It is unfortunate that
18
While the field represents a very interesting strategy for cheap, efficient recycling of CO2,
much remains to be done to achieve the activities required for large scale applications
1.13 Transition-metal free transformations of carbon dioxide
As early as 1950, it was reported that lithium borohydride could reduce carbon dioxide to a formate derivative which could then be hydrolysed to formic acid (HCOOH).83 Shortly after,
it was reported that sodium borohydride could also promote the reduction of CO2 to a mixture
of formate products and that the product distribution was highly dependent on the employed conditions.84,85 In 1967, a similar strategy was employed, but this time for the stoichiometric
reduction of CO2 to formic acid in aqueous conditions.86 Very recently, this aqueous
reduction chemistry was revisited, but the activity could not be expanded beyond stoichiometric transformations.87,88 A drastically different strategy has also been used to
effect stoichiometric CO2 reduction. Indeed, it was recently reported that in the presence of
CO2, highly Lewis acidic silylium ions react to yield a mixture of benzoic acid, formic acid
and methanol after aqueous workup.89
Pushing the silylium ion strategy a step further, a catalytic system for the Lewis acid catalyzed reduction of CO2 could be developed. By using a high loading of AlEt2+ (10 mol%),
the catalytic hydrosilylation of CO2 to methane and several solvent alkylation by products
was carried out by Wehmschulte and co-workers. 90 Using control experiments, the authors
were able to obtain evidence for the catalytic role of the Lewis acidic aluminum species as independently synthesized silylium ions exhibited drastically inferior reactivity. While interesting in concept, the system suffers from a number of drawbacks including slow activity (50% conversion after 14h at 80 °C) and lack of selectivity (Scheme 1-8). A second generation catalyst was developed by the same research group, but the use of a highly sterically congested catalyst [Al(2,6-Mes2C6H3O)2Al]+ (Mes=2,4,6,-trimethylphenyl) still
led to important selectivity problems. 91
19 In contrast to the Lewis acid activation strategy, an antagonistic approach relying on Lewis base activation was developed for the catalytic reduction of CO2 to high energy content
molecules. The use of an N-heterocyclic carbene (NHC) catalyst presumably promoted the hydrosilylation of CO2 to methoxysilanes with over 90% selectivity. The authors proposed
the reduction to occur either through coordination of the NHC moiety to the reducing silane, generating a nucleophilic hydride capable of CO2 reduction (Figure 1-6, pathway A), or
through coordination of the NHC to CO2, rendering the molecule more reactive towards
reduction. (Figure 1-6, pathway B).
Figure 1-6: Proposed mechanistic pathways for the base-catalysed hydrosilylation of CO2.
Wang et al. supported that pathway A was more plausible in a theoretical study,92 arguing
that such a mechanism would account for the experimental evidence that bulkier silanes exhibited lower reactivity. While the record-breaking TON and TOF of 1840 and 25.5 h-1
exceed the performance of most catalysts, the important solvent effects that were observed experimentally remained mostly unexplored. Indeed, a recent report by our group raises certain concerns regarding the reported turnovers. For an in-depth discussion, the reader is directed to chapter 6.
Still, these results suggest that carbon dioxide reduction can be promoted both by Lewis acids and bases. The general strategy that is exploited in this thesis relies on using both of these activations in a synergistic fashion. But before getting into more details, an introduction to Frustrated Lewis Pair (FLP) chemistry is mandatory.
20
1.14 A short history of Frustrated Lewis Pair chemistry
Molecules were first classified as electron donors or acceptors by Gilbert Lewis, thus creating the foundation for the concept of Lewis acids and bases.93 Lewis acids are generally
compounds possessing an empty orbital that can accept electron density from an electron rich molecule. Alternately, a Lewis base will typically possess a free doublet of electrons capable of giving electron density to an electron poor molecule. A more general way of classifying these molecules would be to say that Lewis acids have a low lying LUMO while Lewis bases have a high lying HOMO. In most cases, when combined, Lewis pairs form Lewis adducts, with the electron density of the Lewis base fulfilling the electronic deficiency of the Lewis acid, resulting in significant thermodynamic stabilization (Scheme 1-9).
Scheme 1-9: Formation of a classical Lewis adduct.
In 1942, Brown and co-workers made an interesting observation: lutidine (2,6-dimethylpyridine) forms a Lewis adduct when combined with trifluoroborane (BF3) but does
not react in the presence of trimethylborane (BMe3). The lack of reactivity between lutidine
and BMe3 was attributed to the steric congestion that prevented the formation of a classical
Lewis adduct.94 More than a decade later, Wittig and Benz noted that triphenylphosphine
(PPh3) and triphenylborane (BPh3) showed no evidence of adduct formation when combined.
Nonetheless, when a mixture of the two compounds was exposed to benzyne which was generated in-situ from 2-iodobromobenzene and Mg0, a zwiterrionic phosphonium-borate