Université de Montréal
Importance pathophysiologique de l’hyperendothélinémie
sur la réactivité vasculaire pulmonaire
Par Annik Migneault
Département de physiologie, Université de Montréal Faculté de médecine
Mémoire présenté à la Faculté des études supérieures en vue de l’obtention du grade de
Maître ès sciences (M. Sc.) en physiologie
Avril, 2003
(E)
dh
de Montré al
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal à reproduire et diffuser, en totalité ou en partie, par quelque moyen que ce soit et sur quelque support que ce soit, et exclusivement à des fins non lucratives d’enseignement et de recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le mémoire, ni des extraits substantiels de ce document, ne doivent être imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin de se conformer à la Loi canadienne sur la protection des renseignements personnels, quelques formulaires secondaires, coordonnées ou signatures intégrées au texte ont pu être enlevés de ce document. Bien que cela ait pu affecter la pagination, il n’y a aucun contenu manquant. NOTICE
The author of this thesis or dissertation has granted a nonexciusive license allowing Université de Montréal to reproduce and publish the document, in part or in whole, and in any format, solely for noncommercial educational and research purposes.
The author and co-authors if applicable retain copyright ownership and moral rights in this document. Neither the whole thesis or dissertation, nor substantial extracts from it, may be printed or otherwise reproduced without the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact information or signatures may have been removed from the document. While this may affect the document page count, it does not represent any loss of content from the document.
Ce mémoire intitulé:
Importance pathophysiologique de I’hyperendothélinémie sur la réactivité vasculaire pulmonaire
présenté par Annik Migneault
a été évalué par un jury composé des personnes suivantes:
Dr Éric Thorin DrJocelyn Dupuis, MD
Dr Normand Leblanc Dr Pierre Moreau
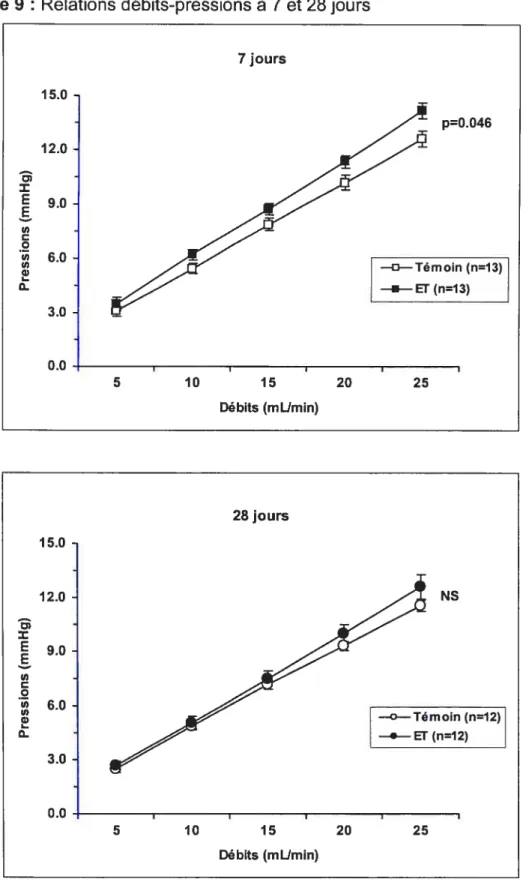
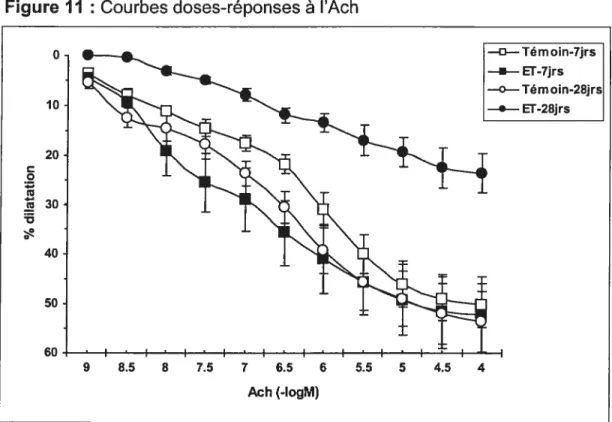
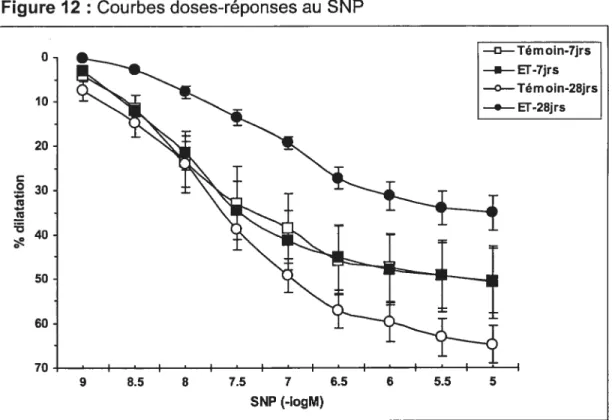
De nombreuses études ont confirmé l’activation du système endothéline (ET) dans l’hypertension pulmonaire (HTP). L’impact pathophysiologique de l’hyperendothélinémie isolée sur la circulation pulmonaire et le ventricule droit (VD) est cependant inconnu. Méthodes Des rats ont reçu pendant 7 ou 28 jours une perfusion constante d’ET-l (10 nglkglmin) par minipompes osmotiques. Ceci résulta en 4 groupes: témoin Zjrs (n=13), ET-7jrs (n=13), témoin-28jrs (n=12) et ET-28jrs (n12). Des mesures hémodynamiques et morphométriques ainsi que des relations débits-pressions pulmonaires furent effectuées. La réactivité isométrique de petites artères pulmonaires (250 pm; n=6 à 10/groupe) a aussi été évaluée. Résultats : Les niveaux veineux et tissulaires pulmonaires d’ET-l ont doublé après perfusion pendant 7 et 28 jours (p<0.05). Les animaux n’ont pas développés d’HTP et d’hypertrophie VD. Seul le groupe ET-7jrs démontre une relation débit-pression pulmonaire plus élevée que son témoin (p<0.05). La sensibilité du groupe ET-Zjrs à l’Ach est augmentée (p<0.05 pour EC5O; EC75). Le groupe ET-28jrs démontre une diminution de sa réponse maximale à l’Ach (p<0.01) ainsi qu’au SNP (p<0.00l). La sensibilité du groupe ET-7jrs à l’ET-l est diminuée de façon significative (p<0.00l pour EC25; EC5O et p<0.05 pour EC75). La vasoconstriction maximale au U-46619 est augmentée dans le groupe ET-28jrs (p<0.O5). Conclusion : La perfusion chronique d’ET-l n’a pas causée d’HTP ou d’hypertrophie ventriculaire droite. Elle résulte cependant en une modification notable de la réactivité vasculaire pulmonaire caractérisée par une diminution de la réponse musculaire lisse au NO.
À
plus long terme, ceci pourrait contribuer au développement de l’HTP.Mots clés acétylcholine, artère pulmonaire, circulation pulmonaire, endothéline, endothélium, hypertension pulmonaire, muscle lisse, oxyde nitrique
Activation of the endothelin (ET) system contributes to the development cf pulmonary hypertension (PH). Although plasma ET levels correlate with the severity of PH, the impact of isolated chronic hyperendothelinemia on the pulmonary circulation and right ventricular (RV) function is unknown. Methods: Mini-osmotic pumps were implanted in rats to deliver ET-1 (longlkglmin) during for 28 days. Four groups were created (1) control f-days (n=13), (2) ET f-days (n=13), (3) control 28-days (n12) and (4) ET 28-days (n=12). After in vivo hemodynamic measurements, the Iungs were isolated to derive pressure-flow relations. Small pulmonary arteries (circa 250 pm, n=6 to 10/group) were mounted on an isometric myograph to study their reactivity. Resuits: Plasma and tissues ET-1 levels approximately doubled (p<0.05) after 7 and 28 days. RV systolic pressure and RV weight were flot affected by the infusions. The pulmonary pressure-flow relation shifted upward (p<0.05) in ET 7-days group, but not in ET 28-days group. ET 7-days displayed increased sensibility to Ach (p<0.05 for EC5O; EC75). Dilatations to Ach (p<0.01) and SNP (p<0.001) in isolated arteries were reduced in the ET 28-days. ET 7-days displayed greatly reduced contractility to ET-1 (p<0.001 for EC25; EC5O, p<0.05 for EC75). U-46619 vasoconstriction was increased in the ET 28-days group (p<0.05). Conclusion: One month of chronic hyperendothelinemia failed to induce PH or RV hypertrophy. However, its causes an important modification cf the pulmonary vascular reactivity characterized by a reduced response of smooth muscle to NO. In the long term, this may contribute to the developpement of PH.
Key words: acetylcholine, endothelin, endothelium, nitric oxide, pulmonary
Liste des tableaux .viii
Liste des figures ix
Liste des abréviations X
Introduction 1
1.1 Biologie de l’endothélium 1
1.2 Les facteurs relaxants dérivés de I’endothélium 3
1.2.1 Le monoxyde d’azote 5
1.2.2 La prostacycline 7
1.2.3 Le facteur hyperpolarisant dérivé de l’endothélium 8
1.2.4 L’adrénomodulline 9
1.2.5 Le facteur natriurétique de type C 10
1.3 Les facteurs contractants dérivés de I’endothélium 12
1.3.1 La thromboxane A2 12
1.3.2 L’angiotensine Il 13
1.3.3 L’anion superoxide 15
1.3.4 L’endothéline 15
1.4 Biologie du système endothéline 16
1.4.1.1 Les enzymes de conversion de l’endothéline-l 19
1.4.1.2 La sécrétion de l’endothéline-l 20
1.4.2 Les récepteurs vasculaires de l’endothéline-l 21
1.4.2.1 Voie de signalisation 22
1.4.2.2 Clairance de l’endothéline-l 24
1.4.3 Interaction monoxyde d’azote-endothéline-l 25 1.4.4 Physiologie et pathologie du système endothéline-1 26
1.4.4.1 Le développement 26
1.4.4.2 Les vaisseaux sanguins 27
1.4.4.3 Le muscle cardiaque 28
1.4.4.5 Le foie .30
1.4.4.6 Le cerveau 31
1.4.4.7 Le système respiratoire 32
1.5 La circulation pulmonaire 33
1.5.1 Réactivité des artères pulmonaires 34
1.5.2 Vasocontriction pulmonaire hypoxique 35
1.5.3 Hypertension pulmonaire 36
1.5.3.1 Classification 38
1.5.3.2 Modèles animaux 42
1.5.4 Traitements de l’hypertension pulmonaire 44
1.5.4.1 Les anticoagulants 44
1.5.4.2 Les bloqueurs des canaux calciques 45
1.5.4.3 Inhalation de monoxyde d’azote 45
1.5.4.4 Les analogues de la prostacycline 46
1.5.4.5 Les antagonistes de I’endothéline-l 48
1.5.4.6 La transplantation d’organes 50
1.6 Évaluation de la circulation pulmonaire 52
1.7 Objectif 55
Matériels et méthodes 57
2.1 Animaux utilisés 57
2.2 Méthologie générale 57
2.3 Mesures hémodynamiques in vivo 58
2.4 Préparation des poumons isolés 59
2.4.1 Relations débits-pressions et mesures morphométriques 59
2.5 Préparation des petites artères isolées 60
2.5.1 Réactivité des petites artères isolées 61
2.6 Homogénisation, extraction et dosage de l’endothéline-l 61
2.7 Composés utilisés 64
2.8 Évaluation statistique des données 64
2.9 Éthique 65
)
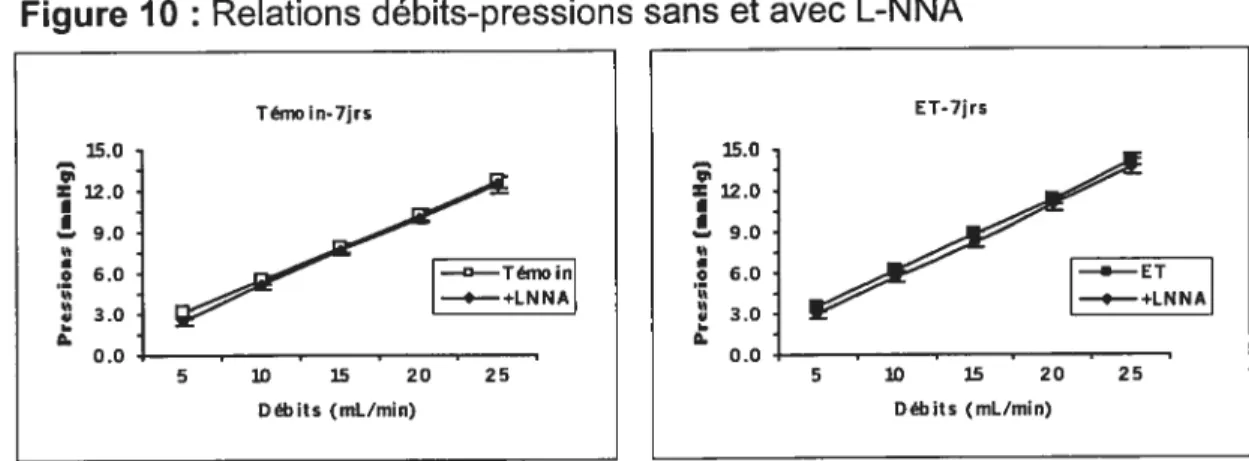
3.1 Dosage de l’endothéline-l 66 3.2 Paramètres hémodynamiques et morphométriques 673.3 Relations débits-pressions 69
3.3.1 Relations débits-pressions avec L-NNA 69
3.4 Réactivité des petites artères pulmonaires isolées 71
3.4.1 Vasodilatation 71
3.4.2 Vasoconstriction 75
Discussion 78
4.1 Dosage de l’endothéline-l 78
4.2 Paramètres hémodynamiques et morphométriques 80
4.3 Relations débits-pressions 81
4.4 Réactivité des artères isolées 83
4.4.1 Relaxation endothélium-dépendante 83 4.4.2 Relaxation endothélium-indépendante 85 4.4.3 Vasoconstriction 87
)
4.4.4 Vasoconstriction à l’endothéline-l 88 Conclusion 90 Remerciements 91 Références 92j
£ite
âs
ta61awc
Tableau I : Classification diagnostique de I’HTP .39 Tableau Il: Antagonistes sélectifs et non-sélectifs des récepteurs à l’ET-l ..49
Tableau III : Paramètres hémodynamiques et morphométriques 68 Tableau IV: Niveau de préconstriction et effet de l’Ach sur la réactivité
vasculaire pulmonaire 73
Tableau V: Niveau de préconstriction et effet du SNP sur la réactivité
vasculaire pulmonaire 74
Tableau VI: Contraction maximale et effet du U-46619 sur la réactivité
vasculaire pulmonaire 76
Tableau VII : Contraction maximale et effet de I’ET-l sur la réactivité
vasculaire pulmonaire 77
J
)
Liste dsfigures
Figure 1: Les facteurs relaxants dérivés de l’endothélium 4 Figure 2 : Les facteurs vasoactifs libérés par l’endothélium 5 Figure 3 : Structure primaire des endothélines-1, -2, et -3 humaines, de VIC
et de la sarafotoxine-6b 17
Figure 4 : Biosynthèse de l’ET-l 18
Figure 5: Mécanisme de la contraction induite par l’ET-l 23
Figure 6: Relation débit-pression typique 52
Figure 7 : Relations débits-pressions diverses 53 Figure 8 : Concentrations d’endothéline-l plasmatiques et tissulaires 66 Figure 9 : Relations débits-pressions à 7 et 28 jours 70 Figure 10: Relations débits-pressions sans et avec L-NNA 71
Figure 11: Courbes doses-réponses à l’Ach 72
)
Figure 12: Courbes doses-réponses au SNP 74Figure 13: Courbes doses-réponses au U-46619 75
Figure 14: Courbes doses-réponses à l’ET-l 77
)
Liste ds a6rbc)iations
(Lorsque l’abréviation réfère à un terme anglais, ce dernier est inscrit en italique)
AA: acide arachidonique AC : adénylate cyclase Ach : acétylcholine
ADP adénosine diphosphate Agil : angiotensine Il
AM adrénomodulline
AMPc: adénosine monophosphate cyclique
ANP : atrial natriuretic factor(facteur natriurétique de l’oreillette) ARNm : acide ribonucléique messager
ATP adénosine triphosphate BK: bradykinine
)
BMPR2: bone morphogenetic protein receptor 2 (récepteur 2 de la protéine morphogénétique osseuse)BNP : brain natriuretic factor(facteur natriurétique du cerveau) CNP: C-type natriuretic factor(facteur natriurétique de type C) COX: cyclooxygénase
DAG: 1,2-diacylglycérol
EC : effective concentration (concentration effective) ECA: enzyme de conversion de I’angiotensine ECE : enzyme de conversion de l’endothéline
EDCF: endothelium-derived contracting factor (facteur contractant dérivé de l’endothélium)
EDE : enzyme de dégradation de l’endothéline
EDCF: endothelium-derived contracting factor (facteur contractant dérivé de I ‘endothéliu m)
)
EDHF: endothelium-derived hyperpolarizing factor (facteur hyperpolarisant dérivé de I’endothélium)EDRF: endothelïum-derived relaxing factor (facteur relaxant dérivé de l’endothéliu m)
ELISA: enzyme Iinked immunosorbent assay (essai immu no-enzymatique) ET: endothéline
ETA: récepteur à l’endothéline de type A ETB: récepteurà l’endothéline de type B GCs: guanylate cyclase soluble
GMPc: guanosine 3’,5’-monophosphate cyclique H PETE : acide hyd ropéroxyeicosatétraénoÏque HPP: hypertension pulmonaire primaire
HPV: hypoxic pulmonary vasoconstriction (vasoconstriction pulmonaire hypoxique)
HTP: hypertension pulmonaire 1P3: inositol I ,4,5-triphosphate
L-NAME: No-nitro-L-arginine méthyl ester L-NMMA: N°-monométhyl-L-arginine L-NNA: Nw-nitro-L-arginine
MAPK: mitogen-activated protein kinase (protéine mitogène kinase activée) NO : nitric oxyde (monoxyde d’azote)
NOS : oxyde nitric synthase (oxyde nitrique synthétase) NS : non-significatif
NSCC : nonselective cation channel (canal non-sélectif pour cation) PAP: pression artérielle pulmonaire
PDE: phosphodiestérase PGH2: prostaglandine H2 PGI2: prostacycline
PIP2: phosphatidylinositol 4,5-biphosphate PKA: protéine kinase A
PKC: protéine kinase C P LA2: phospholipase A2 PLC: phospholipase C
Pc: pression d’ouverture
Pog : pression de l’oreillette gauche RVP: résistance vasculaire pulmonaire SNP : sodium nitroprussiate
TFA: trifluoroacetic acïd (acide trifluoroacétique) Thr: thrombine
TVC : tension veineuse centrale TxA2 : thromboxane A2
U-li : urotensine Il
U-46619 : 9,11 -dideoxy-1 1a,9Œ-epoxymethanoprostaglandin F2Œ VD : ventricule droit
VEGF: vascular endothelial growth factor (facteur de croissance de l’endothélium vasculaire)
VG : ventricule gauche
VIC: vasoactive intestinal constrictor (peptide vasoconstricteur intestinal) VIH : virus d’immunodéficience chez l’humain
VOCC: voltage operated calcium channel (canal calcique voltage-dépendant)
WHO: World Health Organization (Organisation Mondiale de la Santé) 4-AP : 4-aminopyridine
toi, pour ton ai& précieuse, mais surtout pour tes encouragements et ton amoui
J
merci.Les travaux entrepris dans le laboratoire ces dernières années ont visé à explorer et comprendre la biologie du système endothéline, son implication physiologique et pathologique dans l’hypertension pulmonaire afin de permettre le développement et l’optimisation des approches thérapeutiques de cette condition.
Nous évaluerons dans ce mémoire les effets d’une hyperendothélinémie isolée sur la réactivité vasculaire des petites artères pulmonaire chez le rat. Dans un premier temps, nous présenterons la biologie de l’endothélium ainsi que ses différentes composantes et plus précisément l’endothéline, puis nous aborderons la circulation pulmonaire et nous la décrirons en condition physiologique et pathologique ce qui nous permettra de définir la rationnelle de cette étude.
1.1 Biologie de I’endothélium
L’endothélium est composé d’une monocouche cellulaire et forme la surface luminale de tous les vaisseaux du système cardiovasculaire. Il est considéré comme un organe dynamique et multifonctionnel constitué de 1-6x10’3 cellules.1 L’intégrité structurale et fonctionnelle des cellules endothéliales est essentielle pour le maintien de l’homéostasie cardio vasculaire. En effet, elles forment une barrière semi-perméable qui contrôle les échanges entre les composants sanguins et les tissus. Elles participent aussi au contrôle du tonus vasculaire en synthétisant et en libérant de nombreuses substances vasoactives qui influencent le muscle lisse vasculaire. De plus, les cellules endothéliales ont des propriétés
J
prostacycline (PGI2), en 1976, par MoncadaLa première substance endothéliale vasoactive découverte a été laet al.2 Ils ont démontré qu’en plus d’exercer une action vasodilatatrice, la PGI2 inhibe l’agrégation plaquettaire. Quelques années plus tard, en 1980, le groupe de Furchgoff et Zawadski ont démontré que la présence de l’endothélium est essentielle à la réponse vasodilatatrice de l’acétylcholine (Ach) sur des anneaux d’aorte de lapin.3 Ces deux chercheurs ont postulé que cet effet provient de la liaison de Ach à des récepteurs muscariniques situés sur les cellules endothéliales. Cette association provoque la libération d’une ou de plusieurs substances qui agissent sur le muscle lisse. Ces substances vasodilatatrices inconnues ont été qualifiées de facteurs relaxants dérivés de l’endothélium (EDRF). Par la suite, en tenant compte de plusieurs similarités avec les nitrates, Furchgott a identifié la nature de l’EDRF comme étant le monoxyde d’azote (NO).4 Il est à noter que Palmer et Moncada ont présenté les premières évidences de la synthèse de NO, par les cellules mammifères, en 1987 et ont aussi suggéréj
la parité entre l’EDRF et le NO.5 De plus, lors de la même année, Greenberga rapporté les premières évidences d’une dilatation endothélium-dépendantedans les artères pulmonaires humaines.6
Les cellules endothéliales produisent aussi des facteurs contractiles dont l’activité peut être réduite lors de l’endommagement endothélial. Ceci a été prouvé en 1981 par De Mey et Vanhouffe; leurs résultats ont démontré une diminution significative des contractions induites par de grandes concentrations de potassium (K) à la suite de la destruction de l’endothélium.7 Ce ou ces facteurs, par homologie avec l’EDRF, ont été nommés facteur contractant dérivé de l’endothélium (EDCF). En 1985, il fut évident, par l’utilisation d’antagonistes spécifiques, que ce facteur était libéré par les cellules endothéliales en présence de calcium extracellulaire et indépendamment des systèmes a-adrénergiques, cholinergiques, sérotoninergiques et histaminiques.89 Ce facteur a été caractérisé en 1988
)
isolé à partir d’un milieu de culture de cellules endothéliales d’aorte de porc et nommé endothéline (ET).1° Considérée depuis la fin des années 80 comme le plus puissant vasoconstricteur, l’endothéline a récemment été déclassée par l’urotensine Il (U-Il) humaine.11 Toutefois, cette affirmation ne fait pas l’unanimité dans la communauté scientifique puisque les effets de l’U-ll sont variables dans les tissus vasculaires des différentes espèces mammifères.12 Il est à noter que l’endothélium pulmonaire joue un rôle de modulateur. En effet, l’Ach, la bradykinine (BK), l’histamine et l’acide arachidonique (AA) provoquent la vasoconstriction des vaisseaux pulmonaires lorsque celui-ci est absent ou endommagé et une vasodilatation lorsqu’il est intact.3161.2 Les facteurs relaxants dérivés de l’endothélium
Conformément à ce qui a été mentionné dans les paragraphes
)
précédents, l’endothélium vasculaire joue un rôle clé dans la régulation du tonus vasculaire par la libération de substances vasodilatatrices telles que le NO ou EDRF, la PGI2, le facteur hyperpolarisant dérivé de l’endothélium (EDHF), l’adrénomodulline et le facteur natriurétique de type C (CNP).(Figure 1)
Figure I : Les facteurs relaxants dérivés de l’endothéHum
Agonîstes
Cellule
endothélïale
PGI2
NO
EDHF
Résumé schématique de la libération des facteurs relaxants (PGI2, NO et EDHF) par les cellules endothéliales et leurs effets subséquents sur les cellules musculaires lisses. L’adrénomodulline n’est pas représentée. Tiré de « Functions of the healthy endothelium» par Kharbanda, R.K. & Deanfield, ].E..17
5
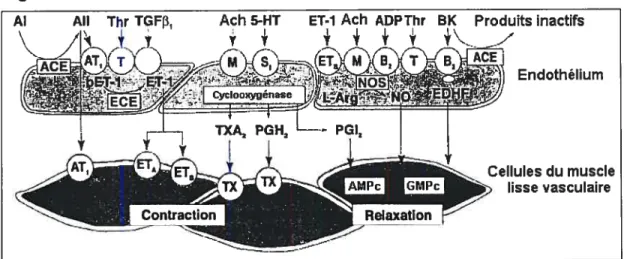
1.2.1 Le monoxyde d’azote
Le NO est libéré par les cellules endothéliales en réponse aux forces de cisaillements produites par le flot sanguin et suite à la liaison de la thrombine (Thr), l’histamine, l’ADP, l’ET, Ach et la BK à leur récepteur tel que
présenté à la droite de la figure 2.18
Le NO a des effets vasodilatateurs et antiprolifératifs sur le muscle lisse
vasculaire et inhibe l’agrégation plaqueffaire et l’adhésion leucocytaire.2° Il est synthétisé à partir de l’acide aminé L-arginine par la NO synthétase (NOS).21
Il existe trois isoformes de la NOS: endothéliale (eNOS ou NOS III),
neuronale (nNOS ou NOS I) et induite par les cytokines (iNOS ou NOS Il).
Deux profils d’expression sont possibles pour ces trois formes : la eNOS et la nNOS sont exprimées de façon constitutive à de faibles niveaux et peuvent
rapidement être activées en présence d’ions calciques (Ca2j et de
calmoduline. En effet, sous sa forme inactive, la eNOS est retrouvée dans
Figure 2 Les facteurs vasoactifs libérés par l’endothélium
Al Ail Thr TGF3, Ach 5-HT ET-1 Ach ADPThr BK Produits inactifs
/‘ ‘r ‘r
+
+
+ +
+
Endothélium
ry PGH2 1 PCI,
Y
1.
Cellules du musclelisse vasculaire
Schéma représentant les différents facteurs vasoactifs produits par l’endothélium suite à la liaison agoniste-récepteur et leurs effets constricteurs (gauche) et relaxants (droite) sur le muscle lisse vasculaire. Tiré et traduit de « Biology of the endothelium» par Lùscher, T.F. & Barton, M..19
J
)
une cavéole attachée à une cavéoline. Une augmentation intracellulaire de Ca2 par des stimuli chimiques et physiques, permet l’activation de la calmoduline qui déplace la calvéoline ce qui favorise l’activation de la eNOS.22 Cette isoforme a été clonée à partir de cellules endothéliales humaines et bovines et elle est responsable de la production du NO nécessaire au tonus vasculaire basal.2324 Pour ce qui est de la nNOS, sa localisation neuronale a été démontrée en 1990 ainsi que la première évidence d’une association entre le NO et les neurones.25 La iNOS, par contre, est induite par les cellules endothéliales ou des macrophages lorsque ceux-ci sont activés par des cytokines lors de réponses inflammatoires et immunitaires. Cette enzyme a été découverte et clonée en 1992, lorsque des macrophages ont été exposés à des cytokines et à des produits bactériens.2627 Elle est Ca2-indépendante et elle est inhibée par les glucocorticoïdes.28Le développement d’analogues de la L-arginine a permis une meilleure connaissance du système du NO ainsi que de son rôle physiologique. En effet, l’activité de la eNOS peut être inhibée pharmacologiquement par l’utilisation de N°-monométhyl-L-arginine (L NMMA) qui compétitionne avec la L-arginine pour le site catalytique de la NOS.29 L’utilisation de L—NMMA en infusion, dans l’artère brachiale de volontaires sains, a permis de révéler le rôle fondamental du NO dans la régulation du tonus vasculaire basal.3° De plus, une étude similaire réalisée sur la circulation pulmonaire confirme l’importance de la production endothéliale de NO pour la régulation de la circulation sanguine chez l’humain.31 Le N°-nitro-L-arginine (L-NNA) et le N°-nitro-L-arginine méthyl ester (L-NAME) sont aussi des inhibiteurs stéréospécifiques de la NOS dont les effets sur le tissu vasculaire sont similaires à ceux observés avec le L NMMA.3233
J
quelques secondes, il diffuse rapidement vers le muscle lisse vasculaire parPuisque la demi-vie biologique du NO est très courte, de l’ordre de la membrane basale de la cellule endothéliale.34 Le NO augmente les concentrations de GMPc par l’activation de l’enzyme guanylate cyclase soluble (GC5).35 Dans plusieurs expériences, le bleu de méthylène est utilisé pour inhiber cette enzyme alors que des composés donneurs de NO comme le sodium nitroprussiate (SNP) la stimulent. Les niveaux élevés de GMPc provoquent une diminution de l’influx calcique par les canaux Ca2 voltage-dépendant de type L et par conséquent, favorise la relaxation du muscle lisse. Le GMP agit de façon directe et indirecte sur les canaux Ca2. Premièrement, une protéine kinase GMPc-dépendante peut phosphoryler le canal Ca2 ou alors une phosphoprotéine associée au canal ce qui cause une diminution de la probabilité d’ouverture de celui-ci indépendamment du voltage.36 Deuxièmement, de façon indirecte, une protéine kinase GMPc dépendante peut causer une augmentation de la probabilité d’ouverture desJ
canaux potassiques (Kj, ce qui hyperpolarise la cellule et provoque une2+37
desactivation voltage-dependante des canaux Ca . De plus, ce nucleotide
stimule aussi l’extrusion du Ca2 cytosolique vers l’extérieur de la cellule. Cet effet semble être causé par une stimulation de la pompe Ca-ATPase de la membrane plasmique.3839 Le NO peut aussi directement activer les canaux K calcium-dépendant (BKca) sans avoir recours au GMPc tel qu’il a été démontré par Bolotina et al. en I99440 Conséquemment, les mécanismes qui favorisent l’activation des canaux K semblent être responsables de la diminution de la concentration intracellulaire de Ca2 et de la relaxation subséquente du muscle lisse.
1.2.2 La prostacycline
L’endothélium vasculaire synthétise et libère de la prostacycline (PGI2)
)
A2 (TxA2), la vasopressine, l’angiotensine Il, l’ET et les forces de cisaillements. Elle est synthétisée à partir de l’AA par la prostaglandine H2 (PGH2) synthétase qui possède deux activités catalytiques: une activité cyclo-oxygénase et une activité peroxydase.2 C’est l’activité cyclo-oxygénase (CCX) qui est inhibée par l’aspirine et l’indométacine.4142 La PGI2 a une courte demi-vie et de faibles niveaux plasmatiques, c’est un autacoïde locale.43 Elle est inactivée par la PG-15 hydroxydéhydrogénase qui est retrouvée en grande quantité dans les artères et les veines.44 En se liant à son récepteur (IP) couplé à une protéine G, la PGI 2 active l’enzymeadénylate cyclase fAC) ce qui augmente les niveaux intracellulaires d’AMP des cellules du muscle lisse et permet une vasodilatation.4546 La PGI2 inhibe aussi l’activation des plaquettes.47 Donc, les effets inhibiteurs de la PGI2 sur l’adhésion leucocytaire et l’agrégation plaqueffaire combinés à ceux du NO, permettent à l’endothélium d’avoir une importante activité antithrombotique.48
)
1.2.3 Le facteur hyperpolarisant derive de l’endotheliumCe ou ces composés, encore non identifiés, produisent une hyperpolarisation endothélium-dépendante des cellules du muscle lisse.49 Ce phénomène présent chez différentes espèces n’est pas influencé par les analogues de la L-arginine ou les inhibiteurs de la COX.5051 L’hyperpolarisation du muscle lisse induit une vasodilatation en diminuant la probabilité d’ouverture des canaux Ca2 voltage-dépendants.52 Le changement de polarité cellulaire est observé sans augmentation intracellulaire des nucléotides cycliques tels que l’AMPc et le GMPc.5° De plus, les inhibiteurs des canaux BKca, des canaux K voltage et ATP dépendants du muscle lisse vasculaire entravent cette hyperpolarisation.5355
L’identité de l’EDHF est toujours controversée, mais un acide
)
proposé.56 De même, en 1996, Randall a postulé que l’EDHF pourrait être un cannabinoïde, l’anandamide, dans les lits mésentériques et coronaires artériels chez le rat.57 Cependant, cette hypothèse a bien vite été rejetée, car l’anandamide ne produit pas d’hyperpolarisation dans les vaisseaux sanguins des autres espèces. De récentes évidences scientifiques démontrent l’ouverture de canaux K sensibles à la charybdotoxine et à l’apamine dans les cellules endothéliales suite à la liaison de substances neurohumorales démontrant par le fait même la possibilité que le K soit l’EDHF. L’augmentation des concentrations K provoquerait l’hyperpolarisation et la relaxation du muscle lisse sous-adjacent par l’activation des canaux sensibles au Ba2 ainsi que de la pompe Na/K ATPase.5859 Finalement, des molécules ayant une courte demie-vie telle que le monoxyde de carbone (CO), les radicaux hydroxyles (H0), le peroxyde d’hydrogène (H202) de même que le NO et la PGI2 produisent une hyperpolarisation et sont considérés comme des EDHF. Néanmoins, celui-ci ou ceux-ci ne sont)
toujours pas identifiés avec certitude. 1.2.4 L’adrénomodullineDécouvert en 1993, ce peptide de 52 acides aminés a été isolé et purifié à partir de cellules tumorales de la médullo-surrénale (phéochromocytome).6° L’expression du gène de l’adrénomodulline (AM), retrouvé sur le chromosome 11, est régulée par une grande variété de stimuli humoraux. L’AM est majoritairement synthétisée par les cellules endothéliales de divers organes dont le poumon et à un moindre degré par différents autres types cellulaires incluant les cellules musculaires lisses.61 Elle produit une vasodilatation puissante et soutenue chez l’homme.62 Les faibles niveaux circulants de celle-ci suggèrent une activité autocrine/paracrine. Toutefois, les concentrations plasmatiques sont
cardiovasculaire, respiratoire, rénal et endocrinien.63 Ceci suggère un rôle hypotenseur en condition pathologique confirmé par des études chez l’homme et le rat où une infusion d’AM à des niveaux élevés provoque une importante chute de la pression artérielle systémique.6465
L’AM exerce ses effets principalement par l’augmentation des niveaux d’AMP dans le muscle lisse et par libération de NO par les cellules endothéliales.6667 Deux types de récepteurs pour l’AM ont récemment été décrits.6869 Ceux-ci sont constitués de deux éléments qui sont essentiels à la liaison agoniste-récepteur. Le premier élément possède sept domaines transmembranaires et se nomme calcitonin receptor-like receptor (CRLR). Il se lie à un deuxième élément, une molécule de chaperronage désignée receptor activity modifying proteins (RAMP1) pour former le récepteur CGRP1. Deux autres molécules, RAMP2 et RAMP3, qui ont aussi un domaine transmembranaire, ont la possibilité de se lier au CRLR pour constituer le deuxième récepteur qui est spécifique à l’AM. Par conséquent, les niveaux tissulaires de ces molécules de chaperronage influencent fortement la densité et la nature des récepteurs de l’AM. L’AM circulante est en majorité liée à une protéine de liaison spécifique l’adrenomedullin binding proteïn (AMBP1) identifiée comme le facteur H du complément.7° Celui-ci agirait à titre de transporteur et de réservoir permettant à l’AM de s’accumuler localement pour stimuler ses récepteurs. En outre, plusieurs études suggèrent un rôle majeur du poumon dans la clairance de l’AM.7172 Cependant, les données publiées demeurent qualitatives et indirectes et des études structurées sont nécessaires afin de bien caractériser ce phénomène.
7.2.5 Le facteur natriurétique de type C
Le CNP originalement mis en évidence dans le cerveau porcin, est le troisième membre de la famille des peptides natriurétiques qui inclut l’ANP et
le BNP; deux peptides vasoactifs essentiels à l’homéostasie cardiovasculai re.73 Quoique structurellement similaire, ce peptide est génétiquement distinct. La forme biologiquement active est composée de 22 acides aminés et elle est produite par les cellules endothéliales de différentes espèces, dont l’humain, le porc, le chien et le rat.74 Sa sécrétion est régulée par les cytokines, les facteurs de croissance, le stress oxidatif et les forces de cisaillements.7475 De plus, l’ANP et le BNP stimulent fortement son expression génique en plus de sa libération dans les cellules endothéliales bovines.76 Le CNP possède plusieurs effets cardiovasculaires tels qu’une diminution des pressions de remplissage et d’éjection cardiaque secondaire à une vasorelaxation et d’une diminution du retour veineux.76 En effet, celui-ci est un venodilatateur endothélium indépendant, il inhibe la production d’aldostérone et il possède également une activité inhibitrice sur la prolifération du muscle lisse vasculaire in vitroet possiblement in vivo.7778
Le CNP exerce ses actions biologiques par trois types de récepteurs NPR-A, NPR-B et NPR-C. Ceux-ci possèdent une protéine kinase intracellulaire liée à un domaine contenant une guanylyl cyclase.79 La formation du GMPc ainsi que l’ouverture des canaux K÷ par l’activation d’une protéine kinase GMPc-dépendante sont responsables des effets vasodilatateurs du CNP.8° Toutefois, ceux-ci dépendent de l’activité de phosphodiestérases intracellulaires qui dégradent le cGMP et préviennent son accumulation.81 Il est à noter que le NPR-C a été identifié comme un récepteur de clairance puisque plusieurs études démontent l’internalisation et le recyclage de celui-ci suite à la dégradation du peptide natriurétique lié dans les lysosomes.82 De plus, le CNP peut être dégradé par une endopeptidase neutre 24.11 (NEP) qui est retrouvée dans les poumons, le coeur, les cellules endothéliales et musculaires lisses vasculaires.
1.3 Les facteurs contractants dérivés de I’endothélium
L’endothélium ne produit pas seulement des substances vasodilatatrices, mais joue plutôt le rôle d’un chef d’orchestre dans l’homéostasie vasculaire en conservant une harmonie fragile entre les composés vasoactifs dilatateurs et constricteurs, II synthétise des facteurs contractants tels que la TxA2, l’ET et l’anion superoxide et régule le système rénine-angiotensine ainsi que celui des kinines.
1.3.1 La thromboxane A2
La TxA2 est produite dans plusieurs types cellulaires, mais plus principalement dans les plaquettes.83 Elle est formée par une enzyme microsomale, la thromboxane synthétase, et sa demi-vie est de l’ordre de
J
quelques secondes.84 De même que pour la PCI2, la TxA2 est un produit de la cascade de I’AA. Chez les humains, il existe deux types de récepteurs pour la TxA2, TPŒ et TPf3, et ils proviennent du même gène.85 Ils sont retrouvés sur les plaquettes et également sur les cellules endothéliales et musculaires lisses.86 Fait intéressant, le récepteur TP peut être désensibilisé et subir une rétroaction négative par l’augmentation d’AMPc induite par la liaison de la PGI2 à son récepteur IP.87La liaison de la TxA2 à son récepteur active la phospholipase C et favorise l’augmentation des concentrations de Ca2 intracellulaire. De plus, il a été démontré que la stimulation des récepteurs TP inactive les canaux BKca et empêchent ainsi l’hyperpolarisation cellulaire.88 La TxA2 stimule fortement l’agrégation plaquellaire et la vasoconstriction du muscle lisse, de même que la mitogénèse et l’hypertrophie des cellules qui le compose.899° Par conséquent, elle joue un rôle majeur dans le développement et le
)
l’ischémie myocardique, l’infarctus du myocarde et l’athérosclérose ainsi que dans plusieurs pathologies rénales.9192 Toutefois, la PCI2 contrebalance ces effets en condition physiologique et un dérèglement de cette homéostasie donne lieu à des conditions propices au développement de pathologies. Le composé U-46619 (9,1 1-dideoxy-1 1Œ,9Œ-epoxymethanoprostaglandin F2) est un analogue mimétique de la TxA2 fréquemment utilisé dans les études scientifiques.Il est à noter que l’AA est aussi transformée par les lipoxygénases de la voie linéaire en plusieurs acides hydropéroxyeicosatétraénoïques (HPETE) et la présence de ce système dans l’endothélium vasculaire a clairement été démontrée.93 Les leucotriènes et les lipoxines, des HPETE, sont majoritairement synthétisées dans les globules blancs. Les lipoxines sont encore peu connues, mais les leucotriènes sont considérées comme des vasoconstricteurs puissants, des activateurs et des chemoaffractants
)
leucocytaires et elles augmentent la perméabilité membranaire. Par conséquent, elles sont des médiateurs importants du choc circulatoire et de l’ischémie en plus de jouer un rôle prépondérant dans les réactions allergiques et inflammatoires.94.1.3.2 L’angiotensine Il
Le système rénine-angiotensine exerce un rôle central dans la physiologie et la pathologie cardiovasculaire. L’Agtl est formée à partir de l’angiotensine I par l’enzyme de conversion de l’angiotensine (ECA) que l’on retrouve à la surface des cellules endothéliales. L’ET-l accélère cette formation dans les cellules endothéliales pulmonaires.95 L’Agll est un vasoconstricteur impliqué dans le contrôle vasculaire local et systémique, dans la filtration glomérulaire et elle a des propriétés prothrombotiques et
)
identifiés.9697 La densité de ceux-ci est régulée par des facteurs hormonaux et leur expression est grandement influencée par les processus pathologiques.98 Le récepteur AT1 se retrouve dans le coeur, les reins et certaines régions du cerveau, Il est présent au niveau des cellules endothéliales et des fibres musculaires lisses de l’ensemble du lit artériel.98 La liaison l’Agll/AT1 permet l’activation des phospholipases A2, C et D, l’ouverture des canaux Ca2 voltage-dépendant et une diminution de l’activité de I’AC. Ces actions se concluent par une augmentation du Ca2 intracellulaire.99 De surcroît, de nombreuses tyrosines kinases sont stimulées et plusieurs radicaux libres sont formés. L’ensemble de ces réactions provoque la croissance et l’hypertrophie des cellules musculaires lisses et endothéliales, la formation d’une matrice extracellulaire, du stress oxidatif et des réactions inflammatoires en plus des effets vasoconstricteurs et de la sécrétion d’ET et de catécholamines.Quant au récepteur AT2, il est exprimé en grande quantité dans les tissus foetaux et son expression diminue rapidement à la naissance. On le retrouve au niveau des cellules endothéliales de la majorité des artères.98 Plusieurs évidences provenant de laboratoires indépendants suggèrent les effets bénéfiques du récepteur AT2 pour contrebalancer les actions pressives et prolifératrices du récepteur AT1.10° Effectivement, la stimulation de l’AT2 est associée à une augmentation de NO, de bradykinine et de GMPc.101102 Toutefois, l’expression cellulaire, la régulation et les actions de celui-ci nécessitent des études plus approfondies.
De plus, l’endothélium libère aussi la bradykinine, une kinine qui produit une réponse hypotensive de faible durée chez l’homme, lorsqu’injectée par voie intraveineuse.103 Ses effets sont causés par un récepteur B2 couplé à une protéine G à la surface des cellules endothéliales. La liaison agoniste-récepteur provoque plusieurs événements intracellulaires
J
Des évidences récentes suggèrent que l’EDHF est beaucoup plus importantque le NO et la PGI2 dans les effets vasodilatateurs induits par la bradykinine dans les vaisseaux de résistance de l’avant-bras humain.104 Cette kinine est presque complètement métabolisée par la kininase Il, identique à la ECA, lors d’un passage unique dans le lit vasculaire pulmonaire.105 Il a été souligné dans une étude intéressante de Horning, que les effets bénéfiques de l’utilisation des inhibiteurs de ECA sont attribués en grande partie à leurs actions sur la bradykinine.°61.3.3 L’anion superoxide
Les cellules endothéliales humaines sont également une source d’anion superoxide dont la formation est stimulée par les cytokines ainsi que par la pulsation sanguine sur la paroi vasculaire.107°9 Celui-ci est considéré
)
comme un facteur contractant dérivé de l’endothélium (EDCF) et il est habituellement inactivé par le NO. Toutefois, la NOS peut également produite ce facteur vasoconstricteur lorsque les concentrations de L-arginine sont faibles.HO Par conséquent, les effets biologiques bénéfiques du NO sont attribuables à son rôle de régulateur de l’anion superoxide en plus de ses effets classiques vasodilatateurs et antiprolifératifs.1.3.4 L’endothéline
La découverte et la caractérisation de l’ET, il y a maintenant 15 ans, ont généré de nombreuses interrogations et études scientifiques qui nous ont permis de mieux comprendre ce système. La biosynthèse, les récepteurs, les rôles physiologiques et pathologiques de ce puissant peptide vasoconstricteur seront discutés de façon détaillée dans la prochaine section
1.4 Biologie du système endothéline
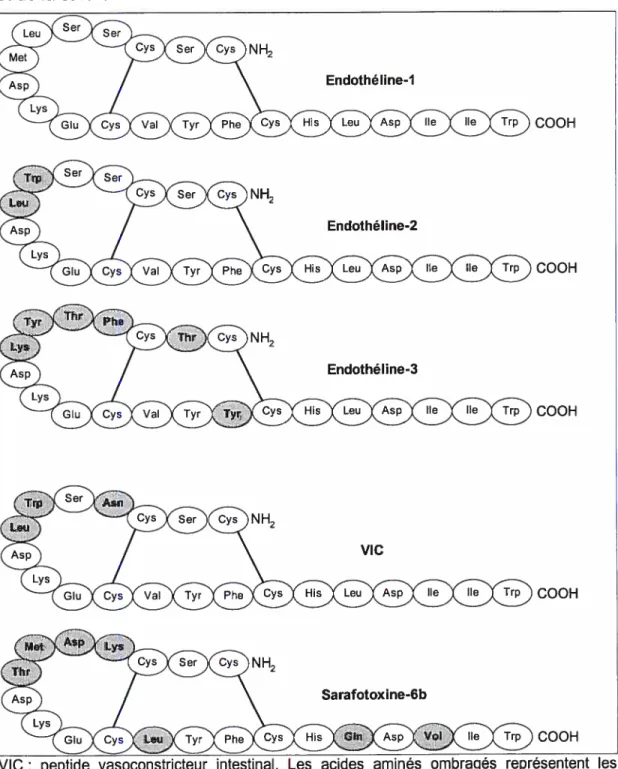
L’endothéline-l (ET-1) est un peptide vasoactif qui possède une activité vasoconstrictrice et des effets mitogènes sur le muscle lisse.101H Elle est composée de 21 acides aminés et caractérisée par deux domaines terminaux et deux ponts disulfures entre les cystéines en position 1-15 et 3-11.(Figure 3) Le domaine N-terminal détermine l’affinité pour le récepteur alors que le domaine C-terminal contient le site de liaison pour l’agoniste. Deux isoformes, ET-2 et ET-3, ayant une structure très similaire et provenant de gènes différents, ont été caractérisées.112 Une quatrième isoforme, ET-4, le peptide vasoconstricteur intestinal (VIC) a aussi été identifié dans le système intestinal de la souris.113 Quoique les trois isoformes humaines soient largement distribuées, l’ET-l est la plus puissante et la plus connue. Elle est la seule présente au niveau des cellules endothéliales du système vasculaire. En outre, il est intéressant de remarquer que la structure des
)
endothélines possède une grande homologie avec une autre famille de peptide, les sarafotoxines (-6a, -6b, -6c et -6d). Celles-ci sont des neurotoxines qui proviennent du venin de serpent (Atractaspis egaddensis) et qui provoquent une vasoconstriction généralisée de la proie de ce reptile.1.4.1 La biosynthèse de l’endothéline
Le précurseur de chaque isoforme est codé par un gène différent: la pré-proendothéline-1 (préproET-1) est retrouvée sur le chromosome humain 6p23-24, la préproET-2 sur le chromosome 1p34 et la préproET-3 sur le chromosome 20q13.2-13.3. L’expression génique de l’ARNm des ETs peut être stimulée par plusieurs facteurs : la Thr, les facteurs de croissance, les cytokines, l’insuline, la vasopressine, l’Agll, les catécholamines, la BK, l’hypoxie et l’ET elle-même. En outre, elle peut aussi être inhibée pat le NO,
J
oestrogènes.114 De plus, l’expression de l’ARNm de l’ET de même que laproduction de ce peptide par les cellules endothéliales, sont régulées par lesforces de cisaillements et l’ischémie.
J
J
Figure 3 Structure primaire des endothélines-1, -2, et -3 humaines, de VIC et de la sarafotoxine-6b.
TyrPheCYSHiSGASPVD11e
peptide vasoconstricteur intestinal. Les acides aminés ombrag différences par rapport à l’endothéline-l•h15
)
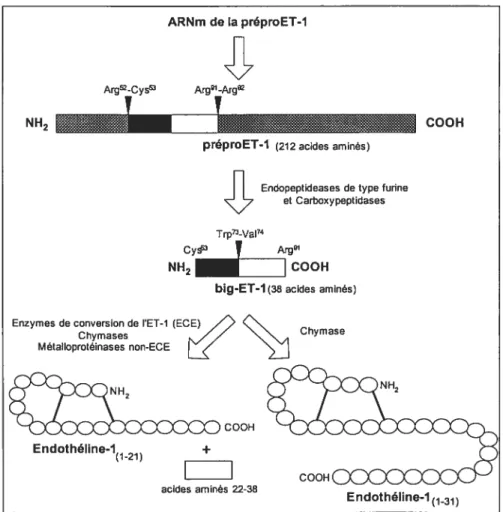
Donc, la préproET-1, une protéine de 212 acides aminés, est clivée protéolytiquement par une endopeptidase entre les résidus 52-53 et par une carboxypeptidase entre les résidus 91-92.’16(Figure 4) Un peptide intermédiaire de 38 acides aminés, la proET-1 plus communément nommée big-ET-1, est ainsi formée. La big-ET-1 est ensuite clivée entre les résidus tryptophane et valine en position 21 et 22 par une autre endopeptidase, l’enzyme de conversion de l’endothéline (ECE), pour engendrer la forme active de l’ET-l. L’ECE est une métalloprotéinase qui est inhibée par le phosphoramidon.’17 Il est à noter que la big-ET-1 peut aussi être clivée par une chymase entre les résidus Try3’ et G1y32 et former une ET de 31 acides aminés qui a aussi des propriétés vasoconstrictrices.118Figure 4: Biosynthèse de l’ET-l
ARNm de la préproET-1
)
Arg-CysNH2 COOH
préproET-1 (212 acides aminés) Endopeptideases de type (urine
et Carboxypeptidases Trp-Val74
Cys5 Arg9’
NH2 COOH
big-ET-1 (38 acides aminés) Enzymes de conversion de CET-1 fECE)
Chymases Chymase Métalloprotéinases non-ECE
acides aminés 22-38
Endotheline-1fi-31)
)
Schéma représentant la voie de la biosynthèse de l’endothéline-l (ET-1) de 21 et de 31 acides aminés; ceux-ci sont représentés par les petits cercles.10)
La conversion de la big-ET-1 en ET-1 est essentielle pour l’activité biologique de celle-ci, car in vivo, la big-ET-1 a seulement 1 % de l’activité contractile du peptide mature.119 La biosynthèse des deux autres isoformes de l’ET se fait exactement de la même façon, mais avec leurs enzymes et précurseurs respectifs. Puisque que l’ensemble de mon travail de maîtrise porte sur la réactivité vasculaire et que l’ET-l est la seule isoforme présente au niveau de l’endothélium vasculaire, les aspects biologiques et physiologiques de ET-1 seront privilégiés dans les prochaines sections.1.4.1.1 Les enzymes de conversion de l’endothéline-l
L’ECE-l a été la première enzyme de cette famille à avoir été identifiée. Elle a initialement été purifiée et clonée à partir du poumon de rat.12021 C’est une métalloendopeptidase zinc-dépendante et elle présente
)
une grande homologie structurale avec l’endopeptidase neutre (NEP)-24.11. Elle est codée par le chromosome humain 1p36 et quatre isoformes (ECE-la, ECE-1 b, ECE-1 c, ECE-ld) ont été découvertes jusqu’à maintenant.122 Celles-ci sont formées par l’utilisation alternative de deux promoteurs et par un épissage complexe. L’ECE-l est non-sélective pour les différentes big-ET et elle agit à pH neutre.123 Elle est omniprésente dans l’endothélium vasculaire humain et particulièrement au niveau pulmonaire où elle catalyse la conversion de la big-ET-1 en ET-1 biologiquemant active. Par contre, l’ECE-l est exprimée de façon faible à modérée dans les cellules musculaires lisses.124 L’immunofluorescence a démontré des différences claires entre les localisations cellulaires des différentes ECE. L’ECE-lb est localisée intracellulairement alors que les deux autres isoformes sont exclusivement (ECE-la) et majoritairement (ECE-1c) retrouvées sur la membrane pI asm iqu e.125)
L’ECE-2, quant à elle, a été clonée à partir de cellules endothéliales bovines et elle possède 59 % d’homologie avec l’ECE-l 126 Elle est expriméedans les cellules endothéliales humaines.127 Le pH optimal pour son activité enzymatique est acide (pHz5.5). Par conséquent, selon certains chercheurs
elle serait localisée dans un compartiment intracellulaire tel que le réseau trans-Golgi ou alors associée à celui-ci.126 Cette enzyme est plus vulnérable au phosphoramidon que l’ECE-l et elle convertit plus efficacement la big-ET I que la big-ET-2 et la big-ET-3. Récemment quatre isoformes de I’ECE-2 (ECE-2a-1, ECE-2a-2, ECE-2b-I, ECE-2b-2) ont été isolées et caractérisées
chez la souris et le bovin.128 Toutefois, leurs fonctions physiologiques sont
toujours inconnues. Un autre type de ECE (ECE-3) a aussi été purifié à partir de microsomes de l’iris bovin. Elle clive sélectivement la big-ET-3 et elle
semble avoir un rôle au niveau neuronal mais celui-ci n’est toujours pas
confirmé.129
1.4.1.2 La sécrétion de l’endothéline-J
Deux théories contradictoires s’affrontent quant au mode de sécrétion de l’ET-I. Dans le premier cas, les cellules productrices d’ET-l telles que les cellules endothéliales vasculaires, ne contiennent pas de granules de
sécrétion dans lesquelles l’ET-l peut être emmagasinée puis relâchée ultérieurement.130 L’ET-l est sécrétée de façon constituve et l’étape limitante
de sa biosynthèse est le niveau de transcription. De nombreux facteurs peuvent stimuler et supprimer la synthèse d’ET-I tel que mentionné dans une précédente section. Plusieurs évidences laissent suggérer que l’ET-l est
libérée constitutivement pour le maintien du tonus vasculaire de base et les
concentrations plasmatiques physiologiques de l’ET-I sont approximativement de I fmol /mL.131 Toutefois, un groupe de chercheurs a émis une hypothèse voulant qu’une seconde voie de sécrétion pour l’ET-l, la
)
existe des vésicules de sécrétion, les vésicules Weidel-Palade, associées à l’ECE et dans lesquelles la big-ET-1 est transformée en ET-1 et où celle-ci est emmagasinée.133Des lésions endothéliales, différents stimuli chimiques ou de faibles forces de cisaillements stimulent la sécrétion d’ET-l et celle-ci est alors relâchée à de fortes concentrations.134 Des études ont démontré que la libération d’ET-l était trop rapide pour être attribuée à une synthèse de novo soutenant par le fait même la présence de granules de sécrétion intracellulaires.135 L’ET-l agit préférentiellement de façon autocrine/paracrine puisqu’environ 80 ¾ de sa synthèse est délivrée vers la membrane basolatérale pour agir sur le muscle lisse.136 La concentration de l’ET-l dans le tissu vasculaire est environ 100 fois celle retrouvée au niveau plasmatique. Toutefois, une très grande proportion du peptide vasoactif est liée à ses récepteurs.137 Il est à noter que la demie-vie de l’ET-l dans le sang est
)
d’environ sept minutes parce qu’elle se lie promptement à ses récepteurs et qu’une enzyme de dégradation (EDE) la métabolise rapidement.1.4.2 Les récepteurs vasculaires de l’endothéline-l
L’ET agit via deux types de récepteurs, ETA et ETB.1394° Ces récepteurs ont sept domaines transmembranaires et sont couplés aux protéines G. Le récepteur ETA humain est codé à partir du chromosome 4, il est formé de 427 acides aminés et lie avec une grande affinité l’ET-l et l’ET-2 et faiblement l’ET-3. Le récepteur ETB, quant à lui, est retrouvé sur le chromosome 13, il est composé de 442 acides aminés et lie indifféremment les trois isoformes de l’ET. Ces deux récepteurs sont distribués en différentes proportions dans une grande variété de cellules et de tissus. Dans le lit vasculaire, le récepteur ETA est situé sur le muscle lisse où il cause la
)
des cellules endothéliales où il favorise la vasorelaxation en permettant la relâche de NO et de PGI2. Certains facteurs peuvent affecter l’expression de ces récepteurs. Par exemple, l’insuline et le NO promeuvent l’expression du récepteur ETA alors que les facteurs de croissances et les cytokines affectent positivement l’ETB.4142 Un récepteur ETc spécifique à l’ET-3 a aussi été cloné, mais il est seulement présent dans les mélanophores de la grenouille Xenopus Ieavis.”43 Jusqu’à maintenant, aucun récepteur homologue n’a été découvert dans les cellules de mammifères.Afin d’élucider les mécanismes d’action de l’ET-l, l’utilisation d’antagonistes sélectifs et non-sélectifs des récepteurs à l’ET-l s’est révélée un outil précieux. Au cours des dernières années, plusieurs d’entre eux ont été développés permettant des découvertes importantes et jouant un rôle essentiel dans le traitement de certaines pathologies. Par exemple, des études pharmacologiques ont confirmé l’existence de deux localisations pour
)
les récepteurs ETB: sur les cellules endothéliales (ETB1) et sur les cellules144 . .
musculaires lisses (ETB2). Toutefois, leurs actions physiologiques sont contradictoires; sur l’endothélium, les récepteurs ETB permettent la libération de facteurs relaxants alors que sur le muscle lisse, ils sont responsables de la vasoconstriction. Les antagonistes des récepteurs à l’ET-l seront discutés plus en détail dans une prochaine section.
1.4.2.1 Voie de signalisation
La liaison de l’ET-l à son récepteur ETA ou ETB sur le muscle lisse stimule une protéine G qui active la phospholipase C (PLC) et augmente la concentration de Ca2 intracellulaire.(Figure 5) La PLC activée hydrolyse le phosphatidylinositol 4,5-biphosphate (PIP2) en deux sous-produits: l’inositol 1,4,5-triphosphate (1P3) et le 1,2-diacylglycérol (DAG).
I
I
Figure 5 : Mécanisme de la contraction induite par l’ET-l
NSCC : canal non-sélectif pour cations; VOCC: canal Ca2 voltage-dépendant; RS: réticulum sarcoplasmique. Tiré de « Subcellular mechanisms of endothelin action in vascular system” par Masaki,T. et al.145
L’1P3 est responsable de l’augmentation initiale du Ca2 intracellulaire par la mobilisation des réserves intracellulaires de Ca2, le réticulum sarcoplasmique. Par contre, l’augmentation soutenue est produite par un influx de Ca2 extracellulaire par différents canaux ioniques. En effet, le DAG active une protéine kinase C (PKC) qui phosphoryle directement ou indirectement les canaux K. Ceci provoque leur inhibition et une dépolarisation de la cellule activant par le fait même les canaux Ca2 voltage-dépendants de type L. De plus, les canaux non-sélectifs pour cations (NSCC) permeffent aussi l’entrée de Ca2.146 L’accumulation de Ca2 peut également stimuler les canaux Cl Ca2-dépendant favorisant ainsi la dépolarisation cellulaire.145 La PKC stimule aussi directement ou indirectement la croissance et la différenciation cellulaire des cellules endothéliales et musculaires lisses
NO
Kca NSCCvocc
‘r jP3 Ca Ca Contraction)
ainsi que des fïbroblastes en activant la voie de la MAP kinase qui stimule à son tour les proto-oncogèges c-Jun et c-Fos.147’48 Ceci induit des changements structuraux tels que la déposition d’une matrice extracellulaire et la synthèse de collagène dans la paroi vasculaire. Ces effets sont généralement associés au récepteur ETA.De surcroît, la liaison de l’ET-l à son récepteur ETB sur la cellule endothéliale active une importante voie de signalisation intracellulaire qui module les effets nets de l’ET-l. Par une protéine G couplée au récepteur, la eNOS est stimulée et le NO formé diffuse vers le muscle lisse vasculaire où il active la GC5.H9 La synthèse de la PGI2 est aussi activée. Celle-ci augmente les concentrations d’AMP qui stimule une protéine kinase A (PKA) et provoque la phosphorylation de la pompe Ca2 interne. Par cette voie, ET-1 stimule elle-même une rétroaction négative sur ses effets constricteurs et mitogènes. De plus, la liaison ET-1/ETB affecte aussi négativement
)
l’expression de l’ARNm de l’ECE-J en modifiant sa transcription et/ou sa stabilité.1491.4.2.2 Clairance de l’endothéline-i
La clairance de l’ET-l est particulièrement importante dans la circulation pulmonaire, mais on la retrouve aussi dans les reins et le foie. Il a été démontré que les antagonistes des récepteurs ET9 réduisent l’élimination de l’ET-l dans ce lit vasculaire établissant ainsi la responsabilité des ETB dans la régulation des niveaux plasmatiques d’ET-1.15051 Chez l’humain sain, l’endothélium pulmonaire élimine, lors d’un passage unique, près de 50 % de l’ET-l contenue dans le sang veineux, Il libère aussi dans la circulation une quantité similaire de ce peptide; il n’y a pas ou alors un très faible gradient artério-veineux d’ET-l dans le poumon.152 Une étude a
)
prédispose au développement de l’hypertension pulmonaire (HTP) caractérisée par une diminution de la production de NO et une augmentation des niveaux d’ET-l circulants.153 De plus, l’expression de l’ARNm des ETB dans les poumons de rats avec HTP est diminuée.154 L’internalisation du complexe ET-1/ETB a été sugéré comme mécanisme de régulation négative suite à une stimulation soutenue de ce récepteur. Après internalisation, le complexe serait séquestré à l’intérieur de lysosomes où l’environnement acide faciliterait sa dissociation.155 L’ET-l serait ainsi récupérée par les cellules endothéliales.156 Néanmoins, le récepteur ETB pourrait aussi agir comme transporteur trans-endothélial et libérer l’ET-l à la surface abluminale de l’endothélium mais cette hypothèse n’est toujours pas confirmée.1.4.3 Interaction monoxyde d’azote-endothéline-i
j
Tel qu’il a été mentionné dans les sections précédentes, en conditionphysiologique, le flot sanguin ainsi qu’une grande variété de substances régulent la production d’ET-l et de NO. Lorsque les forces de cisaillements augmentent, l’expression de la eNOS est induite et les niveaux de GMP augmentent. Celui-ci affecte négativement la production et l’activité de l’ET-J et par conséquent les effets vasodilatateurs et antiproliférateurs du NO l’emportent sur les effets vasoconstricteurs et promitogéniques de l’ET-l. Lorsqu’il y a dysfonction endothéliale, par exemple dans l’HTP, cette balance homéostasique est perturbée et le tonus vasculaire est augmenté. II a été démontré in vivo que l’administration de L-NMMA augmente significativement l’effet vasoconstricteur de I’ET-J Cet effet s’explique par une libération physiologique basale de NO endogène dont les effets vasodilatateurs l’emportent sur les réponses vasoconstrictrices de l’ET-l 158 Effectivement, plusieurs études effectuées au niveau de la circulation brachiale, coronaire et pulmonaire démontrent l’importance de cette production de NO dans la
1.4.4 Physiologie et pathologie du système endothéline-J
L’ET-l est un peptide régulateur et un puissant vasoconstricteur à des concentrations beaucoup moins élevées que la quasi-totalité des autres substances cardiovasculaires. Ses rôles sont ambivalents; en situation physiologique, ils sont bénéfiques alors qu’une perturbation de son homéostasie peut avoir d’importantes conséquences pathologiques dans une très grande variété de tissus.
1.4.4.1 Le développement
L’ET-l joue un rôle sine qua non dans le développement foetal. Son interaction avec le récepteur ETA est essentielle dans le développement postmigratoire des cellules de la crête neurale cardiaque menant à la
)
formation et au développement du coeur, de l’arc aortique ainsi que des structures artérielles.119 De plus, elle joue aussi un rôle dans la croissance des cellules originaires de la crête neurale céphalique qui formeront les structures faciales. De même, l’interaction ET-3/ETB est indispensable à la stimulation des cellules de la crête neurale qui se divisent et se différencient rapidement en mélanocytes et en entéricytes.’6°L’absence d’ET-l et de récepteurs ETA produit des malformations craniales sévères qui affectent la respiration et des anomalies cardiaques souvent non viables.161 Par contre, une carence en ET-3 et en récepteurs ETB empêche le développement fonctionnel du système intestinal (mégacolon aganglionnaire) en plus d’une pigmentation anormale due à une différenciation incorrecte des mélanocytes.
I A.4.2 Les vaisseaux sanguins
Conformément à ce qui a été mentionné précédemment, l’ET-l est la
seule isoforme présente au niveau vasculaire. Le ratio de ses deux
récepteurs ETA:ETB varie selon le lit vasculaire, mais il est généralement plus
faible au niveau des veines. Par contre, l’ET-l provoque une vasoconstriction
autant artérielle que veineuse.10 L’administration d’un bolus intraveineux
d’ET-l génère un effet dépresseur transitoire puis un effet presseur soutenu. La diminution initiale de la résistance vasculaire est attribuée à la libération
de NO et de PGI2 par les récepteurs ETB au niveau des cellules endothéliales. Ceci a été confirmé par l’utilisation d’inhibiteurs de la synthèse de ces deux facteurs vasodilatateurs et par des antagonistes ETB in vivo et in
vitro.152163 Par la suite, l’effet presseur est rattaché au récepteur ETA du muscle lisse vasculaire tel que démontré par l’usage d’antagonistes qui ont
atténué cette réponse pressive.1
Le système ET joue un rôle majeur dans l’athérosclérose.165 Celle-ci
est une maladie inflammatoire de l’endothélium vasculaire caractérisée par une accumulation de lipides sur et à l’intérieur de la paroi artérielle. Des facteurs athérosclérotiques tels que la cigarette, le diabète et l’hyperlipidémie
avivent ce système dans les vaisseaux sanguins. Dans cette pathologie, les cellules endothéliales et les macrophages libèrent de l’ET-I qui stimule les
récepteurs ETA sur les macrophages, les cellules musculaires lisses et les
fibroblastes. L’interaction ET-I/ETA a des effets mitogéniques et prolifératifs
sur les cellules du muscle lisse et les fibroblastes. De plus, elle stimule la synthèse de fibronectine dans le muscle lisse et de cytokines dans les
macrophages.16° La présence des récepteurs à l’ET-l a aussi été confirmée
dans les plaques athérosclérotiques. Tous ces facteurs contribuent à la progression de la lésion athérosclérotique par la formation d’une matrice
extracellulaire, la déposition de collagène et la migration des cellules
1.4.4.3 Le muscle cardiaque
L’ET-l, l’endothéline prédominante au niveau cardiaque, est produite par les cardiomyocytes, les cellules endothéliales et les fibroblastes cardiaques.16° Les deux types de récepteurs de l’ET-l sont retrouvés sur les cardiomyocytes, les fibroblastes et les artères coronaires et les ETA représentent 90 % des récepteurs présents sur les myocytes.166 Ce peptide vasoactif est un puissant constricteur des artères coronaires humaines, c’est un agent ionotrope et chronotrope pour le muscle cardiaque et il a des effets sur la prolongation du potentiel d’action.10167
Le système ET est impliqué dans la pathophysiologie de l’infarctus du myocarde et dans l’insuffisance cardiaque congestive. L’occlusion d’une artère coronaire génère un infarctus et les survivants ont un risque élevé de développer de l’insuffisance cardiaque.168 Plusieurs observations indiquent
)
que l’ET-l a des effets bénéfiques sur la fonction cardiaque et la réparation tissulaire quelques jours après l’infarctus. Toutefois, à long terme, ses effets sont délétères et une insuffisance cardiaque congestive se développe. L’ET-l augmente la masse ventriculaire et stimule le remodelage cardiaque pat ses effets promitogènes sur les cardiomyocytes et les fibroblastes. De fait, les patients ont des niveaux plasmatiques d’ET-l et tissulaires d’ET-l et d’ETA augmentés et ceux-ci reflètent la sévérité des dommages cardiaques et un faible pronostic de survie.16917° De plus, les niveaux plasmatiques d’ET-l corrèlent fortement avec l’augmentation de la pression pulmonaire ainsi que la résistance vasculaire pulmonaire chez les patients qui développent une hypertension pulmonaire secondaire à l’insuffisance cardiaque congestive.171Les antagonistes ETA, par exemple le BQ-123, sont bénéfiques dans ces pathologies. En effet, leur administration dans un modèle canin durant 90 minutes à la suite d’une occlusion coronaire a diminué la taille de l’infarctus
)
et la prévention de l’ischémie cardiaque par la dilatation des vaisseaux collatéraux sont les principaux objectifs des antagonistes ETA. En effet, ceux-ci démontrent d’importants effets bénéfiques quant à une diminution de la résistance systémique et pulmonaire ainsi qu’une augmentation de l’éjection cardiaque.173 Il a également été démontré avec certitude que les antagonistes des récepteurs ETA améliorent le taux de survie des rats avec insuffisance cardiaque chronique.17475 Quoique les antagonistes ETA amméliorent nettement la condition clinique des personnes avec insuffisances cardiaques, plusieurs petites études cliniques dont les résultats ne sont pas connus, s’intéressent présentement à leurs effets à long terme sur la survie des patients.Chez les patients hypertendus avec insuffisance rénale ou maladie vasculaire, on retrouve des niveaux plasmatiques d’ET-l dépassant 10 fois la normale.131 Ceci suggère une diminution de la clairance de l’ET-l et/ou une
)
augmentation de sa production. Comme mentionné plus tôt, ce peptide participe à l’hypertrophie vasculaire et cardiaque associée à l’hypertension. li est de surcroît impliqué par ses effets rénaux et endocriniens. En effet, l’ET-l stimule l’activité sympathique, le système rénine-AglI-aldostérone et la synthèse d’adrénaline ce qui contribue à maintenir la pression artérielle et la résistance vasculaire élevées.176771.4.4.4 Le système rénal
Le système endothéline est intimement lié à la physiologie rénale. Quoique l’ET-l soit un puissant vasoconstricteur de tous les lits vasculaires, un groupe de recherche a constaté que la circulation rénale est 10 fois plus sensible à ce peptide que les autres lits vasculaires.178 L’ET-l est synthétisée par les cellules endothéliales des vaisseaux rénaux et par les différentes cellules qui composent le néphron. Les deux types de récepteurs sont
)
présents, mais lET8 est prédominant.179 Le système ET contrôle le flot sanguin rénal, la réabsorption de l’eau et du sodium (Na) ainsi que la balance acide-base. Ainsi, il est grandement impliqué dans l’homéostasie volumique.180 Ses effets ont été démontrés par l’administration d’ET-l dans les artères rénales chez le lapin anesthésié.181 Le traitement a réduit le débit sanguin rénal, le taux de filtration glomérulaire, le flot urinaire et l’excrétion de Na. Certaines évidences scientifiques suggèrent également que les récepteurs ETB présents dans les reins, régulent la pression sanguineL’ET-l a un rôle actif dans le développement et la progression de l’insuffisance rénale, car la sévérité de cette pathologie corrèle avec ses niveaux rénaux.18283 Elle est responsable des changements structuraux du parenchyme rénal impliquant l’hypertrophie, la prolifération cellulaire et la déposition de collagène. De surcroît, elle diminue la filtration glomérulaire et la réabsorption tubulaire nette, deux indices de la fonction rénale.184
À
vrai)
dire, il y a plusieurs évidences d’un système ET intra-rénal qui contrôle l’homéostasie ionique des fluides corporels ainsi que la pression sanguine.1851.4.4.5 Le foie
L’endothéline est un puissant vasoconstricteur du lit vasculaire hépatique et elle augmente la glycogénolyse c’est-à-dire l’éjection hépatique du glucose.186 De ce fait, l’insuline est augmentée afin de contrer la hausse de glucose dans le sang. Cependant, elle stimule aussi l’expression du gène de l’ET-l et la libération de celle-ci par les cellules endothéliales favorisant ainsi une élévation de ces niveaux plasmatiques chez l’homme.187189 De plus, en présence d’une résistance à l’insuline, l’expression des récepteurs ETA sur les cellules musculaires lisses est augmentés et ceux-ci ont une sensibilité accrue à l’ET-l Par conséquent, cette boucle entre insuline
et
l’ET-J se conclut par une rétroaction positive entre ces deux substances et
par des niveaux plasmatiques de glucose et d’ET-J augmentés.
La cirrhose est aussi une pathologie où les niveaux d’ET-l sont à la hausse.192 L’EV-1 a un rôle très clair dans la vasoconstriction rénale associée au syndrome hépatorénal. Celui-ci est une complication commune et
fréquente de la cirrhose caractérisée par une diminution de la fonction rénale, une perturbation de la circulation artérielle et une activation des systèmes
vasoactifs.193 L’ET-l diminue la filtration glomérulaire et favorise la rétention
sodique et hydrique. En effet, les patients cirrhotiques développent des ascites qui sont des accumulations de fluides qui peuvent atteindre quelques
litres, dans la cavité péritonéale. De plus, la synthèse d’ET-l par les cellules
stellaires situées à l’intérieur du foie, contribue à augmenter la résistance vasculaire intra-hépatique. L’accumulation de collagène provenant de
l’activation des fibroblastes par l’ET-l, s’accompagne d’une perte de
)
fenestrations des cellules endothéliales sinusoïdale. Ceci empêche les échanges entre les hépatocytes et le plasma et favorise la dysfonction hépatique.1.4.4.6 Le cerveau
La présence, au niveau des cellules vasculaires, neuronales et gliales,
de l’ET-l et de l’ET-3 ainsi que des récepteurs ETA et ETB suggère que ceux
ci jouent un rôle important dans le contrôle des actions neuronales. L’ET-l
est la principale endothéline retrouvée dans le cerveau à l’exception de la glande pituitaire où l’ET-3 est majoritaire.194 Une injection intraventriculaire
d’ET-l produit une augmentation transitoire de plusieurs minutes, de la
fréquence cardiaque et respiratoire, de la pression artérielle et de l’activité sympathique rénale.195 Suite à ce changement transitoire, des doses élevées
)
d’ET-l causent une dépression à long terme de ces paramètres. De plus, les)
ETs contrôlent la libération de neurotransmetteurs, par exemple les catécholamines, elles sont impliquées dans la modulation du comportement et du métabolisme et elles influencent le système sympathique.16°Le rôle de I’ET-l dans le vasospasme cérébral à la suite d’une hémorragie subarachnoïdienne est connu ainsi que dans les détériorations neurologiques subséquentes. Ses niveaux plasmatiques sont élevés dans le liquide cérébrospinal des patients atteints.196 Elle est libérée en réponse à l’inflammation et à l’ischémie et provoque une vasoconstriction massive des vaisseaux cérébraux.
I A.4.7 Le système respiratoire
Dans le poumon, l’ET-I est exprimée par les cellules endothéliales, les
j
cellules musculaires lisses des voies respiratoires,endocrines pulmonaires et par les macrophages. Les isoformes ECE-la,les cellules neuroECE-lb, ECE-ic sont présentes et très abondantes dans le poumon comparativement aux autres tissus humains.115 L’expression de l’ET-I dans les voies respiratoires est régulée par les médiateurs de l’inflammation. L’ET I influence le tonus basal des muscles lisses trachéaux et bronchiques.197 Les deux types de récepteurs à l’ET-I sont exprimés au niveau pulmonaire toutefois, les ETB représentent la majorité des récepteurs au niveau du muscle lisse bronchique et sont responsables de la bronchoconstriction.’98
L’ET-l est impliquée dans les diverses formes de l’hypertension pulmonaire ainsi que dans l’inflammation chronique des voies respiratoires; l’asthme. Cette pathologie est caractérisée par une vasoconstriction, une hyperactivité du muscle bronchique à l’ET-I, un influx de cellules inflammatoires, une sécrétion de mucus et un remodelage des voies