Bacterial communities associated with the midgut microbiota of wild Anopheles gambiae complex in Burkina Faso
Texte intégral
Figure

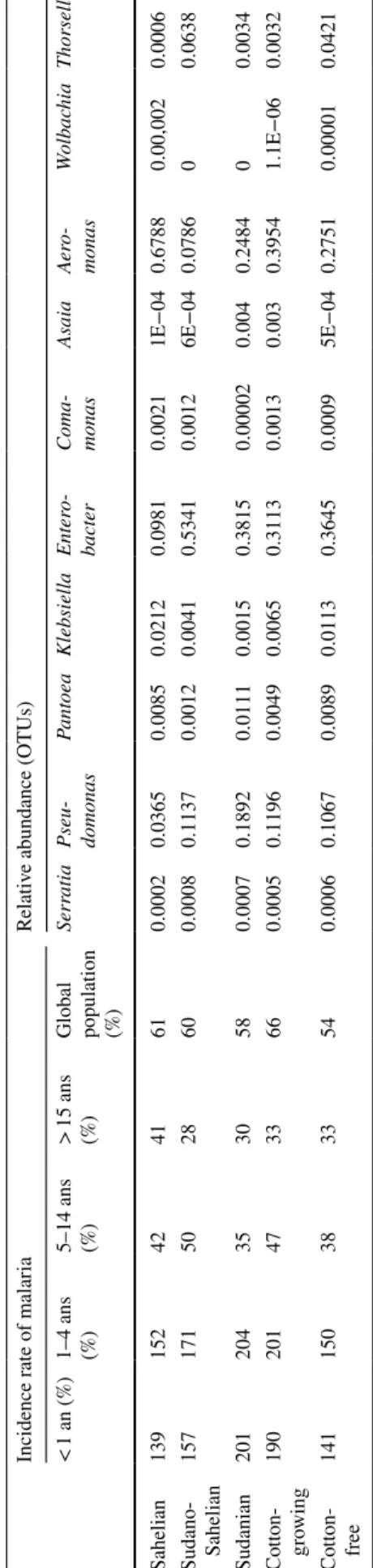
Documents relatifs
Here we demonstrate that bacterial infections of the malaria mosquito Anopheles gambiae are sensed by the orthologous PGRPLC protein which then activates a signaling pathway
Leur comparaison avec les effectifs observés montre que pour les systèmes 2Ra, 2Rb 2Rd’ et 3Ra, les différences ne sont pas significatives et sont compatibles avec
Correspondence analysis (CA) plot generated from the whole non cyanobacterial OTUs obtained with Eub-Pr1 (A) and NC-Pr2 (B) primer set, showing the distribution of the
Table 1 Rotated factor patterns testing for behavioural differences between larval instars (2 nd , 3 rd and 4 th ) of An. High positive scores on PC1 indicated that larvae
gambiae complex is given for all sites, providing a comprehensive description of the extent and diversity of insecticide multiresistance phenotypes and mechanisms in major
Figure 1 Comparison of swarm size, swarming period, height and number of mating couples observed between the swarms of
reported in south-western Burkina Faso, West Africa. Cross-resistance to DDT and pyrethroids was conferred by alterations at site of action in the sodium channel, the Leu-Phe kdr
coluzzii males (Results above, Figure 4A). The criteria used to define the start of activity in the two assays are too different, however, to allow more detailed compari- sons
