RESEARCH OUTPUTS / RÉSULTATS DE RECHERCHE
Author(s) - Auteur(s) :
Publication date - Date de publication :
Permanent link - Permalien :
Rights / License - Licence de droit d’auteur :
Institutional Repository - Research Portal
Dépôt Institutionnel - Portail de la Recherche
researchportal.unamur.be
University of Namur
NFE2L3 Controls Colon Cancer Cell Growth through Regulation of DUX4, a CDK1
Inhibitor
Bury, Marina; Le Calve, Benjamin; Lessard, Frédéric; Dal Maso, Thomas; Saliba, James;
Michiels, Carine; Febeyre, Gerardo; Vloker, Blank
Published in:
Cell Reports
DOI:
https://doi.org/10.1016/j.celrep.2019.09.087
Publication date:
2019
Document Version
Publisher's PDF, also known as Version of record
Link to publication
Citation for pulished version (HARVARD):
Bury, M, Le Calve, B, Lessard, F, Dal Maso, T, Saliba, J, Michiels, C, Febeyre, G & Vloker, B 2019, 'NFE2L3
Controls Colon Cancer Cell Growth through Regulation of DUX4, a CDK1 Inhibitor', Cell Reports, vol. 29, no. 6,
pp. 1469-1481.e9. https://doi.org/10.1016/j.celrep.2019.09.087
General rights
Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain
• You may freely distribute the URL identifying the publication in the public portal ?
Take down policy
If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim.
Article
NFE2L3 Controls Colon Cancer Cell Growth through
Regulation of DUX4, a CDK1 Inhibitor
Graphical Abstract
Highlights
d
NFE2L3 levels are elevated in colon cancer patients
d
NFE2L3 expression is regulated by the RELA subunit of
NF-kB
d
Silencing of NFE2L3 decreases colon cancer cell proliferation
d
NFE2L3 acts as a repressor of DUX4, a direct inhibitor of
CDK1 activity
Authors
Marina Bury, Benjamin Le Calve´,
Fre´de´ric Lessard, ..., Carine Michiels,
Gerardo Ferbeyre, Volker Blank
Correspondence
volker.blank@mcgill.ca
In Brief
Bury et al. show that the expression of the
transcription factor NFE2L3 is regulated
by NF-
kB and that NFE2L3 levels are
elevated in colon cancer patients.
Knockdown of NFE2L3 inhibits colon
cancer cell proliferation through
induction of DUX4, a direct inhibitor of
CDK1, establishing a pathway governing
colon tumor growth.
Bury et al., 2019, Cell Reports29, 1469–1481 November 5, 2019ª 2019 The Authors.
Cell Reports
Article
NFE2L3 Controls Colon Cancer Cell Growth
through Regulation of DUX4, a CDK1 Inhibitor
Marina Bury,1,7,9Benjamin Le Calve´,4,8,9Fre´de´ric Lessard,4Thomas Dal Maso,5James Saliba,1Carine Michiels,6 Gerardo Ferbeyre,4and Volker Blank1,2,3,10,*
1Lady Davis Institute for Medical Research, McGill University, Montreal, QC H3T 1E2, Canada 2Department of Medicine, McGill University, Montreal, QC H3T 1E2, Canada
3Department of Physiology, McGill University, Montreal, QC H3T 1E2, Canada 4Department of Biochemistry, University of Montreal, Montreal, QC H3C 3J7, Canada
5Department of Chemistry, Namur Medicine and Drug Innovation Center (NAMEDIC-NARILIS), University of Namur, 5000 Namur, Belgium 6URBC-NARILIS, University of Namur, 5000 Namur, Belgium
7Present address: De Duve Institute, UCLouvain, 1200 Brussels, Belgium 8Present address: URBC-NARILIS, University of Namur, 5000 Namur, Belgium 9These authors contributed equally
10Lead Contact
*Correspondence:volker.blank@mcgill.ca https://doi.org/10.1016/j.celrep.2019.09.087 SUMMARY
Constitutive nuclear factor
kB (NF-kB) activation is a
hallmark of colon tumor growth. Cyclin-dependent
kinases (CDKs) are critical cell-cycle regulators,
and inhibition of CDK activity has been used
suc-cessfully as anticancer therapy. Here, we show that
the NFE2L3 transcription factor functions as a key
regulator in a pathway that links NF-
kB signaling to
the control of CDK1 activity, thereby driving colon
cancer cell proliferation. We found that NFE2L3
expression is regulated by the RELA subunit of
NF-kB and that NFE2L3 levels are elevated in
pa-tients with colon adenocarcinoma when compared
with normal adjacent tissue. Silencing of NFE2L3
significantly decreases colon cancer cell
prolifera-tion
in vitro and tumor growth in vivo. NFE2L3
knock-down results in increased levels of double homeobox
factor 4 (DUX4), which functions as a direct inhibitor
of CDK1. The discovered oncogenic pathway
gov-erning cell-cycle progression may open up unique
avenues for precision cancer therapy.
INTRODUCTION
Dysfunctional transcriptional and signaling networks have a fundamental role in colorectal cancer (CRC), one of the most common and fatal malignancies worldwide (Kuipers et al., 2015). Different molecular CRC subtypes have been identified, and understanding of the underlying pathogenesis of CRC for-mation is crucial for predicting prognosis and treatment response (De Sousa E Melo et al., 2013). The formation of CRC includes hereditary elements, but, in most cases, is sporadic and forms gradually over several years through the adenoma-carcinoma sequence. Mutations in APC, KRAS, and TP53, in conjunction with chromosomal instability, have been implicated
in CRC development; nevertheless, other pathways can drive tumorigenesis as well (Brenner et al., 2014; Cancer Genome Atlas Network, 2012). Activation of the nuclear factor kB (NF-kB) transcription factor has a critical role in many cancer processes, including inflammation, growth, angiogenesis, inva-sion, metastasis, and resistance (Aggarwal and Sung, 2011). In addition, NF-kB is involved in the initiation and progression of CRC (Karin et al., 2002; Vaiopoulos et al., 2013). Multiple path-ways associated with uncontrolled cell proliferation, including phosphatidylinositol 3-kinase (PI3K)-AKT-mechanistic target of rapamycin (mTOR), have been linked to NF-kB signaling (Dan et al., 2008). NF-kB also controls the levels of key cell-cycle reg-ulators, such as cyclin D1, MYC, and cyclin-dependent kinases (CDKs) (Hinz et al., 1999; Karin et al., 2002; La Rosa et al., 1994; Perkins et al., 1997). Overexpression of NF-kB strongly correlates with worse overall survival of CRC patients (Wu et al., 2015). Moreover, increased levels of tumor necrosis factor (TNF), a cytokine inducing NF-kB activity, have been associated with advanced stages of CRC (Al Obeed et al., 2014), and chronic TNF exposure contributes to a pro-malignant phenotype (Szlosarek et al., 2006).
Cap’n’collar (CNC) transcription factors have crucial roles in a variety of cellular processes, including the stress response and carcinogenesis, with its most extensively investigated family member being the NFE2L2 (NRF2) protein (DeNicola et al., 2011). NFE2L3 (NRF3), a close homolog of NFE2L2, is less well studied and its functions remain largely unknown (Chevillard and Blank, 2011), but some recent findings linked the transcrip-tion factor to apoptosis and different types of cancer ( Chowd-hury et al., 2017; Siegenthaler et al., 2018; Sun et al., 2019; Wang et al., 2017, 2018). Similar to other CNC proteins, NFE2L3 dimerizes with small MAF transcription factors, and the resulting complexes bind to the antioxidant response element (ARE) type of DNA-recognition sites (Che´nais et al., 2005). NFE2L3 transcript and protein levels are induced by TNF (Che´nais et al., 2005). NFE2L3 is a tightly regulated and post-translational-modified protein with a rapid turnover (Nouhi et al., 2007), and FBXW7 ubiquitin ligase and GSK3B have
been shown to control NFE2L3 degradation (Kannan et al., 2015). Subcellular fractionation experiments revealed the presence of three forms of NFE2L3: a primarily endoplasmic re-ticulum (ER)-bound N-glycosylated A form, a cytoplasmic non-glycosylated B form, and a faster migrating, largely nuclear C form (Kannan et al., 2015; Nouhi et al., 2007). In this study, we aimed to unravel the cellular network governing NFE2L3 regula-tion and funcregula-tion. We report that NFE2L3 acts as a central player in an identified NF-kB signaling pathway that controls colon can-cer cell growth.
RESULTS
NFE2L3 Controls Colon Tumor Growth
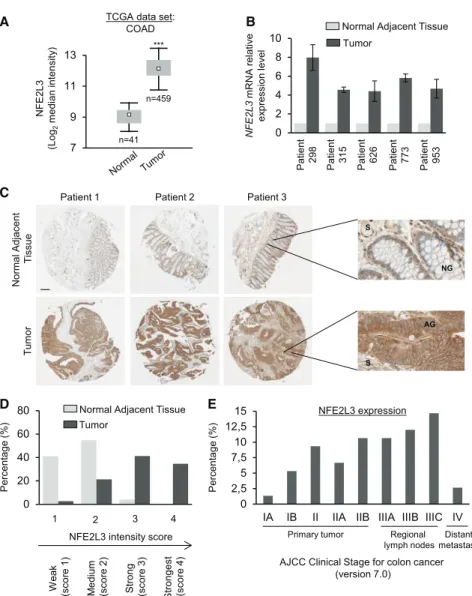
We investigated the mRNA levels of the NFE2L3 transcription factor in normal and tumoral tissues using The Cancer Genome Atlas (TCGA) database (Figure 1A). NFE2L3 transcripts were significantly upregulated in colon adenocarcinoma (n = 459) compared with normal samples (n = 41). Similar results were observed in BioGPS and Oncomine datasets (Figure S1) as well as upon analysis of NFE2L3 mRNA levels in a set of five
co-NFE2L3 (Log 2 median intensity )
TCGA data set: COAD 7 9 11 13 *** n=41 n=459 A B Patient 298 Tumor
Normal Adjacent Tissue
NFE2L3 mRNA relativ e ex pression lev el
Patient 315 Patient 626 Patient 773 Patient 953
0 2 4 6 8 10 0 2,5 5 7,5 10 12,5 15
IA IB II IIA IIB IIIA IIIB IIIC IV
Percentage
(%)
AJCC Clinical Stage for colon cancer (version 7.0)
NFE2L3 expression
E
Primary tumor Regional
lymph nodesmetastasisDistant
Patient 1 Patient 2 Patient 3
Normal A djacent T issue Tu m o r C S AG NG S 0 20 40 60 80 D 1 2 3 4 Percentage (%) Tumor
Normal Adjacent Tissue
We a k (s co re 1 ) M e dium (s co re 2 ) Strong (sco re 3 ) Stronges t (scor e 4 )
NFE2L3 intensity score
Figure 1. Upregulation of NFE2L3 Corre-lates with Poor Prognosis for Patients with Colon Cancer
(A) Colon adenocarcinoma (COAD) cancer gene expression data (mRNA, normalized RNA-seqV2 RSEM) were retrieved from TCGA database using the cBioPortal for cancer genomics. Boxplots represent the first and third quartiles and median values; whiskers represent the 5th and 95th per-centiles; Mann-Whitney U test, ***p < 0.001. (B) Quantitative real-time PCR analysis of NFE2L3 expression in colon cancer tissues and the adja-cent counterpart from five patients with colon adenocarcinoma (means± SD).
(C) Representative IHC images of human colon adenocarcinoma TMA stained for NFE2L3. The scale bar represents 200mm; on the right side, magnification380; S, stroma; NG, normal gland; AG, adenocarcinoma gland.
(D) Classification and quantification of samples according to the intensity of staining of NFE2L3 expression (n = 75).
(E) Distribution of the higher NFE2L3 expression tumor samples (score 3 and 4) using the AJCC Clinical Stage for Colorectal Cancer (Singh, 2017).
lon adenocarcinomas and normal adjacent tissues by quantitative real-time PCR (Figure 1B). To evaluate NFE2L3 protein levels, we performed immunohistochemistry (IHC) staining on a tissue microarray (TMA) of 75 patients, revealing greater expression of NFE2L3 in colon adenocarcinoma compared with matched normal adjacent tissue (Figure 1C). We classified the samples into four groups with increasing staining intensity from weakest (score 1) to stron-gest (score 4) (Figure 1D). We observed that NFE2L3 expression was weak, falling into groups 1 and 2 in most adjacent normal tis-sues (95%). In contrast, NFE2L3 levels were high, falling into groups 2–4 in most colon tumor tissues (97%;Figure 1D). In co-lon adenocarcinoma glands, we observed a strong nuclear and strong rimmed perinuclear pattern for NFE2L3, which diffuses across their cytoplasm, whereas, in normal glands, the staining is mostly perinuclear and cytoplasmic (Figure 1C). We also found that the high expression of NFE2L3 in tumor samples is corre-lated with the advanced stages of the disease, except for the metastasis stage (Figure 1E). Together, these results suggest that NFE2L3 might have a critical role in colon adenocarcinoma development and may be linked to major oncogenic pathways.
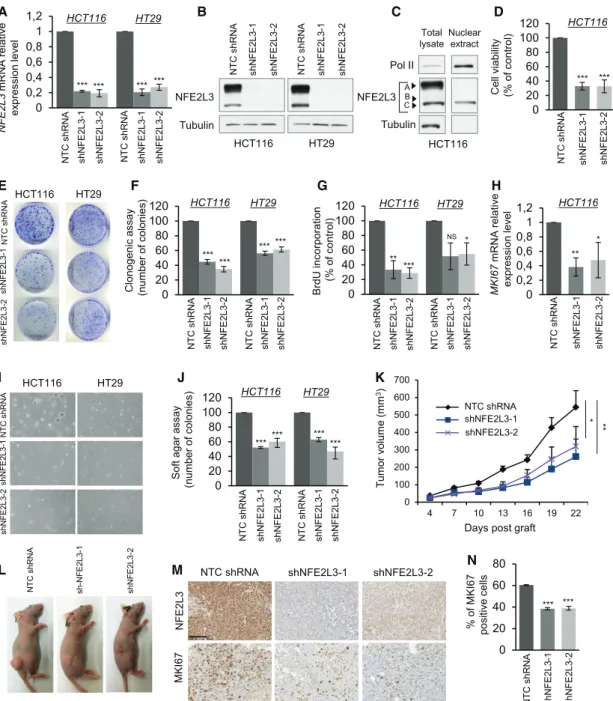
Silencing NFE2L3 Inhibits Colon Cancer Cell Proliferation
To assess the role of NFE2L3 in colon cancer, we depleted NFE2L3 expression with two different lentiviral short hairpin RNAs (shRNAs) in distinct colon cancer cell lines HCT116 and HT29. Efficient knockdown of NFE2L3 was confirmed by quanti-tative real-time PCR and immunoblot (Figures 2A and 2B) and
NFE2L3 HT29 NT C s hRNA sh NFE 2L3-2 sh N F E2 L3 -1 NT C s hRNA sh NFE 2L3-2 sh N F E2 L3 -1 Tubulin HCT116 B HCT116 NT C shRNA shN F E 2L3-1 shN F E 2L3-2 HT29 E I HCT116 HT29 NT C sh RNA shNF E 2L3-1 shN F E 2L3-2 NT C sh RNA sh-N F E 2L3-1 shNF E 2L3-2 L T u mor v o lume (mm 3)
Days post graft
* ** K 0 100 200 300 400 500 600 700 4 7 10 13 16 19 22 NTC shNrf3-1 shNrf3-2 NTC shRNA shNFE2L3-1 shNFE2L3-2 F C lonogenic a ssay (number of colonies) HCT116 HT29 *** *** *** *** NT C s hRNA sh NFE 2L3-1 sh N F E2 L3 -2 NT C s hRNA sh NFE 2L3-1 sh N F E2 L3 -2 0 20 40 60 80 100 120 % of MKI67 positiv e c ells *** *** NT C s hRNA sh N F E2 L3 -1 sh N F E2 L3 -2 N 0 20 40 60 80 0 0,2 0,4 0,6 0,8 1 1,2 NFE2L3 m RNA relativ e ex pression lev el HCT116 HT29 *** *** *** A *** NT C s hRNA sh N F E2 L3 -1 sh N F E2 L3 -2 NT C s hRNA sh N F E2 L3 -1 sh N F E2 L3 -2 0 20 40 60 80 100 120 NT C s hRNA sh N F E2 L3 -1 sh N F E2 L3 -2 C e ll vi ability (% o f c ontrol) *** *** D HCT116 NS 0 20 40 60 80 100 120 BrdU inc o rporatio n (% o f control) ** *** HCT116 HT29 G NT C s hRNA sh NFE 2L3-1 sh NFE 2L3-2 NT C s hRNA sh NFE 2L3-1 sh N F E2 L3 -2 * 0 0,2 0,4 0,6 0,8 1 1,2 MKI67 m RNA relativ e ex pression lev el ** H NT C s hRNA sh NFE 2L3-1 sh N F E2 L3 -2 HCT116 * 0 20 40 60 80 100 120 J ****** *** HCT116 HT29 *** Soft a gar as sa y (number o f colonies) NT C s hRNA sh N F E2 L3 -1 sh NFE 2L3-2 NT C s hRNA sh N F E2 L3 -1 sh N F E2 L3 -2
NTC shRNA shNFE2L3-1 shNFE2L3-2
NFE2L 3 MKI67 M Total lysate A Nuclear extract C NFE2L3 Tubulin Pol II B C HCT116
Figure 2. NFE2L3 Is Required for Colon Cancer Cell GrowthIn Vitro and In Vivo
(A) Quantitative real-time PCR analysis of NFE2L3 mRNA in HCT116 and HT29 cells transduced with NFE2L3-specific shRNAs (shNFE2L3-1 or -2) presented relative to NFE2L3 mRNA expression in cells transduced with a non-targeting control (NTC) shRNA.
(B) Immunoblot analyses of NFE2L3 in the cells described in (A).
(C) Immunoblot analyses of NFE2L3 from total lysate or nuclear extraction in HCT116 cells. Arrows indicate the A, B, and C form of NFE2L3.
(D) Cell viability in HCT116 cells assessed by MTT assay after 72 h. Error bars are means± SEM, n = 3 independent experiments, ***p < 0.001, two-sided Student’s t test.
(E and F) Representative images (E) and quantification (F) of a colony-formation assay of the HCT116 and HT29 cells. For each cell line, all dishes were fixed at the same time, stained, and photographed. Error bars are means± SEM, n = 3 independent experiments, ***p < 0.001, two-sided Student’s t test.
(G) Incorporation of BrdU in HCT116 and HT29 cells as measured by ELISA after treatment for 16 h. Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant; two-sided Student’s t test.
(H) Quantitative real-time PCR analysis of MKI67 mRNA in HCT116 cells. Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, **p < 0.01, two-sided Student’s t test.
(I and J) Representative images (I) and quantification (J) of HCT116 and HT29 cells by soft agar assay. Error bars are means± SEM, n = 3 independent ex-periments, ***p < 0.001, two-sided Student’s t test.
affected the abundance of all three previously characterized NFE2L3 A, B, and C forms (Figure 2C) (Nouhi et al., 2007). We observed a strong reduction in cell numbers three days after plating when NFE2L3 was knocked down, and colony formation was significantly reduced (Figures 2D–2F). No changes in apoptosis were observed by flow cytometry using Annexin V and propidium iodide (PI) staining (Figure S2). In contrast, reduced numbers of bromodeoxyuridine (BrdU)-incorporating cells, as well as a decrease in the mRNA expression of the
MKI67 (Ki67) gene coding for a major cell-proliferation marker,
showed that NFE2L3 functions as a positive regulator of colon cancer cell growth (Figures 2G and 2H). To model physiological conditions, we evaluated anchorage-independent colon cancer cell growth in a soft agar assay (Figures 2I and 2J) and prolifera-tion of HCT116 cells in vivo in a mouse xenograft model ( Fig-ure 2K and 2L). In both settings, NFE2L3 knockdown severely compromised cell growth. Reduced MKI67 staining in xeno-grafts of NFE2L3 knockdown samples confirmed that this effect was largely due to reduced cell proliferation in vivo (Figures 2M and 2N). Altogether, these results establish a crucial role for NFE2L3 as a promoter of colon cancer cell proliferation and tu-mor growth.
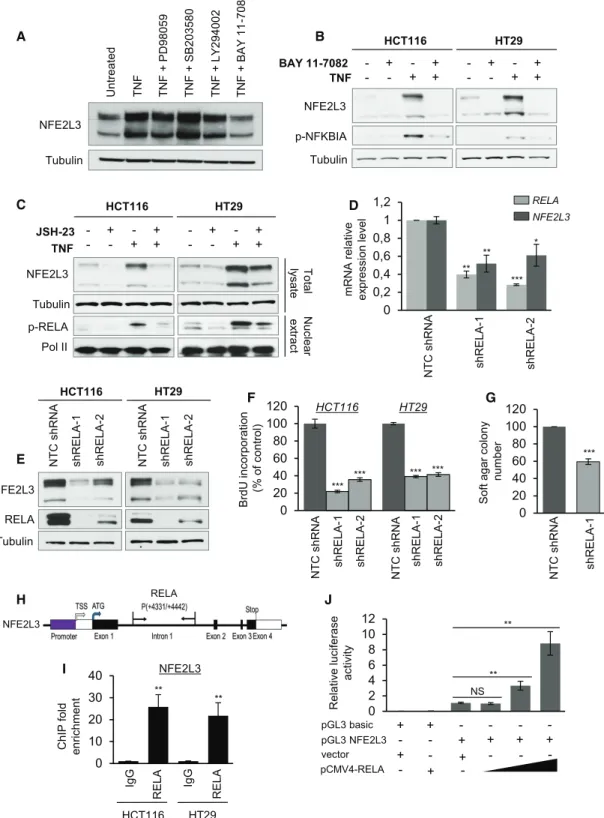
RELA Functions as a Positive Regulator of NFE2L3 Expression
Our previous studies had shown that TNF increases NFE2L3 levels (Che´nais et al., 2005). To determine the molecular mecha-nisms underlying NFE2L3 upregulation in colon cancer cells, we treated the cells with inhibitors of different signaling pathways upon TNF activation and assessed NFE2L3 levels. Specifically, we examined the effect of inhibiting the MEK1/2 (PD98059), p38 (SB203580), PI3K/AKT (LY294002), and NF-kB (BAY 11-7082) pathways because these signaling molecules have been previously linked to colon tumorigenesis (Karin, 2006; Cancer Genome Atlas Network, 2012). Although MEK1/2, p38, and PI3K/AKT inhibitors had no or minimal effect, we observed that the inhibitor of NF-kB signaling strongly reduced NFE2L3 levels (Figure 3A). NF-kB pathway is involved in inflammation, tumor survival, migration, and proliferation of colon cancer cells ( Ben-Neriah and Karin, 2011; Karin, 2006; Wang et al., 2009). In vivo, colorectal cancer cells are exposed to a variety of cytokines released from the tumor stroma, including known activators of NF-kB signaling, such as TNF, which enhances the growth of co-lon cancer cells by the activation of oncogenic pathways ( Ben-Neriah and Karin, 2011; Wang et al., 2009). Treatment of colon cancer cell lines with TNF increased NFE2L3 protein levels; this effect was strongly diminished upon treatment of cells with two mechanistically different inhibitors of NF-kB signaling ( Fig-ures 3B and 3C): BAY 11-7082 blocks TNF-induced NFKBIA (IkBa) phosphorylation, and JSH-23 functions as an inhibitor of NF-kB transcriptional activity (Pierce et al., 1997; Shin et al., 2004). The NF-kB transcription factor family comprises
homo-and heterodimeric complexes that are formed by the combina-tion of five different subunits, including RELA (p65), RELB, REL, NFKB1 (p50), and NFKB2 (p52) (Karin, 2006; Vaiopoulos et al., 2010). We found that shRNA-mediated knockdown of RELA significantly decreased NFE2L3 mRNA and protein expression levels (Figures 3D, 3E, andS3A–S3C), recapitulating the reduced growth phenotype associated with NFE2L3 silencing (Figures 3F and 3G). In contrast, downregulation of the NF-kB subunits REL, RELB, NFKB1, or NFKB2 had no effect on NFE2L3 expression (Figure S3). Consistent with this, data from the ENCODE project consortium showed binding of the RELA subunit of NF-kB to the first intron of NFE2L3 (Figure S4A) (ENCODE Project Consortium, 2012), which we confirmed by chromatin immunoprecipitation (ChIP)-qPCR analyses and lucif-erase reporter assays (Figures 3H–3J). To assess whether low NFE2L3 levels contribute to the inhibition of proliferation observed upon silencing of RELA in colon cancer cell lines, we overexpressed the NFE2L3 full form (generating A, B, and C), the ER/cytoplasmic A–B forms, or the nuclear C form of NFE2L3 and found that both expression of the full or C form of NFE2L3, but not the A–B form, partially rescued the phenotype of reduced colon cancer cell growth upon RELA knockdown, as confirmed by BrdU incorporation (Figures S4B and S4C). We conclude that RELA positively regulates NFE2L3 expression and thus may contribute to its protumoral effects.
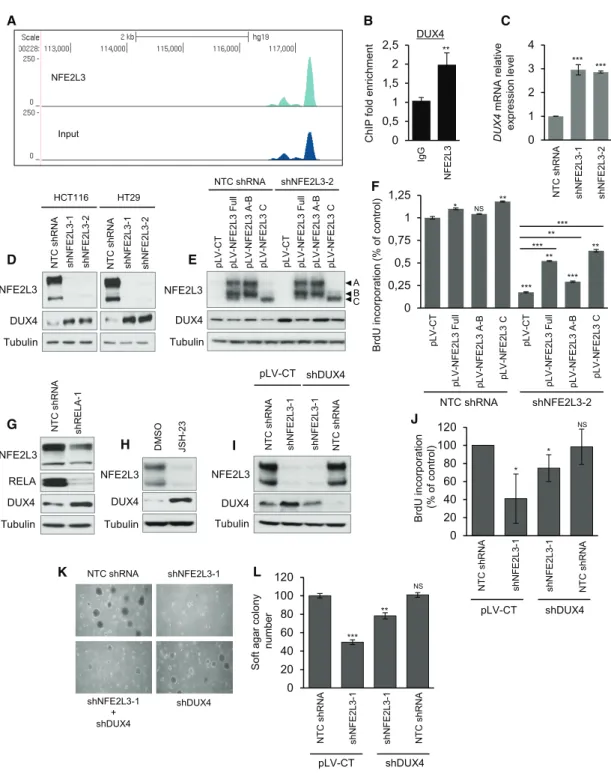
Identification of DUX4 as a Negatively Regulated Target of NFE2L3
To investigate the mechanistic link between NFE2L3 and cell proliferation, we searched for genes controlled by NFE2L3 that may mediate its effect on colon cancer cell proliferation. To iden-tify transcriptional targets, we carried out ChIP-sequencing with NFE2L3-specific antiserum in TNF-treated HCT116 cells. Among the identified potential NFE2L3 target genes, the DUX4 gene emerged as a highly interesting candidate because it had been previously linked to the cell cycle (Bosnakovski et al., 2008). In addition, DUX4 ranked as the top gene to have its NFE2L3 peaks converge when combining both fold enrichment and distance to the transcription start site (TSS) (Figure 4A). The binding of NFE2L3 to the DUX4 locus was corroborated by ChIP-qPCR analysis (Figure 4B), and knockdown of NFE2L3 re-sulted in a significant increase in DUX4 mRNA (Figure 4C) and protein levels (Figure 4D). Reintroduction of the full form and the nuclear C form of NFE2L3 by lentiviral transduction reduced DUX4 levels and partially rescued cellular proliferation (Figures 4E and 4F). Consistent with these data, inhibition of NF-kB by RELA-knockdown or JSH-23 inhibitor treatment reduced NFE2L3 and increased DUX4 protein levels (Figures 4G and 4H). Thus, DUX4 is repressed by NFE2L3 and might have a role in the effects of NFE2L3 on cell proliferation. A gain-of-func-tion phenotype of DUX4 has been linked to facioscapulohumeral muscular dystrophy (FSHD) (Bosnakovski et al., 2008; Gabellini
(K and L) Representative gross images (K) of the xenograft tumors at the endpoint and growth curves (L) of xenograft tumors derived from subcutaneously implanted HCT116 cells expressing a NTC shRNA or NFE2L3-specific shRNAs (shNFE2L3-1 or -2). Error bars are means± SEM, n=9 mice per group, *p<0.05, **p<0.01, Mann-Whitney U test.
(M) Representative images of NFE2L3 and MKI67 analyzed by immunohistochemical staining.
(N) Quantification of MKI67 analyzed by immunohistochemical staining. The scale bar represents 100mm. The bar graph shows the percentage of MKI67-positive cells. Error bars are means± SEM, n = 3 mice per group, ***p < 0.01, two-sided Student’s t test.
H F 0 20 40 60 80 100 120 BrdU inc o rporatio n (% o f c o ntrol) NT C sh RNA shR E LA -1 shR E LA -2 NT C sh RNA shR E LA-1 shR E LA-2 HT29 HCT116 *** *** *** *** I 0 10 20 30 40 ChIP fol d enr ichment IgG
RELA IgG RELA
** ** HCT116 HT29 NFE2L3 NFE2L3 p-NFKBIA Tubulin TNF BAY 11 -7082 HCT116 HT29 B - + - + - - + + -- +- +- ++ C NFE2L3 Tubulin TNF JSH-23 HCT116 HT29 - + - + - - + + -- +- +- ++ p-RELA Pol II To ta l ly sate Nuc le a r ex tract 0 2 4 6 8 10 12 Relativ e luciferase activ ity J pGL3 NFE2L3 pGL3 basic + + - -- - + + pCMV4-RELA vector + - + -- -+ -+ -+ NFE2L3 Tubulin Untreated TN F T N F + L Y 294002 T N F + SB203580 T N F + P D98059 T N F + BAY 11 -7082 A E HCT116 HT29 NFE2L3 Tubulin RELA NT C sh RNA shR E LA-1 shR E LA -2 NT C sh RNA shR E LA-1 shR E LA -2 0 0,2 0,4 0,6 0,8 1 1,2 D NT C sh RNA shR E LA-1 shR E LA-2 mRN A relativ e ex pression lev el * ** ** *** NFE2L3 RELA G Soft a gar colony number NT C sh RNA shR E LA-1 *** 0 20 40 60 80 100 120 RELA NFE2L3 ** ** NS
Figure 3. NFE2L3 Is Regulated by the RELA Subunit of NF-kB
(A) HCT116 cells were pretreated with PD98059 (20mM), SB203580 (20 mM), LY294002 (20 mM), and BAY 11-7082 (10 mM) for 2 h, followed by stimulation with TNF (20 ng/mL) for 6 h. The expression of NFE2L3 was examined by immunoblot.
(B) Immunoblot analysis of NFE2L3 in HCT116 and HT29 cells pretreated with BAY 11-7082 (10mM, 2 h) followed by stimulation with TNF (20 ng/mL, 6 h). (C) Immunoblot analysis of NFE2L3 in HCT116 and HT29 cells pretreated with JSH-23 (50mM, 16 h) followed by stimulation with TNF (20 ng/mL, 6 h).
et al., 2002), but its function in cancer remains unclear, despite some recent data showing that DUX4 is deregulated in acute lymphoblastic leukemia and can control the migration of mesen-chymal stem cells (Dmitriev et al., 2016; Yasuda et al., 2016; Zhang et al., 2016). We found that lentiviral overexpression of DUX4, driven by a strong (cytomegalovirus [CMV]), as well as weak, promoter (ubiquitin C [UBC]), is toxic and leads to the death of most cells within 24–48 h (data not shown). To assess whether elevated DUX4 levels contribute to the inhibition of pro-liferation observed upon silencing of NFE2L3 in colon cancer cell lines, we thus knocked down DUX4 and observed that combined NFE2L3 and DUX4 depletion partially rescued the phenotype of reduced colon cancer cell growth, as confirmed by BrdU incor-poration and soft agar assays (Figures 4I–4L). We also analyzed
DUX4 mRNA levels in colon adenocarcinomas and normal
adja-cent tissue specimens. We found decreased DUX4 transcript levels in four of the five colon adenocarcinomas samples, thus correlating well with NFE2L3 expression (Figures 1B andS5A). Overall, these data suggest that DUX4 mediates in part the pro-liferation phenotype observed upon modulation of RELA and/or NFE2L3 levels.
Role of DUX4 as a Direct Inhibitor of CDK1 Activity
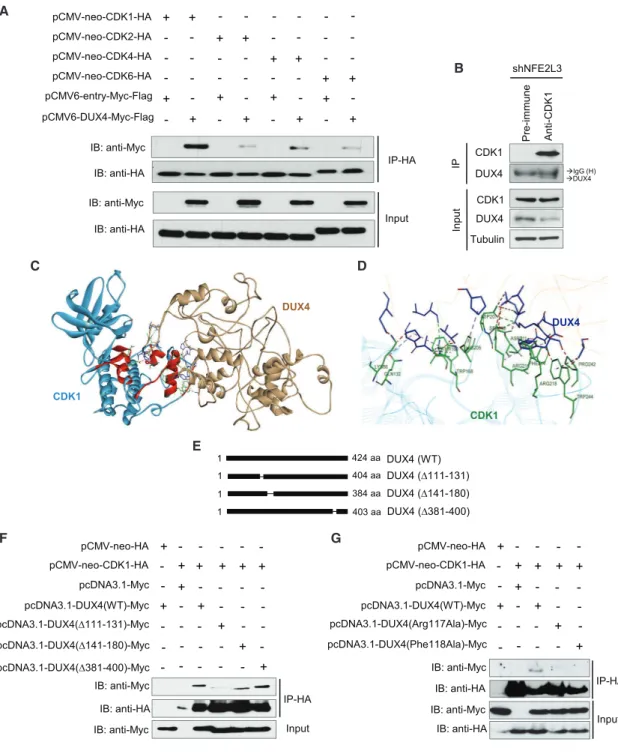
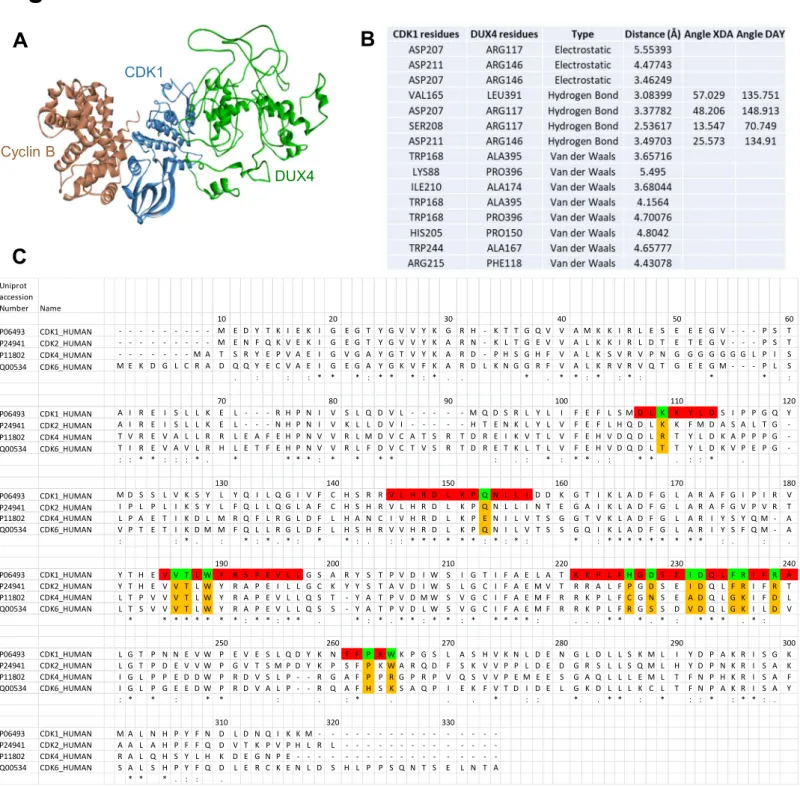
To understand how DUX4 might affect cell proliferation, we searched for DUX4-interacting proteins in colon cancer cells by performing immunoprecipitation followed by mass spectrom-etry analysis in HCT116 cells. Using ReactomePA (Yu and He, 2016), we identified different cell-cycle regulators, including cy-clin-dependent kinase 1 (CDK1), as potential binding partners for DUX4, providing a rationale for the role of NFE2L3 in the control of colon cancer cell proliferation (Figure S5B; Table S1). We confirmed the interaction between DUX4 and CDK1 by co-immunoprecipitation of epitope-tagged CDK1 and DUX4 co-ex-pressed in HCT116 cells (Figure 5A). In parallel, we performed co-immunoprecipitation experiments with three additional CDKs (CDK2, 4, and 6) to verify the specificity of the DUX4-CDK1 interaction (Figure 5A). Clearly, interaction between DUX4 and CDK1 is stronger than that with other members of the CDK family. Immunoprecipitation of endogenous CDK1 from cells expressing NFE2L3 shRNA, which increases endoge-nous DUX4 protein levels, confirmed the interaction between this kinase and the DUX4 protein (Figure 5B). Furthermore, we de-signed an in silico model of the DUX4-CDK1 complex using the ZDOCK server (Figures 5C, 5D, andS6A). Based on the complex CDK1-cyclin B1-CKS2 available on the PDB, we separated the three proteins to generate a model for DUX4-CDK1 interactions.
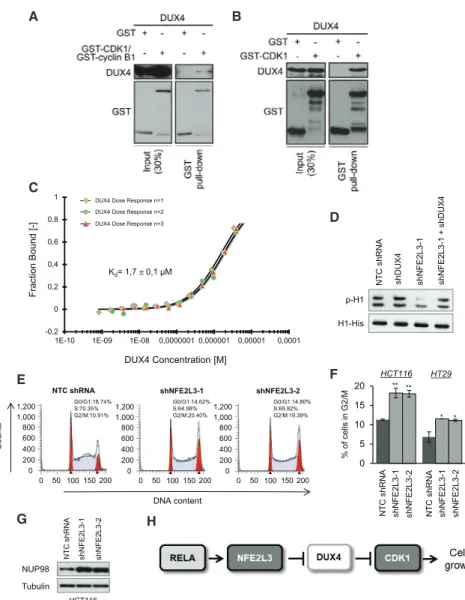
The model predicts that DUX4 binds CDK1 at the same location as CDC28 protein kinase regulatory subunit 2 (CKS2), and we identified the amino acids implicated in this binding (Brown et al., 2015) (Figure S6B). A series of interactions are involved in the formation of the complex, including hydrogen bonds and electrostatic as well as van der Waals interactions. We compared the sequences of CDK1 with CDK2, CDK4, and CDK6 to illustrate the variations among the different members of the CDK protein family and the difference of binding with DUX4. Our model suggests that the binding between DUX4 and CDK1 is mediated by a series of residues present in conserved domains of the CDK family but the variability of some amino acids may explain the difference of affinity observed inFigure 5A (Figure S6C). Based on these in silico models, we generated three different mutants to characterize the domain of DUX4 implicated in the interaction with CDK1 (Figure 5E). Deletion of amino acids from position 111–131 strongly, and deletion from position 141–180 moderately, reduced the interac-tion with CDK1, whereas deleinterac-tion of aa 381–400 had no effect (Figure 5F). We investigated more precisely the amino acids implicated in the interaction between DUX4 and CDK1 ( Fig-ure S6B), and based on those predictions, we generated two mutants, Arg117Ala and Phe118Ala, of DUX4. By immunopre-cipitation, we observed that the interaction between CDK1 and those DUX4 mutants is reduced compared with the wild-type form of the protein (Figure 5G). Next, we performed glutathione S-transferase (GST) pull-down comparing the binding of DUX4 to CDK1 and the CDK1-cyclin B complex (Figures 6A and 6B). Our data showed that DUX4 preferentially interacts directly with CDK1 alone and only minimally with the CDK1-cyclin B complex. In addition, we quantitatively assessed the interaction between DUX4-GST and CDK1-His tag by microscale thermo-phoresis (MST). We determined a KD(dissociation constant) of 1.7± 0.1 mM for the binding of DUX4 to CDK1 (Figure 6C). GST alone was used as negative control. Together, our results show that the DUX4 protein directly interacts with CDK1.
To determine whether DUX4 inhibits the activity of CDK1, we immunoprecipitated CDK1 and assessed its in vitro kinase activ-ity on recombinant histone H1 protein (Ruiz et al., 2010). We showed that CDK1 activity, measured by H1 phosphorylation, was strongly decreased in cells after NFE2L3 knockdown but was not altered in control cells, after a single DUX4 or a com-bined NFE2L3/DUX4 knockdown (Figure 6D). Cell-cycle profiling and high levels of NUP98 also showed that silencing of NFE2L3 leads to an accumulation of cells in the G2/M phase, which is controlled by the CDK1-cyclin B complex (Laurell et al., 2011)
(D) Quantitative real-time PCR analysis of RELA and NFE2L3 mRNA in HCT116 cells transduced with a NTC shRNA or RELA-specific shRNAs (shRELA-1 or -2). Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, two-sided Student’s t test.
(E) Immunoblot analysis of RELA and NFE2L3 in HCT116 and HT29 cells transduced with a NTC shRNA or RELA-specific shRNAs (shRELA-1 or -2). (F) BrdU incorporation measured by ELISA in HCT116 and HT29 cells transduced with a NTC shRNA or RELA-specific shRNAs (shRELA-1 or -2). Error bars are means± SD, n = 2 independent experiments, ***p < 0.001, two-sided Student’s t test.
(G) Quantification of soft agar assay of HCT116 cells transduced with a NTC shRNA or a RELA-specific shRNA (shRELA-1). Error bars are means± SEM, n = 3 independent experiments, ***p < 0.001, two-sided Student’s t test.
(H) Localization of a RELA binding site in the NFE2L3 gene locus.
(I) ChIP analyses were performed in HCT116 and HT29 cells using anti-RELA antibody. An immunoglobulin G (IgG) antibody was used as negative control. Fold enrichment was quantified by qPCR. Error bars are means± SEM, n = 3 independent experiments, **p < 0.01, two-sided Student’s t test.
(J) Luciferase assay: 293T cells were transfected with the indicated plasmids. After incubation for 24 h, luciferase activity was measured and normalized to Renilla. Error bars are means± SEM, n = 3 independent experiments, **p < 0.01, NS, not significant; two-sided Student’s t test.
A NFE2L3 Input B DUX4 0 0,5 1 1,5 2 2,5 ChIP fold enrichment ** Ig G NFE 2L3 0 1 2 3 4 DUX 4 m RNA rel a tive ex pression lev e l *** NT C s hRNA sh NFE 2L3-1 sh NFE 2L3-2 C *** NFE2L3 DUX4 Tubulin HCT116 HT29 D NTC s hRNA sh NFE 2L3-2 sh N F E2 L3 -1 NT C s hRNA sh N F E2 L3 -2 sh NFE 2L3-1 shNFE2L3-2 NTC shRNA NFE2L3 Tubulin DUX4 pLV-CT pLV-NFE 2L3 Full pLV -NFE 2L3 A -B pLV-NFE 2L3 C pLV -CT pLV -NFE 2L3 Full pLV-NFE 2L3 A-B pLV-NFE 2L3 C E A B C 0 0,25 0,5 0,75 1 1,25 BrdU incorporatio n (% o f control) F pLV -CT pLV -NFE 2L3 Full pLV -NFE 2L3 A -B pLV-NFE 2L3 C pLV -CT pLV -NFE 2L3 Full pLV -NFE 2L3 A-B pLV -NFE 2L3 C NTC shRNA shNFE2L3-2 *** ** *** ** *** ***** * NS ** NTC shRNA shNFE2L3-1 shDUX4 shNFE2L3-1 + shDUX4 K 0 20 40 60 80 100 120 Soft a gar co lony number *** ** NS L NT C s hRNA pLV-CT shDUX4 sh N F E2 L3 -1 sh NFE 2L3-1 NT C s hRNA sh NFE 2L3-1 sh N F E2 L3 -1 0 20 40 60 80 100 120 BrdU incorporatio n (% o f c ontrol) NT C s hRNA NT C s hRNA * * NS J pLV-CT shDUX4 DM S O JSH -2 3 Tubulin DUX4 NFE2L3 H I NFE2L3 DUX4 Tubulin NT C s hRNA pLV-CT shDUX4 NT C s hRNA sh NFE 2L3-1 sh NFE 2L3-1 NFE2L3 Tubulin DUX4 NT C s hRNA sh R E LA -1 G RELA
Figure 4. NFE2L3 Functions as a Repressor of DUX4
(A) Example of NFE2L3 ChIP-sequencing traces in HCT116 cells.
(B) ChIP-qPCR validation of NFE2L3 binding to the DUX4 locus using NFE2L3 or IgG control antibody. Error bars are means± SEM, n = 3 independent ex-periments, **p < 0.01, two-sided Student’s t test.
(C) Expression of DUX4 mRNA assayed by quantitative real-time PCR in HCT116 cells. Error bars are means± SEM, n = 3 independent experiments, ***p < 0.001, two-sided Student’s t test.
(D) Immunoblot analysis of NFE2L3, DUX4, and tubulin in HCT116 and HT29 cell lines transduced with a NTC shRNA or NFE2L3-specific shRNAs (shNFE2L3-1 or -2).
(E) Immunoblot analysis of HCT116 cells expressing NFE2L3 shRNA (shNFE2L3-2) or a NTC shRNA upon reexpression of the different forms of NFE2L3 (see
Figure 2C for description of A, B, or C forms).
(Figures 6E–6G). The decrease of CDK1 activity might thus explain the reduced colon cancer cell proliferation upon knock-down of NFE2L3. In conclusion, our data reveal a link between the NF-kB signaling pathway and cell-cycle regulation, with NFE2L3 acting as a repressor of the CDK1 inhibitor DUX4 (Figure 6H).
DISCUSSION
Colorectal cancer ranks as the third most common cancer worldwide (Kuipers et al., 2015). The underlying molecular mech-anisms of CRC pathology are complex, comprising a variety of genetic and epigenetic factors that control the proliferation rate of cells, their differentiation, and proneness to undergo cell death (Cancer Genome Atlas Network, 2012; Vinson et al., 2016). We found that NFE2L3 levels are substantially elevated in human co-lon adenocarcinoma tissue specimens when compared with healthy adjacent tissue, suggesting a possible role for this tran-scription factor in colon tumorigenesis. The signaling cascade discovered in our study comprises the extensively analyzed NF-kB and NFE2L3, a factor of yet largely unknown function. These transcriptional regulators are linked to the central cell cy-cle regulator CDK1, via the control of DUX4, a protein that has previously primarily been analyzed in the context of muscle biology (Chevillard and Blank, 2011; Gabellini et al., 2002; Zhang et al., 2017).
In our earlier studies, we reported that NFE2L3 expression is positively controlled by TNF at both the transcript and protein levels (Che´nais et al., 2005). In addition, many studies have linked the NF-kB pathway to TNF signaling (Li et al., 2015). Our data identified NFE2L3 as a novel downstream effector of RELA, as we found that other NF-kB family members are not involved in the regulation of NFE2L3. As confirmed by ChIP analysis, the control of NFE2L3 transcription by NF-kB is direct. The regulation by RELA is robust because inhibition of the transcriptional activity of NF-kB, NFKBIA phosphoryla-tion, or knockdown of RELA all recapitulated the phenotype observed upon NFE2L3 silencing. This is of interest because RELA is constitutively activated in human CRC tissue ( Reza-pour et al., 2016). Additional studies revealed that NOTCH1 levels, which were found to be an independent predictor of prognosis for patients with CRC, correlated with RELA status (Chu et al., 2011). NF-kB is also known to modulate Wnt signaling, whose activation is a common event in colon cancer initiation: ablation of RELA in intestinal epithelial cells delays the expansion of crypt stem cells, whereas elevated NF-kB signaling results in the dedifferentiation of non-stem cells into
tumor-initiating cells (Schwitalla et al., 2013). These data illus-trate the importance of RELA in CRC development. The identi-fication of NFE2L3 as a RELA target provides novel avenues to dissect the functions of NF-kB signaling pathway in colon can-cer pathology.
NFE2L3 has been first identified almost 20 years ago but its function remains largely unknown (Chevillard and Blank, 2011). Mouse model experiments revealed that NFE2L3 has a protec-tive function in carcinogen-induced lymphomagenesis ( Chevil-lard et al., 2011). Recent studies implicated NFE2L3 in various types of malignancies, including pancreatic, thyroid, and breast cancer (Sun et al., 2019; Wang et al., 2017, 2018). Our results demonstrate a growth-promoting function of NFE2L3 in colon cancer cells, which is likely mediated by the nuclear C form of the transcription factor. Our ChIP-seq analysis, together with molecular analyses, revealed that NFE2L3 serves as a key regu-lator for the control of DUX4 expression. The DUX4 protein has mainly been investigated for its role in FSHD. The DUX4 gene, located in a transcriptionally silent repeat region, is present at high levels in FSHD muscle because of chromatin changes, contributing to the phenotype, but the exact mechanisms remain obscure (Eidahl et al., 2016). Of interest, DUX4 also contributes to leukemia and round-cell sarcoma because of somatic chromosomal rearrangements resulting in DUX4-IGH and
CIC-DUX4 fusions, respectively (Kawamura-Saito et al., 2006; Ya-suda et al., 2016). Overexpression of DUX4 is toxic in many different cellular models and also results in increased sensitivity to oxidative stressors (Bosnakovski et al., 2008). Interestingly, in the context of colon cancer cells, upregulation of endogenous DUX4 protein upon NFE2L3 or RELA knockdown, results in a diminution of cell proliferation, but we did not observe any obvious toxicity to the cells, as measured by Annexin V/PI stain-ing (Figure S2). However, overexpression of exogenous DUX4, even driven by a weak promoter, is highly toxic to the cells in our colon cancer cell model.
How is the function of NFE2L3 transcriptional regulator coupled to cell proliferation in our experimental model? Our studies revealed an unanticipated link, showing that DUX4 is able to interact with CDK1, a crucial cell-cycle regulator, as evidenced by immunoprecipitation, followed by mass spec-trometry analysis. This result was confirmed by co-immunopre-cipitation (coIP) studies of exogenous and endogenous proteins, by performing GST pull-down assays and by measuring the KDof the interaction between DUX4 and CDK1, which is in the low micromolar range. In silico analysis high-lighted amino acids potentially implicated in the interaction be-tween DUX4 and CDK1, and based on these theoretical models,
(F) BrdU incorporation in HCT116 cells treated as in (E) assessed by ELISA. Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant; two-sided Student’s t test.
(G) Immunoblot analysis of NFE2L3, DUX4, and tubulin in HCT116 cells transduced with a NTC shRNA or a RELA-specific shRNA (shRELA-1). (H) Immunoblot analysis of NFE2L3, DUX4, and tubulin in HCT116 cells treated with DMSO or JSH-23 (50mM, 16 h).
(I) Immunoblot analysis of HCT116 cells transduced with a combination of vectors driving non-silencing controls (NTC shRNA or pLV scramble), shNFE2L3-1, or shDUX4.
(J) BrdU incorporation in HCT116 cells treated as in (I) assessed by ELISA. Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, NS, not significant; two-sided Student’s t test.
(K and L) Representative images (K) of soft agar assay of HCT116 cells treated as in (I) and its quantification (L). Error bars are means± SD, n = 2 independent experiments, **p < 0.01, ***p < 0.001, NS, not significant; two-sided Student’s t test.
A
B
C D
E
F G
Figure 5. DUX4 Interacts with CDK1
(A) coIP of pCMV-neo-CDK1-HA, pCMV-neo-CDK2-HA, pCMV-neo-CDK4-HA, pCMV-neo-CDK6-HA, and pCMV6-entry-Myc-Flag or pCMV6-DUX4-Myc-Flag from HCT116-transfected cell lysates. Samples and lysates were analyzed by immunoblot.
(B) Immunoprecipitation of lysates from HCT116 cells transduced with shNFE2L3-1 using anti-CDK1 antibody. Samples and lysates were analyzed by immu-noblot.
(C) Protein structure of the DUX4-CDK1 complex obtained with ZDOCK server. Amino acids implicated in the binding of DUX4 to CDK1 are highlighted and shown in detail inFigure S6B.
(D) DUX4-CDK1 interactions showing DUX4 (blue) and CDK1 (green) amino acids. (E) Representation of the DUX4 mutants and their different regions.
(F) HCT116 cells were transiently transfected with vectors expressing HA-tag or CDK1-HA and Myc-tag, or DUX4(WT)-Myc, or DUX4(D111-131)-Myc, or DUX4(D141-180)-Myc, or DUX4(D381-400)-Myc. Proteins were immunoprecipitated with an anti-HA antibody, and samples were analyzed by immunoblotting. (G) HCT116 cells were transiently transfected with vectors expressing HA-tag or CDK1-HA and Myc-tag, DUX4(WT)-Myc, DUX4(Arg117Ala)-Myc, or DUX4(Phe118Ala)-Myc. Proteins were immunoprecipitated with an anti-HA antibody, and samples were analyzed by immunoblotting.
we found that the region between aa 111 and 180 mediates the DUX4-CDK1 interaction. Our in silico analysis predicted that the DUX4 residues Arg117 and Phe118 are among the key amino acids mediating that interaction (Figure S6). We generated point mutants of DUX4 in these two residues and indeed observed a reduced interaction between these two mutants and CDK1 in coIP studies (Figure 5G). The molecular mechanisms controlling cell-cycle progression are complex, requiring the coordinated action of a series of regulatory proteins, including major tumor suppressors, such as the cyclin-dependent kinase inhibitor 1A (CDKN1A; p21), cyclin-dependent kinase inhibitor 2A (CDKN2A; p16), and retinoblastoma (RB) proteins (Asghar et al., 2015; Lig-gett and Sidransky, 1998). Deficient control of the cell cycle may lead to transformation of normal cells and enhance tumor devel-opment. CDK1-cyclin B1, a serine/threonine kinase complex regulated through protein-protein interactions and post-scriptional modifications, has a major role in G2/M phase tran-sition (Santamarı´a et al., 2007). With respect to the mechanism explaining the effect of NFE2L3 on cell-cycle progression, we hypothesize that DUX4 binds directly to CDK1 and limits the
A B
C
E F
D
G H
Figure 6. DUX4 Interacts Directly with CDK1 and Inhibits Its Activity
(A) In vitro GST pull-down of recombinant GST or GST-CDK1/GST-cyclin B1 and recombinant DUX4-Myc-FLAG using glutathione beads. (B) In vitro GST pull-down of recombinant GST or GST-CDK1 and recombinant DUX4-Myc-FLAG using glutathione beads. Lysate and pull-down were immunoblotted for the indicated proteins (signals shown for each antibody are from the same exposition time).
(C) Microscale thermophoresis analysis for dissociation constant determination between GST-DUX4 and human CDK1. KDvalue is shown. The different-colored lines correspond to the different replicates of the MST dose-response curves (n = 3). GST alone was used as a negative control.
(D) Immunoblots of indicated proteins after in vitro kinase assay containing ATP, His-H1, and immu-noprecipitated CDK1 complexes from HCT116 cells transduced with a NTC shRNA, a NFE2L3-specific shRNA (shNFE2L3-1), a DUX4-NFE2L3-specific shRNA (shDUX4), or both (shNFE2L3-1, shDUX4). (E) Cell-cycle distribution of HCT116 cells trans-duced with a NTC shRNA or NFE2L3-specific shRNAs (shNFE2L3-1 or -2) assessed by flow cytometry after PI staining.
(F) Percentage of HCT116 and HT29 cells treated as in (E) in the G2/M phase. Error bars are means± SEM, n = 3 independent experiments, *p < 0.05, **p < 0.01, two-sided Student’s t test.
(G) Immunoblot analysis of NUP98 and tubulin in HCT116 cells as in (E).
(H) Model for the pathway of the RELA and NFE2L3 transcription factors controlling cell-cycle progression via the regulation of DUX4 and CDK1.
interaction with its targets that are phos-phorylated by the enzyme (Brown et al., 2015). As the overexpression of exoge-nous wild-type DUX4, driven by a regular, and even by a weak, promoter, is highly toxic to the cells, we could not use wild-type DUX4, and in parallel the non-CDK binding mutants, to determine their effect on the activity of CDK1. In addition, our GST pull-down data suggest that DUX4 preferentially inter-acts with CDK1 alone. However, an assay to assess the inhibi-tion of kinase activity with only recombinant CDK1, which is inactive without cyclin B1, is not feasible. However, we have shown in our in vitro kinase assay that inhibition of CDK1 is dependent upon direct binding of DUX4 because the combined NFE2L3/DUX4 knockdown does not lead to a decrease in H1 phosphorylation in contrast to NFE2L3 knockdown alone ( Fig-ure 6D). Nevertheless, complementary experiments are required to determine whether DUX4 is a specific inhibitor of CDK1 or also can inhibit the activity of other CDKs, for example, CDK2, CDK4, or CDK6. Because of their central role in cell-cycle pro-gression, CDKs have been a major target for cancer therapy (Asghar et al., 2015). For example, the CDK4/6 inhibitor palbo-ciclib has been approved recently for use in patients with breast cancer (Finn et al., 2016). Current challenges are in the
development of highly selective agents against specific CDKs, companion diagnostics that will enable the selection of appro-priate patient populations, and a better understanding of the intersection of pharmacology and biology that will provide the basis for rational drug combinations (Asghar et al., 2015). Our finding that DUX4, which is tightly regulated by NFE2L3, func-tions as a direct inhibitor of CDK1, will open novel treatment op-portunities by precision therapy, for example, by screening for molecules that inhibit NFE2L3 activity or promote DUX4 expres-sion in tumors.
Taken together, based on our observations, we propose the existence of an oncogenic pathway, comprising the RELA, NFE2L3, DUX4, and CDK1 proteins that control colon cancer cell growth. Our study establishes a key role for the NFE2L3 tran-scription factor that regulates cell-cycle progression by govern-ing the expression of DUX4, a direct inhibitor of CDK1.
STAR+METHODS
Detailed methods are provided in the online version of this paper and include the following:
d KEY RESOURCES TABLE
d LEAD CONTACT AND MATERIALS AVAILABILITY
d EXPERIMENTAL MODEL AND SUBJECT DETAILS B Tissue Microarray and Patient Samples
B Mouse Xenograft Study
B Cell Lines d METHOD DETAILS
B Reagents
B Lentivirus-Based Transduction of Cells with shRNA
B RNA Extraction and RT-qPCR
B Immunoblot
B Cell Proliferation and Survival Assay
B Clonogenic and Soft Agar Assays
B Apoptosis and Cell Cycle Analysis
B Immunohistochemistry
B ChIP-qPCR and ChIP-Seq
B Dual-luciferase Reporter Assay
B (Co-)Immunoprecipitation
B Mass Spectrometry
B Prediction of DUX4 Structure using Fold Recognition Domain
B Modeling DUX4-CDK1 Complex
B In Vitro Protein Interaction (GST Pull-down Assay) B Purification of GST-DUX4
B Microscale Thermophoresis (MST)
B Kinase Assay
d QUANTIFICATION AND STATISTICAL ANALYSIS B Statistical Methods
d DATA AND CODE AVAILABILITY B ChIP-seq Data Link
B TMA Link
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online athttps://doi.org/10.1016/j. celrep.2019.09.087.
ACKNOWLEDGMENTS
We would like to thank Joo Yeoun Park, Isadore Dodard-Freedman, Meenak-shi Kannan, Yusra Kassim, Ste´phane Richard, Koren Mann, Eric Milot, Colin Crist, Johan Wouters, Guido Bommer, and Alain Israe¨l for reading of the manu-script and/or valuable discussions and/or material support. We would like to thank Alan Spatz and Leon van Kempen (JGH, Montreal) for human colon can-cer samples and Jose Torres for analysis of pathology specimens. We would like to acknowledge the LDI Flow Cytometry Facility (LDI, Montreal) for their flow cytometry technical expertise and the ENCODE project consortium and the ENCODE production laboratory for generating the dataset. Bioinformatics analyses were performed at the Bioinformatics Core Facility at the Institute for Research in Immunology and Cancer (IRIC), Universite´ de Montre´al (Montreal, QC, Canada). Proteomics analyses were performed by the Center for Advanced Proteomics Analyses (IRIC), a Node of the Canadian Genomic Inno-vation Network that is supported by the Canadian Government through Genome Canada. We also would like to thank Le´opold Thabault and Raphae¨l Fre´de´rick for access to the microscale thermophoresis platform (UCLouvain, Brussels) and Jocelyn Eidahl (Nationwide Children’s Hospital, USA) and Fre´d-e´rique Coppe´e (UMons, Mons) for the DUX4-GST plasmid. M.B. was sup-ported by fellowships from McGill Integrated Cancer Research Training Program (MICRTP, Canada), Le Fonds de recherche du Que´bec - Sante´ (FRQS, Canada), and the Cole Foundation (Canada); B.L.C. was supported by Te´le´vie fellowship (FNRS, Belgium); F.L. was supported by FRQS and CRS (Cancer Research Society, Canada) postdoctoral fellowships; T.D.M was supported by Fund for Research training in Industry and Agriculture (FRIA, Belgium); and J.S. was supported by SEG (Lebanon) and MICRTP stu-dentships. The research was supported by grants from the Canadian Institutes of Health Research (CIHR, Canada) MOP-97932 and PJT-152937 to V.B.
AUTHOR CONTRIBUTIONS
Conceptualization, M.B., B.L.C., and V.B.; Methodology, M.B. and B.L.C.; Investigation and Validation, M.B., B.L.C., F.L., T.D.M., and J.S.; Re-sources, C.M., G.F., and V.B.; Writing – Original Draft, M.B., B.L.C., and V.B.; Writing – Review & Editing, M.B., B.L.C., and V.B.; Visualization, M.B.; Supervision, C.M., G.F., and V.B.; Funding Acquisition, V.B.
DECLARATION OF INTERESTS
The authors declare no competing interests. Received: July 12, 2018
Revised: June 26, 2019 Accepted: September 27, 2019 Published: November 5, 2019
SUPPORTING CITATIONS
The following citations appear in the Supplemental Information:McCall et al. (2011); Rhodes et al. (2004); Wu et al. (2009).
REFERENCES
Aggarwal, B.B., and Sung, B. (2011). NF-kB in cancer: a matter of life and death. Cancer Discov. 1, 469–471.
Al Obeed, O.A., Alkhayal, K.A., Al Sheikh, A., Zubaidi, A.M., Vaali-Mohammed, M.A., Boushey, R., Mckerrow, J.H., and Abdulla, M.H. (2014). Increased expression of tumor necrosis factor-a is associated with advanced colorectal cancer stages. World J. Gastroenterol. 20, 18390–18396.
Asghar, U., Witkiewicz, A.K., Turner, N.C., and Knudsen, E.S. (2015). The his-tory and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146.
Ben-Neriah, Y., and Karin, M. (2011). Inflammation meets cancer, with NF-kB as the matchmaker. Nat. Immunol. 12, 715–723.
Bosnakovski, D., Xu, Z., Gang, E.J., Galindo, C.L., Liu, M., Simsek, T., Garner, H.R., Agha-Mohammadi, S., Tassin, A., Coppe´e, F., et al. (2008). An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated mo-lecular pathologies. EMBO J. 27, 2766–2779.
Brenner, H., Kloor, M., and Pox, C.P. (2014). Colorectal cancer. Lancet 383, 1490–1502.
Brown, N.R., Korolchuk, S., Martin, M.P., Stanley, W.A., Moukhametzianov, R., Noble, M.E.M., and Endicott, J.A. (2015). CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat. Commun.
6, 6769.
Cancer Genome Atlas Network (2012). Comprehensive molecular character-ization of human colon and rectal cancer. Nature 487, 330–337.
Che´nais, B., Derjuga, A., Massrieh, W., Red-Horse, K., Bellingard, V., Fisher, S.J., and Blank, V. (2005). Functional and placental expression analysis of the human NRF3 transcription factor. Mol. Endocrinol. 19, 125–137.
Chevillard, G., and Blank, V. (2011). NFE2L3 (NRF3): the Cinderella of the Cap’n’Collar transcription factors. Cell. Mol. Life Sci. 68, 3337–3348.
Chevillard, G., Paquet, M., and Blank, V. (2011). Nfe2l3 (Nrf3) deficiency pre-disposes mice to T-cell lymphoblastic lymphoma. Blood 117, 2005–2008.
Chowdhury, A.M.M.A., Katoh, H., Hatanaka, A., Iwanari, H., Nakamura, N., Ha-makubo, T., Natsume, T., Waku, T., and Kobayashi, A. (2017). Multiple regula-tory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci. Rep. 7, 12494.
Chu, D., Zhou, Y., Zhang, Z., Li, Y., Li, J., Zheng, J., Zhang, H., Zhao, Q., Wang, W., Wang, R., and Ji, G. (2011). Notch1 expression, which is related to p65 Sta-tus, is an independent predictor of prognosis in colorectal cancer. Clin. Cancer Res. 17, 5686–5694.
Coxon, C.R., Anscombe, E., Harnor, S.J., Martin, M.P., Carbain, B., Golding, B.T., Hardcastle, I.R., Harlow, L.K., Korolchuk, S., Matheson, C.J., et al. (2017). Cyclin-Dependent Kinase (CDK) Inhibitors: Structure-Activity Relation-ships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopur-ines. J. Med. Chem. 60, 1746–1767.
Dan, H.C., Cooper, M.J., Cogswell, P.C., Duncan, J.A., Ting, J.P., and Bald-win, A.S. (2008). Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22, 1490–1500.
De Sousa E Melo, F., Wang, X., Jansen, M., Fessler, E., Trinh, A., de Rooij, L.P., de Jong, J.H., de Boer, O.J., van Leersum, R., Bijlsma, M.F., et al. (2013). Poor-prognosis colon cancer is defined by a molecularly distinct subtype and de-velops from serrated precursor lesions. Nat. Med. 19, 614–618.
DeNicola, G.M., Karreth, F.A., Humpton, T.J., Gopinathan, A., Wei, C., Frese, K., Mangal, D., Yu, K.H., Yeo, C.J., Calhoun, E.S., et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109.
Dmitriev, P., Kiseleva, E., Kharchenko, O., Ivashkin, E., Pichugin, A., Dessen, P., Robert, T., Coppe´e, F., Belayew, A., Carnac, G., et al. (2016). Dux4 controls migration of mesenchymal stem cells through the Cxcr4-Sdf1 axis. Oncotarget
7, 65090–65108.
Eidahl, J.O., Giesige, C.R., Domire, J.S., Wallace, L.M., Fowler, A.M., Guckes, S., Garwick-Coppens, S., Labhart, P., and Harper, S.Q. (2016). Mouse Dux is myotoxic and shares partial functional homology with its human paralog DUX4. Hum. Mol. Genet. 25, 4577–4589.
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA ele-ments in the human genome. Nature 489, 57–74.
Finn, R.S., Crown, J.P., Ettl, J., Schmidt, M., Bondarenko, I.M., Lang, I., Pinter, T., Boer, K., Patel, R., Randolph, S., et al. (2016). Efficacy and safety of palbo-ciclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res.
18, 67.
Gabellini, D., Green, M.R., and Tupler, R. (2002). Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystro-phic muscle. Cell 110, 339–348.
Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y.C., Laslo, P., Cheng, J.X., Murre, C., Singh, H., and Glass, C.K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589.
Hinz, M., Krappmann, D., Eichten, A., Heder, A., Scheidereit, C., and Strauss, M. (1999). NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 19, 2690–2698.
Kannan, M.B., Dodard-Friedman, I., and Blank, V. (2015). Stringent Control of NFE2L3 (Nuclear Factor, Erythroid 2-Like 3; NRF3) Protein Degradation by FBW7 (F-box/WD Repeat-containing Protein 7) and Glycogen Synthase Ki-nase 3 (GSK3). J. Biol. Chem. 290, 26292–26302.
Karin, M. (2006). Nuclear factor-kappaB in cancer development and progres-sion. Nature 441, 431–436.
Karin, M., Cao, Y., Greten, F.R., and Li, Z.W. (2002). NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310.
Kawamura-Saito, M., Yamazaki, Y., Kaneko, K., Kawaguchi, N., Kanda, H., Mukai, H., Gotoh, T., Motoi, T., Fukayama, M., Aburatani, H., et al. (2006). Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137.
Kuipers, E.J., Grady, W.M., Lieberman, D., Seufferlein, T., Sung, J.J., Boelens, P.G., van de Velde, C.J., and Watanabe, T. (2015). Colorectal cancer. Nat. Rev. Dis. Primers 1, 15065.
La Rosa, F.A., Pierce, J.W., and Sonenshein, G.E. (1994). Differential regula-tion of the c-myc oncogene promoter by the NF-kappa B rel family of transcrip-tion factors. Mol. Cell. Biol. 14, 1039–1044.
Laurell, E., Beck, K., Krupina, K., Theerthagiri, G., Bodenmiller, B., Horvath, P., Aebersold, R., Antonin, W., and Kutay, U. (2011). Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell 144, 539–550.
Lessard, F., Igelmann, S., Trahan, C., Huot, G., Saint-Germain, E., Mignacca, L., Del Toro, N., Lopes-Paciencia, S., Le Calve´, B., Montero, M., et al. (2018). Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 20, 789–799.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., and Durbin, R.; 1000 Genome Project Data Processing Sub-group (2009). The Sequence Alignment/Map format and SAMtools. Bioinfor-matics 25, 2078–2079.
Li, S., Pinard, M., Wang, Y., Yang, L., Lin, R., Hiscott, J., Su, B., and Brodt, P. (2015). Crosstalk between the TNF and IGF pathways enhances NF-kB activa-tion and signaling in cancer cells. Growth Horm. IGF Res. 25, 253–261.
Liggett, W.H., Jr., and Sidransky, D. (1998). Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 16, 1197–1206.
McCall, M.N., Uppal, K., Jaffee, H.A., Zilliox, M.J., and Irizarry, R.A. (2011). The Gene Expression Barcode: leveraging public data repositories to begin cata-loging the human and murine transcriptomes. Nucleic Acids Res. 39, D1011–D1015.
Mintseris, J., Pierce, B., Wiehe, K., Anderson, R., Chen, R., and Weng, Z. (2007). Integrating statistical pair potentials into protein complex prediction. Proteins 69, 511–520.
Nouhi, Z., Chevillard, G., Derjuga, A., and Blank, V. (2007). Endoplasmic retic-ulum association and N-linked glycosylation of the human Nrf3 transcription factor. FEBS Lett. 581, 5401–5406.
Perkins, N.D., Felzien, L.K., Betts, J.C., Leung, K., Beach, D.H., and Nabel, G.J. (1997). Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 275, 523–527.
Pierce, J.W., Schoenleber, R., Jesmok, G., Best, J., Moore, S.A., Collins, T., and Gerritsen, M.E. (1997). Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103.
Pierce, B.G., Hourai, Y., and Weng, Z. (2011). Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 6, e24657.
Rezapour, S., Bahrami, T., Hashemzadeh, S., Estiar, M.A., Nemati, M., Ravan-bakhsh, R., Feizi, M.A., Kafil, H.S., Pouladi, N., Ghojazadeh, M., and Sakhinia, E. (2016). STC1 and NF-kB p65 (Rel A) is Constitutively Activated in Colorectal Cancer. Clin. Lab. 62, 463–469.
Rhodes, D.R., Yu, J., Shanker, K., Deshpande, N., Varambally, R., Ghosh, D., Barrette, T., Pandey, A., and Chinnaiyan, A.M. (2004). ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6.
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738.
Ruiz, E.J., Vilar, M., and Nebreda, A.R. (2010). A two-step inactivation mech-anism of Myt1 ensures CDK1/cyclin B activation and meiosis I entry. Curr. Biol.
20, 717–723.
Santamarı´a, D., Barrie`re, C., Cerqueira, A., Hunt, S., Tardy, C., Newton, K., Ca´-ceres, J.F., Dubus, P., Malumbres, M., and Barbacid, M. (2007). Cdk1 is suffi-cient to drive the mammalian cell cycle. Nature 448, 811–815.
Schwitalla, S., Fingerle, A.A., Cammareri, P., Nebelsiek, T., Go¨ktuna, S.I., Zie-gler, P.K., Canli, O., Heijmans, J., Huels, D.J., Moreaux, G., et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38.
Shin, H.M., Kim, M.H., Kim, B.H., Jung, S.H., Kim, Y.S., Park, H.J., Hong, J.T., Min, K.R., and Kim, Y. (2004). Inhibitory action of novel aromatic diamine com-pound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 571, 50–54.
Siegenthaler, B., Defila, C., Muzumdar, S., Beer, H.D., Meyer, M., Tanner, S., Bloch, W., Blank, V., Scha¨fer, M., and Werner, S. (2018). Nrf3 promotes UV-induced keratinocyte apoptosis through suppression of cell adhesion. Cell Death Differ. 25, 1749–1765.
Singh, C. (2017). Staging of Colonic Carcinoma, 7th
edition (AJCC).http:// www.pathologyoutlines.com/topic/colontumorstaging.html.
Sun, J., Zheng, Z., Chen, Q., Pan, Y., Lu, H., Zhang, H., Yu, Y., and Dai, Y. (2019). NRF3 suppresses breast cancer cell metastasis and cell proliferation and is a favorable predictor of survival in breast cancer. OncoTargets Ther.
12, 3019–3030.
Szlosarek, P., Charles, K.A., and Balkwill, F.R. (2006). Tumour necrosis factor-alpha as a tumour promoter. Eur. J. Cancer 42, 745–750.
The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45 (D1), D158–D169.
Vaiopoulos, A.G., Papachroni, K.K., and Papavassiliou, A.G. (2010). Colon carcinogenesis: Learning from NF-kappaB and AP-1. Int. J. Biochem. Cell Biol. 42, 1061–1065.
Vaiopoulos, A.G., Athanasoula, K.Ch., and Papavassiliou, A.G. (2013). NF-kB in colorectal cancer. J. Mol. Med. (Berl.) 91, 1029–1037.
van den Heuvel, S., and Harlow, E. (1993). Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054.
Vinson, K.E., George, D.C., Fender, A.W., Bertrand, F.E., and Sigounas, G. (2016). The Notch pathway in colorectal cancer. Int. J. Cancer 138, 1835–1842.
Wang, S., Liu, Z., Wang, L., and Zhang, X. (2009). NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 6, 327–334.
Wang, C., Saji, M., Justiniano, S.E., Yusof, A.M., Zhang, X., Yu, L., Ferna´ndez, S., Wakely, P., Jr., La Perle, K., Nakanishi, H., et al. (2017). RCAN1-4 is a thy-roid cancer growth and metastasis suppressor. JCI Insight 2, e90651.
Wang, H., Zhan, M., Yang, R., Shi, Y., Liu, Q., and Wang, J. (2018). Elevated expression of NFE2L3 predicts the poor prognosis of pancreatic cancer pa-tients. Cell Cycle 17, 2164–2174.
Wu, S., and Zhang, Y. (2007). LOMETS: a local meta-threading-server for pro-tein structure prediction. Nucleic Acids Res. 35, 3375–3382.
Wu, C., Orozco, C., Boyer, J., Leglise, M., Goodale, J., Batalov, S., Hodge, C.L., Haase, J., Janes, J., Huss, J.W., 3rd, and Su, A.I. (2009). BioGPS: an extensible and customizable portal for querying and organizing gene annota-tion resources. Genome Biol. 10, R130.
Wu, D., Wu, P., Zhao, L., Huang, L., Zhang, Z., Zhao, S., and Huang, J. (2015). NF-kB Expression and Outcomes in Solid Tumors: A Systematic Review and Meta-Analysis. Medicine (Baltimore) 94, e1687.
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8.
Yasuda, T., Tsuzuki, S., Kawazu, M., Hayakawa, F., Kojima, S., Ueno, T., Im-oto, N., Kohsaka, S., Kunita, A., Doi, K., et al. (2016). Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 48, 569–574.
Yu, G., and He, Q.Y. (2016). ReactomePA: an R/Bioconductor package for re-actome pathway analysis and visualization. Mol. Biosyst. 12, 477–479.
Zhang, Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40.
Zhang, J., McCastlain, K., Yoshihara, H., Xu, B., Chang, Y., Churchman, M.L., Wu, G., Li, Y., Wei, L., Iacobucci, I., et al.; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project (2016). Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet.
48, 1481–1489.
Zhang, Q., Lenardo, M.J., and Baltimore, D. (2017). 30 Years of NF-kB: a blos-soming of relevance to human pathobiology. Cell 168, 37–57.
STAR+METHODS
KEY RESOURCES TABLE
REAGENT or RESOURCE SOURCE IDENTIFIER
Antibodies
Rabbit anti-NFE2L3 Kannan et al., 2015 N/A
Biotin-SP-conjugated Affinipure Donkey anti-rabbit IgG Jackson Immuno Research Cat# 711-065-152
Rabbit polyclonal anti Pol-II (N-20) Santa Cruz Cat# sc-899 RRID:AB_632359 Lot# G3014
Rabbit polyclonal anti-RELA (A) Santa Cruz Cat# sc-109X RRID:AB_632039 Lot# L0814
Rabbit polyclonal anti-NFKB1 (H-119) Santa Cruz Cat# sc-7178 RRID:AB_650211 Rabbit polyclonal anti-REL (N) Santa Cruz Cat# sc-70 RRID:AB_2178727
Lot# B1814
Rabbit polyclonal anti-RELB (C-19) Santa Cruz Cat# sc-226 RRID:AB_X632341 Mouse monoclonal anti-DUX4 (C-2) Santa Cruz Cat# sc-376490 RRID:AB_11151782
Lot# D1514
Mouse monoclonal anti-CDK1 (17) Santa Cruz Cat# sc-54 RRID:AB_627224 Lot# K1915
Rabbit monoclonal anti-CDK1 (PSTAIRE) Santa Cruz Cat# sc-53 RRID:AB_2074908 Lot# F2714
Mouse monoclonal anti-HA-Tag (F-7) Santa Cruz Cat# sc-7392 RRID:AB_627809 Lot# E1718
Normal rabbit IgG Santa Cruz Cat# sc-2027 RRID:AB_737197
Lot# H2615
Normal mouse IgG Santa Cruz Cat# sc-2025 RRID:AB_737182
Lot# H0615
Rabbit polyclonal anti-NFKB2 Cell Signaling Cat# 4882S RRID:AB_10695537 Lot# 4
Rabbit monoclonal anti-phospho-RELA (93H1) Cell Signaling Cat# 3033S RRID:AB_331284 Lot# 14
Rabbit monoclonal anti-phospho-NFKBIA (14D4) Cell Signaling Cat# 2859S RRID:AB_561111 Lot#14
Rabbit monoclonal anti-Myc-Tag (9B11) Cell Signaling Cat# 2276 RRID:AB_331783 Lot# 24
Rabbit monoclonal anti-NUP98 (C39A3) Cell Signaling Cat# 2598 RRID:AB_2267700 Lot# 4
Mouse monoclonal anti-a-Tubulin (B-5-1-2) Sigma-Aldrich Cat# T6074 RRID:AB_477582 Lot# 075M4823V
Mouse monoclonal anti-His-Tag QIAGEN Cat# 34660 RRID:AB_2619735
Lot# 4
Mouse monoclonal anti-phospho-Histone H1 (12D11) Merck Millipore Cat# 05-1324 RRID:AB_1587115 Lot# 3072319
Rabbit monoclonal anti-MKI67 BioCare Medical Cat# CRM 325 A Bacterial and Virus Strains
E.coli BL21 (DE3) NEB C2527l
Continued
REAGENT or RESOURCE SOURCE IDENTIFIER
Biological Samples
Colon adenocarcinoma, 75 cases, Tumor and Matched NAT (tissue microarray)
Amsbio Col150CS-01
Matched normal colon tissue and colorectal adenocarcinoma tissue samples (FFPE)
Jewish General Hospital Central Biobank (Montreal)
IRB approval number 15-149
Chemicals, Peptides, and Recombinant Proteins
Tumor necrosis factor recombinant human protein Invitrogen Cat# PHC3011
BAY 11-7082 inhibitor Santa Cruz Cat# sc-200615; CAS 19542-67-7
JSH-23 inhibitor Cedarlane Cat# S7351; CAS 749886-87-1
LY294002 inhibitor NEB Cat# 9901
PD98059 inhibitor NEB Cat# 9900
SB-202190 Calbiochem Cat# 559388; CAS 152121-30-7
Recombinant human DUX4-Myc-FLAG protein This paper N/A
Recombinant GST protein Sigma-Aldrich Cat# SRP5348
Recombinant human CDK1/CyclinB1 Sigma-Aldrich Cat# SRP5009 Recombinant full-length human CDK1 protein SignalChem Cat# C22-14G
Recombinant human CDK1 protein Abcam Cat# ab187447
Recombinant human Histone H1 protein Abcam Cat# ab198676
FLAG Peptide Sigma-Aldrich Cat# F3290
Critical Commercial Assays
Amplification kit Roche Cat# 760-080
DAB Map detection kit Roche Cat# 760-124
AllPrep DNA/RNA/Protein Mini kit QIAGEN Cat# 80004
Purelink HiPure Plasmid Maxiprep kit Invitrogen Cat# K210007 QuickChange II XL Site-Directed Mutagenesis kit Agilent Cat# 200522
EasyScript Plus cDNA Synthesis kit Abmgood Cat# G236
BrdU Cell Proliferation ELISA kit Abcam Cat# ab126556
Annexin V-FITC Apoptosis Detection kit Sigma-Aldrich Cat# APOAF Bond Polymer DAB Refine kit Leica Biosystems Cat# DS9800 Dual-Luciferase Reporter Assay System Promega Cat# E1910 Deposited Data
ChIP-seq data This paper https://onedrive.live.com/redir?resid=
3F5A1FD25439B0CE!108&authkey=! APH0PPlEs_AqUw&ithint=folder%2c
TMA data This paper https://onedrive.live.com/redir?resid=
3F5A1FD25439B0CE!109&authkey=! ACVMRj5FWUvVPZg
Mass spectrometry data This paper SeeTable S1
Experimental Models: Cell Lines
Human colorectal carcinoma cells: HCT 116 cells ATCC CCL-247 Human colorectal adenocarcinoma cells: HT-29 cells ATCC HTB-38
Human embryonic kidney cells: HEK293T ATCC CRL-11268
Human embryonic kidney cells: HEK293 ATCC CRL-1573
Experimental Models: Organisms/Strains
Mice: Hsd: Athymic Nude-Foxn1nu Harlan Laboratories N/A
Oligonucleotides
Primers for NFE2L3, seeTable S2 This paper N/A
Primers for MKI67, seeTable S2 This paper N/A
Primers for DUX4, seeTable S2 This paper N/A
Continued
REAGENT or RESOURCE SOURCE IDENTIFIER
Primers for RELA, seeTable S2 This paper N/A
Primers for RELB, seeTable S2 This paper N/A
Primers for REL, seeTable S2 This paper N/A
Primers for NFKB1, seeTable S2 This paper N/A
Primers for NFKB2, seeTable S2 This paper N/A
Primers for ACTINB, seeTable S2 This paper N/A
Primers for GAPDH, seeTable S2 This paper N/A
Primers for CDKN1A, seeTable S2 This paper N/A
Forward primer for NFE2L3-ChIP-qPCR: CCACAGTCATTCACAACGGA
This paper N/A
Reverse primer for NFE2L3-ChIP-qPCR: GCAGCTGTTCCATACGTTTACA
This paper N/A
Forward primer for DUX4-ChIP-qPCR: TCACCTTTGTCATCAGTTCAGG
This paper N/A
Reverse primer forDUX4-ChIP-qPCR: TGATGTAACTCTTGTCTAAGCTCTGC
This paper N/A
Non-target shRNA control Sigma-Aldrich SHC216
shRNA 1 targeting NFE2L3 Sigma-Aldrich TRCN0000430385
shRNA 2 targeting NFE2L3 Sigma-Aldrich TRCN0000013488
shRNA 1 targeting RELA Sigma-Aldrich TRCN0000014684
shRNA 2 targeting RELA Sigma-Aldrich TRCN0000014683
shRNA 1 targeting NFKB1 Sigma-Aldrich TRCN0000006517
shRNA 2 targeting NFKB1 Sigma-Aldrich TRCN0000006518
shRNA 1 targeting NFKB2 Sigma-Aldrich TRCN0000356047
shRNA 2 targeting NFKB2 Sigma-Aldrich TRCN0000355955
shRNA 1 targeting REL Sigma-Aldrich TRCN0000039984
shRNA 2 targeting REL Sigma-Aldrich TRCN0000039983
shRNA 1 targeting RELB Sigma-Aldrich TRCN0000014713
shRNA 2 targeting RELB Sigma-Aldrich TRCN0000014714
Recombinant DNA
pCMV-VSV-G R. Weinberg’s laboratory Addgene plasmid #8454
pCMV-dR8.91 D. Trono’s laboratory Addgene plasmid #12263
pLV-Hygro-CMV-hNFE2L3 Full form NM_004289.6) VectorBuilder N/A
pLV-Hygro-CMV-hNFE2L3 Forms A/B This paper N/A
pLV-Hygro-CMV-hNFE2L3 Form C This paper N/A
pLV-Hygro-CMV-empty VectorBuilder N/A
pLV[shDUX4]-Hygro-U6 (CGAGTGGCTTTGCCCTCCCGA) VectorBuilder N/A pLV[scramble shRNA]-Hygro-U6 (CCTAAGGTTAAGTCGC
CCTCG)
VectorBuilder N/A
pCMV4-RELA W. Greene’s laboratory Addgene plasmid #21966
pCMV6-entry-Myc-Flag Origene PS100001
pCMV6-DUX4-Myc-Flag Origene RC238145
pCMV-neo-CDK1-HA S. Van den Heuvel’s laboratory Addgene plasmid #1888 pCMV-neo-CDK2-HA S. Van den Heuvel’s laboratory Addgene plasmid #1884 pCMV-neo-CDK4-HA S. Van den Heuvel’s laboratory Addgene plasmid #1876 pCMV-neo-CDK6-HA S. Van den Heuvel’s laboratory Addgene plasmid #1868
pCMV-HA-C Clontech 635690
pcDNA3.1-Myc Genscript N/A
pcDNA3.1-DUX4(WT)-Myc Genscript N/A