Biologically Engineering Nanostructures to
Maximize Energy, Electron, and Ion Transport
by
Heechul Park
B.S., Seoul National University (2007)
Submitted to the Department of Materials Science and Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2014
©
Massachusetts Institute of Technology 2014. All rights reserved.
Author………
Department of Materials Science and Engineering
May 12, 2014
Certified by……….
Angela M. Belcher
Professor
Thesis Supervisor
Accepted by………....
Gerbrand Ceder
Chair, Departmental Committee on Graduate Students
To my wife, HanYeaSeul Gweon, with all my love
Biologically Engineering Nanostructures to
Maximize Energy, Electron, and Ion Transport
by
Heechul Park
Submitted to the Department of Materials Science and Engineering on May 12, 2014, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
Abstract
Human intellectual desire inspires recent research to expand to interdisciplinary areas across biology, chemistry, and physics. Interdisciplinary research in unexplored areas is challenging, but holds great promise to elucidate what people did not see before. Scientific discoveries bring us not only intellectual pleasures, but also opportunities to contribute to the advancement of mankind.
Photosynthesis is a representative interdisciplinary research field. Conducting research in photosynthesis requires a collaborative work of biology, photochemistry, and quantum physics. Nature has optimized photosystems in bacteria, algae, and plants over three billion years in an evolutionary fashion to utilize solar energy for their survival. The way nature has mastered such systems can provide insights into designing efficient solar energy conversion applications.
This thesis explores artificial photosystems as proofs of nature’s design concept using a biological scaffold of M13 bacteriophage. The main ideas in the thesis focus on maximizing transport phenomena in the systems, resulting in performance improvements. Genetic engineering of M13 bacteriophage enables nano-scale multi-component assemblies to create tunable, artificial photosystems for solar energy utilization. Artificial photosystems include light-harvesting antenna complexes and oxygen-evolving photocatalytic systems. In particular, a solid collaboration with Seth Lloyd’s theory group inspires me to design a quantum light-harvesting antenna complex. The genetically engineered light-harvesting antenna complex creates a chromophore network interplaying between quantum and semi-classical mechanisms, thus maximizing exciton transport. Thesis Supervisor: Angela M. Belcher
Acknowledgments
My entire Ph.D. course has been full of excitements that I have really enjoyed. Of course, this exciting experience would not have been possible without others.
I am grateful to my advisors. Angela M. Belcher. She is an amazing advisor in both research and life. In particular, she gave me the research freedom that enabled me to design and analyze experiment. Her educational philosophy inspired me to become an independent researcher. Seth Lloyd. He is an insightful advisor. I feel lucky to meet and collaborate with brilliant people like him during the Ph.D. course. Samuel M. Allen. He is a generous advisor. He always gave me considerate advices on my academic career.
I am thankful to all my wonderful collaborators as well. Nimrod Heldman, Hannah C. Johnsen, Yoon Sung Nam, Andrew P. Magyar, and all the others in the Belcher Group. Patrick Rebentrost and Masoud Mohseni from the Lloyd group at MIT. Petra F. Scudo, Roberto Fusco, Laura Meda, Luigi Abbondanza, Mario Salvalaggio, and Andrea Alessi from Eni S.p.A, Italy. Paolo Foggi, Alessandro Iagatti, Barbara Patrizi, Laura Bussotti, and Filippo Caruso from LENS, Italy.
I acknowledge that my thesis was supported from Eni S.p.A, Italy through the MIT Energy Initiative program. I also thank KwanJeong Educational Foundation for its financial support.
I thank my family with all my heart. My parents, sisters, and parents-in-law. Especially, thank my mother for her dedicated prayers over the past many years. HanYeaSeul Gweon; she is an amazing wife. She always supports and encourages me to be who I am.
Contents
1
Introduction
21
1.1 Photosynthesis . . . . 21
1.2 Light-Harvesting Antenna Complexes . . . 26
1.3 Semi-Classical Energy Transport . . . 32
1.4 Quantum Energy Transport . . . 37
1.5 Scope of Work . . . 42
2 Semi-Classical Energy Transport in Porphyrin Light-Harvesting
Antennas
45
2.1 Introduction . . . 45
2.2
Zinc Porphyrin Light-Harvesting Antennas . . . 492.3
Microscopic Characterization . . . 502.4
Steady-State Spectroscopic Results . . . 532.5
Transient Absorption Spectroscopic Results . . . . 562.6
Conclusion . . . 593 Photosynthetic Charge Separation in Biologically Engineered
Antenna-Catalyst Complexes
61
3.1 Introduction . . . . 62
3.2
Genetic Engineering and Catalyst Synthesis . . . 643.3
Zinc Porphyrins as Light-Harvesting Materials . . . 703.4
Photosynthetic Complexes of Light-Harvesting Antenna and Oxygen-Evolving Catalyst. . . . 753.5
Preparation for Performance Measurements . . . 773.7 Microgel Encapulation . . . . 84
3.8 Conclusion . . . 89
4 Quantum Energy Transport in Biologically Engineered
Light-Harvesting Antennas
91
4.1 Introduction . . . . 91
4.2
Biological Engineering . . . 944.3
Chromophore Selection . . . 974.4
Light-Harvesting Antenna Assembly of Donors . . . 994.5
Redshifts in Absorbance . . . . . . . 1034.6 Fluorescence Quenching and Quantum Yield . . . 104
4.7 Förster Radius . . . 108
4.8 Average Inter-Chromophoric Distances . . . 110
4.9 Energy Transport Networks . . . 112
4.10 Both Donors and Acceptors on M13 Virus . . . 113
4.11 Exciton Diffusion Length . . . 118
4.12 Phenomenological Fitting Theory . . . 120
4.13 Diffusion Length by Classical Random Walk . . . 124
4.14 Transient Absorption Spectroscopic Analysis . . . 126
4.15 Exciton Dynamics . . . 131
4.15.1 Classical Master Equation . . . 132
4.15.2 Quantum Master Equation . . . 135
4.16 Conclusion . . . 139
5 Electron and Ion Transport in Electrochromic Hybrid
Nanomaterials
141
5.1 Introduction . . . 141
5.3
Electrochemical Analysis . . . 1475.4
Ion Transport . . . 1495.5
Electron Transport . . . . 1545.6 Conclusion . . . 158
List of Figures
1-1 Schematic diagram of a chloroplast (Nature Education, 2010) . . . 25 1-2 Crystal structure of photosystem II dimer from thermophilic cyanobacteria, T.
vulcanus at a resolution of 1.9 Å (Umena et al., Nature, 2011) . . . 25 1-3 Schematic diagram of a noncyclic electron transfer chain in oxygenic
photosynthetic organisms (Allen et al., Nature, 1999) . . . . 26 1-4 Organization of light-harvesting antenna complexes with reference to the
photosynthetic membrane (Scholes et al., Nature Chemistry, 2011) . . . . 30 1-5 Structures of Fenna-Matthews-Olson (FMO) pigment-protein complexes from
green sulfur bacterium, Prosthecochloris aestuarii (Cheng and Fleming, Annu. Rev. Phys. Chem., 2009) . . . 31 1-6 LH2 antenna complex from purple bacterium, Rhodopseudomonas acidophila
(Cogdell et al., Nature, 2002) . . . 31 1-7 Schematic diagram of energy transfer by classical theories . . . 36 1-8 Two-dimensional electronic spectra of FMO (Fleming and Engel et al., Nature,
2007) . . . 42 2-1 Molecular models of wild type M13 bacteriophage and assembly of zinc
porphyrins on the M13 virus surface (Nam, Park, and Belcher et al., J. Am. Chem. Soc., 2010) . . . 48 2-2 Images of Atomic Force Microscopy (AFM) . . . 51 2-3 Images of Transmission Electron Microscopy (TEM) . . . 52
2-4 Tryptophan fluorescence emission spectra of wild type M13 viruses and
porphyrin light-harvesting antennas . . . . 55
2-5 Absorption spectra of free zinc porphyrin and porphyrin-M13 virus antennas . 55 2-6 Fluorescence spectra of free zinc porphyrin and porphyrin-M13 virus antennas 56 2-7 Experimental set-up for pump-probe transient absorption measurements . . . 57
2-8 Transient absorption spectra . . . . 58
2-9 Transient absorption profiles were fitted by a double exponential decay curve 58 3-1 Synthesis of IrO2 hydrosol clusters (Nam, Park, and Belcher et al., Nature Nanotechnology, 2010) . . . . 67
3-2 Transmission Electron Microscopy (TEM) analysis . . . 68
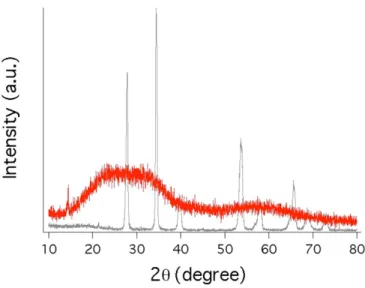
3-3 X-ray Diffraction (XRD) data . . . . 69
3-4 X-ray Photoelectron Spectroscopy (XPS) spectra of IrO2 nanowires . . . 69
3-5 Chemical structure of zinc porphyrin, termed ZnDPEG . . . 72
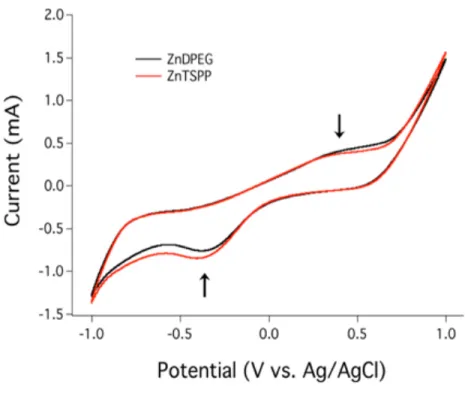
3-6 Cyclic voltammograms of ZnDPEG and ZnTSPP . . . 73
3-7 Absorption spectra of zinc porphyrins . . . . 74
3-8 Fluorescence spectra of zinc porphyrins . . . . 74
3-9 Schematic synthesis diagram of bio-templated IrO2-porphyrin nanowires . . 75 3-10 Photo image of aqueous solutions . . . . 76
3-11 Transmission Electron Microscopy (TEM) images . . . . 76
3-12 Brunauer-Emmett-Teller (BET) isotherm of the IrO2 nanowires . . . 79
3-13 Experimental set-up for real-time oxygen measurements . . . . 80
3-15 Time-course oxygen production profiles . . . 84
3-16 Photo images of the bio-templated IrO2 nanowires . . . 86
3-17 Microgel encapsulation of biologically templated antenna-catalyst complexes using a microfluidic technique . . . 87
3-18 Oxygen evolution profiles from microgel-encapsulated IrO2–ZnDPEG complexes . . . . 88
3-19 Regeneration of catalytic materials . . . 88
4-1 Models of the genetically engineered M13 virus . . . . 91
4-2 Chemical structure of chromophores . . . 99
4-3 Steady-state absorption and emission spectra of free donor and free acceptor . 99 4-4 Photograph of M13SF-DN and M13SF . . . . 102
4-5 Fluorescence Microscopy image of M13SF-DN . . . 102
4-6 Absorbance spectra of donor chromophores . . . 103
4-7 Fluorescence spectra of donor chromophores . . . . 106
4-8 Fluorescence as a function of absorbance . . . . 107
4-9 Fluorescence as a function of absorbance . . . . 107
4-10 Possible distances on the M13 virus models . . . 111
4-11 Energy transport network in M13 viruses . . . . 113
4-12 Absorbance and steady-state fluorescence spectra of the M13CF samples . . 116
4-13 Absorbance and steady-state fluorescence spectra of the M13SF samples . . 117
4-14 Gel images of M13SF . . . 117
4-16 Fluorescence energy normalized by the number of acceptors as a function of
donor-to-acceptor ratio . . . 120
4-17 Free acceptor fluorescence . . . 122
4-18 Fluorescence per acceptor versus donor/acceptor concentration for the M13CF clone . . . 123
4-19 Fluorescence per acceptor for the M13SF clone analogous to Figure 4-18 . . 123
4-20 Transient-absorption (TA) data at room temperature . . . 129
4-21 Transient-absorption (TA) spectra at room temperature . . . . 129
4-22 Transient absorption spectroscopy data for M13SF . . . . 130
4-23 Numerical simulations on the normalized fluorescence data for M13SF . . . 132
4-24 Fluorescence per acceptor versus donor/acceptor concentration for the M13CF clone . . . 134
4-25 Fluorescence per acceptor versus donor/acceptor concentration for the M13SF clone . . . 135
4-26 Fluorescence per acceptor versus donor/acceptor concentration c for the M13SF clone using the decohered quantum walk model . . . 139
5-1 Synthesis of virus-templated IrO2 nanowires (Nam, Park, and Belcher et al., Nanoscale, 2012) . . . 146
5-2 Transmission Electron Microscopy (TEM) images . . . 146
5-3 Electrochromic IrO2 nanowire films on ITO glass slide . . . . 147
5-4 Electrochemical characterization of IrO2 nanowire films . . . 148
5-6 Switching times of the IrO2 nanowires as a function of the ion conductivity of the
electrolyte . . . . 152
5-7 Chronoamperometric profiles of the IrO2 nanowire film as a function of the HClO4 concentration . . . 152
5-8 Cyclic voltammograms of the IrO2 nanowires as a function of the amount of the IrO2 nanowires . . . 153
5-9 Switching times and percent transmission as a function of the amount of the IrO2 nanowires deposited . . . . 153
5-10 Chronoamperometric profiles as a function of the amount of the IrO2 nanowires . . . . 154
5-11 Schematic diagram of assembling Au-IrO2 nanowires . . . 156
5-12 Transmission Electron Microscopy (TEM) images . . . 156
5-13 Comparison between IrO2 and IrO2-Au nanowires . . . 157
List of Tables
2-1 The parameters of fitting profiles to transient absorption data . . 59
4-1 Sample information of M13CF . . . . 115
4-2 Sample information of M13SF . . . . 116
4-3 M13CF clone fit results . . . 124
4-4 M13SF clone fit results . . . . 124
4-5 Diffusion length estimates based on a classical random walk . . 125
4-6 Transient absorption spectroscopy data . . . 130
4-7 Input parameters for the classical random walk exciton simulation . . . 134
Chapter 1
Introduction
1.1 Photosynthesis
Solar energy is the most abundant source of energy and provides sustainable power for life on Earth. The Sun continuously delivers to Earth the tremendous power of 120,000 TW (1 TW = 1012 W) (1, 2). This solar power dramatically exceeds the current annual human global energy consumption rate of about 15 TW. The massive solar energy holds great promise for a clean and sustainable resource as an alternative to finite fossil fuels, whose combustion produces harmful environmental pollutants including greenhouse gases. However, sunlight is a relatively dilute energy source; the energy density is approximately 0.17 kW per square meter (3). Conceiving methods of efficient solar energy conversion and storage is one of significant challenges in order to satisfy human energy needs in our age. Nature has optimized photosynthetic systems in bacteria, algae, and plants over 3 billion years by evolution to use solar energy for their survival (4). Photosynthesis captures solar energy at the average
worldwide power rate of 130 TW, which is 8 times larger than the global energy consumption rate (5).
A complete understanding of photosynthesis can play a key role in solar energy utilization. Photosynthesis naturally captures and stores solar energy as chemical fuels (6, 7). Photosynthesis uses almost unlimited raw materials such as water and carbon dioxide (1). Sunlight triggers photosynthetic systems to split water molecules to produce oxygen gases that are essential for life’s respiration and combustion. The other products, protons, split from water convert carbon dioxide to useful carbohydrates such as sugars and starches that serve as food and fuels.
Photosynthesis takes place in subcellular structures known as chloroplasts, see Figure 1-1. The light-harvesting antennas and reaction centers are located in biological membranes known as thylakoids within the chloroplasts. Stroma is the non-membranous aqueous interior of the chloroplast where carbon dioxides are fixed and reduced to carbohydrates. Overall process of photosynthesis can be divided into four steps for convenience (6): (a) sunlight capture and energy transport by light-harvesting antenna complexes, (b) charge separation and transfer in reaction center, (c) energy stabilization, and (d) the synthesis of stable carbohydrate products.
The first step in photosynthesis is to collect solar energy to be stored. Antenna systems capture sunlight and transport the excitation energy to a reaction center where charge separation occurs. In most cases, light-harvesting antenna systems consist of both pigments and protein subunits that are bound to each other closely, see Figure 1-2 (8). The protein scaffold leads to the highly compact packing of pigments, resulting in effective solar energy capture. The pigment-protein antenna complexes typically
absorb higher photon energy (i.e. short wavelength) on the periphery of the complexes and funnel the higher-energy on the periphery pigments to the lower-energy pigments near the core until the excitation energy reaches a reaction center. The funneling of excitation energy from sunlight absorption to a reaction center shows rapid time scales of 10-100 picoseconds (9).
In the second step, a reaction center transforms pure excited energy to chemical fuels as energy storage in photosynthesis. The reaction center is also a pigment-protein complex carrying electron transfer components such as quinone and iron sulfur (6). Either energy transfer from the light-harvesting antenna or direct sunlight promotes a pigment (P) to an electronic excited state (P*); that is an exciton. The exciton is a very strong reducing species, so rapidly loses an electron to a nearby electron acceptor molecule (A), forming an ion pair of P+A-. The form of energy has changed from electronic excited states to chemical redox potentials. The ion pair of P+A- is very vulnerable to charge recombination because P+ is highly oxidizing and A- is highly reducing. A series of extremely fast secondary reactions successfully win over charge recombination, keeping stored the chemical redox energy. That is because the reactions spatially separate the positive and negative charges by ~ 3 nm, which decreases the chances of recombination by orders of magnitude.
In the third step, slower processes from nanoseconds to milliseconds stabilize stored the redox energy and convert it into the more useful form of energy (6, 9). There are two different ways: a cyclic electron transfer chain and a noncyclic electron transfer chain. Some organisms show the cyclic electron transfer chain which is simply the circulation of electrons from an excited state (P*) to a ground state (P) via
several secondary acceptors, carriers, and donors. In most oxygen-evolving photosynthetic organisms, a noncyclic electron transfer chain shows two photochemical reaction centers of photosystem I (PS I) and photosystem II (PS II) in Figure 1-3 (10). In PS II, two water molecules are split to produce an oxygen gas, four electron, and four protons at an oxygen-evolving reaction center as follows.
2𝐻!𝑂 → 𝑂!+ 4𝑒! + 4𝐻!
The electrons from water are transported across the membrane to PS I and finally reduce NADP+ to NADPH at a light-driven reaction center in PS I. The protons produced from water-splitting are also transferred to thylakoid lumen, leading to a pH difference. The energy in this pH gradient helps to make ATP that is biological phosphate bond energy.
The final step is to produce stable carbohydrates from carbon dioxide using NADPH and ATP generated by the third step in the following equation (6). The carbon assimilation and reduction are reactions catalyzed by soluble enzymes in the stroma of the chloroplast.
𝐶𝑂!+ 4𝑒!+ 4𝐻! → 𝐶𝐻
!𝑂 + 𝐻!𝑂
This thesis focuses on not only the first step of light-harvesting and energy transport, but also the second and third steps of oxygen-evolving charge separation in our artificial systems.
Figure 1-1: Schematic diagram of a chloroplast (Nature Education, 2010).
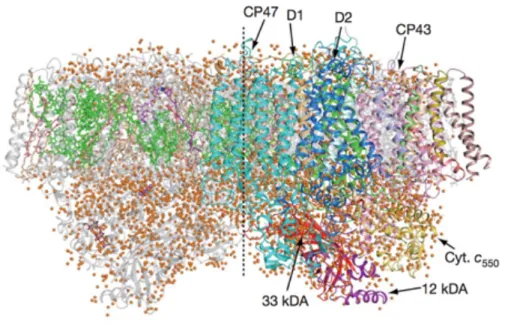
Figure 1-2: Crystal structure of photosystem II dimer from thermophilic cyanobacteria, T. vulcanus at a resolution of 1.9 Å (Umena et al., Nature, 2011). Green, cyan, and blue indicate chlorophylls and protein subunits in light-harvesting complexes that capture solar energy. Red shows an oxygen-evolving center that split water molecules into oxygen gases and protons. Orange circles are water molecules.
Figure 1-3: Schematic diagram of a noncyclic electron transfer chain in oxygenic photosynthetic organisms (Allen et al., Nature, 1999). The diagram shows electron transfer pathways with standard redox potential of electron carriers.
1.2 Light-Harvesting Antenna Complexes
Photosynthesis has been a research subject over a century in order to learn how nature has mastered to capture, transport, and store solar energy for several billion years in an evolutionary manner (6, 7). One of the remarkable components in photosynthesis is light-harvesting pigment-protein complexes. When he was a Ph.D. student in the Netherlands in early 1950’s, L.N.M. Duysens confirmed that the light-harvesting
antennas capture sunlight and transport the energy with nearly 100 % quantum efficiency to a reaction center where the energy is converted into chemical fuels
(11-13). Since then, both experimental and theoretical efforts have been made to elucidate
the energy transfer mechanism of such a high efficiency in light-harvesting complexes.
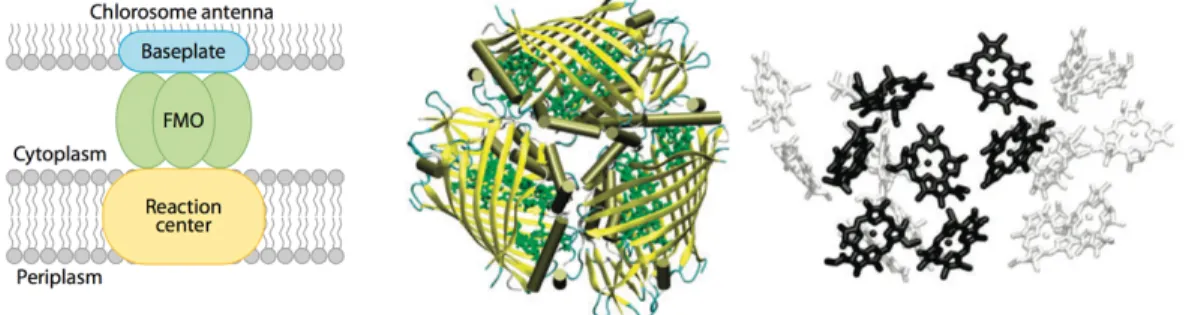
Our understanding of the energy transfer mechanism in light-harvesting complexes comes with unraveling structures in major photosynthetic complexes. Figure 1-4 shows overall structural organization of light-harvesting antenna complexes with respect to the thylakoid membrane in plants, green algae, red algae, cyanobacteria, and cryptophytes. Green algae shows similar properties compared with higher plants and are evolutionary ancestors to plants (6). Light-harvesting complexes of higher plants and green algae are located within the membrane, whereas phycobilisomes, the light-harvesting antennas found in red algae and cyanobacteria, are located on the aqueous stroma side of the membrane within the chloroplast (9). Similarly, the light-harvesting antenna, chlorosome, in green sulfur bacteria is arranged on the stroma side of the membrane. Cryptophyte, a kind of algae, show that phycobiliprotein antenna complexes are located on the opposite side of the membrane in the thylakoid lumen.
The high-resolution crystal structures of major photosynthetic complexes provide insights into our understanding of light-harvesting antenna systems (9, 14). The well-characterized structure of light-harvesting complexes is the Fenna-Matthews-Olson (FMO) of green sulfur bacteria. The FMO protein was named after it’s discoverers; Olson et al. discovered the water-soluble FMO protein in early
1960’s, and Fenna and Matthews proposed first the FMO structure at an atomic resolution level from Chlorobium tepidum in mid 1970’s by using X-ray crystallography (15, 16). Blankenship et al. solved the FMO pigment-protein from
Prosthecochloris aestuarii at a high resolution of 2.2 Å in early 2000’s (17). As shown
in Figure 1-5, the FMO antenna trimer is located between the large chlorosome antenna and the reaction center and directs excitation energy from the antenna to the reaction center (16, 18). The FMO consists of three identical subunits that each comprises seven bacteriochlorophyll (BChl) molecules and proteins. The closest center-to-center distance between nearest BChl molecules in a monomer subunit is 12 Å, while that between BChl molecules in neighboring subunits is 24 Å (19).
Purple bacteria are another example that is well studied in their structure. Purple bacteria have two types of antenna complexes: light-harvesting complexes 1 (LH1) and light-harvesting complexes 2 (LH2). The crystal structure of the LH2 were finally obtained in mid 1990’s at a resolution of 2.5 Å from Rhodopseudomonas
acidophila by using X-ray crystallography (20); the LH1 and a reaction center in early
2000’s at a 4.8 Å resolution from Rhodopseudomonas palustris by using a molecular replacement method and an electron density map (21). The LH2 are located around the perimeter of the LH1 ring, whereas the LH1 surround the reaction center in a two-dimensional structure (22). Upon sunlight, the LH2 at the periphery transfer excitation energy towards center to LH1, and subsequently to the reaction center. The LH2 show a cylindrically symmetric structure and comprises two pigments of 27 BChl a and 18 carotenoid molecules non-covalently bound to a protein matrix in Figure 1-6. The BChl a molecules are arranged in two distinct binding sites in concentric rings within
the protein matrix. They are labeled according to their absorption maxima as B800 and B850. The B800 ring consists of 9 well-separated BChl a molecules, whereas the B850 ring features a group of 18 closely interacting BChl a molecules (20). The closest center-to-center distance between BChl a molecules of the B850 is ~ 9 Å (20,
23). The carotenoid pigments capture sunlight shorter wavelength (i.e. higher photon
energy) in the range of 450 nm to 540 nm. Energy from sunlight is transported from the carotenoid to B800, and subsequently to B850. That is, energy flows from higher- to lower- photon energy.
Figure 1-4: Organization of light-harvesting antenna complexes with reference to the photosynthetic membrane (Scholes et al, Nature Chemistry, 2011). Images are representative of: A, Higher plants and green algae. B, Cyanobacteria and red algae. C, Cryptophytes.
Figure 1-5: Structures of Fenna-Matthews-Olson (FMO) pigment-protein complexes from green sulfur bacterium, Prosthecochloris aestuarii (Cheng and Fleming, Annu. Rev. Phys. Chem., 2009). (Left) The FMO is located between chlorosome antenna and the reaction center. (Middle) Top view of the FMO trimer. The protein is in yellow and the bacteriochlorophyll (BChl) molecules are depicted in green. (Right) Side view of the BChl arrangement in the FMO trimer. Seven black BChl belong to one of the monomeric subunits.
Figure 1-6: LH2 antenna complex from purple bacterium, Rhodopseudomonas
acidophila (Cogdell et al., Nature, 2002). (Left) Structural arrangement of the
carotenoid (Car) and BChl molecules. The Car molecules are depicted in blue, B800 BChls are in orange, and B850 BChls are in red. (Right) Steady-state absorption spectrum of Car, B800, and B850 molecules.
1.3 Semi-Classical Energy Transport
Theoretical efforts have been made to untangle the energy transfer mechanism of light-harvesting antenna complexes in addition to the experimental efforts in revealing the high-resolution structures of photosystems.
Independent of photosynthesis research, fluorescence quenching observations since 1920’s had led to the development of classical theories: one is Förster theory, and the other is the Dexter mechanism (24). A German physical chemist, Theodor Förster, developed an energy transfer theory of fluorescence in late 1940’s, and D. L. Dexter in Washington DC developed another fluorescence theory in early 1950’s (25,
26). That is the same era when Duysens in the Netherlands confirmed the high transfer
efficiency of photosystems. The mechanisms were named after the developers as Förster and Dexter theories, respectively. The Förster theory is often called as Förster resonance energy transfer (FRET). The classical theories show energy transfer processes from an excited donor to a ground state acceptor, denoted by D* + A → D + A*, in Figure 1-7. The Förster theory is a non-radiative energy transfer mechanism by induced dipole-dipole interaction, which explains long-range energy transfer between chromophores from a nanometer up to a few tens of nanometers (25, 27). It is an incoherent hopping mechanism (28). Non-radiative Dexter mechanism is a direct electron exchange between chromophores by orbital overlap where it becomes dominant in the short range of closer than 1 nm (27, 29). Both Förster and Dexter are semi-classical theories because they use quantum mechanical treatments in their formulations in order to bridge macro- and microscopic worlds (24).
𝑘!"(𝑅!") = 𝐾𝐽𝑒𝑥𝑝(−2𝑅!"
𝐿)
where K represents specific orbital interactions, J is a spectral overlap integral, and 𝑅!" is a donor-to-acceptor distance relative to their van der Waals radii, L (27). J is normalized for the extinction coefficient of the acceptor. Note that the rate of exchange mechanism depends on both spectral overlap and the exponential decay of the donor-to-acceptor distance, and is independent of the oscillator strength of the D* → D and A → A* transitions (27). Since the Dexter exchange becomes significant only at a short range of less than 1 nm, long-range Förster energy transfer theory is the dominant mechanism in a variety of fluorescence energy transfer experiments that range from biological use to optoelectronic devices (30-34).
Förster theory is based on the weak electronic coupling limit of Fermi Golden Rule given by
𝑘!" =2𝜋
ℏ 𝜌 𝜓! 𝐻 𝜓!
!
where 𝜓! and 𝜓! denotes the wave function of an initial and a final sate, respectively,
H is a perturbing Hamiltonian, 𝜌 is the density of final states, and ℏ is the Planck constant (24, 27). The transition rate of ET is influenced by the equilibrium perturbation magnitude that changes the positions or motions of particles from the initial state to the final state. From the Fermi Golden Rule, the observed transition rate is divided into a theoretical maximum rate constant and three components that possibly act as rate-limiting steps to inhibit the maximum rate constant: orbital interaction, spin-orbit interaction, and Franck-Condon factors. The Franck-Condon principle separates a spectral overlap into electronic coupling and nuclear overlap factors by the Born-Oppenheimer approximation. The spectral overlap between donor
emission and acceptor absorption describes both energy conservation and nuclear overlap factors.
The power of Förster theory comes from that it has been related to and well established with experiments (35, 36). Experimental quantities include the inter-chromophoric distance, the fluorescence quantum yield, a spectral overlap, and the fluorescence lifetime. The dipole-dipole interaction between chromophores can be seen as an interaction between charged particles. The electric field near an electronically excited chromophore behaves like a field generated by an oscillating charged particle of dipole moment 𝜇, as expressed in 𝜇 = 𝜇!cos (2𝜋𝜈𝑡)*. The
dipole-dipole interaction energy E between a donor and an acceptor chromophore depends on the magnitude of two chromophore dipole moments (𝜇! and 𝜇!) and the distance (R) between them as !!!!
!!"! . Förster identified the dipole moments of chromophores with
the experimentally measurable oscillating strength for radiative transitions. Förster demonstrated that the transition rate of D* + A → D + A* depends on the square of the interaction energy and further simply a pure electronic coupling given by
𝑘!" ∝ 𝐸! = 𝜇!𝜇! 𝑅!"! ! 𝑘!" = 2𝜋 ℏ 𝑉 ! 𝑑𝜀 𝐽(𝜀) ! !
where V denotes the electronic coupling between the donor and acceptor, 𝜀 is energy, and 𝐽(𝜀) is the spectral overlap. (24, 27). The electronic coupling V is given by
𝑉 = 1
4𝜋𝜀!
𝜅𝝁!𝝁! 𝑹!" !
* 𝜇 represents an instantaneous dipole moment, 𝜇
!is the maximum magnitude of
where an orientation factor 𝜅 is defined as 𝜅 = 𝝁! ∙ 𝝁!− 3(𝝁!∙ 𝑹!")(𝝁! ∙ 𝑹!") and 𝜀! is vacuum permittivity. Förster showed this equation in a practical fashion given by
𝑘!" 𝑅!" = 1 𝜏! 𝑅! 𝑅!" ! 𝑅! = 0.211×(𝜅!𝑛!!𝛷 !𝐽 𝜆 )! ! in Å
where 𝑅! is defined as a Förster radius where transfer efficiency is 50 %, 𝜏! is a fluorescence lifetime of donor, 𝑅!" is a center-to-center distance in Å between a donor and an acceptor, 𝜅! is a relative orientation factor between the dipoles of a donor and
an acceptor, n is a refractive index of solvent, 𝛷! is a fluorescence quantum yield of a donor, and 𝐽 𝜆 is the spectral overlap integral* between donor emission and acceptor
absorption in cm-1M-1 (37). Note that the rate of energy transfer depends on the inverse
sixth power of the inter-chromophore distance and on the oscillating strength of the radiative transitions (27). One of the useful aspects in Förster theory is that we can precisely monitor the distance or conformational changes of specific sites in a biological molecule such as a protein and RNA by labeling each site with fluorescent dyes (30, 33).
From 1990’s through early 2000’s, the high-resolution structure of photosystems and vibrational information caused by environmental fluctuations enabled detailed theoretical modeling. Cumulative studies in both non-linear laser spectroscopic measurements and structure-based dynamical modeling revealed that Förster theory cannot be applied to certain light-harvesting complexes in
* 𝐽 𝜆 = 𝑓
! !
! (𝜆)𝜀!(𝜆)𝜆!𝑑𝜆. 𝜆 is the wavelength in nm, 𝑓! 𝜆 is the fluorescence
intensity of the donor in the absence of the acceptor at 𝜆 in dimensionless normalized such that 1 = !!𝑓!(𝜆)𝑑𝜆, and 𝜀!(𝜆) is the molar extinction coefficient at 𝜆 in units
photosynthesis (19, 22, 28, 38-40). Förster theory assumes that electronic couplings between chromophores are weak and molecular vibrational relaxation is fast compared to the timescale given by the electronic coupling. However, those assumptions are no longer valid in the light-harvesting complexes, so other theories such as excitonic delocalization based on stronger electronic couplings and environment-vibrational states have to be taken into account.
Figure 1-7: Schematic diagram of energy transfer by classical theories. The signs of e- represent active electrons that take part in interaction and move to other states. (Upper) Förster resonance energy transfer by dipole-dipole interaction. (Bottom) Dexter exchange mechanism by orbital overlap.
1.4 Quantum Energy Transport
Recent advances in spectroscopic measurements and theoretical modeling deepen our understanding of excitation energy transport dynamics in light-harvesting antenna complexes. Conducting research in photosynthesis becomes an emerging area where biology, photochemistry, and quantum physics overlap. Fleming and Engel et al. at UC Berkeley verified experimentally first in 2007 long-lasting quantum-coherent dynamics (> 600 fs) in the FMO antenna complexes at cryogenic temperature of 77 K using two-dimensional electronic spectroscopy, in contrast to incoherent semi-classical transport (41). Fleming et al. also probed coherent dynamics in purple bacteria reaction center in the same year and quantified dephasing dynamics in the system, suggesting that protein environment protects electronic coherences (42). The discovery of the long-lived quantum coherence in photosynthesis has attracted a broad community of quantum physicists and physical chemists to investigate the energy transfer mechanism. In 2008, Lloyd and Aspuru-Guzik et al. at MIT and Harvard developed a quantum walk theory with environmental fluctuations in the FMO and demonstrated that the environmental vibration assists the excitation energy transfer rather than destroys it (43). Lloyd and Aspuru-Guzik et al. in 2009 even quantified the contributions of quantum coherence and environmental relaxation to the overall energy transfer efficiency in the FMO (44, 45). Scholes et al. at University of Toronto in 2010 observed long-lasting coherent oscillations (> 400 fs) even at room temperature in the light-harvesting antenna proteins in cryptophyte marine algae (46). In the same year, Engel et al. at the University of Chicago experimentally suggested
long-lived quantum coherence (> 300 fs) in the FMO complexes at an elevated temperature of 277 K (47).
Quantum theory provides deeper insights into the dynamics of energy transport in light-harvesting antenna complexes. There are many ways to derive the equations of motion for the quantum energy transport. The starting point is the Hamiltonian (operator for the energy of a quantum system) for the electronic and vibrational degrees of freedom. It is given by
𝐻!"! = 𝐻!+ 𝐻!"+ 𝐻!
where the total Hamiltonian sums up system, system-bath coupling, and bath Hamiltonian (18, 43-45). The Frenkel exciton model describes light-driven excitation in pigment-protein antenna complexes. The system Hamiltonian made of interacting N chromophores by a single excitation is given by
𝐻! = 𝜀! 𝑛⟩⟨𝑛
! !!!
+ 𝑉!"( 𝑛⟩⟨𝑚 + 𝑚⟩⟨𝑛 )
!!!
where ∣ 𝑛⟩ denotes a molecular excited state at site n, 𝜀! is the site energy* of ∣ 𝑛⟩, and
𝑉!" is the excitonic coupling between two chromophores (18, 45). Defining the exciton states requires information on the site energies and the excitonic couplings. Exciton states ∣ 𝜓!⟩ is obtained from diagonalization of 𝐻!; that is 𝐻! ∣ 𝜓!⟩ = 𝐸! ∣ 𝜓!⟩. Delocalized exciton states are described as the following linear combination of molecular excited states:
∣ 𝜓!⟩ = 𝜙!! ! !!!
∣ 𝑛⟩.
* The site energy is defined as the transition energy of a chromophore from a ground
Interactions between the transition dipoles of the chromophores give rise to the excitonic coupling. For example, the linear combination of molecular excited states describes the transition dipole moments of excitons, denoted as 𝝁𝜶 = 𝜙!!𝝁
𝒏
! .
High-resolution structural information in the pigment-protein complexes helps us to estimate excitonic couplings, but precise numbers of them remain elusive because protein environment screens the dipole-dipole interactions and subsequently modifies the excited states. The site energy is also differed by the interactions with environment. The proteins as well as long-range electrostatic interactions affect the site energies.
The multi-chromophoric exciton system interacts with the environmental bath. The environment consists of the local molecular vibrations of chromophores, the vibration of proteins, and solvent molecules. A general assumption is that bath fluctuations are not spatially correlated with different chromophores. The system-bath coupled Hamiltonian 𝐻!" denotes independent phonon-induced fluctuations of the site energies on each chromophore, so the dynamics of 𝐻!" is fully characterized with the spectral density 𝐽 𝜔 . The spectral density carries the coupling strength of system-bath and the density of states of the phonon system-bath (48). Spectroscopic analysis can provide the strength of system-bath coupling and further the spectral density (19, 49). The bath reorganization energy 𝜆! measures the strength of system-bath coupling as
𝜆! = !!𝑑𝜔𝐽!(𝜔)/𝜔.
There are three regimes to describe the transport dynamics of excitation energy: weak electronic coupling limit, strong electronic coupling limit, and intermediate regime. If the electronic coupling is much weaker than the bath
reorganization energy, i.e. 𝑉 ≪ 𝜆, excitons are localized on each chromophore. In this weak electronic coupling limit, the incoherent Förster hopping theory describes well the energy transport dynamics. If the electronic coupling is stronger than the bath reorganization energy, i.e. 𝑉 ≫ 𝜆, exciton delocalization becomes important. The total density matrix evolves as
𝑑
𝑑𝑡𝜌!"! 𝑡 = − 𝑖
ℏ 𝐻!"!, 𝜌!"! 𝑡 .
In the limit of the stronger electronic coupling, the various perturbation approaches can be embedded in 𝐻!"! such as a Redfield model and a quantum-walk based dephasing model, and a pure-dephasing model derived from Haken-Strobl-Reineker equation (18, 43, 45). For example, the dynamics of the electronic degrees of freedom are described by the pure-dephasing master equation:
𝑑
𝑑𝑡𝜌! 𝑡 = − 𝑖
ℏ 𝐻!, 𝜌! 𝑡 + ℒ𝜌! 𝑡 where the Lindblad operator is given by
ℒ𝜌! 𝑡 = 𝛾! 𝐿!𝜌 𝑡 𝐿!!−1
2𝐿!𝐿!!𝜌 𝑡 − 1
2𝜌 𝑡 𝐿!𝐿!!
!
where 𝐿! = 𝑛⟩⟨𝑛 denotes generators and 𝛾! is a pure dephasing rate (45). Interestingly, natural light-harvesting complexes are found to reside in the intermediate regime where coherent dynamics and environmental vibrations collaborate for maximizing energy transport efficiency (9, 18, 43-45, 50). The intermediate regime is the region where the electronic coupling strength is comparable to the bath reorganization energy, i.e. 𝑉 ≈ 𝜆. In the FMO complexes, the Förster coupling strength between nearest chromophores is 100 cm-1. This coupling strength is
enhanced by the electrostatic protein environment, leading to the site-dependent chromophoric coupling up to 300 cm-1 (45).
Research in light-harvesting complexes promises to elucidate the way nature has devised over 3 billion years to efficiently transport solar energy; finding nature’s design principles is a stepping-stone to future efficient solar energy conversion applications. Recent efforts in both quantum theory and spectroscopic experiments have shown evolutionarily chosen major design principles: chromophore selection, the structural arrangement of chromophores, chromophore interactions, and protein environment. Chromophores with strong dipole moments have been naturally chosen and have good spectral overlaps between their emission and absorption for energy transfer. The protein scaffold helps the chromophores to be organized with right distances and orientations, so the chromophores can strongly interact with each other. The proteins provide hydrophobic environment, which protects the coherent interactions of chromophores, as well. Thus, exciton delocalization over several chromophores plays a remarkable role in energy transport.
Figure 1-8: Two-dimensional electronic spectra of FMO (Fleming and Engel et
al., Nature, 2007). Selected two-dimensional spectra show the evolution of quantum
coherence at population times from 0 to 600 fs at 77 K. The off-diagonal peak (white arrow) exhibits the electronic couplings of the chromophores. The symmetric diagonal peak denotes energy fluctuations induced by system-bath coupling.
1.5 Scope of Work
Our understanding of natural photosynthesis inspires us to extend our expertise to artificial photosystems. This thesis demonstrates that engineering M13 bacteriophage as a biological scaffold enables nano-sized multi-component assemblies to create
artificial photosystems for solar energy utilization. The main ideas of the thesis focus on boosting energy, electron, and ion transport, leading to the achievement of maximum performance in our systems. Chapter 2 describes porphyrin light-harvesting antenna assemblies showing semi-classical energy transport. Chapter 3 continues to assemble oxygen-evolving catalysts to the porphyrin antenna for photosynthetic charge separation. Chapter 4 demonstrates that biologically controlled distances create energy transport network interplaying between quantum and semi-classical transport. Chapter 5 discusses electron and ion transport in hybrid electrochromic nanomaterials.
Chapter 2
Semi-Classical Energy Transport
in Porphyrin Light-harvesting
Antennas
*
M13 viruses can be used as a biological scaffold to assemble molecular chromophores to build light-harvesting nanoantennas. This chapter demonstrates that energy is transported along the highly ordered chromophore arrays by semi-classical energy transfer mechanisms.
2.1 Introduction
Energy transport is of essence in the natural light-harvesting complexes in order to maximize the exciton flux into the reaction center, where the electron transfer is
* Reproduced with permission from Nam, Y. S., Shin, T., Park, H., Magyar, A. P.,
Choi, K., Fantner, G., Nelson K.A. & Belcher, A. M. (2010). Virus-templated assembly of porphyrins into light-harvesting nanoantennae. Journal of the American
triggered to drive photosynthetic reactions (51). Natural photosystems provide insight into sophisticated self-organization as a model for artificial photosynthetic systems that require efficient energy and electron transfers (52-56). The synthesis and supramolecular self-assembly of a variety of pigments have been widely explored for photochemical and photoelectronic applications such as photovoltaics, non-linear optical materials, and photo-switched conductors (55-58). Dendritic systems have been widely studied for multi-pigment arrays; however, it is challenging and time-consuming to produce such complex molecules in a reasonable yield. Recent research has shown that biological materials including DNA and viruses can be used as templates for guiding nano-scale organization of pigments via chemical linkage or electrostatic interactions (59-62). Viruses provide attractive scaffolds because of their highly ordered coat protein structures (59, 63). M13 virus has a long and skinny shape whose length is ~ 880 nm and diameter is 7 nm in Figure 2-1A. This filamentous structure comprises 2,700 copies of the α-helical major coat proteins, termed pVIII. The major pVIII proteins are arrayed along the single-stranded DNA in a symmetrical fashion; that is, a five-fold rotational symmetry with a two-fold screw axis of 3.2 nm pitch. The chemical modification of the M13 coat proteins is straightforward because of the exposed N-terminus and lysine residue on the viral surface, which can be pigment-binding sites; both of them are indicated as arrows in Figure 2-1B. The full amino acid sequence of the pVIII protein is AEGDDPAKAAFNSLQASAT EYIGYAWAMVVVIVGATIGIKLFKKFTSKAS. The total number of the primary amines available for conjugation is approximately 5,400 per virus. In Figure 2-1C, the locations of primary amines are indicated as letters, a, b, c, and d for the N-termini,
and o for the lysine. The calculated average distances between the N-termini and the lysine are oa ≈ 1.0 nm, ob ≈ 1.6 nm, oc ≈ 2.4 nm, and od ≈ 2.4 nm. Thus, it is highly probable that such close distances between the primary amines on the viral surface allow the energy transfer to occur between neighboring pigments attached to the virus, as demonstrated below. Moreover, the flexible N-terminus of pVIII may allow the attached pigments to have considerable freedom in their orientation and thus facilitate interactions between pigments.
Figure 2-1: Molecular models of wild type M13 bacteriophage and assembly of zinc porphyrins on the M13 virus surface (Nam, Park, and Belcher et al., J. Am. Chem. Soc., 2010). The pVIII coat protein assemblies were constructed from the structural model (Protein Data Bank number 1IFJ). A, A filamentous M13 virus is approximately 880 nm in length, ~ 7 nm in diameter, and composed of 2,700 copies of the major coat protein pVIII. B, A single pVIII protein and a zinc porphyrin pigment, denoted by ZnDPEG. In the pVIII protein, negatively charged amino acids are depicted in red; positively charged ones are in blue; neutral, hydrophilic ones are in yellow; and hydrophobic ones are in white. The arrows show the binding sites of primary amines which ZnDPEG are conjugated to. C and D, Side view and cross section of the assembled pVIII proteins, respectively. In the side view, letters a, b, c, and d denote the locations of N-termini, and a letter o indicates the location of lysine.
2.2 Zinc Porphyrin Light-Harvesting Antennas
We chose Zn(II) deuteroporphyrin IX 2,4 bis ethylene glycol (ZnDPEG, Figure 2-1B) as a model pigment because of its well-known optical properties, water solubility, and pendant carboxylic groups. The zinc porphyrins were conjugated to pVIII via an amide bond formation reaction. Carboxylic acids in ZnDPEG (0.5 mM, Frontier Scientific, Inc.) were pre-activated by incubating with N,N'-dicyclohexylcarbodiimide (1 mM, Sigma-Aldrich) and N-hydroxysuccinimide (1 mM, Sigma-Aldrich) in anhydrous dimethyl sulfoxide for 1 h at room temperature. The activated ZnDPEG was then added to a virus suspension in 1 mM phosphate-buffered saline (PBS, pH 7.4) and incubated for 15 h with magnetic stirring at about 100 rpm. The virus concentration was 1010 pfu/ml as determined by the standard bacteriophage titer analysis. The detailed procedures are available at the New England Biolabs website
(http://www.neb.com). Two samples, ZP-M13-1 and ZP-M13-2, were prepared using
two different molar ratios of ZnDPEG to NH2-: 1:1 for M13-1 and 3:1 for
ZP-M13-2. The reaction mixture was centrifuged at 8,000 rpm for 2 min to remove the insoluble urea byproducts. The unreacted ZnDPEG was removed via dialysis (Spectra/Por® membrane 4, molecular cut-off: 12 ~ 14 kDa, Spectrum, Rancho
Dominguez, CA) against excess amount of deionized water in the dark for 2 days. Approximately, 1,564 and 2,900 porphyrins were conjugated per virus for ZP-M13-1 and ZP-M13-2, respectively.
The number of ZnDPEG conjugated to the virus was determined by measuring the zinc concentration using inductively coupled plasma-atomic emission spectrometry (ICP-AES, ACITVA, Horiba Jobin Yvon, Edison, NJ). Dilute HCl (3%)
was prepared by diluting HCl (reagent grade, Mallinckrodt Chemicals) with reagent grade water (Ricca Chemical Company) and used as a blank solution. A series of zinc solutions at different concentrations (10.0, 20.0, 40.0 and 80.0 ppm) was prepared by diluting a commercial zinc standard solution (Ultra Scientific, Inc.) with the prepared 3% dilute HCl. The porphyrin-virus conjugates were also diluted with HCl (the final concentration = 3%). The zinc calibration curve was validated within a 1% relative standard deviation, which is defined as the ratio of the standard deviation to the average multiplied by 100.
2.3 Microscopic Characterization
Atomic Force Microscopy (AFM) and Transmission Electron Microscopy (TEM) enabled us to observe structural changes. The AFM images were obtained using a Nanoscope IV (Digital Instruments, Sterling Heights, MI) in tapping mode under ambient conditions using etched silicon cantilever tips. The TEM images of porphyrin-M13 nanoantennae were taken on a 200CX electron microscope (JEOL, Akishima, Japan) at 200 kV after negatively staining with 1 %-w/v uranyl acetate. M13 viruses are relatively flexible and thus easily contorted on a flat surface (64). Interestingly, ZP-M13 exhibited a straight structure in Figure 2-2 and 2-3. Such a morphological change in ZP-M13 is thought as a result of the interactions of the hydrophobic, π-conjugated macrocycles of the porphyrin with the viral coat protein.
Figure 2-2: Images of Atomic Force Microscopy (AFM). The samples were prepared on mica. A, wild type M13 viruses. B, Porphyrin light-harvesting antenna, ZP-M13-2. The height of ZP-M13 is measured to be 2.7 ± 1.2 nm, which is considerably smaller than 4.7 ± 1.6 nm of the wild type M13 viruses.
Figure 2-3: Images of Transmission Electron Microscopy (TEM). The samples were negatively stained with 1 wt% uranyl acetate and prepared on carbon-coated copper grid. A, Wild type M13 viruses. B, Porphyrin light-harvesting antennas, ZP-M13-2.
2.4 Steady-State Spectroscopic Results
Spectroscopic data analysis reveals the semi-classical energy transfer along the M13 virus antenna. Fluorescence emission spectra were recorded on a FluoroLog-3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ). Figure 2-4 shows the tryptophan fluorescence of ZP-M13 at an excitation wavelength, 295 nm, is considerably lower than that of wild type M13 viruses. This change indicated the conformational change of pVIII; that is, the tryptophan residues were exposed to the outer aqueous environment presumably through their interaction with the attached porphyrins. Protein structures in the natural light harvesting complexes play a critical role in modulating the site energy levels of individual pigments via interactions with protein residues and neighboring pigments, which in turn affect the pathway of excitation energy transfer (65).
Such protein matrix effects could be observed in the absorption spectral changes of ZP-M13. Absorption spectra were obtained on a DU 800 spectrometer (Beckman Coulter) at ambient temperature with a 1-cm path-length quartz cuvette. Figure 2-5 shows the absorption spectrum of monomeric ZnDPEG monomers exhibited a strong Soret band at 406 nm and two weak Q bands at 538 nm and 574 nm. Compared to free ZnDPEG, the porphyrins assembled on the virus had a considerably broader Soret band. This spectral broadening can be explained by the diverse pigment-protein interactions, which generate different site energies of the excited states. Such heterogeneous microenvironments of the viral surface are illustrated by the various surface functional groups, as shown in Figure 2-1C, rendering diverse local effects of dipole-dipole interactions and hydrogen bonding to the attached porphyrins.
The most dramatic optical variation of ZP-M13 was the quenching of the fluorescence emission in Figure 2-6. The excitation wavelength was set to be 400 nm. At this wavelength each sample showed very similar absorbance: 9.5 x 103 m2/mol for ZnDPEG, 10.0 x 103 m2/mol for ZP-M13-1 and 10.5 x 103 m2/mol for ZP-M13-2. The quenching effect can be explained by a small redshift found in the Q bands of the absorption spectrum of ZP-M13 (538 → 543 nm and 574 → 577 nm, respectively) in Figure 2-5. It is known that the redshift of the Q bands reflects the formation of the electronic coupling between pigments. However, such interaction may be not very strong in the ZP-M13 presumably because ZnDPEG lacks of meso-substituents, which are known to stabilize the J-type aggregates (66). Accordingly, the circular dichroism spectra of the ZP-M13 exhibited no distinctive peaks in the Soret region, indicating the formation of H-type coupling between the conjugated pigments (data not shown). The orbital disordering caused by the pigment-pigment interactions may induce a significant change in the nuclear coordinates of the excited state relative to the ground state. This change can result in the formation of trap sites where the radiative transition becomes much slower than the non-radiative relaxation. This explanation is supported by the result that the fluorescence quenching increases with increasing degree of conjugation, which implies that the Dexter-type electronic interactions between porphyrins are essential for the efficient quenching process (67).
Figure 2-4: Tryptophan fluorescence emission spectra of wild type M13 viruses and porphyrin light-harvesting antennas. The samples were excited at 295 nm.
Figure 2-5: Absorption spectra of free zinc porphyrin and porphyrin-M13 virus antennas. ε denotes a molar extinction coefficient. The inset is the magnified spectra near the Q bands.
Figure 2-6: Fluorescence spectra of free zinc porphyrin and porphyrin-M13 virus antennas. The samples were excited at 400 nm. The inset shows a magnified spectrum of ZP-M13-2.
2.5 Transient-Absorption Spectroscopic Results
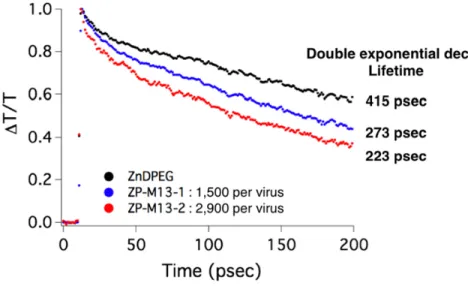
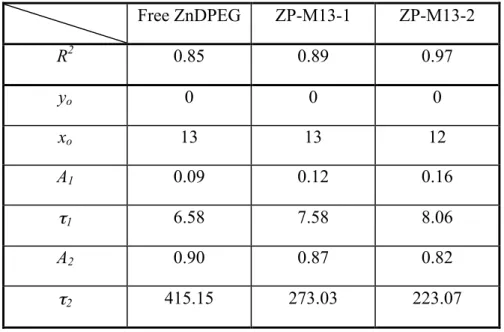
In order to verify the exciton migration along the pigments assembled on the virus, pump-probe transient absorption spectroscopy was used in Figure 2-7. The transient absorption change of the Soret band in Figure 2-8 indicates that the excited state of ZP-M13 is relaxed significantly faster than that of ZnDPEG. The transient absorption profiles were fitted using a double exponential decay curve given by y = yo + A1 exp
{-(x-xo)/t1} + A2 exp {-(x-xo)/t2}, where yo is the baseline, An (n = 1 or 2) represents
the magnitude, and tn (n = 1 or 2) is the lifetime. A two-parameter fit in Figure 2-9 and
Table 2-1 indicates that the dominant relaxation pathway has a lifetime of 273 ± 4 psec for ZP-M13-1 and 223 ± 5 psec for ZP-M13-2, while free ZnDPEG has a lifetime of 415 ± 3 psec. These data imply the excitonic transfer through the pigments
assembled on the virus. The relatively long distance between the pigments assembled on the virus (about 3.5 nm in average for ZP-M13-2) and no significant shift of the Soret band in the absorption spectrum of ZP-M13 suggest that such exciton transport is driven via the Förster resonance energy transfer, though the Dexter mechanism seems to contribute to fluorescence quenching process as described above.
Figure 2-7: Experimental set-up for pump-probe transient absorption measurements. A 1 kHz Ti:sapphire regenerative amplifier system produces 70 fsec optical pulses centered at 800 nm. The frequency of such pulses was doubled using a 0.1 mm- thick type-I BBO, and the optical beams were split into a pump and probe arm. The pump beam was time-delayed with a motorized translation stage. The energies of pump and probe were adjusted to 60 nJ and 10 nJ, respectively, and focused at the sample in a cuvette of 500 mm-path-length. To supply a fresh sample for each laser shot, the sample solution was flowed through the cuvette at a constant flow rate of about 50 mm/msec. The pump beam was chopped at a rate of 500 Hz and the modulated probe beam intensity was recorded with a lock-in amplifier via a photodiode.
Figure 2-8: Transient absorption spectra. T in y-axis denotes the transmission of light.
Figure 2-9: Transient absorption profiles were fitted using a double exponential decay curve.
Table 2-1: The parameters of fitting profiles to transient absorption data. Free ZnDPEG ZP-M13-1 ZP-M13-2 R2 0.85 0.89 0.97 yo 0 0 0 xo 13 13 12 A1 0.09 0.12 0.16 τ1 6.58 7.58 8.06 A2 0.90 0.87 0.82 τ2 415.15 273.03 223.07
2.6 Conclusion
Our work has shown that molecular pigments can be assembled into a light-harvesting antenna using M13 viruses as a biological template through chemical linkage. We observed distinctive spectroscopic changes such as a small redshift and line-broadening in absorbance, fluorescence quenching, and faster kinetics of the excited states. These spectral changes suggest that the excitons in the porphyrin light-harvesting antennas are in the limit of weak electronic couplings, so travel through semi-classical energy transport theories: long-range Förster dipole-dipole interactions and short-range Dexter quenching mechanism. This porphyrin system inspires us to co-assemble catalysts and electron transport mediators with pigments on M13 viruses through genetic engineering. Next chapter exhibits that such co-assembled
nanostructures can be applied to photochemical systems which often require a large charge flux to catalytic sites via energy transfer. Moreover, the porphyrin antenna system implies that the energy transfer in the assembled pigments is controllable by genetically inserting or deleting amino acids on pVIII, which can modify the site energies and the pigment-pigment distances. In chapter 4, we will address this issue for showing quantum-enhanced energy transport, mimicking natural light-harvesting complexes.
Chapter 3
Photosynthetic Charge Separation
in Biologically Engineered
Antenna-Catalyst Complexes
*
Genetically engineering M13 viruses enables us to co-assemble both catalysts and chromophores into an artificial photosystem for efficient oxygen-evolving charge separation. The high proximity between the catalysts and the chromophores enhances the large charge flux to the catalytic sites via energy transfer, leading to improved oxygen evolution. Moreover, polymer microgels encapsulates the biologically templated nanowires of both the catalysts and the chromophores. This encapsulation increases structural stability and shows recyclable capability, resulting in sustained water oxidation.
* Nam, Y. S., Magyar, A. P., Lee, D., Kim, J. W., Yun, D. S., Park, H., Pollom TS,
Weitz D. A. & Belcher, A. M. (2010). Biologically templated photocatalytic nanostructures for sustained light-driven water oxidation. Nature nanotechnology,
3.1 Introduction
Over several billion years, highly organized photosynthetic systems in cyanobacteria, algae, and plants have evolved to shuttle both electronic and chemical species for the oxidation of water (6, 8, 9). Similar to reaction centres in natural photosystems, molecular and metal oxide catalysts have been used to photochemically oxidize water as a charge separation process; however, various approaches involving the molecular design of ligands, surface modification, and immobilization still show limitations in catalytic efficiency and sustainability (68-73). Here we demonstrate a biologically templated nanostructure for visible light-driven water oxidation that employs a genetically engineered M13 virus scaffold to mediate the co-assembly of zinc porphyrins (photosensitiser) and iridium oxide hydrosol clusters (catalyst) (74). Porous polymer microgels are used as an immobilization matrix to improve the structural durability of the assembled nanostructures and to allow the materials to be recycled. Our results suggest that biotemplated nano-scale assembly of functional components is a promising route to significantly improve photocatalytic water splitting systems.
One of the most fascinating aspects of natural photosynthesis is the sophisticated self-organization of light harvesting complexes, electron transfer mediators, and oxygen evolving complexes (6, 8, 9). In such photosystems, dozens of chlorophyll molecules within the light-harvesting complex are responsible for the primary light absorption event. This energy is efficiently transferred to the reaction centre, where it is transformed into a pair of spatially separated charge carriers. Sunlight-driven water splitting for hydrogen generation has received increasing
attention as a means of storing solar energy in chemical bonds (75, 76). However, to evolve hydrogen efficiently in a sustainable manner, it is pre-requisite to develop a stable and efficient catalytic system for water oxidation, which is the more challenging half reaction of photo-driven water splitting (77-80). Multiple attempts to model water oxidation systems on photosynthesis have had limited success; most Mn-oxo based catalysts require oxygen-transfer oxidants to catalyse oxygen formation (81). Until now, the most promising catalytic systems are based on metal oxide colloids, which drive the photochemical oxidation of water with several different components cooperatively: a photosensitiser absorbs visible light and subsequently transfers an electron to an electron acceptor, stimulating the transfer of a hole from the photosensitiser to the catalyst (82). Repeated cycles of light absorption lead to the accumulation of four holes in the catalyst and the holes drive the evolution of a molecule of oxygen from water. As in photosynthesis, distances between functional components are critically important in these colloidal systems; non-optimal spacing between photosensitiser, electron acceptor, and catalyst can kinetically promote corrosive mechanisms, leading to the degradation of catalytic materials (83).
Various approaches to developing efficient sustainable metal oxide catalysts driven by visible light have been undertaken; these approaches include the polyelectrolyte-mediated complexation of catalysts and photosensitisers, ligand design, and heterogeneous catalysts (69, 71, 73, 84). However, it is challenging to design multi-component systems with controlled structural arrangement at the molecular level; the lack of this precise arrangement can hinder the appropriate trafficking of chemical and electron species between individual active components