Unexpected ancestry of Populus seedlings from a hybrid zone implies a large role for postzygotic selection in the maintenance of species
40
0
0
Texte intégral
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(38)
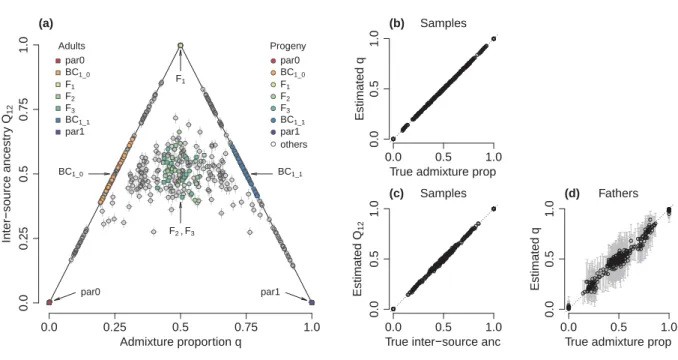
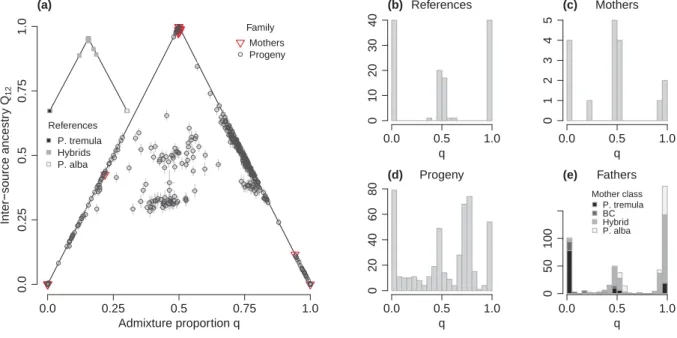
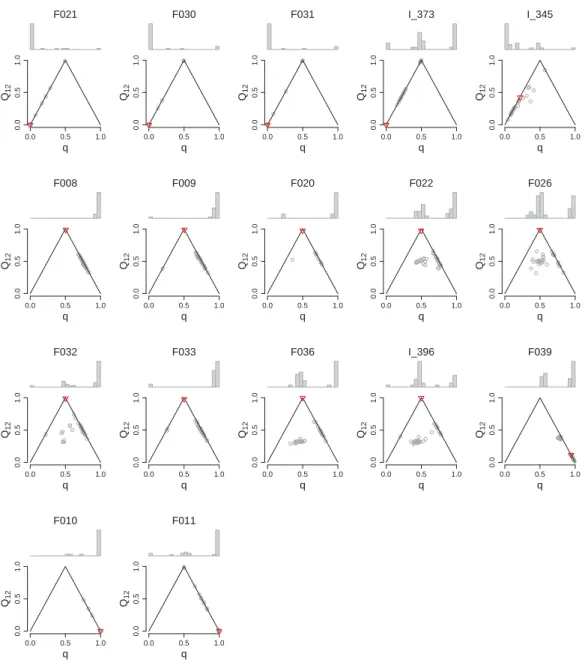
Figure

+5
Documents relatifs
Generally, investigating the macronutrient profiles such as protein, lipids, and moisture content is necessary to evaluate if the overlooked (non-targeted for human
In most specimens of Sayimys sivalensis the hypolophid is directed obliquely forward and it is continuous with the posterior arm of the protoconid (Munthe, 1980; Wessels et al.,
(1997)’s ABX task again in which participants heard three words produced by three different speakers and had to decide whether X was similar to A or to B. Two types of
angustifolia (physical dormancy); (iii) seed source was a key factor in the germination response of all three species, either by having a significant overall effect or by having
Our models were used under three di fferent cir- cumstances: three data sets and conditions: (a) all individuals alive in 1999 to evaluate mortality risk at the onset of the study,
To clarify the role of large trees in biomass production of heterogeneous forest, we used data of tree growth, mortality and recruitment monitored during 20 years in 10×4-ha plots in
albilineans is a highly distinctive xanthomonad harbouring specific cell wall degrading enzymes with long linkers and CBD suggesting its adaptation for the degradation of
In the case of an inductor, we know that if there is a steady, con- stant current flowing through it, then the magnetic field is constant, and so is the amount of energy stored;
