HAL Id: hal-01396539
https://hal.archives-ouvertes.fr/hal-01396539
Submitted on 4 Feb 2020HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Theoretical mechanistic study of self-sensitized
photo-oxygenation and singlet oxygen thermal release in
a dimethyldihydropyrene derivative
Martial Boggio-Pasqua, Marta López Vidal, Marco Garavelli
To cite this version:
Martial Boggio-Pasqua, Marta López Vidal, Marco Garavelli. Theoretical mechanistic study of self-sensitized photo-oxygenation and singlet oxygen thermal release in a dimethyldihydropyrene deriva-tive. Journal of Photochemistry and Photobiology A: Chemistry, Elsevier, 2017, 333, pp.156 - 164. �10.1016/j.jphotochem.2016.10.020�. �hal-01396539�
Theoretical mechanistic study of self-sensitized photo-oxygenation and singlet oxygen thermal release in a
dimethyldihydropyrene derivative
Martial Boggio-Pasquaa,*, Marta López Vidala, Marco Garavellib
a Laboratoire de Chimie et Physique Quantiques (UMR 5626), CNRS et Université Toulouse III, 118 route de Narbonne, 31062 Toulouse, France b Dipartimento di Chimica “G. Ciamician”, Universita’ degli Studi di Bologna, Via Selmi 2, I-40126 Bologna, Italy * Corresponding author: martial.boggio@irsamc.ups-tlse.fr Abstract The self-sensitized photo-oxygenation and singlet oxygen thermal release mechanisms in a pyridinium-substituted dimethyldihydropyrene (DHP) derivative have been investigated using quantum chemical calculations. First, the main photophysical pathway for intersystem crossing was identified, allowing the production of a DHP triplet state. Second, the energy transfer pathway between this triplet state and the oxygen triplet ground state was computed revealing a very efficient route for the photosensitized generation of singlet oxygen. Finally, the thermal pathway for the formation of a metacyclophanediene endoperoxide and the singlet oxygen release was characterized. A concerted and a stepwise mechanism were identified, the first one being lower in energy. All these results are consistent with recent experimental results and confirm that this type of system could be attractive oxygen carriers and singlet oxygen delivery agents. Keywords: Computational chemistry Density functional theory calculations Dimethyldihydropyrene Energy transfer Photosensitizer Endoperoxide 1. Introduction
Singlet oxygen (1O2) is one of the most important reactive oxygen species. Due to its
remarkable reactivity and oxidizing property, it can be exploited in various applications ranging from chemical synthesis, waste water treatment, atmospheric chemistry, materials science, optical imaging and therapy [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. Not surprisingly then, the control of trapping and releasing 1O2 has become a very important topic of
1O2 is the first electronic excited state of molecular oxygen and the direct transition from
its triplet ground state 3O2 is spin forbidden. To facilitate this transition, an indirect
mechanism can be achieved using photosensitizers, which are light-absorbing molecules capable of transferring a part of the photonic energy to another molecule (e.g., 3O2) to
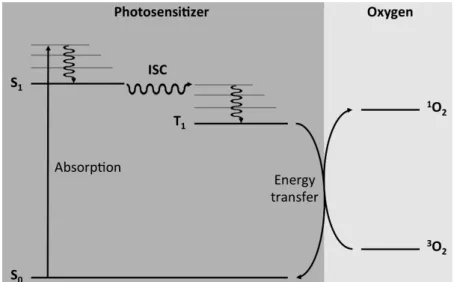
induce an electronic transition (e.g., 3O2→1O2). As schematized in Fig. 1, in the present
context, the photosensitizer in its ground electronic state S0 absorbs light to produce a
singlet excited state S1, which can decay non-radiatively by intersystem crossing (ISC) to a
triplet state T1. Provided that this triplet state is sufficiently high in energy and that the
photosensitizer is physically close enough to 3O2, an energy transfer process can occur
producing 1O2 and regenerating the photosensitizer in its original ground state. Fig. 1. Schematic diagram depicting the photosensitized excitation of oxygen in its ground triplet state (3O2) to its lowest singlet excited state (1O2).
One way to control the release of 1O2 is to produce chemical species incorporating
molecular oxygen and capable of releasing 1O2 in a controlled way. This aim can be achieved
by the use of specific aromatic organic compounds [8,9,15,16,17,18,19,20,21,22,23,24] that can chemically trap 1O2 in the form of endoperoxides (EPO) following a cycloaddition
reaction. These EPOs exhibit the exceptional feature of releasing oxygen, often in its singlet excited state 1O2, under heating or UV irradiation.
The properties of molecular switches can also be used to regulate the production of 1O2
[25,26]. Supramolecular species and solid materials based on spiropyran [27] and dithienylethene [11,28,29] photochromic compounds associated with an external 1O2
photosensitizer (e.g., a metal complex or a porphyrin) were used for the reversible control of 1O2 generation. In these systems, the production of 1O2 is governed by the state (“on” or
“off”) of the photochromic unit. Very recently, photochromic dimethyldihydropyrene [30,31,32,33,34,35] (DHP) derivatives were also found to be efficient singlet oxygen carriers and releasing agents [36,37]. The main advantages of these DHPs over the spiropyran and dithienylethene compounds is that they do not need an external 1O2
photosensitizer, as they photosensitize oxygen themselves, and they work using low energy (red) light. As illustrated in Scheme 1, a pyridinium-appended DHP 1 can be switched to its open-ring cyclophanediene (CPD) isomer 2 by irradiation at λ ≥ 630 nm [38]. The reverse conversion can be achieved either by irradiation in the UV range or thermally. A solution of
1 in the absence or presence of air and exposed to such an irradiation produces compounds 2 and 2-O2 quantitatively. Upon heating at 35 °C the thermal release of 1O2 was observed
from 2-O2.
Δ
Scheme 1. Conversion processes between 1, 2 and 2-O2. Adapted from ref. [36].
These experimental observations were interpreted with the mechanism proposed in Eqs. (1)-(4). Red visible irradiation of the closed isomer 1 in its ground state S0 produces its
singlet excited state 1(S1), which isomerizes to 2 (Eq. (1)). However, the excited state 1(S1)
can also non-radiatively decay by ISC to produce the triplet 1(T1) (Eq. (2)). The DHP in its
lowest triplet state can then undergo an energy transfer process with molecular oxygen O2(T0) producing singlet oxygen O2(S1). Thus, 1 also plays the role of O2 photosensitizer (Eq.
(3)). The photogenerated 1O2 rapidly reacts with 2 to form the corresponding EPO 2-O2,
which can release singlet oxygen upon warming (Eq. (4)). 1(S0) + hν → 1(S1) → 2 (1) 1(S1) → 1(T1) (2) 1(T1) + O2(T0) → 1(S0) + O2(S1) (3) O2(S1) + 2 2-O2 (4)
The purpose of the present study is to bring some mechanistic information on this mechanism using theoretical chemistry. While the photoswitching mechanism between the closed-ring DHP and the open-ring CPD isomers (Eq. (1)) has been investigated from a computational point of view for a number of DHP derivatives [39,40,41], including the pyridinium-appended DHP shown in Scheme 1 [38], no theoretical study has focused on the self-sensitized photo-oxygenation and singlet oxygen thermal release of dimethyldihydropyrene derivatives (Eqs. (2-4)). In this article, we report a theoretical study based on density functional theory (DFT) of the singlet to triplet intersystem crossing pathway of compound 1 (Eq. (2)), the subsequent energy transfer process occurring with oxygen (Eq. (3)), and the thermal pathway for the corresponding EPO formation (Eq. (4)).
2. Computational details
DFT and time-dependent DFT (TD-DFT) have been used to perform calculations on the ground and first excited singlet and triplet states of the isolated system 1. A model system was used by simply replacing the bulky tert-butyl groups by hydrogen atoms in order to reduce the computational cost. The B3LYP functional [42] was used throughout along with the 6-311G(d,p) basis set [43], unless otherwise specified. Open-shell singlet biradical structures were computed at the broken-symmetry unrestricted B3LYP level. To account for the spin contamination, spin-projected energies have been calculated with the approximate spin-correction procedure proposed by Yamaguchi and coworkers [44,45]. Broken-symmetry DFT calculations combined with such a spin-correction procedure was successfully used to describe the 1O2 thermal release pathway in a model EPO [46]. The
spin-orbit coupling (SOC) between a pair of intersecting singlet and triplet states was computed at the complete active space self-consistent field (CASSCF) level using the four most relevant orbitals involved in the main electronic transitions (Fig. S1 in Supplementary material). All the optimized Cartesian coordinates and energies are collected in Table S1 in Supplementary material. All calculations were performed with Gaussian 09 [47]. 3. Results and discussion 3.1 Electronic transitions to singlet and triplet excited states The nature and vertical transition energies of the lowest singlet and triplet excited states of compound 1 are first described in this subsection. The optimized structure of compound
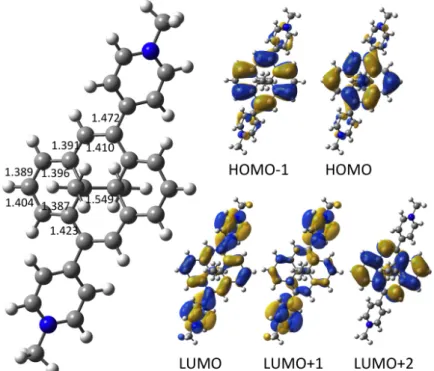
1, which belongs to the Ci symmetry point group, is presented in Fig. 2 along with the
molecular orbitals involved in the main electronic transitions describing these excited states. Table 1, which collects the TD-DFT results, shows that the first two singlet excited states S1 and S2 correspond mainly to a HOMO→LUMO and a HOMO–1→LUMO transition,
respectively. These states display substantial charge transfer character due to the electron withdrawing character of the pyridinium substituents and are located respectively at 1.720 and 2.122 eV vertically above the ground state S0. The corresponding absorption
wavelengths are 721 nm for S1 and 584 nm for S2, in good agreement (error < 0.3 eV) with
the experimental first absorption bands centered at 680 and 528 nm [38]. These results are also in good agreement with previous TD-DFT results performed with a range-separated CAM-B3LYP functional [48], for which absorptions at 657 and 548 nm were computed for these two singlet states [38]. Two triplet states were located below S1. T1 corresponds to a
HOMO→LUMO transition like S1, whereas T2 is mainly described by a HOMO–1→LUMO
transition like S2. The next two triplet states T3 and T4 are intercalated between S1 and S2
and correspond respectively to HOMO→LUMO+2 and HOMO→LUMO+1 transitions.
Fig. 2. Optimized structure of model compound 1 and relevant molecular orbitals. Bond lengths are in Å. Table 1 TD-DFT lowest energy electronic transitions in model compound 1. State Nature Excitation
energy /eV λabs /nm Oscillator strength (f) Main transitions and amplitudesa
T1(Au) (π,π*) 1.039 n.r. 0.000 0.60(H→L) T2(Au) (π,π*) 1.474 n.r. 0.000 0.55(H–1→L) S1(Au) (π,π*) 1.720 721 0.101 0.66(H→L) T3(Au) (π,π*) 1.831 n.r. 0.000 0.56(H→L+2) T4(Ag) (π,π*) 1.919 n.r. 0.000 0.70(H→L+1) S2(Au) (π,π*) 2.122 584 0.344 0.62(H–1→L) a H and L represent the HOMO and LUMO, respectively. Amplitudes > 0.5 are given. 3.2 Singlet to triplet intersystem crossing pathway Upon visible irradiation, DHPs are excited to singlet excited states, which decay either by fluorescence or internal conversion, formation of the corresponding CPD, and ISC to the triplet state [33]. The formation of a triplet excited state is necessary for the photo-sensitization of oxygen. Thus, we have investigated the main photophysical pathway for ISC in compound 1. In Fig. 3a, we report the potential energy profiles of the first three singlet and four triplet electronic states computed at the TD-DFT level along the main relaxation coordinate of the S1 state initially populated. This coordinate mainly involves a stretching of the transannular central C–C bond along with a loss of the planarity of the DHP core. The minimum on the S1
potential energy surface (PES) corresponds to a CPD precursor for CPD formation, thus associated with the initial part of the photoisomerization mechanism [38]. Because the ISC probability will mainly depend on the energy gap between this singlet state and the triplet states, and the associated SOCs, the search for singlet/triplet crossings is interesting as they provide efficient ISC funnels provided that the SOC is non negligible. In Fig. 3a, it is clear
that the T1 state remains energetically well below the S1 state at all relaxation geometries.
However, the T2 state increases slightly in energy to become close to the S1 state (~0.1 eV)
at the S1 minimum. As a consequence, the T2-S1 energy gap is reduced along the S1
relaxation coordinate and this behavior opens the opportunity to find a T2/S1 crossing in
the vicinity of the S1 minimum. Indeed, following the T2 gradient, the T2 state crosses the S1
state only 0.04 eV above the S1 minimum, as shown in Fig. 3b, making this funnel for ISC
easily accessible energetically.
Fig. 3. Potential energy profiles for (a) the first three singlet (S0, S1 and S2) and four triplet
(T1, T2, T3, T4) electronic states along the S1 relaxation coordinate, and for (b) the first two
singlet (S0 and S1) and second triplet (T2) electronic states along the S1 relaxation
coordinate and along the T2 gradient (beyond the vertical dotted line).
CASSCF calculations of the SOC between the S1 and T2 states at the T2/S1 crossing
geometry reveals that it is small (2 cm–1) but sufficient to induce the expected ISC between
singlet excited state 1(S1), which can undergo an ISC to 1(T2). Further relaxation by internal
conversion to the lowest T1 state will then proceed efficiently, as this non-radiative
transition is not spin-forbidden. Thus, the photophysical pathway for triplet DHP formation can be summarized as follows: 1(S0) + hν → 1(S1) → 1(T2) → 1(T1) (5) 3.3 Photosensitized generation of singlet oxygen
As observed experimentally, 1(T1) can be quenched by molecular oxygen to produce
singlet oxygen (Eq. (3)). This quenching process requires an energy transfer between the triplet excited state of DHP and the triplet ground state of molecular oxygen so that DHP is deactivated back to its singlet ground state and oxygen is excited to its singlet state. To be efficient, this energy transfer step will need to be favorable both thermodynamically and kinetically. Moreover, the two reactants (i.e., 1(T1) and O2(T0)) will need to approach each
other close enough, as the energy transfer efficiency decreases upon increasing the distance between the two partners.
Our DFT study provides information only on the energetics of a possible physical quenching pathway (Eq. (3)), its potential energy profile and the activation energy involved. Other factors governing the energy transfer such as vibronic couplings are not considered here. Thus, our study gives an indication of the feasibility of the process based on the existence of an energetically favorable route connecting the triplet–triplet (1(T1) and
O2(T0)) starting reactants to the singlet–singlet (1(S0) and O2(S1)) products.
To determine a possible energy transfer path, we have performed a linear interpolation between two optimized electrostatic complexes, the relaxed triplet–triplet complex [3O2•••1(T1)], which represents the starting reactant, and the relaxed singlet–singlet
complex [1O2•••1(S0)], which represents the final product. These optimized electrostatic
complexes are shown in Fig. 4 and provide stable structures allowing the two partners (i.e., DHP and molecular oxygen) to be in close contact to proceed with the energy transfer. The initial position of the oxygen molecule in the two electrostatic complexes has been chosen to be consistent with the observed EPO 2-O2 where the oxygen is only observed on one exo
side of the cyclophane for steric reasons [36]. The structure of the DHP core in the triplet– triplet [3O2•••1(T1)] complex is consistent with the optimized structure of 1(T1) (compare
with Fig. S2 in Supplementary material), where the transannular bond length is increased by 0.035 Å compared to the ground-state structure. On the other hand, the transannular bond length in the singlet–singlet [1O2•••1(S0)] complex is similar to the one in 1(S0), as
expected (compare with Fig. 1). Note that the relaxed structures of 3O2 and 1O2 remain
almost identical, reflecting the fact that the interatomic distance in both states is very similar, but the distance and orientation between the oxygen molecule and the DHP core is very different in both complexes.
Fig. 4. DFT optimized structures for (a) the triplet–triplet [3O2•••1(T1)] complex and (b) the
singlet–singlet [1O2•••1(S0)] complex. Bond lengths are in Å.
Single-point energy calculations have been performed along the interpolated energy transfer pathway. Spin-projection on the singlet–singlet 1(1(S0) + 1O2) wavefunction is
required to obtain a correct energy profile but for the triplet–triplet coupling (globally a singlet state wavefunction), no decontamination is needed. Due to the abrupt change in the singlet state wavefunction passing from a triplet–triplet coupling 1(1(T1) + 3O2) to a singlet–
singlet 1(1(S0) + 1O2) coupling description, it was possible to follow each coupling situation
all along the linearly interpolated pathway. In this way, we have determined the diabatic components of the path as well as the singlet ground-state and the singlet excited-state potential energy profiles along the energy transfer path. The results are collected in Fig. 5 and suggest that the energy transfer process (physical quenching) is energetically favorable, as the product complex is located 0.091 eV below the reactant complex. In addition, the energy transfer occurs along an almost barrierless pathway with an upper limit to the barrier of only about 0.03 eV (a simple linear interpolation provides an upper boundary to the energy). Along the linear interpolation path, going from a triplet–triplet to a singlet–singlet coupling through the diabatic state crossing point, a change in the spin densities of the system occurs (see Fig. 5). Of course, our linear interpolation provides only an approximate pathway that connects reactants to products. However, the fact that we have obtained a very small barrier is a demonstration of the efficiency of such a process (at least from an energetic point of view). Thus, we can expect a very fast process for the singlet oxygen production.
Fig. 5. Potential energy profiles along the energy transfer process. The energy transfer
coordinate has been approximated by linear interpolation between the optimized structures of the triplet–triplet [3O2•••1(T1)] reactant complex and the singlet–singlet
[1O2•••1(S0)] product complex. The blue and red curves correspond to the triplet–triplet
coupling 1(1(T1) + 3O2) and to the singlet–singlet coupling 1(1(S0) + 1O2) diabatic states,
respectively. The spin densities are given for each diabatic state (positive spin density in blue and negative spin density in cyan).
3.4 Thermal pathway for endoperoxide formation and singlet oxygen release
Experimentally, the DHP compound 1 irradiated with red light (λ ≥ 630 nm) under aerobic conditions is quantitatively converted into an EPO derivative, which can then thermally release singlet oxygen to regenerate the starting compound (Scheme 1) [36]. In this subsection, we investigate the thermal pathway for EPO formation between the photoproduced CPD isomer 2 and singlet oxygen (Eq. (4)). It is important to note that 2 does not absorb the irradiation red light and thus can only react in its ground electronic state with 1O2. This is confirmed by our TD-DFT calculation of the electronic transitions in 2, as the lowest energy π→π* bright transition is computed at 438 nm with CAM-B3LYP
(Table S2 in Supplementary material) [49], in good agreement with the experimental data [38].
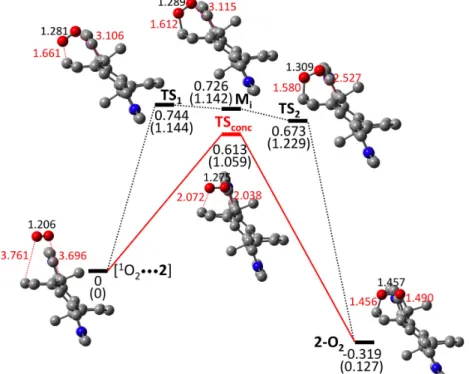
To investigate the cycloaddition reaction of 1O2 with CPD, an electrostatic complex
[1O2•••2] was first optimized (Fig. 6 and Table S1 in Supplementary material). This complex
represents the starting reactant and provides a stable structure allowing the two partners (i.e., CPD and singlet oxygen) to be in close contact to proceed with the EPO formation. The CO bond lengths are ca. 3.7 Å and the OO bond length is 1.2 Å corresponding to that of 1O2.
The 2-O2 EPO product has also been optimized (Fig. 6 and Table S1 in Supplementary
material). The two CO bond lengths are 1.46 and 1.49 Å. The X-ray structure gives bond lengths of 1.51 and 1.79 Å, respectively [36]. The much longer distance for the CO bond adjacent to the methyl group is not reproduced by the DFT calculation. The OO bond length is shortened to 1.46 Å in fairly good agreement with the 1.50 Å distance found experimentally.
As shown in Fig. 6, two pathways that connect [1O2•••2] with 2-O2 were identified
corresponding to a stepwise and to a concerted mechanism. This is similar to what was found on a simple benzene EPO model system [46]. The lowest energy path involves the concerted mechanism in which the transition state (TSconc) displays two simultaneously
amounts to 0.61 eV, and the 2-O2 EPO product generated is lower in energy (by 0.32 eV)
than the reactant [1O2•••2] complex. The second mechanism involving a stepwise process
lies slightly higher in energy. In this case, the two CO bonds are broken sequentially. In a first step, one CO bond is formed (1.61 Å), leading to a biradical intermediate denoted Mi
lying 0.73 eV above the reactant. The transition state involved along this step (TS1) lies
close in energy to the Mi intermediate and corresponds to an energy barrier of 0.74 eV. In
the second step, the other CO bond is being formed (2.5 Å at TS2). Note however that upon
applying the spin-correction procedure, the energy barrier along this step disappears indicating that the second CO bond could be formed in a barrierless manner. Considering the lowest energy concerted mechanism, the cycloreversion process from the 2-O2 EPO will
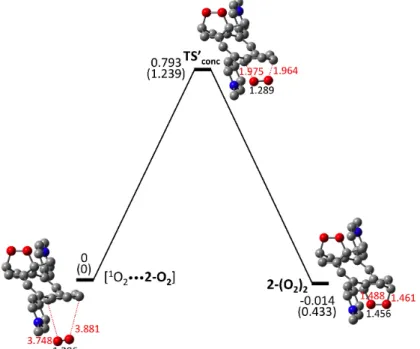
require an energy barrier of 0.93 eV, which accounts for the thermal release of singlet oxygen upon heating. Fig. 6. DFT energies (eV) for stepwise (black) and concerted (red) thermal pathways of endoperoxide 2-O2 formation. Spin-corrected energies in normal font and non-corrected energies in parentheses. Energies given relative to the starting [1O2•••2] complex. CO and OO bond lengths (in Å) are in red and black font, respectively. The further addition of singlet oxygen on the endoperoxyde 2-O2 could potentially form a bis-endoperoxyde 2-(O2)2. Experimentally, no such bis-endoperoxyde was identified. The
cycloaddition reaction of 1O2 with 2-O2 was investigated considering the lowest energy
concerted pathway, as shown in Fig. 7. First, an electrostatic complex [1O2•••2-O2] was
optimized (Fig. 7 and Table S1 in Supplementary material) to provide the starting reactant. The CO bond lengths are ca. 3.8 Å and the OO bond length is 1.2 Å corresponding to that of
1O2. The transition state found along the concerted path (TS’conc) displays two
simultaneously elongated CO bonds (~2 Å). The energy barrier of this synchronous activated process amounts to 0.79 eV, which is 0.18 eV higher than the cycloaddition barrier of 1O2 with 2 (compare with Fig. 6). In addition, the 2-(O2)2 bis-EPO product
generated is almost isoenergetic with the reactant [1O2•••2-O2] complex. Thus, the
calculations show that the cycloaddition of a second singlet oxygen molecule is energetically less favorable than the first one. However, when it is compared with the
thermally activated 1O2 release pathway from 2-O2, this second cycloaddition is a
competitive process from an energetic point of view. The fact that the 2-(O2)2 bis-EPO
product is not observed is certainly due to kinetic reasons, as the 1O2 thermal release is a
unimolecular process, while the bis-EPO formation results from a bimolecular reaction. Note also that to undergo the cycloaddition 1O2 has only one reactive site left in 2-O2, while
two sites were present in 2.
Fig. 7. DFT energies (eV) for the concerted thermal pathway of bis-endoperoxide 2-(O2)2
formation. Spin-corrected energies in normal font and non-corrected energies in parentheses. Energies given relative to the starting [1O2•••2-O2] complex. CO and OO bond
lengths (in Å) are in red and black font, respectively.
4. Conclusions
In this contribution, we have investigated theoretically the mechanisms of the self-sensitized photo-oxygenation and singlet oxygen thermal release in a dimethyldihydropyrene derivative following the recent experimental work of Royal and coworkers [36]. These authors reported that red light irradiation (λ ≥ 630 nm) of a colored pyridinium-substituted DHP photochromic compound readily leads to the quantitative formation of the thermodynamically less stable colorless CPD form following the opening of the central C–C bond [38]. In the presence of molecular oxygen, this DHP derivative also plays the role of an O2 sensitizer. The photogenerated 1O2 reacts with the CPD isomer to
afford the corresponding EPO-CPD species. The EPO-CPD then releases 1O2 thermally with a
high yield of ca. 85% [36].
Our study is based on the computation of the relevant photophysical and chemical pathways using DFT-based methods. The first step involves the formation of a DHP triplet excited state (Eq. (2)), which is required for the photo-sensitization of oxygen. The main photophysical pathway for ISC was identified and involves the non-radiative decay at an easily accessible crossing between the initially excited S1 state with the T2 state. This state
can then rapidly decay to the lowest T1 state by efficient internal conversion. The resulting
T1 state can then be quenched by molecular oxygen to produce singlet oxygen. This second
ground state of molecular oxygen so that DHP is deactivated back to its singlet ground state and oxygen is excited to its singlet state (Eq. (3)). The potential energy profile computed for this process shows that it is energetically favorable, as it involves a quasi-barrierless energy transfer. Finally, the thermal pathway for the EPO-CPD formation between the photoproduced CPD isomer 2 and singlet oxygen (Eq. (4)) has been computed. Two different pathways were identified, one corresponding to a concerted mechanism, while the other involves a stepwise process. The lowest energy synchronous pathway involves a barrier of 0.61 eV for the EPO formation, while a 0.93 eV barrier needs to be overcome for the thermal release of singlet oxygen by this EPO. These results are consistent with an efficient EPO-CPD formation at low radiant energies and singlet oxygen production upon warming [36] and they confirm that this type of system could be attractive oxygen carriers and singlet oxygen delivery agents.
Acknowledgements
This work was granted access to the HPC resources of CALMIP under the allocation
2016-[12158]. The corresponding author thanks G. Royal for fruitful discussions on this topic.
Supplementary material
Fig. S1 for a comparison between TD-DFT and CASSCF electronic structures at the T2/S1
crossing. Table S1 for Cartesian coordinates and energies of all the optimized structures. Fig. S2 for the DFT geometry of 1(T1). Table S2 for the TD-DFT lowest energy singlet
electronic transitions in 2 using CAM-B3LYP.
References
[1] M. Prein, W. Adam, The Schenck ene reaction: Diastereoselective oxyfunctionalization with singlet oxygen in synthetic applications, Angew. Chem. Int. Ed. Engl. 35 (1996) 477– 494. [2] P. Esser, B. Pohlmann, H.-D. Scharf, The photochemical synthesis of fine chemicals with sunlight, Angew. Chem. Int. Ed. Engl. 33 (1994) 2009–2023. [3] M.C. DeRosa, R.J. Crutchley, Photosensitized singlet oxygen and its applications, Coord. Chem. Rev. 233–234 (2002) 351–371. [4] C. Schweitzer, R. Schmidt, Physical mechanisms of generation and deactivation of singlet oxygen, Chem. Rev. 103 (2003) 1685–1758. [5] a) P.R. Ogilby, Singlet oxygen: there is indeed something new under the sun, Chem. Soc. Rev. 39 (2010) 3181–3209. b) P.R. Ogilby, Singlet oxygen: there is still something new under the sun, and it is better than ever, Photochem. Photobiol. Sci. 9 (2010) 1543–1560. [6] W. Fudickar, A. Fery, T. Linker, Reversible light and air-driven lithography by singlet oxygen, J. Am. Chem. Soc. 127 (2005) 9386–9387.
[7] W. Fudickar, T. Linker, Imaging by sensitized oxygenations of photochromic anthracene films: Examination of effects that improve performance and reversibility, Chem. Eur. J. 12 (2006) 9276–9283.
[8] D. Zehm, W. Fudickar, T. Linker, Molecular switches flipped by oxygen, Angew. Chem. Int. Ed. 46 (2007) 7689–7692.
[ 9 ] D. Zehm, W. Fudickar, T. Linker, 9,10-Diarylanthracenes as molecular switches. Syntheses, properties, isomerisations and their reactions with singlet oxygen, Chem. Eur. J. 14 (2008) 11429–11441.
[10] W. Fudickar, T. Linker, Photoimaging with singlet oxygen at the solid-air interface, Langmuir 25 (2009) 9797–9803.
[11] J. Park, D. Feng, S. Yuan, H.-C. Zhou, Photochromic metal-organic frameworks: Reversible control of singlet oxygen generation, Angew. Chem. Int. Ed. 54 (2015) 430–435. [12] A. Dewilde, C. Pellieux, S. Hajjam, P. Wattré, C. Pierlot, D. Hober, J.-M. Aubry, Virucidal activity of pure singlet oxygen generated by thermolysis of a water-soluble naphthalene endoperoxide, J. Photochem. Photobiol. B 36 (1996) 23–29. [13] A.P. Castano, P. Mroz, M.R. Hamblin, Photodynamic therapy and anti-tumour immunity, Nat. Rev. Cancer 6 (2006) 535–545. [14] L.F. Agnez-Lima, J.T.A. Melo, A.E. Silva, A.H.S. Oliveira, A.R.S. Timoteo, K.M. Lima-Bessa, G.R. Martinez, M.H.G. Medeiros, P. Di Mascio, R.S. Galhardo, C.F.M. Menck, DNA damage by singlet oxygen and cellular protective mechanisms, Mutat. Res.–Rev. Mutat. Res. 751 (2012) 15–28.
[15] J.-M. Aubry, B. Mandard-Cazin, M. Rougee, R.V. Bensasson, Kinetic studies of singlet oxygen [4+2]-cycloadditions with cyclic 1,3-dienes in 28 solvents, J. Am. Chem. Soc. 117 (1995) 9159–9164.
[16] V. Nardello, J.-M. Aubry, Synthesis and properties of a new cationic water-soluble trap of singlet molecular oxygen, Tetrahedron Lett. 38 (1997) 7361–7364.
[17] C. Pierlot, J.-M. Aubry, First evidence of the formation of 5,8-endoperoxide from the oxidation of 1,4-disubstituted naphthalene by singlet oxygen, Chem. Commun. (1997) 2289–2290.
[18] J.-M. Aubry, C. Pierlot, J. Rigaudy, R. Schmidt, Reversible binding of oxygen to aromatic compounds, Acc. Chem. Res. 36 (2003) 668–675.
[19] W. Fudickar, T. Linker, Remote substituent effects on the photooxygenation of
9,10-diarylanthracenes: Strong evidence for polar intermediates, Chem. Commun. (2008) 1771–1773.
[20] W. Fudickar, T. Linker, Novel anthracene materials for applications in lithography and reversible photoswitching by light and air, Langmuir 26 (2010) 4421–4428.
[21] W. Fudickar, T. Linker, Reversible photooxygenation of alkynylanthracenes: Chemical generation of singlet oxygen under very mild conditions, Chem. Eur. J. 17 (2011) 13661– 13664.
[ 22 ] W. Fudickar, T. Linker, Why triple bonds protect acenes from oxidation and decomposition, J. Am. Chem. Soc. 134 (2012) 15071–15082.
[23] M. Klaper, T. Linker, Evidence for an oxygen anthracene sandwich complex, Angew. Chem. Int. Ed. 52 (2013) 11896–11899.
[24] J. Zhang, Z.C. Smith, S.W. Thomas, Electronic effects of ring fusion and alkyne substitution on acene properties and reactivity, J. Org. Chem. 79 (2014) 10081–10093. [25] V. Balzani, M. Venturi, A. Credi, Molecular devices and machines, Wiley-VCH, Weinheim, 2008.
[26] B.L. Feringa, W.R. Browne, Molecular switches: Second, completely revised and enlarged edition, Wiley-VCH Verlag GmbH & Co. KGaA, 2011.
[27] S. Silvi, E.C. Constable, C.E. Housecroft, J.E. Beves, E.L. Dunphy, M. Tomasulo, F.M. Raymo, A. Credi, Photochemical switching of luminescence and singlet oxygen generation by chemical signal communication, Chem. Commun. (2009) 1484–1486.
[28] L. Hou, X. Zhang, T.C. Pijper, W.R. Browne, B.L. Feringa, Reversible photochemical control of singlet oxygen generation using diarylethene photochromic switches, J. Am. Chem. Soc. 136 (2014) 910–913.
[29] G. Liu, X. Xu, Y. Chen, X. Wu, H. Wu, Y. Liu, A highly efficient supramolecular photoswitch for singlet oxygen generation in water, Chem. Commun. 52 (2016) 7966–7969.
[ 30 ] H.-R. Blattmann, D. Meuche, E. Heilbronner, R.J. Molyneux, V. Boekelheide, Photoisomerization of trans-15,16-dimethyldihydropyrene, J. Am. Chem. Soc. 87 (1965) 130–131.
[31] R.H. Mitchell, The metacyclophanediene-dihydropyrene photochromic π switch, Eur. J. Org. Chem. (1999) 2695–2703.
[32] R.S. Murphy, Y. Chen, T.R. Ward, R.H. Mitchell, C. Bohne, Photophysical studies on the photochromism of trans-10b,10c-dimethyldihydropyrene, Chem. Commun. (1999) 2097– 2098.
[ 33 ] M.A.L. Sheepwash, R.H. Mitchell, C. Bohne, Mechanistic insights into the photochromism of trans-10b,10c-dimethyl-10b,10c-dihydropyrene derivatives, J. Am. Chem. Soc. 124 (2002) 4693–4700.
[34] M.A.L. Sheepwash, T.R. Ward, Y. Wang, S. Bandyopadhyay, R.H. Mitchell, C. Bohne, Mechanistic studies on the photochromism of [e]-annelated dimethyldihydropyrenes, Photochem. Photobiol. Sci. 2 (2003) 104–112.
[35] C. Bohne, R.H. Mitchell, Characterization of the photochromism of dihydropyrenes with photophysical techniques, J. Photochem. Photobiol. C 12 (2011) 126–137.
[36] S. Cobo, F. Lafolet, E. Saint-Aman, C. Philouze, C. Bucher, S. Silvi, A. Credi, G. Royal, Reactivity of a pyridinium-substituted dimethyldihydropyrene switch under aerobic conditions: self-sensitized photo-oxygenation and thermal release of singlet oxygen, Chem. Commun. 51 (2015) 13886–13889.
[37] A. Bakkar, S. Cobo, F. Lafolet, E. Saint-Aman, G. Royal, A new surface-bound molecular switch based on the photochromic dimethyldihydropyrene with light-driven release of singlet oxygen properties, J. Mater. Chem. C 3 (2015) 12014–12017.
[38] D. Roldan, S. Cobo, F. Lafolet, N. Vilà, C. Bochot, C. Bucher, E. Saint-Aman, M. Boggio-Pasqua, M. Garavelli, G. Royal, A multi-addressable switch based on the dimethyldihydropyrene photochrome with remarkable proton-triggered photo-opening efficiency, Chem. Eur. J. 21 (2015) 455–467.
[39] R.V. Williams, W.D. Edwards, R.H. Mitchell, S.G. Robinson, A DFT study of the thermal, orbital symmetry forbidden, cyclophanediene to dihydropyrene electrocyclic reaction. Predictions to improve the dimethyldihydropyrene photoswitches, J. Am. Chem. Soc. 127 (2005) 16207–16214.
[40] M. Boggio-Pasqua, M.J. Bearpark, M.A. Robb, Toward a mechanistic understanding of the photochromism of dimethyldihydropyrenes, J. Org. Chem. 72 (2007) 4497–4503.
[41] M. Boggio-Pasqua, M. Garavelli, Rationalization and design of enhanced photoinduced cycloreversion in photochromic dimethyldihydropyrenes by theoretical calculations, J. Phys. Chem. A 119 (2015) 6024–6032.
[42] A.D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys. 98 (1993) 5648–5652.
[43] R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys. 72 (1980) 650–654. [44] K. Yamaguchi, F. Jensen, A. Dorigo, K.N. Houk, A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals, Chem. Phys. Lett. 149 (1988) 537–542.
[45] S. Yamanaka, T. Kawakami, H. Nagao, K. Yamaguchi, Effective exchange integrals for open-shell species by density functional methods, Chem. Phys. Lett. 231 (1994) 25–33. [46] M. Boggio-Pasqua, J.-L. Heully, Thermolysis biradical mechanisms in endoperoxides: A challenge for density functional theory?, Theor. Chem. Acc. 135 (2016) 9.
[47] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P.
Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009.
[48] T. Yanai, D.P. Tew, N. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett. 393 (2004) 51–57.
[49] The TD-DFT calculation of the lowest π→π* electronic transition in 2 at the B3LYP level is inaccurate with a corresponding absorption wavelength computed at 576 nm. This can be explained by the strong charge transfer character of the HOMO→LUMO transition mainly describing the S1 state of 2.

![Fig. 4. DFT optimized structures for (a) the triplet–triplet [ 3 O 2••• 1(T 1 )] complex and (b) the singlet–singlet [ 1 O 2••• 1(S 0 )] complex](https://thumb-eu.123doks.com/thumbv2/123doknet/14318122.496530/9.892.206.690.90.410/optimized-structures-triplet-triplet-complex-singlet-singlet-complex.webp)
![Fig. 5. Potential energy profiles along the energy transfer process. The energy transfer coordinate has been approximated by linear interpolation between the optimized structures of the triplet–triplet [ 3 O 2••• 1(T 1 )] reactant complex and the singlet](https://thumb-eu.123doks.com/thumbv2/123doknet/14318122.496530/10.892.191.701.96.393/potential-profiles-transfer-coordinate-approximated-interpolation-optimized-structures.webp)