HAL Id: hal-00407786
https://hal.archives-ouvertes.fr/hal-00407786
Submitted on 1 Oct 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Distributed under a Creative Commons Attribution| 4.0 International License
recombination signal sequences: a critical control point
in TCRbeta gene assembly
Don-Marc Franchini, Touati Benoukraf, Sébastien Jaeger, Pierre Ferrier,
Dominique Payet-Bornet
To cite this version:
Don-Marc Franchini, Touati Benoukraf, Sébastien Jaeger, Pierre Ferrier, Dominique Payet-Bornet.
Initiation of V(D)J recombination by Dbeta-associated recombination signal sequences: a critical
control point in TCRbeta gene assembly. PLoS ONE, Public Library of Science, 2009, 4 (2), pp.e4575.
�10.1371/journal.pone.0004575�. �hal-00407786�
Recombination Signal Sequences: A Critical Control Point
in TCRb Gene Assembly
Don-Marc Franchini1,2,3, Touati Benoukraf1,2,3, Se´bastien Jaeger1,2,3, Pierre Ferrier1,2,3, Dominique Payet-Bornet1,2,3*
1 Centre d’Immunologie de Marseille-Luminy, Universite´ Aix Marseille, Marseille, France, 2 CNRS, UMR6102, Marseille, France, 3 Inserm, U631, Marseille, France
Abstract
T cell receptor (TCR) b gene assembly by V(D)J recombination proceeds via successive Db-to-Jb and Vb-to-DJb rearrangements. This two-step process is enforced by a constraint, termed beyond (B)12/23, which prohibits direct Vb-to-Jb rearrangements. However the B12/23 restriction does not explain the order of TCRb assembly for which the regulation remains an unresolved issue. The initiation of V(D)J recombination consists of the introduction of single-strand DNA nicks at recombination signal sequences (RSSs) containing a 12 base-pairs spacer. An RSS containing a 23 base-pairs spacer is then captured to form a 12/23 RSSs synapse leading to coupled DNA cleavage. Herein, we probed RSS nicks at the TCRb locus and found that nicks were only detectable at Db-associated RSSs. This pattern implies that Db 12RSS and, unexpectedly, Db 23RSS initiate V(D)J recombination and capture their respective Vb or Jb RSS partner. Using both in vitro and in vivo assays, we further demonstrate that the Db1 23RSS impedes cleavage at the adjacent Db1 12RSS and consequently Vb-to-Db1 rearrangement first requires the Db1 23RSS excision. Altogether, our results provide the molecular explanation to the B12/ 23 constraint and also uncover a ‘Db1 23RSS-mediated’ restriction operating beyond chromatin accessibility, which directs Db1 ordered rearrangements.
Citation: Franchini D-M, Benoukraf T, Jaeger S, Ferrier P, Payet-Bornet D (2009) Initiation of V(D)J Recombination by Db-Associated Recombination Signal Sequences: A Critical Control Point in TCRb Gene Assembly. PLoS ONE 4(2): e4575. doi:10.1371/journal.pone.0004575
Editor: Wasif N. Khan, University of Miami, United States of America
Received October 3, 2008; Accepted January 15, 2009; Published February 24, 2009
Copyright: ß 2009 Franchini et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was supported by institutional grants from Inserm and the CNRS, and by specific grants from the ‘Fondation Princesse Grace de Monaco’, the ‘Association pour la Recherche sur le Cancer’ (ARC 3275XA0331F), the ‘Agence Nationale de la Recherche’ (ANR-06-BYOS-0006) and the Commission of the European Communities (MRTN-CT-2006-035733). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist. * E-mail: payet@ciml.univ-mrs.fr
Introduction
Immunoglobulin (Ig) and T-cell receptor (TCR) genes are assembled from separate variable (V), diversity (D) and joining (J) gene segments via a series of site-specific events of DNA rearrangement, termed V(D)J recombination. This process requires the binding of the lymphocyte-specific recombination activating gene 1 and 2 (RAG1/2) protein complex to recombi-nation signal sequences (RSSs) flanking the rearranging sides of individual V, D and J gene segments [1]. These RSSs consist of conserved heptamer and nonamer sequences, separated by a spacer of 12 or 23 base pairs (bp) of relatively non-conserved DNA. Efficient recombination involves pairs of gene segments flanked by dissimilar 12- and 23RSSs (the 12/23 rule) [2].
The molecular mechanism of V(D)J recombination has been described in great detail [3–5]. Upon binding, the RAG1/2 recombinase introduces a single-strand nick at the border between the RSS heptamers and adjacent coding sequences, thus exposing a 39-hydroxyl (OH) group on each coding flank. The 39-OH then attacks the opposite DNA strand in a direct transesterification reaction producing a hairpin-sealed coding end (CE) and blunt phosphorylated signal end (SE). Transesterifications occur simul-taneously at complementary RSSs paired within a synaptic or paired complex (PC), a property referred to as coupled cleavage.
The processing and joining of CEs and SEs, mediated by DNA repair factors of the nonhomologous end-joining (NHEJ) machin-ery [6], yield one signal joint and one coding joint as the final products of recombination. The critical event of PC formation likely proceeds via a capture mode in which RAG1/2 complex assembles on one RSS and then captures the second RSS as recombinase-free DNA (Figure 1A) [7–9].
A tight regulation of V(D)J recombination ensures proper lymphocyte development and eludes lymphoid malignancy-causing chromosomal translocations [3,5,10,11]. Regulated control of V(D)J rearrangement during lymphoid cell ontogeny includes, (i) cell lineage specificity (with for example TCR gene rearrangement occurring in T lymphocytes only); (ii) developmental specificity (with for example TCRb gene rearrangement occurring prior to that of TCRa); and, at some loci, (iii) allele specificity (to mediate allelic exclusion). By and large, these controls are thought to involve lineage- and developmentally-regulated changes in chromatin structure that precisely modulate the accessibility of individual Ig/ TCR gene loci and/or segments, with their associated RSSs, to the unique RAG1/2 recombinase [3,5,10].
Beyond the chromatin barrier, individual 12- and 23RSS-flanked gene segments can still display high disparity in recombination frequency, mainly due to nucleotide variations in their RSSs and/or adjacent coding flanks [12–14]. In fact, RSS
heterogeneity is a major reason for non-random usage in V(D)J recombination. Moreover RSSs can impose significant constraints on antigen receptor gene assembly beyond enforcing the 12/23 rule [15]. Revealed at the TCRb locus, this B12/23 restriction allows Db 12RSSs but not Jb 12RSSs, to efficiently target Vb 23RSSs for rearrangement. With unique dependence on the RAG1/2 apparatus and no other lymphoid-specific factors, B12/ 23 relies on the RSS nucleotides structure and occurs at or prior to coupled cleavage [15–24]. However, this phenomenon, which in preserving Db gene segment utilization contributes to the optimal generation of a functionally diverse repertoire, remains incom-pletely understood at the molecular level [22]. Furthermore, while both Vb-to-Db and Db-to-Jb are allowed by B12/23 restriction, an additional level of regulation ensures an ordered V(D)J recombination at the TCRb locus, with Db-to-Jb joining occurring before Vb-to-DJb gene assembly [25] (Figure 1B). Although differential chromatin accessibility of TCRb gene segments may control the rearrangement order, the molecular basis of this process remains however unclear (reviewed in [26]). In this regard, we wondered whether TCRb RSSs could also organize ordered recombination by orchestrating synaptic com-plex nucleation in a sequential manner. By investigating RAG1/2-dependent DNA cleavages in vivo and in vitro, we provide evidence that, at the TCRb locus, Db-flanking 23- and 12RSSs constitute primary anchoring sites for PC formation for D-to-J and V-to-DJ rearrangements respectively. Most importantly, we found that the Db1 23RSS also prohibits RAG1/2-mediated nicking at the
adjoining 59Db1 12RSS. These data elucidate the mechanism of B12/23 and reveal a role for the Db1 23RSS in imposing ordered (‘D-J prior to V-DJ’) rearrangement at the Db1 locus.
Results
Nicking products preferentially accumulate at Db-associated RSSs in vivo
The oligo-capture assay, initially described by Curry et al. [9] (Figure 2A), uncovers RAG1/2-mediated nicks generated at a given RSS site(s) in the genome. When applied to the analysis of nicking profiles within the Igk, IgH and TCRa loci from RAG1/ 2-expressing cells, this methodology provided evidence that 12RSSs represent initial nicking targets, nucleating synaptic complex formation and the capture of a 23RSS partner [9].
We used the oligo-capture approach to probe RSS nicks associated with rearranging TCRb gene segments in early developing T lymphocytes. Briefly, genomic DNA from cell-sorted CD42CD82 double-negative (DN) thymocytes of a WT mouse was oligo-captured using heptamer-specific oligonucleotides, T4 DNA ligase and proper restriction enzymes. Next, the digested DNAs were fractionated using streptavidin-conjugated magnetic beads and the captured DNAs tested for the presence of TCRb sequences of interest using PCR and Southern blotting (Figure 2 A–B and see materiel and methods for details). Among all Vb, Db and Jb RSSs tested, we only detected signal for 59Db1, 39Db1 and Db2 captures (Figure 2C; nicking at the two neighboring Db2
Figure 1. Initial steps of V(D)J recombination and structure of mouse TCRb locus. (A) According to the capture model initially proposed by Jones and Gellert [8], RAG1/2 complex binds to one RSS (step 1) and then captures the second RSS to form the PC (step 3). Within the PC, pairwise double-strand breakages occur via coupled transesterification reactions, thus leading to the production of SE and CE (step 5). Within this reactions pathway Curry et al. [9] proposed the order of the two nicking reactions; the first one occurs at the initiating RSS (black triangle) (step 2), the second one occurs at the captured RSS (white triangle) (step 4). An alternative model in which the first nick would occur at the captured RSS was considered in theSupplementary Text S1. (B) Schematic depiction of the TCRb locus. 12- and 23RSSs are represented by black and white triangles, respectively. Gene segments are figured by grey rectangles. TCRb locus rearrangements are ordered (Db-to-Jb occur before Vb-to-DJb). The B12/23 constraint prohibits direct Vb-to-Jb rearrangements.
12- and 23 RSSs cannot be distinguished due to the presence of identical heptamers). These signals were above the background level and were specific from WT DN cells. As a negative control, we used genomic DNA from RAG1-deficient (RAG12/2) thymocytes. We also assessed background level from DNA samples treated in parallel but omitting the heptamer oligonucleotide. Finally, each captured DNA at Jb, Db or Vb gene segments were compared with that at a Cb2 gene fragment lacking RSS sequences.
According to the previous study suggesting that the 12RSS initiates V(D)J recombination and captures the 23RSS, we expected to observe some nicks at Jb 12RSS. However, we didn’t detect any oligo-captured Jb1 or Jb2 DNA. Of note, a greater number of Jb gene segments cannot explain the difference between the amounts of Db versus Jb capture since we investigated all segments together within each Jb1 or Jb2 genomic cluster (see legend toFigure 2 and Table S1). As expected, nicks at Vb 23RSSs were not detected (Figure 2C). Outnumbered targets is also unlikely to account for Vb vs. Db differential recovery since when focusing on the Vb8.1/8.2/8.3 segments (also analyzed together) representing ,20% of total Vb rearrangements [27], we still could not detect amplification signals upon using 5 fold more captured DNA (data not shown). We tested the ability of the consensus heptamer CACAGTG (used for the capture of endogenous Vb2, Vb6, Vb8, Vb15 and Jb1.1 gene segments) to capture DNA which was previously nicked in vitro. The results indicate that the pCACAGTG-biotin heptamer can capture an RSS carrying a RAG1/2-mediated nick (Figure S1) and thus does not present any inherent problem. The oligocapture assay appears to be not sensitive enough to detect Vb or Jb nicks, mainly two explanations can be considered, either the amount of Vb or Jb RSS nicks is underneath the detection threshold or, as discussed below, these nicks exist only transiently.
To verify that RAG1/2 cleavage activity is primarily dependant on RSS accessibility, we used DNA from TCRb enhancer-deleted (Eb2/2) thymocytes in which Db-Jb clusters display a hetero-chromatin structure [10,28]. In contrast to the WT situation, we could not detected any 39Db1 capture (Figure 2D), confirming that nicking at the Db1 23RSS depends on Eb-mediated modulation of chromosomal accessibility at this site.
Altogether, our data clearly indicate that rearranging Db gene segments in vivo contain precisely positioned nicks at their 12- and/or 23RSSs, whereas their potential Vb and Jb partners still carry intact
complementary RSSs. These profiles argue for a capture mode of PC formation in vivo in which Db 12- and 23RSSs capture Vb 23RSS and Jb 12RSS respectively. The in vivo assay failed to detect Vb or Jb RSS nicks which likely occur upon formation of the PC (Figure 1A, step 4). As discussed by Curry et al., this may signify that nicking at the paired RSS exists only transiently in PCs in vivo due to the quasi-instant nucleophilic attack in direct transesterification [9]. The oligo-capture assay uncovers RAG1/2-mediated nicks and is not a direct measure of RAG1/2 binding to DNA. Therefore we cannot state about the RAG1/2 binding pattern. Hence we cannot exclude that RAG1/2 initially binds to Jb or Vb RSS and that the resulting complex synapses with a Db RSS which is next nicked. This alternative scenario is considered in the supplementary textS1.
In conclusion, the in vivo nicking pattern of the TCRb locus strengthens the capture model for synapsis. However, our data suggest that the 12RSS nick leading to the 23RSS capture is not the only order of event; alternatively the initial RAG1/2–mediated cleavage can occur onto a 23RSS such as the Db 23RSSs during Db-to-Jb rearrangements. Furthermore, since neither the Vb 23RSSs nor the Jb 12RSSs efficiently anchor RAG1/2 cleavage activity, direct Vb-to-Jb recombination is prohibited. This anchoring hierarchy represents very likely the molecular basis of the B12/23 restriction at the TCRb locus.
B12/23 restriction results from the inefficiency of Vb 23RSS and Jb 12RSS to form functional single complex
Previous studies have demonstrated that the B12/23 restriction can be recapitulated in vitro with chromatin-free substrates [17– 19,24]. Thus, we undertook to use an in vitro RAG1/2-mediated DNA cleavage system to validate our proposition that Vb and Jb RSSs are captured by Db RSSs and therefore cannot recombine together.
As a source of recombinase activity, we used a cellular extract prepared from the D10 cell line [29] after heat-shock induced expression of core RAG1/2 proteins. This extract (hereafter RAG1/ 2 extract) has been shown to enforce the 12/23 rule in vitro [30]. Our various attempts to perform cleavage assays with an in vitro system using purified core RAG1/2 and HMGB1 proteins were unsuccess-ful. This observation is consistent with a previous study in which the Db2 23RSS was replaced by the Jk1 23RSS because the level of recombination of the natural Db 23RSS-Jb 12RSS pair was too low to be properly investigated [14]. The necessity to use crude extracts may suggest that RAG-mediated cleavages on TCRb RSS-based
Figure 2. RSS nicks at the TCRb locus in mouse developing T cells. (A) Strategy to detect RSS nicks in vivo using oligo-capture as described by [9]. Vertical and horizontal arrows schematize, respectively, sites for restriction enzyme digestion and primers for PCR amplification. The bar schematizes the hybridization probe used for Southern blot analysis. (B) Schematic view of the TCRb regions analyzed in this study (not drawn to scale); 12- and 23RSSs are figured by black and white triangles, respectively; the single strand nick positions are indicated by vertical arrows. The locations of the PCR primers and hybridization probes are shown; H (HindIII); G (BglII); Ss (SstI); S (SphI); RV (EcoRV); RI (EcoRI). (C, D) Autoradiographs of Southern blots of oligo-captured DNAs. Total genomic DNA from WT, RAG12/2or Eb2/2DN thymocytes was investigated for single strand nicks at
TCRb RSSs or Cb2 sequences (used as a negative control). Nicks at Vb4 and Vb16 genes were analyzed together; Vb8 and Vb5 corresponded to RSSs from three (Vb8.1, Vb8.2 and Vb8.3) and two (Vb5.1 and Vb5.2) genes, respectively; single-strand nicks at Db2 12- and 23RSSs were analyzed conjointly as these two RSSs possess identical heptamers. Jb1 and Jb2 corresponded to all functional Jb1 and Jb2 12RSSs, respectively. They were analyzed in one single round using a mixture of specific heptamers followed by PCR amplification of a genomic fragment located at the 39 end of the Jb1 (or Jb2) cluster. PCR reactions were carried out using increasing amounts of template DNA from the bead release (0.5, 1 and 2% of captured DNA) or the flow through (10, 25 and 50 ng of non-captured DNA). Additional controls used 2% of captured (C) and 50 ng of non captured (NC) fractions from genomic DNA treated in parallel except that the biotinylated oligonucleotide was omitted (No 7mer). Estimation of the amount of captured DNA. Assuming that the amount of genomic DNA is 6 pg per cell, the cellular equivalent of 10 ng of genomic DNA is 1650 cells or 3300 alleles. For the less efficient Db RSS (59Db1 12RSS) the intensity of the band (when 2% of captured DNA is analyzed) is tenfold lower than the band of non-captured DNA (10 ng of DNA analyzed). Hence, for 2% of non-captured 59Db1, we estimated that 330 Db1 alleles were amplified and that the total amount of captured 59Db1 DNA is around 16500 Db1 alleles. We observed that the signal is well detected when approximately 80 copies were analyzed (0.5% of 59Db1 captured DNA). Conversely, nicked Vb and Jb RSSs were not detected (even with an input of 10% of captured DNA, not shown), suggesting that there is less than 80 copies of Vb or Jb DNA in the PCR tube. If we considered that the efficiency of the oligocapture assay is similar for all DNA targets and that only the amount of nicked DNA varies, we estimated that the amount of nicked Vb or Jb RSSs is at least twentyfold lower than the amount of nicked Db.
substrates require, besides RAG1/2 and HMGB1, additional factors. This suggestion is consistent with a recent study indicating that c-Fos would be involved in RAG deposition on Db 23RSS [31]. Western blot analysis revealed that our cell-free system supplies the c-Fos protein (not shown).
In addition to the 12- and 23RSSs flanking each Db1 and Db2 gene segments, we tested the frequently used Jb1.1- and Jb2.5 12RSSs [32]; the Vb2 23RSS, comprised of genuine heptamer and nonamer consensus motifs; and the Vb14 23RSS, used previously to define and analyze the B12/23 constraint [16,20]. Sequences of the RSSs analyzed in this study are shown inTable S2.
To test our in vitro system, we investigated the RAG1/2-mediated DNA coupled cleavage using various pair-wise RSS combinations. As shown in Figure S2, this system faithfully reproduced B12/23 restriction and our results are consistent with published data (reviewed in [22]).
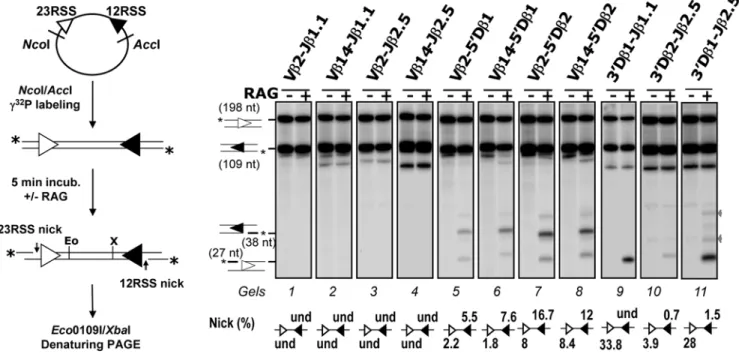
Next, we adapted this in vitro system to investigate the earliest catalytic phase (RAG1/2-mediated nicking) of V(D)J recombina-tion and especially the aptitude of RSSs to form a funcrecombina-tional RAG:RSS single complex, visualized by the production of single-strand nicks. To do so, the incubation with the RAG1/2 extract was limited to 5 min, and the two 38 and 27 nucleotides (nt) fragments corresponding to respectively RAG1/2-mediated 12-and 23RSS nicks, were monitored (Figure 3). When testing Vb-Jb substrates, nicking products were not detected (Figure 3, gels 1–4). By contrast, we found nicking products from the rearranging Vb-Db substrates, with nicks at the Db 12RSSs (38 nt) prevailing over nicks at the Vb 23RSSs (27 nt) (Figure 3, gels 5–8). The detection of higher amounts of the 38 nt fragment complies with
our suggestion that Db 12RSSs are targeted first for RAG1/2 nicking and PC nucleation. Moreover, we observed that the amount of nicked Vb 23RSS rose from undetected, for Vb-Jb substrates, to ,2–8% for Vb-Db substrates. The capture model implies that synapsis precedes nicking at the captured RSS (Figure 1A, step 3). Therefore, we reasoned that if Vb 23RSS has to be captured to form the synapse, such capture is dependent on the 12RSS partner. If the 12RSS partner (for instance Jb 12RSS) cannot initiate the formation of the synapse, Vb 23RSS would not be nicked, while a 12RSS competent for synapse nucleation would induce Vb 23RSS nicking. Our observation that nicks are increased at Vb 23RSSs when associated with Db (in comparison with Jb) 12RSSs thus supports the capture model of Figure 1A and confirms that Db 12RSSs represent the platforms of choice for PC nucleation in Vb/Db partnership.
Strikingly, when testing Db1-Jb substrates, we detected large amounts of nicked products at the 23RSS while nicks at the 12RSS were either not detected (39Db1-Jb1.1 substrate) or quite low (,1.5% for 39Db1-Jb2.5 substrates). This outstanding asymmetry is consistent with a model of PC nucleation whereby the RAG1/2 proteins first react with the Db1 23RSS before the capture of a free Jb 12RSS. Nicking profiles of Db2-Jb2 and Db1-Jb substrates are qualitatively similar. However, Db1 and Db2 23RSS yielded different amount of single strand nicks, respectively ,30% and 4% of input material (Figure 3, gels 9 to 11), implying that the Db1 23RSS surpasses the Db2 ortholog as a nicking target (hence PC nucleating site) in vitro.
Altogether, our in vitro data using non chromatinized templates shows that RAG1/2 catalysis preferentially targets the Db 12- and
Figure 3.In vitroRAG1/2-mediated nicking assays. As described on the left panel, the recombination substrate was first digested with NcoI and AccI restriction enzymes and the resulting 12/23 RSS-containing fragment was radio-labeled at the 59ends (indicated by a star), then incubated for 5 min without (2) or with (+) the RAG1/2 extract. DNA samples were further digested with Eco0109I (Eo) and XbaI (X) enzymes and separated by denaturing PAGE. (Right) Autoradiographs of nicking assay analysis of the indicated recombination substrates. 12/23 RSS substrates were named according to the gene segments flanking the 23- and 12RSS (seeFigure S6 for the construction of recombination substrates). The sizes of the intact 23- and 12RSSs (198 and 109 nt) and of the corresponding nicking products (27 and 38 nt) are indicated. Percentages of 12- and 23RSS single strand nicks (i.e., scanning intensity of individual nicked products vs. that of the corresponding original fragments) are shown below the gel image (und: undetected). For some substrates, two additional products of ,35 and ,45 nt in length (indicated by grey arrows) were detected in the RAG positive lane, they may correspond to non-hairpin CE DNA breaks (i.e., processed products of RAG1/2-generated hairpins) [46]. All results shown are representative of at least three separate experiments.
23RSSs, likely nucleating the formation of Db/Vb and Db/Jb PCs, respectively. This conclusion is consistent with the in vivo nicking pattern of the TCRb locus and confirms our proposition that B12/23 restriction results from the inability of Vb 23RSSs and Jb 12RSSs to focus the initial RAG1/2 cleavage activity (nicking), leading to a defect of Vb/Jb PC formation.
Db1 12RSS nicks are not detected at germline Db1 alleles
Throughout, our data suggest that Db-flanking 23- and 12RSSs represent initial RAG1/2-entry sites in Db/Jb and Vb/Db PC formation, respectively. This prompted us to investigate whether the two Db-flanking RSSs could be differentially nicked. For this purpose, we used in vivo oligo-capture assay at the Db1 locus, since (conversely to Db2 locus) nicks at Db1 12- and 23RSSs can be distinguished due to their divergent heptamers. In our previous oligo-capture assay (Figure 2) the 59Db1 and 39Db1 captured DNA were PCR-amplified using primers localized upstream Db1 gene segment. For 59Db1 capture, this PCR approach does not differentiate 12RSS nicks at germline Db1 and Db1Jb rearranged alleles. Conversely, in the context of 39Db1 capture, this approach detects only 23RSS nicks at germline Db1 allele. To detect specifically 12RSS nicks at non rearranged Db1 locus we carried out further PCR amplifications from the 59Db1 captured DNAs using primers hybridizing to Db1-Jb1.1 intervening sequences. In this condition, no signal was detected using the 59Db1-captured DNA. When applied to the 39Db1-captured DNA, as expected, this PCR approach (supposed to detect all Db1 23RSS nicks, independently to Db1 allele configuration) led to the clear detection of the downstream Db1 sequence (Figure 4). These results clearly show that Db1 12RSS nicks are not formed at non-rearranged Db1 alleles in vivo, while Db1 23RSS nicks are produced (Figure 2). We deduced that nicking at 59Db1 12RSS occurs after removal of the downstream 23RSS via Db-Jb recombination, only when the allele is in Db1Jb configuration. Here again, the preferential RAG1/2-targeting of 39Db1 23RSS over 59Db1 12RSS would provide an explanation to the ordered rearrangement at the TCRb locus.
The Db1 23RSS blocks RAG1/2-mediated cleavage at the adjacent Db1 12RSS
In vivo, in the context of an intact chromatin structure, we showed that nicking of the Db1 12RSS (and thus initiation of
Vb-to-Db1 rearrangement) requires the previous elimination of the Db1 23RSS. To test if the inhibition of RAG1/2 cleavage activity on the Db1 12RSS is mediated by the neighboring Db1 23RSS and not by the chromatin structure, we performed in vitro cleavage assays. We first carried out in vitro nicking assays using Db-based substrates. As previously shown, nicking at a single RSS can occur in presence of Mg2+ions in the buffer [33]. Substrates containing Db1 coding sequence flanked by either the Db1 12- or 23RSS (59Db1 and 39Db1, respectively) were cleaved in the presence of the RAG1/2 extract to produce the corresponding nicking product (gels 1 and 2, Figure 5A). However, a substrate containing the Db1 coding sequence flanked by both RSS mostly produced the 23RSS-derived fragment (gel 3) indicating prefer-ential nicking at the Db1 23RSS. We observed no such bias towards the 23RSS when using a modified substrate (D1V14), in
which the Db1 23RSS is replaced by the Vb14 23RSS (gel 4). On the contrary, preferential cleavage fell on the 12RSS. These data therefore suggest a regulatory function unique to the Db1 23RSS which, in the germline situation, might anchor RAG1/2 catalytic activity at the expense of the neighboring Db1 12RSS.
To further assess the possibility that the Db1 23RSS impairs Vb-to-59Db1 cis-rearrangement, we next performed in vitro RSS coupled-cleavage assays using various forms of recombination substrates (Figure 5B). As demonstrated by the formation of significant amounts of Vb-59Db1 SE products, coupled cleavage readily occurred using a Vb14/Db1-containing substrate lacking the Db1 23RSS (pV59D1) and a related substrate carrying the Vb14
23RSS at the 39side of Db1 gene segment (pVDv). Conversely, cleavage was severely reduced when using a substrate carrying the Db1 23RSS (pVD1). Additional experiments demonstrated coupled
cleavage within Db1/Jb1-containing substrates whether the Db1 12RSS was present or not (pD1J1 and p39D1J1, respectively),
arguing that this site has no detrimental effect on PC nucleation involving the downstream 39Db1-/Jb1-associated RSSs.
Overall, our in vitro data using non chromatinized templates demonstrate that RAG1/2 catalysis preferentially targets the Db1 23RSS instead of Db1 12RSS. Since Db1 23RSS mediates the inhibition of the adjacent Db1 12RSS nicking, the nucleation of Db/Vb synaptic complex formation is impeded. Thereby, this ‘Db1 23RSS-mediated restriction’ provides a potential mechanism to direct ordered rearrangements (‘D-J prior V-DJ’) at the Db1 locus.
Figure 4. Db1 12RSS nicks are not detected at germline Db1 locus. Oligocapture assays were performed as described in Figure 2, except that the PCR primers and hybridization probe were specific for sequences in Db1-Jb1 intervening DNA.
Replacement of the Db1 23RSS alters the rearrangement order
In order to explore further the possibility that an RSS could orchestrate the sequence of VDJ recombination events, we used the transgenic VbDbJbECm (hereafter TCRbwt) minilocus system. This system has been shown to recapitulate the main features of endogenous TCRb gene assembly, including B12/23 restriction and ordered TCRb assembly (i.e. D-J and V-DJ detected in transgenic T cells, but not V-D) [20,34]. In vitro results, using D1V14 and pVDv substrates, have shown that Db1 12RSS
cleavage is not impaired when the Vb14 23RSS (instead of the Db1 23RSS) lies at the 39side of Db1 (Figure 5). Since we expected that our in vitro system mirrors the in vivo situation, we constructed an altered version of the TCRbwtminilocus in which we replaced the Db1 23RSS by the Vb14 23RSS and the Jb1.2 12RSS by the Db1 12RSS, this yielded the TCRbDMFminilocus (Figure 6A). Theoretically, in this configuration the various VJ1.2, DJ1.2, VD or VDJ1.2 rearrangements are possible since they all comply both with the B12/23 and ‘Db1 23RSS-mediated’ restrictions. We generated the TCRbDMF transgenic mice and
Figure 5. The Db1 23RSS impairs RAG1/2-mediated cleavages at the adjacent Db1 12RSSin vitro. (A) RAG1/2-mediated nicking assays using substrates comprised of the Db1 gene segment flanked by various combinations of 59 and/or 39 RSSs. The various Db-containing fragments were radio-labeled at the 59 ends and incubated for 5 min without (2) or with (+) the RAG1/2 extract. (B) RAG1/2-mediated coupled cleavage assays of the substrates illustrated on the left. Depending on the substrate, Southern blot analysis used probes A or B, as indicated. (A and B) 12- and 23RSSs are depicted as black and white triangles respectively. Db1 RSSs are highlighted by a dot within the triangle. All results shown are representative of at least three separate experiments.
Figu re 6. Analy sis of TCR b DMF minil ocus rearr angemen t. (A ) TCR b wt con tains germl ine V b 14, D b 1, Jb 1.1 and Jb 1. 2 gene segm ents linke d to the IgH intronic enhancer (E m ) and con stant regio n g ene (C m ). The loca tions of the V b 14 prob e (P ), Bgl II (G), Bam HI (B) and Hind III (H) sites are indi cated. 23RS Ss (whit e triangle ) and 12RS Ss (black triangle ) are shown ; dotted triangle s correspo nd to the D b 1 12-and 23RSSs. The TCR b DMF is similar to the TCR b wt excep t that the D b 1 23RSS and the Jb 1.2 12RSS were replaced wit h the V b 14 23RSS and the D b 1 12RSS ,res pectively. Minil oci were not d rawn to scal e. (B ) Sou thern blot analys is of Bgl II-di geste d genomic DNA. DNA was isola ted fro m wild-ty pe thy mocytes (wt) and TCR b wt or TCR b DMF transge nic thymo cytes. The exp ected size band s fro m the no n rea rrange d endogen ous TCR b locus (end) and the TCR b wt or TCR b DMF mini locus in the no n rearranged (GL ), DJ, V D and VDJ/VJ conf iguratio ns are indicat ed. The 2, 4, 6 and 9 k b marke rs are shown . (C ) Seque nces of some VDJ/VJ coding join ts fro m T C R b DMF rearrangemen ts. Germli ne codin g seq uences of V b 14, D b 1 and Jb 1.2 are indicated on the top . Nucl eotid e inser tions (P /N) are indicated by capital letters. Pres umptive P nucleo tides are und erline d. doi:10.137 1/journal.po ne.0004 575.g00 6
then analyzed the genomic DNA isolated from thymocytes by Southern blotting using BglII restriction enzyme and a probe that spans the Vb14 gene segment (Figure 6). As previously published [20,34], we observed that the TCRbwtminilocus undergoes DJ and VDJ rearrangements, but not VD rearrangement. By contrast and as anticipated, VDJ/VJ and VD rearrangements were readily found within the TCRbDMFminilocus, indicating that the rearrangement order of TCRbDMFis altered compared to endogenous TCRb locus or TCRbwtminilocus (Figure 6B). VDJ/VJ joints specific for the TCRbDMFtransgene were analyzed by PCR and some of them were cloned and sequenced (Figure 6C). We did not detect any VDJ1.1/VJ1.1 rearrangement in agreement with the B12/23 restriction. Only the Jb1.2 segment flanked with the Db1 12RSS was used either for direct VJ rearrangements (,40%) or, for VDJ rearrangements (,50%) (the remaining 10% could not be clearly assigned to either VDJ or VJ joints). This result reproduced previous data indicating that the substitution via knock-in of the Jb1.2 12RSS by the Db1 12RSS results in direct Vb-Jb1.2 rearrangements [15]. As DJ joints were not detected by Southern blotting, we deduced that the stepwise order of VDJ assembly for the TCRbDMFis mainly
V-to-D rearrangement, followed by VD-to-J rearrangement. As such, RSS can interfere with the sequential steps of TCRb gene assembly and the Db1 23RSS is crucial for the proper ordered ‘D-J prior V-DJ’ rearrangement. This conclusion is consistent with previous results showing that the mutation of the Db1 23RSS leads to the formation of V-D joints [20]; however in this study, the Db1 23RSS mutation prevents Db-to-Jb rearrangements consequently no VDJ joints were formed. Thus, an alternative scenario would be that V-D rearrangements occur because the D-J rearrangements are inefficient. In TCRbDMF transgenic mice the Db1 23RSS is replaced by the functional Vb14 23RSS and as expected we detect some VDJ joints. Moreover, in vitro coupled cleavage assays using transgene-based substrates showed that D-J coupled cleavage is not particularly slowed down with pTCRbDMFsubstrate compared to pTCRbwtsubstrate. On the other hand, with pTCRbDMFsubstrate, the V-D coupled cleavage is more efficient than D-J cleavage (Figure S3). Therefore this data support our initial scenario; the formation of VD joints in TCRbDMFminilocus likely results from the inability of the Vb14 23RSS to restrain the V-D cleavage but not from a defect in D-J cleavage.
Altogether our in vivo and in vitro data converge towards a model in which the Db1 23RSS not only represents a preferential target for RAG1/2 nicking at germline Db1 alleles but also prohibits nicks at the adjacent 12RSS, unless removed via 39 rearrange-ment. Consistent findings in both in vitro and in vivo assays strongly suggest that these properties do not rely on a function of the non-core domains of the RAG1/2 or on the selective tuning of chromosomal accessibility on both sides of the Db1 segment.
Discussion
This study shows that TCRb RSSs, regardless of their structural (12/23) type, display broad disparities in their overall ability to undertake the first catalytic step of V(D)J recombination, RAG1/ 2-mediated nicking. Within the limits of sensitivity of single strand nick assays, these range from a relatively high potential (Db1 23RSS) to lower aptitude (Db1 12RSS, Db2 12- and 23RSSs) to near ineffectiveness (Vb 23-, Jb 12RSSs). The proficiency of the Db1 23RSS to undergo RAG1/2-mediated nicking activity is coupled with an inhibition of that at the 59 adjoining 12RSS. These data have a number of implications for the biology of V(D)J recombination and the control of TCRb gene assembly. Notably, the emerging picture that nicks preferentially accumulate at Db segments strengthens the model that recombination synapsis
proceeds via the capture of a free RSS by a RAG1/2-loaded partner [7–9]. However, the nucleating site is not necessarily the 12RSS; at the Db-Jb clusters, the Db 23RSSs assume this function. At the TCRb locus, the pattern of nucleating and captured RSS provides an explanation for the B12/23 restriction and reveals how the capture mechanism for PC formation contributes to V(D)J recombination regulation.
We observed an ineffective RAG1/2-mediated nicking of Vb/ Jb substrates in vitro, with complete absence at the endogenous TCRb locus in WT DN cells. These data strongly argue that one aspect of the B12/23 constraint results from the inability of Vb 23-and, especially, Jb 12RSSs to initiate PC assembly, and therefore to form a synapse together. Our data do not establish where RAG1/2 proteins bind; therefore they don’t discriminate between two possible hypotheses to explain the scarce nicking at Vb and Jb RSSs: these RSSs are poor substrates for either RAG binding or for the RAG nicking reaction per se. The first hypothesis is not supported by previous EMSA studies showing that RAG binding to Db1 12RSS and to Jb 12RSSs was equivalent [18,24]. Moreover, it was proposed that the scarce nicking of Jb 12RSSs (compared to Db 12RSS) results from a slow nicking rate [24]. We note that these EMSA were performed with purified RAG1/2 in Ca2+ buffer, thus it cannot be excluded that DNA binding properties of RAG1/2 proteins in Ca2+and Mg2+buffers differ slightly. Also, if we considered that some additional proteins could be involved in RAG binding to RSS [31], the DNA binding properties of RAG1/2 may well vary depending on the system used (purified RAG or cell-free system). Certainly, at one stage of V(D)J recombination Vb and Jb are bound and nicked by RAG1/ 2. Thus, we suggest that during the PC formation the RAG1/2-loaded Db 12- or 23RSS locks the RAG1/2 multimers in a conformation [8] favoring either binding or nicking reaction at the captured (Vb or Jb) RSS (see Supplementary Text S1).
A previous study has stressed the usual proficiency of 12RSSs to capture their 23RSS partner [9]. This ‘12RSS anchoring model’ is challenged by our suggestion that Jb 12RSSs are captured by Db 23RSSs. We attempted to understand this atypical situation by analyzing DNA sequences. This analysis showed that Jb RSSs are heterogeneous within each cluster, only few nt are conserved (Figure S3A). Notably, Jb 12RSS nonamers tend to deviate strongly from the consensus hallmark (more significantly at the Jb1 cluster). As previously proposed RAG:RSS complexes may contain two types of interactions: ‘digital’ which involve critical nt residues absolutely required for RSS function and ‘analog’ (or ‘multiplicative’) which involve non critical nt residues that modulate the activity of the RAG:RSS complex [14]. Probably, Jb RSSs contain the critical nt (which are well-conserved, for instance d(TGTG) at the 39end of the heptamer) but do not possess nt residues required for optimal analog contact, thus explaining the atypical inefficiency of Jb 12RSSs to form functional single complex. It is tempting to speculate that due to such suboptimal function, Jb RSS sequences may have been selected in order to maintain the B12/23 constraint (i.e., avoid PC nucleation at Jb RSSs). In contrast heptamers and nonamers of Db RSSs are close to the consensus sequences. Additionally, Db RSSs display high conservation across distant species (Figure S4B). Especially the spacer/heptamer and the spacer/nonamer boundaries are well conserved in Db 23RSSs. We performed further in vitro cleavage analysis using Db1 23RSS carrying mutations in the spacer. The results showed that some mutations in the spacer/heptamer boundary (which comprises a putative d(TGATTCA) AP-1 binding site), affect both nicking and coupled cleavages of 39Db1-Jb1.1 substrates and also partially abolish the ‘Db1 23RSS-mediated restriction’ (Figure S5). These results are
consistent with the suggestion that the AP-1 site may be crucial for Db 23RSS function [31].
The coding sequence affects V(D)J recombination, thus if the heptamer is flanked by a ‘‘bad’’ coding sequence (such as T or A stretch) the recombination efficiency may decrease [13,35,36]. Also, it has been demonstrated that d(TTT) coding flank slows down nicking rate but does not interfere with RAG binding [37]. Db RSSs are flanked by ‘‘good’’ coding sequences which could account for their higher efficacy to focus RAG-mediated cleavage, compared to Jb or Vb RSSs [17]. However this could not explain why Db1 23RSS is more efficiently nicked than the Db2 23RSS since Db1 and Db2 RSSs possess identical coding flanks. Thus, the difference in performance of these two RSSs lies in their sequence variation that could be further investigated by RSS mutagenesis. Despite this difference, the coupled cleavages of Db1-Jb1 and Db2-Jb2 substrates are similar and are both weak compared to Db1-Jb2 substrates, suggesting that Jb2 RSSs are better partners than Jb1 RSSs (Figure S2 gels 13 to 18); Jb2 RSSs may counterbalance the low efficiency of Db2 23RSS to focus RAG activity whereas Jb1 RSSs (likely because of an unfavorable nonamer) may restrain Db1 23RSS performance to assemble a functional PC. Thus the likely efficiency of coupled cleavage of a given RSS pair would first depend, on the proficiency of the nucleating RSS to focus RAG activity (such as Db1 23RSS.Db2 23 RSS and Db2 12RSS.Db1 12RSS) and then for some RSS pairs, on the aptitude of the captured RSS to be bound by the RAG complex and to possibly undertake the nicking reaction. Further-more, besides the individual features of the RSS, we should also consider the possibility that depending on the type of nucleating site (12- or 23RSS), the PC assembly could slightly differ which may, to some extent, account for the pair-wise modulation of RAG-mediated cleavages. Indeed, Jones and Gellert have previously pointed out that initial binding onto a 12RSS leads to a more faithful adherence to the 12/23 rule and in explaining this observation, they proposed that the RAG1/2 multimers could be differentially locked depending on the initial binding RSS [8].
Our genome is scattered with sequences akin to RSS (the so-called cryptic RSS), but surprisingly, these cryptic RSSs are rarely mis-targeted by the recombinase (reviewed in [38]). In addition to possessing the critical nucleotides required for RSS function and to be accessible at the time of RAG1/2 expression, the cryptic RSS must find a suitable RSS partner in order to recombine. Such pair-wise modulation of the RAG1/2-mediated coupled-cleavage represents an additional constraint that safeguards the genome against illegitimate recombinations. Indeed, as shown in this study, amongst the TCRb RSSs tested, only the four Db-associated RSSs are competent for the initiation of V(D)J recombination. We hypothesize that most of the cryptic RSS may belong to the captured RSS category, and therefore a productive reaction with RAG1/2 would rely on their RSS partner. Up to now, the fully-characterized V(D)J-mediated translocations resulting from a targeting mistake of the recombinase involving the TCRb and various oncogenes (Lck, Tal2 or Lmo2) occur between the Db1 23RSS and cryptic 12RSS [38]. This observation complies with our scenario; the functional single complex RAG:Db1 23RSS could capture a cryptic 12RSS which may well be (as Jb1 12RSS) suboptimal, leading to translocation.
According to RAG1/2-mediated cleavage analysis, the Db1 23RSS blocks concurrent processing of the cis-linked 59 12RSS and consequently is likely to be essential for the proper ‘D-J prior V-DJ’ rearrangement order at the TCRb locus. Our data strongly supports the model in which removal of Db1 23RSS through Db1-to-Jb rearrangement is an essential step to eliminate the impediment to Vb-to-Db1 rearrangement. Consistent with this
model, if the Db1 23RSS is replaced by the functional Vb14 23RSS (Figure 6) or a mutated Db1 23RSS [20] VDb1 joints are then detected. Our data indicate that the Db1 23RSS (compared to all other Db RSSs) focuses RAG1/2 activity with a greater effectiveness and likely this mediates the inhibiting role of Db1 23RSS on the Db1 12RSS nicking. Footprinting analysis have shown that in the single RSS:RAG complex few nt adjacent to the heptamer are protected by RAG1/2, however a much larger region in the coding sequence, at least 12 bp, is protected in the synaptic complex [39]. Therefore as the Db1 coding sequence is only 12 bp long, it seems consistent that the RAG:Db1 23RSS complex sterically hinders the formation of a PC involving the Db1 12-RSS. Nevertheless, further molecular studies are necessary to clearly define the mechanics of this Db1 23RSS-mediated restriction.
Similarly to other DNA transactions, V(D)J recombination is prominently regulated by chromatin structure and modifications [10]. In this context, recent reports showed that RAG2 interacts with histone H3 hypermethylated at lysine 4, an epigenetic mark usually associated with active chromatin [40,41]. In addition to help RAG1/2 to target loci poised to undergo rearrangement, the authors proposed that this interaction, through allosteric activation of the recombinase, is directly involved in V(D)J recombination reaction. Concerning the TCRb locus, an increasing body of evidence argues in favor of at least two types of cis-acting regulatory elements, the transcriptional enhancer (Eb) and the germline promoter pDb1, controlling the initiation of V(D)J recombination [10,26]. Eb alone supports chromatin opening along the Db-Jb clusters while an interaction with pDb1 converts the Db1 segment into an accessible site. As shown inFigure 2D, the Db1 23RSS is not nicked in the Eb2/2thymocytes confirming that RSS accessibility is a prerequisite for RAG1/2 cleavage activity. Therefore chromatin structure and epigenetic marks, by modulating appropriately RSS accessibility (or inaccessibility) of the various TCRb gene segments, could be sufficient for a tight regulation of V(D)J recombination. Nonetheless, mechanisms distinct from RSS accessibility exist to ensure B12/23 restriction [22], allelic exclusion [42] and, as shown herein, ordered rearrangement. What is the purpose of such additional regulation mechanisms? As a minimum, they may represent security systems that guarantee proper V(D)J recombination in cases where RSSs are untimely accessible. However a previous report demonstrated that, within CD4+CD8+T-cells undergoing V(D)J recombination, Vb gene segments upstream of a functional VDJb1 rearrangement are maintained in an active chromatin environment but were still restricted from further rearrangement despite the proximity of Db2 gene [43]. This study highlights the possibility that, during normal T lymphocytes development, Vb, Db and Jb RSSs can be concomitantly accessible; this would therefore justify the existence and preservation of regulation systems operating beyond chroma-tin accessibility.
Materials and Methods Cells and mice
The D10 cell line [29] was provided by Dr. D.G. Schatz (Yale University School of Medicine, New Haven, CT). Cells were cultured in DMEM supplemented with 10% FCS, 100 U/mL penicillin, 100 mg/ml streptomycin, 50 mM 2-mercaptoethanol; and incubated at 37uC in a humidified chamber containing 5% CO2. C57BL/6J wild-type (WT), RAG1-deficient (RAG12/2) [44], Eb-deleted (Eb2/2) [45], TCRbWT [34] and TCRbDMF mice were housed under specific pathogen-free conditions, and handled in accordance with French and European directives.
Isolation of CD42CD82double-negative (DN) thymocytes and DNA purification
Total thymocytes were incubated 1 h at 37uC in the presence of rabbit complement (Low-Tox, Cederlane) and rat IgM anti-mouse-CD4 (RL172.4) and -CD8a (3.155) antibodies. Living cells were collected on a ficoll gradiant (Ficoll-Paque Plus, GE-Healthcare). Cell preparations were .95% DN as determined by flow cytometric analysis. Genomic DNA from purified DN WT, RAG12/2or Eb2/2thymocytes was prepared as previously described [28].
Oligo-capture assays
Analysis of single strand nicking products by oligo-capture assays was performed according to [9], using genomic DNA from DN thymocytes, 59 phosphorylated, 39 biotinylated oligonucleo-tides specific to RSS heptamers within the TCRb locus, and appropriate restriction enzymes (Table S1). Detection of the oligo-captured DNA fragment(s) was carried out by PCR. Briefly, PCR reactions (25 ml in 16 PCR buffer; 3 min at 94uC, followed by 28 cycles of 30 sec/94uC, 60 sec/60uC, 30 sec/72uC, and 7 min at 72uC) contained increasing amounts of either the captured (0.5%, 1% and 2%) or non-captured (10 ng, 25 ng and 50 ng) DNA, specific primers (5 pmol each), 0.2 mM dNTP, 2.5 mM MgCl2 and 1 U Taq DNA polymerase (Invitrogen).
Amplified DNAs were separated through a 1% agarose gel, transferred onto a Biodyne B membrane, and hybridized using a 59 end32P radio-labeled specific probe (sequences of PCR primers and hybridization probes listed inTable S3).
Plasmid constructs
Substrates for DNA cleavage were constructed using PCR amplified fragments from various genomic DNA regions within the mouse TCRb locus and standard molecular cloning procedures. PCR amplifications (30 sec/94uC; 30 sec/59uC; 45 sec/68uC; 32 cycles, with a final amplification step at 68uC for 7 min) were performed using PlatinumH Taq DNA Polymerase High Fidelity (Invitrogen) and appropriate oligonucleotide primers (Table S4). PCR products were purified following electrophoresis through a 1% agarose gel and subcloned into the pGEMT-easy or pGEM-7Zf vectors (Promega). In all constructs, RSSs and adjacent flanking sequences were checked by DNA sequencing (MWG Biotech). In total, three groups of substrates were used. The first two groups comprised DNA plasmids that were derived from either a construct containing a 657 bp Db1-Jb1.1 insert (group I; Figure S6) or a construct containing a 580 bp Db1 overlapping insert (group II;Figure S7). A third group comprised four DNA fragments (59Db1, 39Db1, Db1, and D1V14),
individually produced by PCR amplification using a plasmid from group II as template and oligonucleotide primers #181 and #318 (respectively, templates p59Db1, p39Db1, pDb1 and pDv).
Protein extracts
The RAG1/2-containing extract was prepared from heat-shocked D10 cells according to a published protocol [29]. Protein contents were determined using the Bio-Rad Protein Assay (Bio-Rad).
RAG1/2-mediated DNA cleavage in vitro assays
RAG1/2-mediated coupled cleavage was performed for 3 h at 30uC in a final volume of 25 mL of cleavage reaction buffer (50 mM Hepes-KOH pH 7.5, 73 mM KCl, 2 mM NaCl, 10 mM MgCl2, 1 mM DTT) supplemented with the RAG1/2 extract (20–
30 mg), 1.5 mM rATP, and proper recombination substrate
(0.3 pmol). To increase cleavage efficiency, 6 mg of a nuclear extract prepared from mouse WT thymocytes were also added. Negative controls were carried out using similar conditions without addition of the RAG1/2 extract. After phenol extraction and ethanol precipitation, the DNA samples were electrophoresed through a 1% agarose gel and analyzed by Southern blot using a Biodyne B transfer membrane (Pall Corporation). Membranes were hybridized with TCRb-specific, radio-labeled probes A GAGAAGAGTAGAGGACTGTGGGCCTTGG-39) or B (59-GACTTGAATCATGTTGTTTTCC-39). For RAG1/2-mediat-ed nicking assays, the substrate was first digestRAG1/2-mediat-ed by restriction enzymes AccI and NcoI. The resulting 700 bp fragment was gel purified (Wizard SV Gel and PCR Clean-Up System, Promega) and radio-labeled at 59 ends using T4 polynucleotide kinase (Invitrogen) and c32P-[ATP] (GE-Healthcare). The labeled substrate (,0.1 pmol) was used for DNA cleavage as described above, except that incubation was for 5 min. The DNA samples were then deproteinized and further digested by restriction enzymes EcoO109I/XbaI (that cut within RSS-intervening se-quences) to ensure proper quantification of nicked vs. intact RSSs. Formamide loading buffer was added to the digests and samples were heated at 95uC then separated by 15% polyacrylamide gel electrophoresis (PAGE) under denaturing conditions (7 M urea). Nicking assays of the 59Db1, 39Db1, Db1 or D1V14 amplified
products used similar conditions except that the deproteinized samples were electrophoresed directly without further restriction enzyme treatment.
TCRbDMFminilocus
To generate the TCRbDMFminilocus, the Db1 23RSS and Jb1.2 12RSS of the TCRbwtminilocus described in [34] were replaced by the Vb14 23RSS and Db1 12RSS respectively. Briefly, the TCRbwt HindIII/BamHI fragment containing the germline Db1, Jb1.1 and Jb1.2 gene segments was first subcloned in pGEMT-7zf, thereby generating pTgDJ. The Db1 23RSS mutation was introduced by replacing the pTgDJ EcoO109I fragment by the one from the pDV
substrate, thus generating pTgDVJ. The 59Jb1.2 12RSS mutation
was introduced by a two-steps PCR approach. First, pTgDVJ was
amplified using primers 213/208 and 207/214 to generate the fg1 and fg2 fragments, respectively. Then, a second PCR using fg1, fg2 and 213/214 primers was performed to produce the Jb1.2 mutated fragment. This latter PCR product was digested with EcoRV/BamHI and subcloned into EcoRV/BamHI-digested pTgDVJ construct to
produce the pTgDVJ25D vector. Finally the HindIII/BamHI
fragment of pTgDVJ25D was inserted into the
HindIII/BamHI-digested TCRbwtto produce the TCRbDMFminilocus. Microinjec-tion of TCRbDMFinto fertilized eggs, production of transgenic mice lines and analysis by southern blot of the rearrangement specific to the minilocus were conducted as previously described [34].
Supporting Information
Figure S1 Oligocapture mediated by p-CACAGTG-biotin.
Found at: doi:10.1371/journal.pone.0004575.s001 (0.06 MB PDF)
Figure S2 Pairwise modulation of RAG1/2-mediated coupled
cleavage.
Found at: doi:10.1371/journal.pone.0004575.s002 (0.20 MB PDF)
Figure S3 RAG1/2-mediated coupled cleavage of pTCRbWT
and pTCRbDMF substrates;
Found at: doi:10.1371/journal.pone.0004575.s003 (0.12 MB PDF)
Figure S4 Phylogenetic profiles of Db and Jb RSSs
Found at: doi:10.1371/journal.pone.0004575.s004 (0.15 MB PDF)
Figure S5 Effect of Db1 23RSS spacer mutations on
RAG1/2-mediated cleavages
Found at: doi:10.1371/journal.pone.0004575.s005 (0.16 MB PDF)
Figure S6 Cloning strategy to construct recombination
sub-strates
Found at: doi:10.1371/journal.pone.0004575.s006 (0.02 MB PDF)
Figure S7 Cloning strategy to construct additional
recombina-tion substrates
Found at: doi:10.1371/journal.pone.0004575.s007 (0.03 MB PDF)
Table S1 59-phosphorylated 39-biotinylated oligonucleotides
(7-mers) and restriction enzymes used in the oligo-capture assays to displace and ligate the nicked strand and to restrict the genomic DNA before purification on streptavidin-conjugated magnetic beads.
Found at: doi:10.1371/journal.pone.0004575.s008 (0.05 MB PDF)
Table S2 DNA sequences of the RSSs (plus the three proximal
nucleotides from coding flanks) used in this study.
Found at: doi:10.1371/journal.pone.0004575.s009 (0.01 MB PDF)
Table S3 Oligonucleotide primers and hybridization probes
used in the oligo-capture assays for PCR amplification and Southern blotting identification of the captured DNAs
Found at: doi:10.1371/journal.pone.0004575.s010 (0.05 MB PDF)
Table S4 Oligonucleotides used in the construction of the
various DNA cleavage substrates analyzed in this study.
Found at: doi:10.1371/journal.pone.0004575.s011 (0.03 MB PDF)
Text S1 Consideration of an Alternative capture model.
Found at: doi:10.1371/journal.pone.0004575.s012 (0.07 MB PDF)
Acknowledgments
We thank Dr. S. Spicuglia and Dr. B. Nadel for critical reading of this manuscript and members of the PF lab for helpful discussions. The D10 cell line was kindly provided by Dr. D.G. Schatz (Yale University, New Haven, Connecticut, USA). DMF and TB are fellows from the ARC and ANR, respectively. TCRbDMF transgenic mice were generated with the
help of the mouse functional genomics Platform of the Marseille-Nice GenopoleH.
Author Contributions
Conceived and designed the experiments: DMF PF DPB. Performed the experiments: DMF TB SJ. Analyzed the data: DMF TB SJ PF DPB. Wrote the paper: PF DPB. Did all the biological experiments: DMF. Did the phylogenetic analyses: TB SJ.
References
1. Jung D, Alt FW (2004) Unraveling V(D)J recombination; insights into gene regulation. Cell 116: 299–311.
2. Tonegawa S (1983) Somatic generation of antibody diversity. Nature 302: 575–81.
3. Bassing CH, Swat W, Alt FW (2002) The mechanism and regulation of chromosomal V(D)J recombination. Cell 109 Suppl: 45–55.
4. Gellert M (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 71: 101–132.
5. Hesslein DG, Schatz DG (2001) Factors and forces controlling V(D)J recombination. Adv Immunol 78: 169–232.
6. Lieber MR, Ma Y, Pannicke U, Schwarz K (2004) The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst) 3: 817–826.
7. Mundy CL, Patenge N, Matthews AG, Oettinger MA (2002) Assembly of the RAG1/RAG2 synaptic complex. Mol Cell Biol 22: 69–77.
8. Jones JM, Gellert M (2002) Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J 21: 4162–4171.
9. Curry JD, Geier JK, Schlissel MS (2005) Single-strand recombination signal sequence nicks in vivo: evidence for a capture model of synapsis. Nat Immunol 6: 1272–1279.
10. Cobb RM, Oestreich KJ, Osipovich OA, Oltz EM (2006) Accessibility control of v(d)j recombination. Adv Immunol 91: 45–109.
11. Roth DB (2003) Restraining the V(D)J recombinase. Nat Rev Immunol 3: 656–666.
12. Feeney AJ, Goebel P, Espinoza CR (2004) Many levels of control of V gene rearrangement frequency. Immunol Rev 200: 44–56.
13. Gerstein RM, Lieber MR (1993) Coding end sequence can markedly affect the initiation of V(D)J recombination. Genes Dev 7: 1459–1469.
14. Lee AI, Fugmann SD, Cowell LG, Ptaszek LM, Kelsoe G, et al. (2003) A functional analysis of the spacer of V(D)J recombination signal sequences. PLoS Biol 1: 056–068.
15. Bassing CH, Alt FW, Hughes MM, D’Auteuil M, Wehrly TD, et al. (2000) Recombination signal sequences restrict chromosomal V(D)J recombination beyond the 12/23 rule. Nature 405: 583–6.
16. Hughes MM, Tillman RE, Wehrly TD, White JM, Sleckman BP (2003) The B12/23 restriction is critically dependent on recombination signal nonamer and spacer sequences. J Immunol 171: 6604–10.
17. Jung D, Bassing CH, Fugmann SD, Cheng HL, Schatz DG, et al. (2003) Extrachromosomal recombination substrates recapitulate beyond 12/23 re-stricted VDJ recombination in nonlymphoid cells. Immunity 18: 65–74. 18. Olaru A, Patterson DN, Villey I, Livak F (2003) DNA-Rag protein interactions
in the control of selective D gene utilization in the TCR beta locus. J Immunol 171: 3605–11.
19. Olaru A, Patterson DN, Cai H, Livak F (2004) Recombination signal sequence variations and the mechanism of patterned T-cell receptor-beta locus rearrangement. Mol Immunol 40: 1189–201.
20. Sleckman BP, Bassing CH, Hughes MM, Okada A, D’Auteuil M, et al. (2000) Mechanisms that direct ordered assembly of T cell receptor beta locus V, D, and J gene segments. Proc Natl Acad Sci U S A 97: 7975–7980.
21. Tillman RE, Wooley AL, Khor B, Wehrly TD, Little CA, et al. (2003) Cutting edge: targeting of V beta to D beta rearrangement by RSSs can be mediated by the V(D)J recombinase in the absence of additional lymphoid-specific factors. J Immunol 170: 5–9.
22. Tillman RE, Wooley AL, Hughes MM, Khor B, Sleckman BP (2004) Regulation of T-cell receptor beta-chain gene assembly by recombination signals: the beyond 12/23 restriction. Immunol Rev 200: 36–43.
23. Wu C, Bassing CH, Jung D, Woodman BB, Foy D, et al. (2003) Dramatically increased rearrangement and peripheral representation of Vbeta14 driven by the 39Dbeta1 recombination signal sequence. Immunity 18: 75–85.
24. Drejer Teel AH, Fugmann SD, Schatz DG (2007) The beyond 12/23 restriction is imposed at the nicking and pairing steps of DNA cleavage during V(D)J recombination. Mol Cell Biol 27: 6288–6299.
25. Born W, Yagu¨e J, Palmer E, Kappler J, Marrack P (1985) Rearrangement of T-cell receptor b-chain genes during T-T-cell development. Proc Natl Acad Sci U S A 82: 2925–2929.
26. Jackson AM, Krangel MS (2006) Turning T-cell receptor beta recombination on and off: more questions than answers. Immunol Rev 209: 129–141. 27. Wilson A, Marechal C, MacDonald HR (2001) Biased Vb usage in immature
thymocytes is independent of DJb proximity and pTa pairing. J Immunol 166: 51–57.
28. Mathieu N, Hempel WM, Spicuglia S, Verthuy C, Ferrier P (2000) Chromatin remodeling by the T cell receptor (TCR)-beta gene enhancer during early T cell development: Implications for the control of TCR-beta locus recombination. J Exp Med 192: 625–636.
29. Leu TM, Schatz DG (1995) rag-1 and rag-2 are components of a high-molecular-weight complex, and association of rag-2 with this complex is rag-1 dependent. Mol Cell Biol 15: 5657–5670.
30. Eastman QM, Leu TM, Schatz DG (1996) Initiation of V(D)J recombination in vitro obeying the 12/23 rule. Nature 380: 85–8.
31. Wang X, Xiao G, Zhang Y, Wen X, Gao X, et al. (2008) Regulation of Tcrb recombination ordering by c-Fos-dependent RAG deposition. Nat Immunol 9: 794–801.
32. Livak F, Burtrum DB, Rowen L, Schatz DG, Petrie HT (2000) Genetic modulation of T cell receptor gene segment usage during somatic recombina-tion. J Exp Med 192: 1191–1196.
33. van Gent DC, Ramsden DA, Gellert M (1996) The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell 85: 107–113.
34. Ferrier P, Krippl B, Blackwell TK, Furley AJ, Suh H, et al. (1990) Separate elements control DJ and VDJ rearrangement in a transgenic recombination substrate. Embo J 9: 117–25.
35. Boubnov NV, Wills ZP, Weaver DT (1995) Coding sequence composition flanking either signal element alters V(D)J recombination efficiency. Nucleic Acids Res 23: 1060–1067.
36. Ezekiel UR, Engler P, Stern D, Storb U (1995) Asymmetric processing of coding ends and the effect of coding end nucleotide composition on V(D)J recombination. Immunity 2: 381–389.
37. Yu K, Lieber MR (1999) Mechanistic basis for coding end sequence effects in the initiation of V(D)J recombination. Mol Cell Biol 19: 8094–102.
38. Marculescu R, Vanura K, Montpellier B, Roulland S, Le T, et al. (2006) Recombinase, chromosomal translocations and lymphoid neoplasia: targeting mistakes and repair failures. DNA Repair (Amst) 5: 1246–1258.
39. Nagawa F, Hirose S, Nishizumi H, Nishihara T, Sakano H (2004) Joining mutants of RAG1 and RAG2 that demonstrate impaired interactions with the coding-end DNA. J Biol Chem 279: 38360–8.
40. Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S (2007) A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is
necessary for efficient antigen-receptor-gene rearrangement. Immunity 27: 561–571.
41. Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, et al. (2007) RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450: 1106–1110.
42. Jackson A, Kondilis HD, Khor B, Sleckman BP, Krangel MS (2005) Regulation of T cell receptor beta allelic exclusion at a level beyond accessibility. Nat Immunol 6: 189–197.
43. Jackson AM, Krangel MS (2005) Allele-specific regulation of TCR beta variable gene segment chromatin structure. J Immunol 175: 5186–5191.
44. Spanopoulou E, Roman CA, Corcoran LM, Schlissel MS, Silver DP, et al. (1994) Functional immunoglobulin transgenes guide ordered B-cell differentia-tion in Rag-1-deficient mice. Genes Dev 8: 1030–1042.
45. Bouvier G, Watrin F, Naspetti M, Verthuy C, Naquet P, et al. (1996) Deletion of the mouse T-cell receptor beta gene enhancer blocks alphabeta T-cell development. Proc Natl Acad Sci U S A 93: 7877–7881.
46. Schlissel MS (1998) Structure of nonhairpin coding-end DNA breaks in cells undergoing V(D)J recombination. Mol Cell Biol 18: 2029–2037.