Development of Novel Chemical Biology Tools to Probe Malaria Parasite
Physiology and Aid in Antimalarial Drug Discovery
by
James R. Abshire
B.S., University of Maryland – College Park (2008) Submitted to the Department of Biological Engineering in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN BIOLOGICAL ENGINEERING
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2015
© 2015 Massachusetts Institute of Technology. All rights reserved.
Signature of Author ... James R. Abshire Department of Biological Engineering
Certified by ... Jacquin C. Niles Associate Professor, Department of Biological Engineering Thesis Supervisor
Accepted by... Forest M. White Associate Professor, Department of Biological Engineering Co-Chair, Course XX Graduate Program Committee
This doctoral thesis has been examined by a committee of the Department of Biological Engineering as follows:
Certified by ... K. Dane Wittrup Professor, Departments of Chemical Engineering and Biological Engineering
Thesis Committee Chair
Certified by ... Jacquin C. Niles Associate Professor, Department of Biological Engineering Thesis Supervisor
Certified by ... Peter C. Dedon Professor, Department of Biological Engineering Thesis Committee Member
Development of Novel Chemical Biology Tools to Probe Malaria Parasite
Physiology and Aid in Antimalarial Drug Discovery
by
James R. Abshire
Submitted to the Department of Biological Engineering on April 14, 2015 in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biological Engineering
ABSTRACT
Malaria remains a major burden to global public health. Antimalarial drugs are a mainstay in efforts to control and eventually eradicate this disease. However, increasing drug resistance threatens to reverse recent gains in malaria control, making the discovery of new antimalarials critical. Antimalarial discovery is especially challenging due to the unique biology of malaria parasites, the scarcity of tools for identifying new drug targets, and the poorly understood mechanisms of action of existing antimalarials. Therefore, this work describes the development of two chemical biology tools to address unmet needs in antimalarial drug discovery.
A particular challenge in antimalarial development is a shortage of validated parasite drug targets. Potent antimalarials with demonstrated clinical efficacy, like the aminoquinolines and artemisinins, represent a promising basis for rational drug development. Unfortunately, the molecular targets of these drugs have not been identified. While both are thought to interact with parasite heme, linking in vitro heme binding with drug potency remains challenging because labile heme is difficult to quantify in live cells. This work presents a novel genetically-encoded heme biosensor and describes its application to quantify labile heme in live malaria parasites and test mechanisms of antimalarial action.
Another challenge is posed by the widespread malaria parasite Plasmodium vivax, which, unlike P. falciparum, cannot be propagated in vitro, hindering research into parasite biology and drug target identification. P. vivax preferentially invades reticulocytes, which are impractical to obtain in continuous supply. The basis for this invasion tropism remains incompletely
understood, mainly because current tools cannot directly link molecular binding events to invasion outcomes. This work presents novel methods for immobilizing synthetic receptors on the red blood cell surface. These receptors are used in proof-of-concept experiments to
investigate requirements for efficient invasion via a well-characterized P. falciparum invasion pathway, suggesting this method can be used to elucidate molecular mechanisms underlying parasite invasion tropisms. Future receptor designs could promote the invasion of P. vivax into mature red blood cells and potentially facilitate practical in vitro culture. Taken together, these tools present new opportunities for drug discovery to aid efforts in malaria control and eventual eradication.
Thesis Supervisor: Jacquin C. Niles
ACKNOWLEDGEMENTS
This thesis represents roughly six years of focused effort, during which I have benefited from the guidance and support of mentors, colleagues, friends, and family.
I would first like to thank Doug Lauffenburger and the Department of Biological Engineering for the opportunity to pursue my graduate education at MIT. The BE department has a unique blend of exciting, diverse research and a collegial, supportive environment fostered by both the faculty and the students. I am grateful to have started my scientific career as a part of this community. I would especially like to thank my advisor, Jacquin Niles, for his support and mentorship over the past six years. During my time in his lab, Jacquin has been consistently attentive,
encouraging, and thoughtful in helping me approach challenging scientific questions, design and execute research plans, and think critically about results. I would also like to thank my thesis committee, Dane Wittrup, Manoj Duraisingh, and Pete Dedon for their feedback and suggestions along the way.
Additionally, I would like to acknowledge several other students, postdocs, and core facility personnel for their assistance. In particular, Ceth Parker, Helena de Puig Guixé, Prabhani Atukorale, Matthew Wohlever, Charlie Knutson, Koli Taghizadeh, Wendy Salmon, and Glenn Paradis have provided technical help in various aspects of my thesis work. Matthew Edwards and Hunter Elliott also provided valuable technical feedback. In addition, I would like to thank Professors Steven Tannenbaum, John Essigmann, Leona Samson, Darrell Irvine, Robert Sauer, Kim Hamad-Schifferli, and Lee Gehrke for use of their equipment and facilities.
I would also like to extend my gratitude to the other members and alumni of the Niles Lab. Brian Belmont, Steve Goldfless, and Jeff Wagner were instrumental in much of my day-to-day training as I started working in the lab. Erika Bechtold, Chris Birch, Sumanta Dey, Suresh Ganesan, Sebastian Nasamu, Bridget Wall, and Daiying Xu all provided helpful discussions and feedback. In addition to the technical help, everyone in the lab contributed to a working environment that was both unique (with our diverse musical tastes) and enjoyable. I am also grateful to this group for their friendship both inside and outside the lab. Additionally, I would like to thank Denise MacPhail for her enthusiastic work behind the scenes helping our lab run smoothly.
I would also like to thank my BE classmates and as well as my Boston-area friends for helping make my time here so enjoyable. I will always be grateful for their camaraderie and friendship during the challenging and celebratory moments of graduate school.
I was fortunate to receive several fellowships that funded my education and research, including the DuPont Presidential Fellowship and the NIGMS Biotechnology Training Program
Fellowship. I would especially like to acknowledge the opportunity provided by the BTP fellowship to pursue an industrial internship during my graduate studies. To that end, I would like to thank Eugene Antipov of Amyris, Inc. for his guidance as my mentor during my internship.
I would also like to acknowledge some of my early mentors for fostering my interests in science and engineering. I am especially grateful to Professors Daniel Stein, Ann Smith, and William Bentley for the opportunity to conduct research as an undergraduate student at the University of Maryland. Colin Hebert, a graduate student at the time, was instrumental in my early training as my mentor in the Bentley Lab. I would also like to extend my gratitude to Professor Anne Simon and Dr. Bonnie Dixon, whose engaging Biology and Organic Chemistry lectures inspired me to continue my education and explore a career in these fields. I am also grateful to Dr. Xufeng Wu and Dr. John Hammer at the National Institutes of Health for introducing me to molecular biology research and for the opportunity to learn in the Hammer Lab.
I am also incredibly grateful to my family, especially to my parents, for their love and support throughout all my endeavors. Having their advice, encouragement, and perspective “just a phone call away” has been truly indispensible. Finally, I would like to thank my wonderful girlfriend Cheryl for her strength, optimism, and good humor, all of which have helped immensely.
ATTRIBUTIONS
In addition to the general acknowledgements described above, I would like to detail several specific contributions to the work described in this thesis.
The fluorescence lifetime measurements described in Chapter 2 were performed in collaboration with Professor Peter So and his postdoctoral fellow Christopher Rowlands. In addition, Suresh Ganesan built and tested several P. falciparum strains that helped inform my strain construction efforts.
The text of Chapter 2 represents a collaborative writing effort between Jacquin Niles, Christopher Rowlands, and me, with input from Peter So.
Finally, Professor Carolyn Bertozzi and her graduate student Jason Hudak synthesized and provided the aminooxy-functionalized reagents for several of the experiments described in Chapter 3.
TABLE OF CONTENTS
CHAPTER 1: INTRODUCTION . . . 11
1.1 Malaria burden and pathogenesis . . . 11
1.2 Malaria chemotherapy and resistance . . . 13
1.3 Challenges and opportunities in antimalarial drug development . . . . 15
1.4 Heme metabolism in malaria parasites . . . 16
1.4.1 Degradation of host cell hemoglobin . . . 16
1.4.2 Heme biosynthesis and utilization . . . 18
1.4.3 Other potential sources of parasite heme . . . 19
1.4.4 Role of heme in antimalarial potency . . . 20
1.5 In vitro culture of P. vivax . . . . . . . . . . . . . 23
1.6 Invasion of red blood cells by malaria parasites . . . 24
1.6.1 Overview of the invasion process . . . 24
1.6.2 Ligand-receptor interactions governing invasion . . . 26
1.6.3 Linking ligand-receptor interactions to invasion outcomes . . 27
1.7 Summary of rationale and work presented . . . 28
1.8 References . . . 30
CHAPTER 2: DEVELOPMENT OF A NOVEL GENETICALLY-ENCODED FRET BIOSENSOR AND QUANTIFICATION OF A LABILE CYTOSOLIC HEME POOL IN LIVE MALARIA PARASITES . . 38
2.0 Note . . . 38
2.1 Abstract . . . 38
2.3 Methods . . . 42
2.3.1 Molecular cloning . . . 42
2.3.2 Protein expression and purification . . . 42
2.3.3 Absorbance titrations . . . 43
2.3.4 Fluorescence titrations and FRET efficiency calculations . . 44
2.3.5 Fluorescence lifetime spectroscopy . . . 44
2.3.6 Malaria parasite culture . . . 46
2.3.7 Preparation of giant multilamellar vesicles . . . 46
2.3.8 In situ FRET analysis . . . . 47
2.3.9 Western immunoblotting . . . 48
2.4 Results . . . 49
2.4.1 Design and characterization of initial FRET-based heme biosensor 49 2.4.2 Optimization of initial heme biosensor design . . . 54
2.4.3 Correlating FRET efficiencies determined by imaging microscopy and fluorimetry for calibrating heme concentrations . . . . 60
2.4.4 Measuring labile heme in live malaria parasites . . . 61
2.4.5 Quantitative analysis of perturbed heme homeostasis by heme-interacting antimalarials . . . 65
2.5 Discussion . . . 68
2.6 References . . . 72
2.7 Appendix: MATLAB Scripts . . . 75
2.7.1 Calculate heme concentration and confidence intervals from image data . . . 75
2.7.2 Calculate average heme concentration and confidence intervals
from bootstrapping data obtained from multiple experiments . 77
CHAPTER 3: USING SYNTHETIC RECEPTORS TO ELUCIDATE HOST CELL REQUIREMENTS
FOR PARASITE INVASION . . . 78
3.1 Abstract . . . 78
3.2 Introduction . . . 79
3.3 Methods . . . 83
3.3.1 Malaria parasite culture . . . 83
3.3.2 Neuraminidase treatment . . . 83
3.3.3 Sialyltransferase treatment . . . 83
3.3.4 Oxime ligation . . . 84
3.3.5 Flow cytometry . . . 84
3.3.6 Fluorimetric sialic acid quantitation . . . 85
3.3.7 HPLC sialic acid quantitation . . . 86
3.3.8 Glycophorin extraction and biotinylation . . . 86
3.3.9 Glycophorin immobilization . . . 87
3.3.10 Invasion assay . . . 88
3.4 Results . . . 89
3.4.1 Effect of surface receptor density on parasite invasion rates . . 89
3.4.2 Enzymatic restoration of sialic acid receptors . . . 91
3.4.3 Enzymatic attachment of sialic acid receptors with an alternate terminal linkage . . . 93
3.4.4 Synthetic glycan receptor construction using
aminooxy-functionalized reagents . . . 95
3.4.5 Synthetic glycoprotein receptor construction using biotin-NeutrAvidin interactions . . . 97
3.5 Discussion . . . 101
3.6 References . . . 104
CHAPTER 4: CONCLUSIONS AND FUTURE WORK . . . 107
4.1 Parasite heme biology . . . 107
4.2 Antimalarial drug action . . . 108
4.3 Antimalarial drug discovery . . . 109
4.4 Heme sensing in other biological systems . . . 111
4.5 Synthetic receptor development . . . 112
4.6 Towards synthetic receptor use for in vitro culture of P. vivax . . . . 114
4.7 Conclusions . . . 115
CHAPTER 1: INTRODUCTION
1.1 Malaria burden and pathogenesis
Malaria is an ancient parasitic disease that remains a major burden to global public health. In 2013, there were an estimated 198 million cases of malaria worldwide, which led to an estimated 584,000 deaths, mostly in young children living in sub-Saharan Africa1. Nearly half the world’s
population is at risk for malaria infection, with active disease transmission occurring in 97 countries1
. While a single malaria infection can be effectively treated and cured with modern antimalarial drugs, the widespread distribution of the disease, the possibility for repeat infections, the limited infrastructure in the most severely affected countries, and the lack of an effective vaccine currently preclude malaria eradication.
Malaria in humans is caused by five species of the eukaryotic parasite genus Plasmodium – P. falciparum, P. vivax, P. knowlesi, P. malariae, and P. ovale. Of these, P. falciparum and P. vivax are responsible for the vast majority of malaria morbidity, while P. falciparum infection accounts for most malaria-associated deaths1. These parasites are transmitted by the bite of an infected
female Anopheles mosquito, where haploid sporozoites are injected from the mosquito’s salivary glands, and travel to the liver of the host. Sporozoites then multiply within hepatocytes and are released into the bloodstream as merozoites, which invade and replicate inside red blood cells. P. vivax infections retain a population of quiescent parasites in the liver, termed hypnozoites, which can reactivate and cause disease relapse after the blood-stage infection has been cleared2
.
The blood stage of infection is solely responsible for the symptoms of malaria, which include a recurring high fever and anemia9
. During this stage, the infective merozoites bind to and invade the red blood cell, and begin digesting the contents of the red blood cell cytosol. Parasites of this stage, termed trophozoites, consume more than 75% of the hemoglobin from the host red blood
cell3
before undergoing schizogeny to produce daughter merozoites. Rupture of the schizont releases the daughter merozoites, which can then infect other red blood cells. Once released, merozoites are only viable for a few minutes, and typically reinvade new host red blood cells within 90 seconds4
. Haploid blood-stage parasites can also differentiate into sexual-stage gametocytes, through a process that is not fully understood, but appears to involve epigenetic regulation5
of gametocyte-specific transcription factors6,7
. These gametocytes can then be taken up by a mosquito during a blood meal, where fertilization and oogenesis lead to the production of new sporozoites8
(Fig. 1-1).
In addition to fever and anemia, P. falciparum infections often lead to further complications due to the sequestration of infected red blood cells in the host microvasculature. Blood-stage P. falciparum parasites extensively remodel the surface of their red blood cell hosts, expressing proteins that adhere the infected red blood cell to endothelial cells9. Presumably, this enables
infected red blood cells to avoid clearance by the host spleen, but can lead to coagulation, breakdown in blood vessel structure, and inflammation in the host10-12
, with further complications in individual organs13
. Cerebral malaria has a high mortality rate in children and can lead to permanent neurological impairment14
. Sequestration can also occur in the placenta during pregnancy, leading to anemia in the mother and reducing fetal birth weight15, thereby increasing
the risk of infant mortality16
Figure 1-1. Malaria parasite life cycle in both the human host and the mosquito vector. After inoculation by an
infected mosquito, sporozoites invade and replicate inside liver cells (A). Rupture of infected liver cells releases merozoites into the bloodstream, where parasites infect red blood cells (B). Blood-stage parasites can differentiate into gametocytes, which are acquired by the mosquito during a blood meal. Gametocyte fertilization occurs in the mosquito and produces new sporozoites (C). Figure from [17].
1.2 Malaria chemotherapy and resistance
Global efforts to control malaria rely on a combination of approaches. Vector control methods, through both physical barriers (e.g. bed nets) and insecticide spraying, aim to reduce disease transmission by preventing mosquito bites. While these methods can be effective, bed nets must be replaced regularly, and mosquito populations can develop resistance to
insecticides18
. Development is also ongoing on a variety of malaria vaccines, with over 40 candidates reaching clinical trials19
targets the circumsporozoite protein20
, has achieved only partial protection with approximately 31% efficacy in Phase III trials21
. Therefore, chemotherapy remains a mainstay in combating malaria.
Two of the most important classes of antimalarial drugs, the aminoquinolines and the artemisinins, have formed the backbone of modern efforts against malaria. The potent
antimalarial activity of chloroquine was first highlighted by clinical trials in the United States during World War II22
, and chloroquine quickly became the most extensively-used antimalarial drug23
. In addition to its rapid activity against blood-stage malaria parasites, chloroquine was easily administered, safe, and inexpensive24
. The extraordinary success of chloroquine, along with the insecticide DDT, generated optimism in the 1950s and 1960s that malaria would soon be eradicated25
. However, after extensive use as a monotherapy26
, widespread resistance to chloroquine emerged in the 1960s and 1970s, leading to a devastating resurgence of morbidity and mortality, especially in sub-Saharan Africa27,28
. Today, chloroquine is no longer
recommended to treat P. falciparum malaria due to the high rates of resistance in endemic areas29
. Interest in chloroquine remains, however, due to its unmatched combination of safety, and
affordability, and historical efficacy30
.
Artemisinins, in combination with other drugs, have become the standard-of-care in treating chloroquine-resistant malaria. Artemisinins rapidly kill all blood stages of the parasite (including gametocytes, making them active against transmission), and exhibit the most rapid clearing of malaria-induced fever of any antimalarial drug class31
. However, artemisinin resistance, first noted as increased parasite clearance times among patients in Cambodia32
, now appears to be spreading across Southeast Asia33
effective in these regions, likely due to action of their partner drugs, rates of treatment failure are increasing33
. Therefore, new antimalarial drugs are urgently needed34
.
1.3 Challenges and opportunities in antimalarial drug development
Antimalarial drug discovery is especially daunting due a unique combination of scientific and public health challenges. While an in vitro culture system for P. falciparum malaria developed in the 1970s35
has revolutionized our understanding of parasite biology, such a system does not exist for P. vivax. This has hindered efforts to measure efficacy of current antimalarial drugs, and identify new drug targets34
. Attempts at P. vivax culture have met with only limited success (discussed below). Therefore, developing a practical method for in vitro culture of P. vivax is a major priority for malaria research34,36,37
.
Even with a practical in vitro culture system, drug development in P. falciparum remains challenging. Although sequenced in 200238
, the genome of P. falciparum remains poorly understood, as the functions of many predicted gene sequences have not been determined34
. In addition, its extreme A-T richness and sparse toolkit for gene manipulation have hindered drug development efforts. While exciting new technologies promise to accelerate this process39-41
, identifying promising drug targets remains a top priority42. In addition, public health challenges
in endemic areas place additional constraints on drug development. Future drugs must be well tolerated when given in combination with other drugs to minimize the need for follow-up care, which is often limited, and delay the development of resistance. Additionally, drugs must also be orally bioavailable and rapidly cure the underlying disease to enable practical mass
administration and maximize patient compliance. Finally, drugs must be especially inexpensive to be broadly accessible to populations in endemic areas43,44
Taken together, the scarcity of validated drug targets and the stringent requirements for successful drug candidates suggest that understanding the mechanisms of action of existing antimalarial drugs is critical. Antimalarials with demonstrated clinical efficacy like the aminoquinolines and artemisinins represent a promising basis for rational drug development45
. However, the molecular targets of aminoquinoline and artemisinin antimalarials remain
controversial, which precludes broader efforts to exploit these targets. Both classes of drugs have been shown to interact with heme in vitro, but connecting this in vitro interaction to a mechanism of parasite toxicity has proven difficult, partly due to a limited understanding of heme
metabolism in the malaria parasite.
1.4 Heme metabolism in malaria parasites
1.4.1 Degradation of host cell hemoglobin
Blood-stage malaria parasites ingest roughly 75% of the hemoglobin from the host red blood cell into the lysosome-like digestive vacuole3
(Fig. 1-2). Here, the polypeptide chains of
hemoglobin are cleaved into short peptides and individual amino acids by the concerted action of multiple classes of proteases3
. Proteolytic degradation products are then transported into the parasite cytoplasm, where the individual amino acids are used by the parasite in protein translation46
. In addition to liberating peptides and amino acids, hemoglobin proteolysis releases large amounts of heme. Given that the digestive vacuole represents only 3-5% of the parasite’s total volume47
, heme liberated from hemoglobin digestion could reach concentrations up to 500 mM in this compartment absent sequestration or destruction of the excess heme48
. High
concentrations of free heme are cytotoxic, due to its affinity for lipids in cellular membranes and its ability to generate reactive oxygen species49
To prevent vacuolar damage from free heme accumulation, the parasite sequesters liberated heme into inert crystals of heme dimers termed hemozoin50,51
. However, the mechanism of hemozoin crystallization in the parasite is not completely understood. Crystallization of β-hematin, a synthetic analogue of hemozoin, propagates readily from seed crystals in vitro, suggesting hemozoin crystallization may be autocatalytic52
. In vitro, several parasite proteins localized to the digestive vacuole, namely histidine-rich proteins 2 and 3 (PfHRP2, 3) and heme detoxification protein (PfHDP) have been shown to expedite the formation of hemozoin53,54
. Interestingly, while knockouts of PfHRP2 and PfHRP3 still form hemozoin55
, PfHDP appears to be essential54
. Crystallization can also be nucleated by parasite-derived lipids in vitro56
, which corroborates electron microscopy data showing hemozoin crystals localized near membrane structures in the digestive vacuole57
.
Importantly, heme sequestration into hemozoin is the only known method by which parasites can detoxify surplus heme. Recent studies in P. falciparum showed that parasites lack heme oxygenase activity, and that the heme oxygenase-like enzyme encoded in the parasite genome appears not to degrade heme58
. Others have proposed non-enzymatic degradation pathways for heme in the food vacuole59
and cytosol60
based on in vitro experiments, but it remains unknown whether these reactions contribute appreciably to heme degradation in the parasite61. Finally, it is
not known whether heme liberated from hemoglobin degradation is able to escape the digestive vacuole. The acidic pH of the digestive vacuole would tend to protonate the propionate groups of free heme molecules, perhaps allowing them to diffuse across the vacuolar membrane48,61
. Heme may also exit the digestive vacuole via specific transporters, although none have been
definitively identified61
1.4.2 Heme biosynthesis and utilization
In addition to large-scale hemoglobin degradation and crystallization of the liberated heme, malaria parasites also contain a complete pathway for heme biosynthesis. Heme biosynthesis in the parasite spans three organelles – the cytosol, mitochondrion, and apicoplast62,63
(Fig. 1-2). This is likely a result of endosymbiotic events that left the parasite with two complete heme biosynthesis pathways, where redundant functions were eliminated over time64
. Heme biosynthesis begins in the parasite mitochondrion, where a condensation reaction combines succinyl-CoA and glycine to form δ-aminolevulinic acid (ALA). Next, ALA is converted in a series of steps to coproporphyrinogen III in the parasite apicoplast65,66
, and is then oxidized to protoporphyrinogen IX in the cytosol67
. The final conversion steps involving further oxidation and loading with iron occur in the mitochondrion68,69
. Presumably, trafficking of heme biosynthesis intermediates between the mitochondrion, apicoplast, and cytosol relies on transporters or specific binding proteins, as cellular membranes are generally impermeable to these compounds70
. How these intermediates are trafficked between parasite organelles remains to be elucidated62
.
The parasite genome encodes only a small number of known hemoproteins38,63,71
. Multiple cytochromes are present in the parasite mitochondrion and function in the electron transport chain, which appears to be essential for parasite survival. Atovaquone, which binds to cytochrome b and inhibits electron transport72
is toxic to the parasite73
, while certain mutations in cytochrome b can render parasites atovaquone-resistant74
. The electron transport chain is required, however, for regenerating ubiquinone, the electron acceptor for dihydroorotate dehydrogenase (DHOD) during pyrimidine biosynthesis. Expressing a yeast DHOD, which is cytosolic and operates independently of ubiquinone, renders parasites insensitive to atovaquone75
suggesting that other functions of mitochondrial electron transport (such as ATP generation) are dispensable. While the parasite genome encodes orthologues of cytochrome b5, the functions of
these proteins have not been determined63
. Binding to heme has also been demonstrated with other recombinantly-expressed parasite proteins76
, but the physiological relevance of these interactions has not been specifically addressed61
.
1.4.3 Other potential sources of parasite heme
Recent evidence suggests that blood-stage parasites can meet their metabolic needs without synthesizing heme de novo. First, the penultimate enzyme of the heme biosynthesis pathway, protoporphyrinogen IX oxidase, requires an electron acceptor coupled to the mitochondrial electron transport chain68
. Given that parasites expressing yeast DHOD survive electron transport inhibition with atovoquone75, protoporphyrinogen IX oxidase activity appears not to be required
for growth. Other steps in the heme biosynthesis pathway appear dispensable, as well. Double-crossover knockouts of the first and last enzymes in the heme biosynthesis pathway
(δ-aminolevulinic acid synthase and ferrochelatase, respectively) have been successfully generated in blood-stage P. berghei77
and P. falciparum78
parasites, which grew normally but were unable to progress to the mosquito stages, suggesting that heme biosynthesis is only required for the exoerythrocytic stages of the parasite life cycle.
Therefore, blood-stage parasites are likely able to obtain heme from other sources. In a recent study, radiolabeled hemoglobin-heme (obtained by incubating mouse reticulocytes with 14
C-ALA) was found in the mitochondrial cytochromes of ferrochelatase-null P. berghei parasites77
, suggesting that hemoglobin-heme can be trafficked outside the digestive vacuole. Additionally, a micromolar pool of “free” heme has been indirectly measured in the cytosol of erythrocytes79
which may be accessible to the parasite63
. Studies with zinc protoporphyrin IX suggest that parasites in culture can accumulate protoporphyrins added to the extracellular media80
, which may represent another pathway for scavenging. Finally, heme escape from the digestive vacuole has been suggested, based on the ability of several cytosolic parasite proteins (especially
glyceraldehyde-3-phosphate dehydrogenase [GAPDH] and thioredoxin reductase [TrxR]) to bind and be regulated by heme76
. However, mechanistic details regarding these proposed trafficking pathways remain to be elucidated.
1.4.4 Role of heme in antimalarial potency
Multiple classes of antimalarial drugs are known to interact with heme. Artemisinin and its derivatives are potent drugs extensively used for treating P. falciparum malaria. Artemisinin activity requires an endoperoxide moiety81 which is thought to undergo iron-assisted reductive
cleavage in the parasite to form damaging radicals82
. Recent experiments have shown that inhibiting hemoglobin degradation attenuates artemisinin toxicity83
, as does iron chelation84
, implicating heme and ferrous iron as potential activators in the parasite. However, the
mechanism of artemisinin activation remains incompletely understood, partly due to an inability to quantify pools of labile heme or labile iron in the parasite. Elucidating this mechanism is particularly critical given the potency of the artemisinins, which is not well understood, and the recent emergence of artemisinin resistance33
. Mechanistic study of artemisinin action could aid in developing additional artemisinin derivatives and identify validated molecular targets for new antimalarials.
Chloroquine, in particular, has been one of the most potent and successful drugs ever developed against an infectious disease25
1960s and 1970s85
. However, the mechanism(s) of chloroquine action remain controversial. Chloroquine has been shown to accumulate significantly in the digestive vacuoles of treated parasites86
. In vitro, chloroquine binds both free87
and exposed heme on growing hemozoin crystals88
and inhibits crystal growth. Additionally, chloroquine-treated parasites contain less detectable hemozoin89-91
, and higher amounts of heme unassociated with hemozoin or hemoglobin91
. Recently, increased extravacuolar iron density has also been observed in chloroquine-treated parasites91
. Taken together, these results support a model where the heme-chloroquine complex blocks heme detoxification to hemozoin in the digestive vacuole and causes free heme to accumulate in the parasite. However, a direct link between chloroquine and toxicity through accumulation of unbound heme has not been demonstrated, partly due to the inability to reliably quantify unbound heme in live cells.
Other effects of chloroquine treatment have been observed in a series of in vitro and in situ experiments. Chloroquine has been proposed to target polyamine biosynthesis based on its activity against ornithine decarboxylase in cultured parasites92
. Chloroquine has also shown inhibitory activity against protein synthesis both in cell-free extracts and in cultured parasites93
. In vitro, chloroquine has been shown to inhibit proteases involved in hemoglobin degradation94
, and to inhibit proposed heme degradation pathways involving hydrogen peroxide59 and
glutathione95
. However, the extent to which these interactions contribute to parasite toxicity are not known. Furthermore, recent studies with chloroquine analogues found that the inhibition of hemozoin formation was correlated with cytostatic but not cytocidal activity96
, suggesting that chloroquine toxicity may be the result of multiple mechanisms. Further studies are needed to dissect the mechanism of chloroquine cytotoxicity and elucidate the role(s) played by heme.
Resistance to chloroquine has been mapped to mutations in the P. falciparum chloroquine resistance transporter (PfCRT)97,98
, a transmembrane protein associated with the digestive vacuolar membrane99
. While the native role of PfCRT is not known, bioinformatic studies have suggested a possible function as a transporter of small molecules100,101
. Similarly, the role of PfCRT in determining chloroquine resistance has not been fully defined. Less chloroquine appears to accumulate in chloroquine-resistant parasites expressing mutant PfCRT than in sensitive strains102
, and similar results have been obtained in experiments using isolated digestive vacuoles103
. This suggests that resistance may be mediated primarily by reducing parasite exposure to chloroquine, and therefore chloroquine’s mechanism(s) of action may hold promise for future antimalarial development.
Figure 1-2. Summary of heme metabolism in blood-stage parasites, depicting hemoglobin digestion, hemozoin
formation, interactions with antimalarial drugs, and heme biosynthesis. Abbreviations used: amino acids (AA), heme detoxification protein (HDP), chloroquine (CQ), artemisinin (ART), and activated artemisinin (ART**). Figure from [61].
1.5 In vitro culture of P. vivax
A landmark 1976 study established a method for continuously propagating P. falciparum cultures using human red blood cells35
, which are readily available. This discovery was critical in subsequent research into P. falciparum biology, and as of early 2015, had been cited in over 5,000 articles (statistic from Web of Science). However, such a system does not exist for P. vivax, which is currently impractical to culture in vitro because it preferentially invades reticulocytes (immature red blood cells)104
, which constitute between 0.5% and 1.5% of the circulating cells in human peripheral blood105
. In contrast, P. falciparum invades both reticulocytes and mature red blood cells efficiently106
. Furthermore, reticulocytes mature rapidly into normocytes in vitro, with a half-life of approximately 30 hours107
. Therefore, propagating P. vivax relies on a continuous supply of a rare and transient blood component to maintain an adequate population of invadable cells. Studies reporting P. vivax propagation without enriched reticulocytes were either of very short duration108-112
, could not maintain high parasitemia113
, or could not be reproduced114,115
. Obtaining enriched reticulocytes for P. vivax culture is difficult and costly. In one study, reticulocytes were supplied from the blood of a hemochromatosis patient being treated by therapeutic phlebotomy and enriched by centrifuging the blood cells in homologous plasma. While this technique resulted in stable P. vivax propagation over a two-week period, total reticulocyte yields were low (< 20%)105
, and these results have not been replicated in other groups115
. Human umbilical cord blood, which is also naturally enriched in reticulocytes, has also been used for continuous P. vivax culture up to two months116-118
. However, these techniques were unable to maintain high parasitemia. In both cases, these techniques required continuous access to patient-derived samples that are not readily available. Reticulocytes can also be
HSC-derived reticulocytes have been shown to be usable in P. vivax culture119,120
. However, this
method is labor-intensive, as HSCs require two weeks of culture to mature into reticulocytes, and expensive, due to the mixture of growth factors and cytokines required121
. Parasites can be obtained for brief ex vivo studies from infected research animals, as P. vivax also infects New World monkeys of the Saimiri and Aotus genera105,122
. However, maintaining infected research animals (especially primates) is often cost-prohibitive and raises ethical concerns. In contrast to these existing methods, an ideal culturing system would propagate this parasite continuously using human normocytes and other reagents that are readily and inexpensively available. However, this would depend on overcoming the P. vivax preference for invading reticulocytes, the basis of which is only partially understood.
1.6 Invasion of red blood cells by malaria parasites
1.6.1 Overview of the invasion process
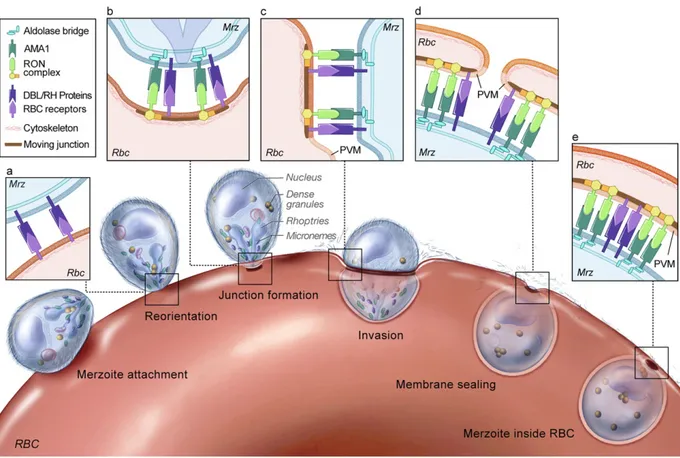
During the blood stage of malaria infection, parasites bind to and invade red blood cells in a multi-step process (Fig. 1-3)123
. Merozoites released from a bursting schizont quickly associate with erythrocytes, averaging less than 40 seconds between schizont rupture and contact with a potential host cell4. This initial contact is mediated by long distance and relatively low-affinity
interactions and can occur with the merozoite in any orientation124
. Invasion proteins are then released to the merozoite surface from secretory organelles termed rhoptries and micronemes, which are located at the apical end of the merozoite125
. Secretion of invasion proteins occurs shortly before the proteins are needed, which minimizes exposure to the host’s immune system and slows the development of an immune response126
The parasite then repositions to place its apical end in contact with the red blood cell127
, and through a series of ligand-receptor interactions, attaches irreversibly to the red cell membrane126
. By electron microscopy, the interface between parasite and host cell shows the electron density and close contact between the two cells typical of a tight junction128
. Finally, the parasite enters the red blood cell by moving the junction along its length, effectively pushing itself into the host cell and sealing the membrane closed behind it. Shortly after invasion, the red cell undergoes morphological changes induced by ion fluxes but then quickly returns to its previous shape4
.
Figure 1-3. Overview of the red blood cell invasion process. Merozoites (Mrz) initially attach to the red blood cell
(RBC) surface in any orientation through low-affinity interactions. After attachment, the merozoite reorients to place its apical end in close proximity to the RBC surface, where a series of ligand-receptor interactions (a) stabilize the formation of a tight junction (b). The parasite then moves the junction along its length and sheds its protein coat (c-d), creating the parasitophorous vacuole (PVM) and sealing the RBC membrane closed (e). Figure from [129], copyright © the authors.
1.6.2 Ligand-receptor interactions governing invasion
Ligand-receptor interactions that precede tight junction formation define the cell types that can be invaded by the merozoite and irreversibly begin the invasion process126,130,131
(Fig. 1-3). Two families of parasite ligands have been identified: the Duffy-binding like (DBL) and reticulocyte-binding like homologues (Rh), which originate in micronemes132,133
and rhoptries134
, respectively, before release to the merozoite surface. DBL-family proteins are homologous to P. vivax proteins that bind to the Duffy antigen and mediate invasion135,136
. The Rh proteins are homologous to a family of P. vivax proteins that bind to reticulocytes137,138
, and are believed to underlie the parasite’s reticulocyte-specific invasion tropism, although their cognate receptors have not yet been identified.
Like P. vivax, P. falciparum expresses ligands from both the DBL and Rh families. Ligand expression varies by strain, allowing strain-specific differences in host cell preference for invasion139
. The DBL-family proteins EBA-175 (for Erythrocyte Binding Antigen, 175 kDa), EBA-140, and EBA-181 mediate interactions with sialylated receptors on the red blood cell surface140
. EBA-175 binding to glycophorin A is likely the dominant interaction, as deleting EBA-175 from a sialic acid-dependent strain results in a switch to sialic acid-independent pathways for invasion139. Structural data showing EBA-175 co-crystallized with sialyllactose
demonstrates glycan contacts with two DBL domains, suggesting that dimerization of EBA-175 is also important for receptor binding141
. Additionally, inhibition studies with glycophorin A peptides demonstrate that the glycophorin A protein backbone participates in binding, either by direct contacts to EBA-175 or by maintaining a specific conformation of sialic acid residues140
P. falciparum can also invade host red blood cells through sialic acid-independent pathways mediated by Rh-family proteins. Of these, only the receptors for PfRh4 (complement receptor 1)142
and PfRh5 (basigin)143
have been identified. PfRh5 appears unique in that it cannot be disrupted and has limited homology to other proteins in the Rh family, suggesting that it may have an unrelated function144,145
After engaging receptors on the red cell surface through DBL and Rh-family proteins, parasites then secrete additional rhoptry proteins into the membrane of the host red blood cell146
. These proteins (termed RONs, for their apparent origin in the rhoptry neck) form a complex that provides a high-affinity anchor for the merozoite147
. The merozoite protein AMA-1 (for Apical Membrane Antigen-1) then binds to RON2 to form the tight junction and initiate invasion129
.
1.6.3 Linking ligand-receptor interactions to invasion outcomes
While multiple ligand-receptor interactions have been identified, it has remained challenging to link individual binding events to invasion outcomes. Although P. vivax expresses ligands that preferentially bind reticulocytes, it is unclear whether this preferential binding is solely
responsible for the inability of P. vivax to efficiently invade mature red blood cells. Current techniques to link ligand-receptor interactions with invasion outcomes rely on protease or glycosylase treatments that cleave necessary receptors from the host cell surface, or inhibit binding interactions with soluble competitors. These interventions are limited in their specificity: protease and glycosylase treatments remove broad classes of receptors from the host cell surface, and soluble competitors are often added at high concentrations in order to inhibit invasion.
Synthetic receptors represent a promising potential strategy for linking binding events to invasion outcomes. Providing a specific receptor in trans and promoting invasion into an
otherwise-refractory cell type would provide conclusive evidence that particular ligand-receptor interaction(s) are necessary and/or sufficient for a given invasion tropism. In the case of P. falciparum, synthetic receptors could demonstrate causal links between engaging various DBL or Rh proteins and strain-specific invasion preferences. In the case of P. vivax, synthetic receptors could demonstrate whether engaging the reticulocyte-binding proteins is sufficient to promote invasion into Duffy-positive mature red blood cells, and potentially facilitate culture system development.
1.7 Summary of rationale and work presented
This thesis documents the development of novel chemical biology tools to address critical needs in antimalarial drug discovery. Among these are validated molecular targets to guide drug discovery efforts. Some of the most potent and successful antimalarial drugs are thought to interact with parasite heme, although their mechanisms of action remain controversial.
Elucidating the mechanisms of action of these drugs, in the context of a broader understanding of parasite heme metabolism, would identify validated targets for future drug development efforts. Currently, heme metabolism and the action of heme-binding drugs are poorly understood because heme is difficult to quantify in situ. Chapter 2 describes the development of a novel genetically-encoded biosensor for quantifying labile heme in live cells, and applications in P. falciparum to derive new insights about parasite heme metabolism and antimalarial drug action.
Another critical need is a method for in vitro propagation of blood-stage P. vivax, which is currently impractical due to its preference for invading reticulocytes. The molecular basis for this preference is incompletely understood, precluding culture system development. Chapter 3
describes a chemoenzymatic toolkit for displaying synthetic receptors on the surface of the red blood cell, which can be used to link molecular interactions with invasion outcomes.
Proof-of-concept experiments in P. falciparum demonstrate the utility of this approach for elucidating structural requirements in a well-understood invasion pathway, and define rules for developing future synthetic receptor technologies geared towards facilitating in vitro culture of P. vivax parasites in mature erythrocytes.
1.8 References
1. WHO Global Malaria Programme. World Malaria Report 2014. (World Health Organization).
2. Krotoski, W. A. et al. Demonstration of hypnozoites in sporozoite-transmitted Plasmodium vivax infection. Am J Trop Med Hyg 31, 1291–1293 (1982).
3. Goldberg, D. E. Hemoglobin degradation. Curr Top Microbiol Immunol 295, 275–291 (2005).
4. Gilson, P. R. & Crabb, B. S. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int J Parasitol 39, 91–96 (2009).
5. Brancucci, N. M. B. et al. Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe 16, 165–176 (2014).
6. Kafsack, B. F. C. et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 507, 248–252 (2014).
7. Sinha, A. et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507, 253–257 (2014).
8. Ghosh, A., Edwards, M. J. & Jacobs-Lorena, M. The journey of the malaria parasite in the mosquito: hopes for the new century. Parasitol. Today (Regul. Ed.) 16, 196–201 (2000).
9. Miller, L. H., Baruch, D. I., Marsh, K. & Doumbo, O. K. The pathogenic basis of malaria. Nature 415, 673–679 (2002).
10. Francischetti, I. M. B., Seydel, K. B. & Monteiro, R. Q. Blood coagulation, inflammation, and malaria. Microcirculation 15, 81–107 (2008).
11. Moxon, C. A., Heyderman, R. S. & Wassmer, S. C. Dysregulation of coagulation in cerebral malaria. Mol Biochem Parasitol 166, 99–108 (2009).
12. Miller, L. H., Ackerman, H. C., Su, X.-Z. & Wellems, T. E. Malaria biology and disease pathogenesis: insights for new treatments. Nature medicine 19, 156–167 (2013).
13. Smith, J. D., Rowe, J. A., Higgins, M. K. & Lavstsen, T. Malaria's deadly grip:
cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell Microbiol 15, 1976– 1983 (2013).
14. Idro, R., Marsh, K., John, C. C. & Newton, C. R. J. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr. Res. 68, 267– 274 (2010).
15. Steketee, R. W., Nahlen, B. L., Parise, M. E. & Menendez, C. The burden of malaria in pregnancy in malaria-endemic areas. Am J Trop Med Hyg 64, 28–35 (2001).
16. McCormick, M. C. The contribution of low birth weight to infant mortality and childhood morbidity. N. Engl. J. Med. 312, 82–90 (1985).
17. da Silva, A. J. & Moser, M. Malaria. (2002). ID #3405 at <http://phil.cdc.gov/> 18. Liu, N. Insecticide resistance in mosquitoes: impact, mechanisms, and research
directions. Annu. Rev. Entomol. 60, 537–559 (2015).
19. Schwartz, L., Brown, G. V., Genton, B. & Moorthy, V. S. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar J 11, 11 (2012).
20. Vekemans, J., Leach, A. & Cohen, J. Development of the RTS,S/AS malaria candidate vaccine. Vaccine 27 Suppl 6, G67–71 (2009).
in African infants. N. Engl. J. Med. 367, 2284–2295 (2012).
22. Loeb, F. et al. ACTIVITY OF A NEW ANTIMALARIAL AGENT, CHLOROQUINE (SN 7618): Statement Approved by the Board for Coordination of Malarial Studies. JAMA 130, 1069–1070 (1946).
23. Slater, A. F. Chloroquine: mechanism of drug action and resistance in Plasmodium falciparum. Pharmacol. Ther. 57, 203–235 (1993).
24. Sá, J. M., Chong, J. L. & Wellems, T. E. Malaria drug resistance: new observations and developments. Essays Biochem 51, 137–160 (2011).
25. Wellems, T. E. & Plowe, C. V. Chloroquine-resistant malaria. J Infect Dis 184, 770–776 (2001).
26. D'Alessandro, U. & Buttiëns, H. History and importance of antimalarial drug resistance. Trop. Med. Int. Health 6, 845–848 (2001).
27. Trape, J. F. et al. Impact of chloroquine resistance on malaria mortality. C. R. Acad. Sci. III, Sci. Vie 321, 689–697 (1998).
28. Marsh, K. Malaria disaster in Africa. Lancet 352, 924 (1998).
29. Wells, T. N. C., Alonso, P. L. & Gutteridge, W. E. New medicines to improve control and contribute to the eradication of malaria. Nat Rev Drug Discov 8, 879–891 (2009). 30. Ecker, A., Lehane, A. M., Clain, J. & Fidock, D. A. PfCRT and its role in antimalarial
drug resistance. Trends Parasitol 28, 504–514 (2012).
31. Meshnick, S. R., Taylor, T. E. & Kamchonwongpaisan, S. Artemisinin and the
antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol. Rev. 60, 301–315 (1996).
32. Noedl, H. et al. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359, 2619–2620 (2008).
33. Ashley, E. A. et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371, 411–423 (2014).
34. malERA Consultative Group on Basic Science and Enabling Technologies. A research agenda for malaria eradication: basic science and enabling technologies. PLoS Medicine
8, e1000399 (2011).
35. Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
36. Mueller, I. et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. The Lancet Infectious Diseases 9, 555–566 (2009).
37. Price, R. et al. Vivax Malaria: Neglected and Not Benign. American Journal of Tropical Medicine and Hygiene 77, 79 (2007).
38. Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002).
39. Armstrong, C. M. & Goldberg, D. E. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nature Methods 4, 1007–1009 (2007).
40. Goldfless, S. J., Wagner, J. C. & Niles, J. C. Versatile control of Plasmodium falciparum gene expression with an inducible protein-RNA interaction. Nat Commun 5, 5329 (2014). 41. Wagner, J. C., Platt, R. J., Goldfless, S. J., Zhang, F. & Niles, J. C. Efficient
CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nature Methods 11, 915–918 (2014).
42. Wells, T. N. & Poll, E. M. When is enough enough? The need for a robust pipeline of high-quality antimalarials. Discov Med 9, 389–398 (2010).
43. Nwaka, S. & Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov 5, 941–955 (2006).
44. Gelb, M. H. Drug discovery for malaria: a very challenging and timely endeavor. Current Opinion in Chemical Biology 11, 440–445 (2007).
45. O'Neill, P. M. et al. Isoquine and related amodiaquine analogues: a new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 46, 4933–4945 (2003). 46. Rosenthal, P. J. & Meshnick, S. R. Hemoglobin catabolism and iron utilization by
malaria parasites. Mol Biochem Parasitol 83, 131–139 (1996).
47. Yayon, A., Timberg, R., Friedman, S. & Ginsburg, H. Effects of chloroquine on the feeding mechanism of the intraerythrocytic human malarial parasite Plasmodium falciparum. J. Protozool. 31, 367–372 (1984).
48. Becker, K. et al. Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int J Parasitol 34, 163–189 (2004).
49. Jeney, V. et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood 100, 879– 887 (2002).
50. Pagola, S., Stephens, P. W., Bohle, D. S., Kosar, A. D. & Madsen, S. K. The structure of malaria pigment beta-haematin. Nature 404, 307–310 (2000).
51. Egan, T. J. et al. Fate of haem iron in the malaria parasite Plasmodium falciparum. Biochem J 365, 343–347 (2002).
52. Dorn, A., Stoffel, R., Matile, H., Bubendorf, A. & Ridley, R. G. Malarial
haemozoin/beta-haematin supports haem polymerization in the absence of protein. Nature 374, 269–271 (1995).
53. Sullivan, D. J., Gluzman, I. Y. & Goldberg, D. E. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 271, 219–222 (1996).
54. Jani, D. et al. HDP-a novel heme detoxification protein from the malaria parasite. PLoS Pathog 4, e1000053 (2008).
55. Sullivan, D. J. Theories on malarial pigment formation and quinoline action. Int J Parasitol 32, 1645–1653 (2002).
56. Bendrat, K., Berger, B. J. & Cerami, A. Haem polymerization in malaria. Nature 378, 138–139 (1995).
57. Hempelmann, E., Motta, C., Hughes, R., Ward, S. A. & Bray, P. G. Plasmodium falciparum: sacrificing membrane to grow crystals? Trends Parasitol 19, 23–26 (2003). 58. Sigala, P. A., Crowley, J. R., Hsieh, S., Henderson, J. P. & Goldberg, D. E. Direct Tests
of Enzymatic Heme Degradation by the Malaria Parasite Plasmodium falciparum. J Biol Chem 287, 37793–37807 (2012).
59. Loria, P., Miller, S., Foley, M. & Tilley, L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem J
339 ( Pt 2), 363–370 (1999).
60. Atamna, H. & Ginsburg, H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J Biol Chem 270, 24876–24883 (1995).
61. Sigala, P. A. & Goldberg, D. E. The peculiarities and paradoxes of Plasmodium heme metabolism. Annu. Rev. Microbiol. 68, 259–278 (2014).
62. Lim, L. & McFadden, G. I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 365, 749–763 (2010).
63. van Dooren, G. G., Kennedy, A. T. & Mcfadden, G. I. The use and abuse of heme in apicomplexan parasites. Antioxid Redox Signal 17, 634–656 (2012).
64. Ralph, S. A. et al. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2, 203–216 (2004).
65. Sato, S., Clough, B., Coates, L. & Wilson, R. J. M. Enzymes for heme biosynthesis are found in both the mitochondrion and plastid of the malaria parasite Plasmodium falciparum. Protist 155, 117–125 (2004).
66. Nagaraj, V. A. et al. Localisation of Plasmodium falciparum uroporphyrinogen III decarboxylase of the heme-biosynthetic pathway in the apicoplast and characterisation of its catalytic properties. Int J Parasitol 39, 559–568 (2009).
67. Nagaraj, V. A., Prasad, D., Arumugam, R., Rangarajan, P. N. & Padmanaban, G. Characterization of coproporphyrinogen III oxidase in Plasmodium falciparum cytosol. Parasitology International 59, 121–127 (2010).
68. Nagaraj, V. A., Arumugam, R., Prasad, D., Rangarajan, P. N. & Padmanaban, G. Protoporphyrinogen IX oxidase from Plasmodium falciparum is anaerobic and is localized to the mitochondrion. Mol Biochem Parasitol 174, 44–52 (2010). 69. Nagaraj, V. A., Prasad, D., Rangarajan, P. N. & Padmanaban, G. Mitochondrial
localization of functional ferrochelatase from Plasmodium falciparum. Mol Biochem Parasitol 168, 109–112 (2009).
70. Severance, S. & Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem Rev
109, 4596–4616 (2009).
71. Kořený, L., Oborník, M. & Lukeš, J. Make it, take it, or leave it: heme metabolism of parasites. PLoS Pathog 9, e1003088 (2013).
72. Kessl, J. J. et al. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem 278, 31312–31318 (2003).
73. Srivastava, I. K., Rottenberg, H. & Vaidya, A. B. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272, 3961–3966 (1997).
74. Srivastava, I. K., Morrisey, J. M., Darrouzet, E., Daldal, F. & Vaidya, A. B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol Microbiol 33, 704–711 (1999).
75. Painter, H. J., Morrisey, J. M., Mather, M. W. & Vaidya, A. B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446, 88–91 (2007).
76. Campanale, N. et al. Identification and characterization of heme-interacting proteins in the malaria parasite, Plasmodium falciparum. J Biol Chem 278, 27354–27361 (2003). 77. Nagaraj, V. A. et al. Malaria parasite-synthesized heme is essential in the mosquito and
liver stages and complements host heme in the blood stages of infection. PLoS Pathog 9, e1003522 (2013).
78. Ke, H. et al. The heme biosynthesis pathway is essential for Plasmodium falciparum development in mosquito stage but not in blood stages. Journal of Biological Chemistry
289, 34827–34837 (2014).
79. Liu, S. C., Zhai, S. & Palek, J. Detection of hemin release during hemoglobin S denaturation. Blood 71, 1755–1758 (1988).
80. Sartorello, R. et al. In vivo uptake of a haem analogue Zn protoporphyrin IX by the human malaria parasite P. falciparum-infected red blood cells. Cell. Biol. Int. 34, 859–
865 (2010).
81. Brossi, A. et al. Arteether, a new antimalarial drug: synthesis and antimalarial properties. J. Med. Chem. 31, 645–650 (1988).
82. Meshnick, S. R., Thomas, A., Ranz, A., Xu, C. M. & Pan, H. Z. Artemisinin
(qinghaosu): the role of intracellular hemin in its mechanism of antimalarial action. Mol Biochem Parasitol 49, 181–189 (1991).
83. Klonis, N. et al. Artemisinin activity against Plasmodium falciparum requires
hemoglobin uptake and digestion. Proc Natl Acad Sci USA 108, 11405–11410 (2011). 84. Stocks, P. A. et al. Evidence for a common non-heme chelatable-iron-dependent
activation mechanism for semisynthetic and synthetic endoperoxide antimalarial drugs. Angew Chem Int Ed Engl 46, 6278–6283 (2007).
85. Payne, D. Spread of chloroquine resistance in Plasmodium falciparum. Parasitol. Today (Regul. Ed.) 3, 241–246 (1987).
86. Ginsburg, H. & Geary, T. G. Current concepts and new ideas on the mechanism of action of quinoline-containing antimalarials. Biochem. Pharmacol. 36, 1567–1576 (1987).
87. Bachhawat, K., Thomas, C. J., Surolia, N. & Surolia, A. Interaction of chloroquine and its analogues with heme: An isothermal titration calorimetric study. Biochem Biophys Res Commun 276, 1075–1079 (2000).
88. Sullivan, D. J., Matile, H., Ridley, R. G. & Goldberg, D. E. A common mechanism for blockade of heme polymerization by antimalarial quinolines. J Biol Chem 273, 31103– 31107 (1998).
89. Slater, A. F. & Cerami, A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature 355, 167–169 (1992).
90. Zhang, J., Krugliak, M. & Ginsburg, H. The fate of ferriprotorphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol Biochem Parasitol 99, 129–141 (1999).
91. Combrinck, J. M. et al. Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem Biol 8, 133–137 (2013).
92. Königk, E. & Putfarken, B. Inhibition of ornithine decarboxylase of in vitro cultured Plasmodium falciparum by chloroquine. Tropenmed Parasitol 34, 1–3 (1983).
93. Surolia, N. & Padmanaban, G. Chloroquine inhibits heme-dependent protein synthesis in Plasmodium falciparum. Proc Natl Acad Sci USA 88, 4786–4790 (1991).
94. Gluzman, I. Y. et al. Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J Clin Invest 93, 1602–1608 (1994).
95. Ginsburg, H., Famin, O., Zhang, J. & Krugliak, M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 56, 1305–1313 (1998).
96. Gorka, A. P. et al. Cytostatic versus cytocidal activities of chloroquine analogues and inhibition of hemozoin crystal growth. Antimicrob. Agents Chemother. 57, 356–364 (2013).
97. Fidock, D. A. et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861– 871 (2000).
98. Wellems, T. E., Walker-Jonah, A. & Panton, L. J. Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proc Natl Acad Sci USA 88,
3382–3386 (1991).
99. Cooper, R. A. et al. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol 61, 35–42 (2002).
100. Martin, R. E. & Kirk, K. The malaria parasite's chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol. Biol. Evol. 21, 1938–1949 (2004).
101. Tran, C. V. & Saier, M. H. The principal chloroquine resistance protein of Plasmodium falciparum is a member of the drug/metabolite transporter superfamily. Microbiology (Reading, Engl.) 150, 1–3 (2004).
102. Chinappi, M., Via, A., Marcatili, P. & Tramontano, A. On the mechanism of chloroquine resistance in Plasmodium falciparum. PLoS ONE 5, e14064 (2010). 103. Saliba, K. J., Folb, P. I. & Smith, P. J. Role for the plasmodium falciparum digestive
vacuole in chloroquine resistance. Biochem. Pharmacol. 56, 313–320 (1998).
104. Kitchen, S. The infection of reticulocytes by Plasmodium vivax. Am J Trop Med Hyg 1, 347 (1938).
105. Golenda, C. F., Li, J. & Rosenberg, R. Continuous in vitro propagation of the malaria parasite Plasmodium vivax. Proc Natl Acad Sci USA 94, 6786–6791 (1997).
106. Wilson, R. J., Pasvol, G. & Weatherall, D. J. Invasion and growth of Plasmodium falciparum in different types of human erythrocyte. Bull World Health Organ 55, 179– 186 (1977).
107. Skadberg, O., Brun, A. & Sandberg, S. Human reticulocytes isolated from peripheral blood: maturation time and hemoglobin synthesis. Lab Hematol 9, 198–206 (2003). 108. Bass, C. C. & Johns, F. M. THE CULTIVATION OF MALARIAL PLASMODIA
(PLASMODIUM VIVAX AND PLASMODIUM FALCIPARUM) IN VITRO. J Exp Med 16, 567–579 (1912).
109. Brockelman, C. R., Tan-Ariya, P. & Laovanitch, R. Observation on complete schizogony of Plasmodium vivax in vitro. J. Protozool. 32, 76–80 (1985).
110. Sutar, N. K. & Renapurkar, D. M. Effect of liver extract on growth of Plasmodium vivax in vitro. Indian J. Exp. Biol. 29, 286–287 (1991).
111. Zhou, W. Z. & Hu, L. Q. [Erythrocytic schizogony of Plasmodium vivax under various conditions of in vitro cultivation]. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 9, 258–260 (1991).
112. Chotivanich, K. et al. Ex-vivo short-term culture and developmental assessment of Plasmodium vivax. Trans. R. Soc. Trop. Med. Hyg. 95, 677–680 (2001).
113. Larrouy, G., Magnaval, J. F. & Moro, F. [Obtaining intraerythrocytic forms of
Plasmodium vivax by in vitro culture]. Comptes rendus des séances de l'Académie des sciences. Série III, Sciences de la vie 292, 929–930 (1981).
114. Renapurkar, D. M., Pradhan, V. R. & Sutar, N. K. The continuous in vitro cultivation of Plasmodium vivax. IRCS Medical Science 11, 7–8 (1983).
115. Noulin, F., Borlon, C., Van Den Abbeele, J., D’Alessandro, U. & Erhart, A. 1912-2012: a century of research on Plasmodium vivax in vitro culture. Trends Parasitol 29, 286– 294 (2013).
116. Borlon, C. et al. Cryopreserved Plasmodium vivax and cord blood reticulocytes can be used for invasion and short term culture. Int J Parasitol 42, 155–160 (2012).