Publisher’s version / Version de l'éditeur:
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca.
Questions? Contact the NRC Publications Archive team at
PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca. If you wish to email the authors directly, please see the first page of the publication for their contact information.
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
ACS Sustainable Chemistry & Engineering, 7, 1, pp. 1512-1523, 2018-12-14
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
NRC Publications Archive Record / Notice des Archives des publications du CNRC :
https://nrc-publications.canada.ca/eng/view/object/?id=4b1a63b9-c0a2-46f6-8a59-9ec675f354e1 https://publications-cnrc.canada.ca/fra/voir/objet/?id=4b1a63b9-c0a2-46f6-8a59-9ec675f354e1
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous.
https://doi.org/10.1021/acssuschemeng.8b05272
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Mechanistic investigation on catalytic deoxygenation of phenol as a
model compound of biocrude under methane
Wang, Aiguo; Austin, Danielle; He, Peng; Ha, Michelle; Michaelis, Vladimir
K.; Liu, Lijia; Qian, Hui; Zeng, Hongbo; Song, Hua
Mechanistic Investigation on Catalytic Deoxygenation of Phenol as
a Model Compound of Biocrude Under Methane
Aiguo Wang,
†Danielle Austin,
†Peng He,
†Michelle Ha,
‡Vladimir K. Michaelis,
‡Lijia Liu,
§Hui Qian,
∥Hongbo Zeng,
⊥and Hua Song
*
,††Department of Chemical and Petroleum Engineering, University of Calgary, 2500 University Drive, NW, Calgary, Alberta T2N
1N4, Canada
‡Department of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta T6G 2G2, Canada §Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Institute of Functional Nano and Soft Materials
(FUNSOM), Soochow University−Western University Centre for Synchrotron Radiation Research, Soochow University, Suzhou, Jiangsu 215123, China
∥National Institute for Nanotechnology, National Research Council, Edmonton, Alberta T6G 2M9, Canada
⊥Department of Chemical and Materials Engineering, University of Alberta, 9211-116 Street NW, Edmonton, Alberta T6G 1H9,
Canada
*
S Supporting InformationABSTRACT: A novel hydrogen-free approach is developed to effectively convert phenolic compounds into aromatics including benzene, toluene, ethylbenzene, and xylene (BTEX), a very important and widely used class of petrochemical intermediates, and naphthalene. High yield and selectivity of BTEX are achieved over Zn modified zeolite catalyst during phenol deoxygenation under a methane environment at 400 °C and 2 MPa. Methane can greatly enhance the liquid yield and selectivity of BTEX, but also improve the catalyst activity. Experimental analysis and mechanistic studies reveal that methane is incorporated into both the methyl group and aromatic ring. The embedding of methane into aromatic compounds is achieved via
C-alkylation of phenyl ring, coaromatization with hydrocarbon radicals, and a ring-expansion/contraction mechanism. Liquid-and solid-state NMR investigations make evident the phenyl ring-opening in the reaction. Our mechanistic understLiquid-anding of this process will provide valuable insights into the chemistry of phenolic deoxygenation, and open perspectives for valorization of biomass-derived oil with cheaper alternatives.
KEYWORDS: Phenol, Methane, Zeolite, Deoxygenation, Aromatics
■
INTRODUCTIONCatalytic conversion of biomass into fuels and valuable chemicals has the potential of substantially alleviating the dependence on fossil fuels.1−6 However, the liquid products from biomass are highly oxygenated (35−40 wt %1) and disadvantageous to be directly used as a transportation fuel.7,8 Removing oxygenates from biomass-derived products to improve the quality is necessary. In addition to alcoholic and carboxylic compounds as investigated in our previous studies,9,10 phenolic compounds are the major recalcitrant oxygenated species in the liquid products from biomass conversion, particularly lignocellulosic biomass.11 Catalytic transfer hydrogenation is an efficient strategy to selectively convert phenolics into corresponding arenes or naphthenes under the facilitation of a H-donor, such as 2-propanol,12 tetralin,13 and hydrogen.14,15 Extensive research efforts focus on hydrodeoxygenation (HDO) of phenolic compounds,16−18
including phenol,19,20 cresol,21−26 anisole,13,27 and guaia-col.28,29 Some attempts are made to eliminate the hydroxyl group of phenolics via the derivatization using an electron-withdrawing group such as triflate or mesylates;30 however, they produce stoichiometric quantities of nonrecyclable waste, which is understandably difficult for commercial applications on a large scale as required in lignin or bio-oil upgrading. Therefore, a hydrogen-lean or hydrogen-free approach to remove the phenolic compounds is desirable.31
Methane (CH4), the principal constituent of natural gas, can
be an ideal alternative to hydrogen as a H-donor for removing phenolic components because of its low cost and high hydrogen to carbon effective (H/Ceff) ratio. Higher H/Ceff
Received: October 12, 2018
Revised: December 6, 2018
Published: December 14, 2018
pubs.acs.org/journal/ascecg
Cite This:ACS Sustainable Chem. Eng. 2019, 7, 1512−1523
ratio is beneficial for the conversion of biomass derivatives into olefins and aromatics with less coke.32 It is reported that methane can be efficiently activated and converted into aromatics when cofed with higher hydrocarbons33−35 or oxygenated hydrocarbons.9,10,36In addition to the promotion effect of methane on the improvement of oil quality by reducing the oxygen content, methane can be incorporated into the liquid products to increase the yield.37−39Hence, it is promising and economically attractive to develop a technology that can utilize methane as a H-donor for the removal of phenolic compounds to produce high quality fuels or high value-added chemicals.
Methane deoxygenation of phenolic compounds is a considerably challenging chemical transformation. Needless to say, methane is not highly reactive and quite difficult to be activated to participate in the chemical reaction. Moreover, the dissociation energy of C−O bond in phenol is 465 kJ/mol, which is even higher than that of a C−H bond in methane (439 kJ/mol).12 Current approaches available for activating C−O bond mainly involve three reaction pathways depending on the used catalyst. Direct deoxygenation is proposed for the oxophilic metals such as Fe−Pd,40 Ru,41 and CoMoS2,24 in which C−O bond is weakened by the interaction between −OH and the oxophilic metal species or oxygen vacancy sites. HDO (hydrogenation followed by deoxygenation) is appli-cable to the low-oxophilicity metals such as Pt42 supported over acidic materials, and tautomerization is proposed for the non-acid-based supported catalysts such as Ni−Fe,43and Pt/ SiO2.44Both undergo hydrogenation of phenyl ring to reduce the electrophilicity of aromatic ring, thus lowering the activation energy barrier for the C−O cleavage. The most energetically favorable reaction mechanism for methane deoxygenation of phenolics is direct deoxygenation. The work performed by Yang et al.28,45demonstrated that methane could exhibit as good deoxygenation performance as hydrogen in terms of conversion and product distribution during guaiacol deoxygenation, which implies methane could be an alternative to hydrogen for phenolics deoxygenation. Addi-tionally, based on the thermodynamic analysis, provided that toluene is expected to be the major product when using methane as a coreactant, Gibbs free energy (ΔrG) of phenol deoxygenation, C6H6O(g) + CH4(g)⇋ C7H8(g) + H2O(g), is
calculated to be −25.5 kJ/mol at 400 °C and 1 atm. The negative ΔrG of this reaction indicates that this route is
thermodynamically feasible.
In this work, phenol is selected as the model compound to investigate the feasibility of phenolic deoxygenation with methane. A variety of supported metal catalysts are screened in terms of phenol conversion, BTEX selectivity, and aromatics yield at 400 °C and 2 MPa (BTEX: benzene, toluene, ethylbenzene, and xylene). The positive effect of methane on phenol deoxygenation is observed, and methane incorporation into liquid products is made evident by 13C NMR
investigations. The reaction pathway and mechanism are investigated by conducting pseudo-situ experiments combined with solid-state NMR analysis. The engaged catalyst is characterized including powder X-ray diffraction (XRD), X-ray absorption spectroscopy (XAS), X-X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), diffuse reflectance infrared transform spectroscopy (DRIFT), and temperature-programmed desorption (NH3
-TPD) to understand the intrinsic relationship between the physicochemical properties and the catalytic performance on
phenol deoxygenation. This novel process provides a more economical way for the conversion of phenolic compounds to high quality biofuels and valuable chemicals.
■
EXPERIMENTAL SECTIONCatalyst Preparation. The ammonium ZSM-5 zeolite with SiO2/ Al2O3of 30 (Zeolyst, CBV3024E, 405 m2/g) was calcined at 600°C for 5 h in air to attain HZSM5. Various metals such as Zn, Ag, Ga, Pt, Pd, Ru, and Ir were loaded as 1 wt % onto HZSM-5 via incipient wetness impregnation (IWI). Metal precursors were Zn(NO3)2·6H2O (99%, Alfa Aesar), AgNO3 (99.0+%, Sigma-Aldrich), Ga(NO3)3· xH2O (99.9%, Alfa Aesar), Pt(NH3)4(NO3)2 (≥50.0% Pt basis, Sigma-Aldrich), Pd(NO3)2·2H2O (∼40% Pd basis, Sigma-Aldrich), RuCl3·xH2O (Sigma-Aldrich), and IrCl3 (≥62% Ir, Alfa Aesar), respectively. IWI was conducted by dissolving corresponding amounts of metal precursors in DI water (10.0 g), followed by dropwise impregnation of 8.0 g of HZSM5 support. The material was subsequently dried at 92 °C overnight, followed by calcination at 550 °C (10 °C/min heating rate) and a holding time of 3 h in ambient air. The resulting catalysts were then stored in a sealed and moisture proof container for later use.
Catalytic Performance Evaluation. The phenol deoxygenation reaction was conducted in a batch Parr reactor with 100 or 300 mL. Typically, a 10:1 weight ratio of catalyst to phenol (0.1 g) was applied to the reactor (300 mL) and well mixed without any further solvent addition. The reactor was completely purged of air and pressurized (7 bar) with CH4or N2followed by heating to 400°C at a ramp rate of 20°C/min and subsequent holding for 1 h. Postreaction, the system was naturally cooled to room temperature. The incondensable gas products were analyzed before depressurizing using a four-channel micro-GC (Agilent 490). Equipped with thermal conductivity detectors (TCD’s), quantification of H2, O2, N2, CH4, and CO was conducted in thefirst channel (10 m molecular sieve 5A column and Ar carrier gas); CO2, C2H2, C2H4, and C2H6in the second channel (10 m PPU column and He carrier gas); and C3−C6 paraffin and olefins in channels 3 and 4 which are equipped with 10 m alumina (He carrier gas) and 8 m CP-Sil 5CB (He carrier gas) column, respectively. After releasing the gas in the reactor, CS2(GC grade, EMD chemicals) was used as an extractant to obtain liquid product embedded into the catalyst. Postextraction using 10:1 weight ratio of CS2to catalyst, the resulting diluted liquid product was applied to a carefully precalibrated GC-MS provided by PerkinElmer (GC Claus 680 and MS Clarus SQ 8T). The column was provided by Agilent (HP-PONA) and is capable of quantifying paraffins, olefins, naphthenes, and aromatics. The system was programmed as follows: 35°C hold for 15 min, ramp to 70 °C at a rate of 1.5 °C/min, ramp to 150°C at a rate of 3 °C/min, and hold for 30 min. The program was finally ramped to 250 °C at a rate of 3 °C/min and held for 2 min. A split ratio of 27.6 was used for the GC-MS analysis. The mass detector was set to scan the m/z range from 10 to 400. Identification of the compounds was achieved by comparing the mass spectra obtained with those in the system’s database (NIST).
Isotopic labeling experiments were conducted in a similar manner with some minor alterations. The reactor used was a 100 mL Parr reactor;13CH
4 was obtained from Cambridge Isotope Laboratories Inc. (99.9%13C), and CD
4 (99%2H) was obtained from the same provider. Phenol-1-13C was provided by Sigma-Aldrich (99% 13C). Time sensitive experiments were conducted by holding the final reaction temperature (400°C) for 5, 10, 30, 60, 90, and 120 min. Same reactions were also conducted at different temperatures (300, 350, and 400°C) and held for 1 s. Upon reaction completion, the reaction was quenched in ice cold water.
Calibrations of GC-MS and micro-GC responses were conducted using the standard solutions (benzene, toluene, xylene, naphthalene, and 2-methyl naphthalene) and standard gas mixtures with known concentrations. The mass of total gas is recorded by noting the weight difference after discharging gas from the reactor, and then the moles of each gas component are calculated using the ideal gas law based on gas composition determined from micro-GC. The moles of coke are
calculated by measuring the weight loss during the calcination of spent catalyst from 200 to 600°C assuming the coke is composed of pure carbon. All yields are reported in terms of carbon yield (moles of carbon in the products relative to those in the reactants). The gas, coke, and aromatic yields, as well as selectivity and methane conversion are given by the following equations:
= ×
gas product yield (C mol %)
moles of carbon in a gas product
total moles of carbon in the reactants 100% (1)
=i ×
k
jjj y{zzz
coke yield (C mol %)
moles of carbon in coke
total moles of carbon in the reactants 100% (2)
=i − ×
k
jjjj y{zzzz
aromatics yield (C mol %)
1 moles of carbon in gas and coke
total moles of carbon in the reactants 100% (3)
= ×
selectivity of aromatic products (C mol %) moles of carbon in a product
moles of carbon in identified aromatic products 100% (4)
=i − ×
k
jjjj y{zzzz
methane conversion (%)
1 moles of methane in gas products
moles of methane before reaction 100% (5) Generally, the aforementioned calculation error was measured within 5%.
Catalyst Characterization. The 1H,2H, and 13C solution-state NMR experiments were conducted on a Bruker Avance 400 NMR spectrometer equipped with a BBFO probe (9.4 T,ν0(1H) = 400.1 MHz;ν0(2H) = 61.4 MHz; andν0(13C) = 100.6 MHz).1H and13C chemical shifts were referenced to CDCl3 at 7.24 and 77.3 ppm, respectively.1H NMR spectra were obtained at a spectral width of 2.5 kHz, pulse delay of 2 s, and 64 scans per spectrum.13C NMR spectra were obtained at a spectral width of 26 kHz and 1000 scans. The unoptimized recycle delays for solution-state NMR were set at 2 s (less than the full relaxation time of carbon-13 nuclei). Thus, peak areas may not be proportional to the concentration of13C species in the products.
Solid-state1H and13C magic-angle spinning (MAS) NMR spectra were acquired at 7.05 T (v0(1H) = 300.37 MHz, v0(13C) = 75.54 MHz) on a Bruker Avance 300 NMR spectrometer equipped with a 4 mm double channel (H-X) Bruker probe. All samples were packed into 4 mm outer diameter zirconia rotors and acquired with a spinning frequency of 12 kHz.1H MAS NMR data were acquired using a Bloch pulse with a 4.0μs pulse width (90° pulse, γB1/2π = 62.5 kHz), 3.0 s recycle delay, and 64 scans.13C MAS NMR data were acquired using a cross-polarization46 pulse sequence with a 4.0 μs (90° pulse, γB1/2π = 62.5 kHz), contact time of 3 ms, a recycle delay of 3.0 s, and between 20 480 and 61 400 scans. All spectra were acquired with TPPM high power 1H decoupling (γB
1/2π = 62.5 kHz).47 1H data were referenced to TMS (δ = 0) by setting the residual1H peak of D2O, 99.9%2H, to 4.80 ppm, and the 13C data were referenced to TMS (δ = 0) by setting the high frequency adamantane peak to 38.56 ppm.48
XAS measurements were obtained from the synchrotron at the Canadian Light Source located at the University of Saskatchewan. Spectra were obtained at the Zn L3-edge influorescence mode with 50−60 scans followed by subsequent normalization using the incoming synchrotron beam flux signal (I0). A high resolution spherical grating monochromator (SGM) and beamline 1ID-1 (240− 2000 eV) were used.
More characterizations, such as XRD, TEM, DRIFT, NH3-TPD, H2-TPR, and XPS, are provided in theSupporting Information.
■
RESULTS AND DISCUSSIONCatalyst Screening. Various supported metal catalysts (1%Zn/ZSM5, 1%Ga/ZSM5, 1%Ag/ZSM5, 1%Pt/ZSM5, 1% Pd/ZSM5, 1%Ru/ZSM5, 1%Ir/ZSM5) are prepared by IWI. XRD patterns (shown in Figure S1) of these catalysts demonstrate that the doped metal species are well dispersed on the support and cause nondetectable distortion of the hierarchical framework pre- and postreaction. The catalytic performances on phenol deoxygenation over the catalysts are evaluated at 400°C and 2 MPa under a methane environment, and the corresponding results are presented inTable 1. Almost complete conversion of phenol is achieved over all the catalysts. BTEX and naphthalene are the major aromatic products, accounting for more than 98% of liquid composition, while CO and CO2 are the dominant components in gas products accompanied by a small amount of H2(not shown) and incondensable hydrocarbons (CxHy). Phenol conversion is
Table 1. Catalytic Performance on Phenol Deoxygenationaover Supported Various Metal Catalysts under a CH4Environment
entry 1, HZSM5 2, 1%Zn/ZSM5 3, 1%Ag/ZSM5 4, 1%Ga/ZSM5 5, 1%Pt/ZSM5 6, 1%Pd/ZSM5 7, 1%Ru/ZSM5 8, 1%Ir/ZSM5 phenol conversion (%) 99.8 99.9 99.8 100.0 100.0 99.8 100.0 100.0 methane conversion (%) 0.9 4.8 1.7 2.4 5.4 1.4 3.6 5.0 C balance (%) 98.3 106.8 112.0 112.4 105.5 106.7 112.9 103.1
Product Yields (C mol %)
CO 5.6 1.7 4.2 4.2 3.6 5.1 3.9 3.0
CO2 4.1 4.0 2.7 3.0 2.4 3.7 2.7 2.1
CxHy 2.4 0.4 1.1 0.9 1.2 1.3 0.1 0.8
aromatics 68.5 85.1 62.9 68.0 84.8 66.5 72.3 83.9
coke 19.5 8.8 29.2 24.0 8.0 23.5 20.2 10.2
Selectivity of Aromatic Products (C mol %)
BTEX 64.8 82.0 75.2 76.8 68.0 69.9 66.6 63.6
naphthalene 34.4 17.3 24.3 23.1 31.8 29.9 33.1 35.9
others 0.8 0.7 0.5 0.1 0.2 0.2 0.3 0.5
aReaction conditions:∼1.0 g of catalyst with ∼0.1 g of phenol, 400 °C, 60 min. C
xHy: incondensable hydrocarbons including C2H4, C2H6, C3H8, and C4H10. Aromatics: all identified aromatic compounds by GC-MS. BTEX: benzene and toluene with tiny proportion of ethylbenzene and xylene. Naphthalene: naphthalene and alkyl naphthalene. Others: C9aromatics, biphenyl, and biphenyl ether.
probably initiated by the generation of aryl, hydroxyl, phenoxy, and hydrogen radicals over the acidic sites of zeolite catalyst.49 A portion of these radicals recombine to yield trace quantities of biphenyl and biphenyl ether observed in GC-MS chromato-gram of liquid products. Most aryl radicals might react with hydrogen or methyl radicals derived from methane to form BTEX as the major monoaromatics in the condensable products. Phenoxy radicals may undergo the ring-opening defragmentation via decarbonylation, releasing carbon mon-oxide and producing the short-lived hydrocarbon radicals (such as •C5H6•, •C4H4•, •C2H2•, •CH2•).49−52 The
aromatic-ring-opening of phenol is also supported by the formation of alkyl aromatics (toluene, xylene, and other aromatics) over HZSM5 (entry 9) and 1%Zn/ZSM5 (entry 10) inTable 2, when phenol is the sole carbon source under a N2environment. The hydrocarbon radicals generated from the ring-opening are eventually converted into BTEX and naphthalene via aromatization or reactions with phenyl radicals. The formation of CO2might involve the secondary
reactions of CO such as water−gas shift (WGSR) and Boudouard reactions.49The small portion of hydrogen in the gas products may be derived from the aromatization of radicals and methane or the recombination of hydrogen radicals, while light hydrocarbons (CxHy) may originate from the
hydro-carbon radicals.
As shown in Table 1, the aromatics yield and methane conversion are significantly enhanced when Zn (entry 2), Pt (entry 5), and Ir (entry 8) species are introduced on the zeolite. This is understandable, because higher methane conversion could offer more hydrogen or methyl moieties for the formation of aromatic products, thus resulting in higher yields of aromatics. The addition of Ag (entry 3), Ga (entry 4), Pd (entry 6), or Ru (entry 7) leads to more coke when compared to the bare support (entry 1). Further comparison of aromatic products shows that 1%Zn/ZSM5 catalyst has the best selectivity of BTEX (entry 2) among them, suggesting that Zn addition favors aromatization or radical recombination to form BTEX rather than naphthalene. Lower yield of naphthalene is favorable, which would result in less formation of coke precursors (i.e., polyalkylaromatics and polyaromatics), and thus reduce carbonaceous residues53 and extend the catalyst lifetime. The good catalytic performance on phenol deoxygenation over 1%Zn/ZSM5 may be attributed to more Lewis acidic sites54(seen by the appearance of an additional peak at 1619 cm−1in DRIFT spectra upon pyridine adsorption inFigure S2) available and higher ratio of weak to strong acidic sites (determined by the characterization of acidic sites upon NH3-TPD in Table S1) created by Zn loading.55 Therefore, 1%Zn/ZSM5 is chosen for further investigations due to higher methane conversion, higher aromatics yield with better selectivity of BTEX, and lower coke production over other catalysts.
Methane Participation. Influence of Methane Presence. The effect of methane on phenol deoxygenation is evaluated by conducting equivalent reactions over HZSM5 and 1%Zn/ ZSM5 under CH4and N2environments. As presented inTable
2, the significant increase in aromatics yield along with the noticeable decrease in coke formation are visualized when the reactions are carried out under a CH4 environment.
Particularly, the aromatics yield is improved more pronoun-cedly than that from its N2 counterpart over 1%Zn/ZSM5.
Higher aromatics yield with an approximately 4.8% of methane conversion over 1%Zn/ZSM5 demonstrates that methane is activated and incorporated into aromatic products. When comparing aromatic products from the runs under CH4and N2
environments, the presence of methane greatly enhances BTEX selectivity by reducing the formation of naphthalene. The positive effect of methane on increasing the yield of aromatics and the selectivity of BTEX may be attributed to its potential to donate a hydrogen or methyl group, which can facilitate the formation of monoaromatic hydrocarbons such as BTEX while effectively preventing overaromatization for polyaromatics minimization.
Besides, methane promotes Zn dispersion over the catalyst as observed in XRD patterns (Figure S4), TEM-EDX (Figure S5), and XPS (Figure S6). The intensity of peaks derived from (101) and (020) crystal planes of ZSM5 in XRD patterns in Figure S4are well maintained under a CH4environment, but
significantly reduced under a N2environment probably due to
agglomeration of Zn on these crystal planes to a certain degree. This promoting effect is seen from the analysis of TEM-EDX in Figure S5. There is no signal of Zn species on the EDX Table 2. Catalytic Performance from the Control
Experiments over HZSM5 and 1%Zn/ZSM5 under CH4and
N2Environments HZSM5 1%Zn/ZSM5 CH4 N2 CH4 N2 entry 1 9 2 10 phenol conversion (%) 99.8 99.9 99.9 99.5 C balance (%) 98.3 98.9 106.8 104.2
Yields of Products (C mol %)
CO 5.6 4.9 1.7 3.3
CO2 4.1 2.8 4.0 6.8
CxHy 2.4 5.8 0.4 6.6
aromatics 68.5 51.0 85.1 56.2
coke 19.5 35.5 8.8 27.1
Selectivity of Aromatic Products (C mol %)
BTEX 64.8 48.8 82.0 66.7
naphthalene 34.4 50.6 17.3 32.8
others 0.8 0.6 0.7 0.5
Figure 1.Zinc L3-edge XAS spectra collected influorescence mode from metal Zn, ZnO references, fresh catalyst, and spent catalysts under CH4/N2 environment at room temperature. The spectra are normalized by subtracting a straight linefitted to zero before L3-edge.
spectra of the fresh catalyst (Figure S5d) and spent catalyst (Figure S5e) collected under a CH4environment, which may
be due to relatively low concentration of Zn caused by high dispersion, while the detectable Zn species on the spent catalyst (Figure S5f) collected under a N2 environment is indicative of slightly higher concentration of Zn species probably due to Zn agglomeration. These observations coincide with XPS analysis at Zn 2p region (Figure S6), in which Zn 2p peak intensity is slightly reduced under a CH4
environment but increases a little after reaction under a N2 environment.
The chemical environment of Zn species on the catalyst is explored by XAS. The XAS of Zn L3-edge probes the unoccupied Zn s and d derived states.56As shown inFigure 1, the Zn L3-edge is typically composed of three peaks. The peak at 1028 eV is ascribed to Zn 4s components, while the other peaks (at 1032 and 1037 eV) originate mainly from Zn 4d and O 2p hybridized band components. Higher Zn L3-edge absorption intensity is indicative of larger density of Zn 4s and 4d states due to the presence of oxygen defect.57 Compared with absorption intensity from the N2run, higher intensity of
Zn L3-edge observed in the CH4run suggests that methane is beneficial for the formation of unoccupied Zn s and d derived states by creating oxygen vacancy sites, which render a better activity of 1%Zn/ZSM5 on phenol deoxygenation. Interest-ingly, Zn species on fresh and spent catalysts exhibits the identical sharp L3-edge (E0= 1028.45 eV) located between the sharp edges of metal Zn (E0= 1026.95 eV) and ZnO (E0=
1030.15 eV) references. The variation in sharp L3-edge implies that Zn species on the zeolite catalyst are in a local chemical environment different from both metallic Zn and ZnO. This can be explained by the fact that Zn species can be chemically bonded to the zeolite support by replacement of H+ to form the coordinated Zn active sites (as shown in theSupporting Information). H2-TPR (temperature-programmed reduction)
(Figure S7) and ammonia adsorption measurements (Table S2) provide further evidence for the presence of coordinated Zn active sites.
Methane Incorporation. The direct sign of methane participation into phenol deoxygenation is made evident from the NMR spectra of aromatic products using 13CH
4
and CD4. The molecular ion region of the mass spectra of major products (benzene, toluene, naphthalene, and methyl naphthalene) formed from 13CH
4 and CH4 is presented in
Figure 2. The appreciable excess of the mole fraction of13C 1
(13.4%) singly and13C
2(7.4%) doubly labeled molecules for
toluene, as well as the presence of singly and doubly labeled molecules in other products from the13CH
4run (Figure 2b),
provides supplementary evidence for the incorporation of13
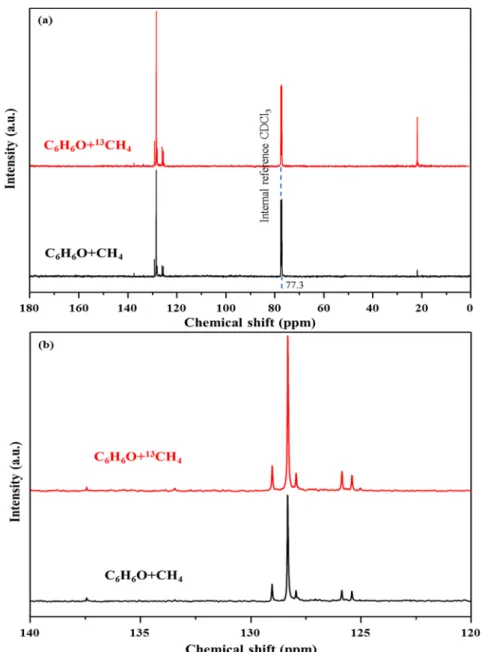
C-labeled methane into the aromatic products. More solid evidence is witnessed from liquid13C NMR spectra inFigure 3
and a semiquantitative comparative analysis in Table S3. Comparing with 13C signals originating from aromatic
products collected from the non-isotopic-labeled counterpart, the nonuniform increase in 13C signals from the 13CH
4 run
strongly supports the involvement of methane into phenol deoxygenation. The intensity of the peak at 21.70 ppm due to the benzylic carbon significantly increases (>10 times) when
13CH
4is employed, implying that methane is incorporated into
the methyl groups. The signals at the region 120−140 ppm in Figure 3b correspond to the resonances of carbons within aromatic ring. Combining with the product analyzed by GC-MS (entry 2 inTable 1), the peak at 137.55 ppm is attributed to the phenyl carbon of toluene or xylene attached with a methyl group, and the signal at 134.30 ppm is due to the carbons shared by two aromatic rings of naphthalene. The peaks at 129.05 and 125.42 ppm are assigned to ortho- and para-carbons of toluene or xylene, respectively. The chemical shifts at 127.96 and 125.88 ppm are derived from the carbons atα and β positions of naphthalene. The peak at 128.34 ppm with the largest intensity originates from benzene or the meta-carbon of toluene and xylene.58,59 The relatively high increment of the signals at 129.05 and 128.34 ppm indicates that methane is also incorporated into the phenyl ring of BTEX, preferentially ortho- and meta-carbons of toluene and
Figure 2.Mass spectra of major products benzene, toluene, naphthalene, methyl naphthalene: (a) with the natural abundance of 13C and (b) formed from13C-labeled methane and phenol at 400°C (60 min) on 1%Zn/ZSM5 with the estimated isotopic composition (mol %).
Figure 3.(a)13C liquid NMR spectra and (b) its zoom-in views in the 120−140 ppm chemical shift region of the liquid products collected from the reaction between phenol and13CH4/CH4. The triplet peak centered at 77.3 ppm belongs to CDCl3which serves as the internal reference for more accurate comparison.
Table 3. Yields of Aromatic, Gas, and Coke Products, and Phenol and Methane Conversion Collected in the Course of Phenol Deoxygenation over 1%Zn/ZSM5 under a CH4Environment
entry 11 12 13 14 15 16 17 18 19
temperature (°C) 300 350 400 400 400 400 400 400 400
time (min) 1sa 1s 1s 5 10 30 60 90 120
phenol conversion (%) 2.7 16.7 52.2 76.4 91.9 97.7 99.5 99.7 99.9
methane conversion (%) <0.1 <0.1 0.2 2.0 2.1 2.4 4.4 2.4 1.7
Yield of Products (C mol %)
CO 0.0 0.0 1.8 2.5 2.9 2.9 2.0 2.3 2.0 CO2 0.0 0.2 0.8 1.9 2.4 4.0 4.2 5.6 5.8 CxHy 0.0 0.3 0.4 0.5 0.7 0.6 0.7 0.6 0.7 BTEX 2.0 12.5 33.6 51.9 59.4 59.9 69.2 67.9 63.8 naphthalene 0.7 2.5 12.2 20.6 22.7 22.8 16.1 13.0 14.4 alkylphenol 0.0 1.0 0.7 0.4 0.2 0.0 0.0 0.0 0.0 coke 0.4 1.7 4.3 4.1 5.1 7.5 6.9 10.2 13.1
aThe initial stage of the reaction. Once temperatures reaches 300, 350, and 400°C in the ramp, respectively, the reactions are terminated.
Alkylphenol: cresol and xylenol.
xylene, and benzene ring. Therefore, methane tends to be incorporated into both the methyl group and aromatic ring.
The results of1H and2H solution-state NMR investigations also support the conclusion drawn from liquid 13C NMR spectra.1H (Figure S8a,b) and2H solution-state NMR (Figure S8c,d) spectra of aromatic products prepared using CD4are acquired and compared with those of natural abundance products. As shown in Table S4, the intensity of peaks originating from protons on phenyl rings (7.0−7.8 ppm) and benzylic hydrogens (2.2−2.8 ppm) is significantly reduced, which suggests that hydrogen atoms in methane favor the aromatic and benzylic hydrogen sites rather than the alkyl hydrogen sites. This matches the observation in 2H NMR spectra (Figure S7d) that two strong NMR resonances arise in the aromatic (∼7.6 ppm) and benzylic (∼2.6 ppm) regions.
Reaction Pathway Investigation. Pseudo-Situ Reac-tions. The evolution of phenol deoxygenation with methane is studied pseudo-situ by quenching the reaction at different temperatures (300, 350, and 400 °C) and terminating the reaction with various reaction times (5, 10, 30, 60, 90, and 120 min) at 400 °C. The results obtained from these trials are tabulated in Table 3. As the reaction proceeds, phenol is gradually consumed and converted into BTEX and naph-thalene accompanied by the increased formation of CO and CO2. The yield of BTEX reaches a maximum after the reaction
of 60 min at 400°C (entry 17). Further reaction reduces the formation of aromatic hydrocarbons and has an insignificant influence on the gas yield, which implies that the formed aromatics are partially converted into coke because of excessive aromatization, which leads to more coke deposited on the catalyst with the reaction (entry 18 and 19). A reasonable mechanism could be that most of the active sites are initially occupied for the conversion of phenol into aryl and/or phenoxy radicals when phenol is abundant. With the consumption of phenol, these active sites are free and catalyze the polymerization of naphthalene and BTEX to yield the coke.60 Meanwhile, a part of phenol is probably directly attacked by methane to form cresol or xylenol (not shown) via C-alkylation over the acidic catalyst at the initial stage,61which are observed in liquid products collected from the trials of 350 °C 1 s (entry 12), 400 °C 1 s (entry 13), 400 °C 5 min (entry 14), 400°C 10 min (entry 15). The formed cresol and xylenol are gradually converted into toluene and xylene as the final
products or phenoxy to participate in the subsequent reactions. The formation of CO is related to the aromatic-ring-opening followed by decarbonylation of phenoxy radicals derived from phenol. These phenoxy radicals are witnessed in the form of the trace of diphenyl ether detected by GC-MS (not shown) in aromatic products. The formation of CO2 involves the
secondary reactions of CO (i.e., WGSR, Boudouard reaction). The intermediate radicals, such as methylene (•CH2•), ethylene (•CHCH•), and diene (•CHCHCH CH•) radicals, are generated from phenyl ring fragmentation and react with aryl or activated methane to ultimately produce aromatics by means of recombination, aromatization, and Diels−Alder reaction. Light hydrocarbons (CxHy) in the gas
products are formed in the reactions between these radicals and hydrogen atoms or methane. Based on the above analysis, a hypothetical reaction network involved in phenol deoxyge-nation with methane over 1%Zn/ZSM5 is proposed inFigure 4. Aromatic-ring-opening reaction and methane activation might be the limiting steps because of higher energy barrier associated with these two reactions, slowing down the splitting rate of phenoxy moiety and the supply rate of methyl or proton species to react with aryl radicals. Therefore, surplus phenoxy and aryl moieties would recombine to form the trace of biphenyl and diphenyl ether (not shown) in the products.
Additionally, it is interesting to notice that methane conversion exhibits the same trend as the yield of aromatic products. Lower methane conversion after 60 min of reaction (entry 18 and 19) is due to the demethylation, which removes the methyl group of alkyl aromatics (toluene, ethylbenzene, xylene, and methyl naphthalene) to generate methane. Coincidentally, the intensity of Zn L3-edge at 1028 eV (Figure S9) displays the similar tendency with methane conversion during the reaction. The initially observed reduction in peak intensity is due to the interaction between phenol and catalyst, which decreases the number of unoccupied 4sp and 3d states. This hypothesis is quite reasonable, because the pseudo- first-order rate constant of phenol (6.7× 10−2gcat−1min−1) is much
higher (124 times) than that of methane (5.4 × 10−4 gcat−1 min−1) as shown in Figure S10. It is speculated that, at the initial stage, most of the active sites are occupied by phenol molecules, and limited active sites are used for methane activation (low conversion of methane). The positive effect of methane becomes more dominant with phenol consumption,
which results in the increase of Zn L3-edge intensity. The Zn L3-edge intensity reaches the highest level after 60 min, suggesting that the catalyst has the optimal activity, which contributes to the highest aromatics yield and methane conversion (entry 17), upon which, more coke formed over
the catalyst decreases the density of unoccupied 4sp and 3d states, deactivating the catalyst.
Validation with Solid-State NMR. The formation of hydrocarbon radicals in phenol deoxygenation mentioned above is further confirmed by solid-state NMR investigations. As illustrated inFigure 5, the signals around 129.1 and 19.6 ppm are assigned to13C-labeled carbon atoms of aromatic ring
and the methyl group,34,62−65respectively. This points to the formation of benzene and methyl-substituted aromatic hydro-carbons (i.e., toluene and xylenes) as observed inTable 3. The signal at 140 ppm belongs to the phenyl carbon bound to a methyl group. The peak at 154 ppm is attributed to the phenyl carbon attached by the hydroxyl group (phenol),66,67and the small shoulder at 158.7 ppm is associated with phenol derivatives. Compared to the unlabeled counterpart (Figure 5a), the significant increase in the signal at 19.6 ppm along with similar peak intensity around 129.1 ppm in Figure 5b implies that methane is activated and initially attached to phenyl ring as a methyl group through C-alkylation. As the reaction proceeds, more methane is incorporated into aromatics ring as verified by the notable increase in the signal intensity at 129.1 ppm in Figure 5e. Namely, methane is incorporated into both the methyl group and aromatic ring in the reaction, which is consistent with the observation in 1H MAS NMR in Figure S11 and liquid 13C NMR spectra in
Figure 3. It is worth noting that the signal at 19.6 ppm due to the methyl group is enhanced significantly after 1 h of the reaction between phenol-1-13C and methane as shown in
Figure 5. Solid-state 13C CP/MAS NMR investigation into the reaction of methane and phenol over Zn/ZSM-5 zeolite. The13C CP/ MAS NMR spectra were recorded upon the reaction of phenol and CH4for (a) 1 s and (d) 1 h, phenol and13CH4for (b) 1 s and (e) 1 h, phenol-1-13C and CH
4for (c) 1 s and (f) 1 h at 400°C. * denotes the spinning side bands.
Figure 6.(a) 13C liquid NMR spectra and (b) an expansion of the 120−140 ppm chemical shift region of the liquid products collected from the reactions between phenol/phenol-1-13C and CH
4. The triplet peak centered at 77.3 ppm belongs to CDCl3which serves as the internal reference for more accurate comparison.
Scheme 1. Mechanism of the Transfer from Phenol-1-13C
(Phenyl Ring) into the Benzylic Carbon Site (Side Chain) during Phenol Deoxygenation
Figure 5f, and the increase in this peak intensity is also observed in Figure 5c (compared with that in Figure 5a). These observations demonstrate that phenol-1-13C also contributes to the formation of the methyl group. The transfer of phenol-1-C from phenyl carbon site to benzylic carbon site is achieved through a ring-expansion/contraction mechanism as reported previously.10,65The resonance at 115.6 ppm is the result of the ortho-carbon of phenol or the formation of π-complexes absorbed on the zeolite catalyst.63,68,69 The π-complex intermediates are derived from the hydrocarbon radicals (i.e., •CHCH•, •CHCHCHCH•), which could be produced by methane or phenol fragmentation during the reaction. These hydrocarbon radicals are stabilized by hydrogen or methyl moieties to form olefin intermediates, and interact with active sites (such as Zn, Al) to formπ-complex intermediates on the catalyst surface.68,69 Interestingly, the intensity of peak at 115.6 ppm inFigure 5a,b is quite similar, but it becomes stronger inFigure 5c when using phenol-1-13C as feedstock, suggesting that the increase in peak intensity in Figure 5c is attributed to phenol-1-13C rather than methane.
However, the13C-labeled phenol (phenol-1-13C) cannot lead to the increase in signal unless the phenyl ring (or aryl radicals derived from phenol) is fragmented into π-complex intermediates, and thus increase the intensity of peak at 115.6 ppm. After 1 h of reaction, all these radicals and most of phenol are converted into aromatics as the final product as made evident by the disappearance of the signal at 115.6 ppm, the small broad peak at 154 ppm, as well as significant growth of the signal at 129.1 ppm inFigure 5f.
Phenyl Ring-Opening Reaction. The formation of inter-mediateπ-complexes due to the fragmentation of phenyl ring provides a proof for phenyl ring-opening reaction. The results of liquid13C NMR investigations listed inTables S3 and S5
show the slight loss of phenol-1-C, which may be caused by aromatic-ring-opening reaction that removes phenol-1-C in the form of CO or CO2. This assumption is verified by GC-MS
analysis of gas products obtained from the reactions between phenol/phenol-1-13C and methane as shown in Figure S12.
The significant increase in the mole fraction of 13C-labeled molecules for CO (31.11%) and CO2(76.95%) indicates that a part of carbon in the gas products is derived from the
phenol-1-13C of phenol. Namely, the aromatic ring of phenol is cleaved via decarbonylation to form the primary gas product CO and secondary product CO2. Additionally, the phenyl ring-opening reaction is also made evident by the significant increase in the formation of alkyl benzene (inTable S6) due to the cracking of the benzene ring when exclusively using benzene as the feedstock under a N2atmosphere. The dissociation energy of
CC bond (near phenol-1-C site) in phenol is theoretically lower than that in benzene due to the hydroxyl group; therefore, the phenyl ring of phenol is more readily split at phenol-1-C site.
Mechanism for Methane Incorporation.Figure 6displays
13C solution-state NMR spectra and an enlarged region
between 120 and 140 ppm of the aromatic products from the reactions between phenol/phenol-1-13C and methane. The peak signals, especially those due to aromatics (inFigure 6b), are significantly increased in the13C NMR spectrum collected
from the isotopic-labeled sample, indicating that most of phenol-1-C atoms remain in the phenyl ring of aromatic products. In other words, direct deoxygenation of phenol is prevailing over decarbonylation. Interestingly, the intensity of the peak at 21.72 ppm (Figure 6a) increases greatly, demonstrating that a fraction of phenol-1-C atoms are integrated into the methyl groups through a ring-expansion/ contraction mechanism as described in Scheme 1. This observation is in reasonable agreement with the results of
13C CPMAS NMR presented inFigure 5. These carbons in the
side chain derived from phenol-1-C, in turn, mutually explain why some carbons from methane show the preference for the ortho-substituted positions of aromatics as shown inFigure 3a andTable S3.
Based on solution and solid-state13C NMR analysis, as well as the proposed reaction pathway above, methane incorpo-ration into aromatic products during phenol deoxygenation is achieved through three mechanisms as illustrated inScheme 2. The primary reaction for the involvement of methane into aromatic products is C-alkylation of phenyl ring over acidic sites of zeolite catalyst, in which methane reacts with phenol and phenol derivative aryl to form cresol and toluene. The intermediate cresol is eventually converted into toluene via Scheme 2. Possible Mechanisms Involved in Phenol Deoxygenation for13C Label of Methane Embedding into Aromatics
direct deoxygenation. This may be the main route for methane incorporation into the aromatic branch as a methyl group. The aromatization of methane and hydrocarbon radicals formed in phenyl ring-opening followed by fragmentation is likely to be the governing pathway for the embedding of 13C-labeled
methane into the aromatic ring. Certainly, the intramolecular scrambling of ortho-carbon in toluene via a ring-expansion/ contraction mechanism also contributes to the slight enrich-ment of the13C atoms of aromatic ring from methane.
■
CONCLUSIONThis work presents the selective transformation of phenol into aromatic hydrocarbons under methane over 1%Zn/ZSM-5. In addition to promoting the dispersion of Zn species, the coreactant methane significantly improves the selectivity of BTEX and the yield of aromatics. The loaded Zn species not only appropriately modulates the acidity of catalyst, but also enhances methane activation which could provide more hydrogen atoms and methyl groups for phenol deoxygenation. The investigations by liquid- and solid-state NMR spectrosco-py demonstrate that methane tends to be incorporated into the phenyl rings and benzylic sites of aromatic compounds. The transfer of methane-13C carbon atoms into aromatic molecules is achieved via C-alkylation of phenyl ring, coaromatization with hydrocarbon radicals, and a ring-expansion/contraction mechanism. Apart from direct deoxygenation of phenol, the phenyl ring is cleaved to generate hydrocarbon radicals, which undergo recombination, aromatization, and Diels−Alder reaction to form BTEX and naphthalene as thefinal products. Our work may provide valuable insights into phenolic deoxygenation mechanism and open perspectives for exploring the heterogeneous catalysts for valorization of biomass-derived oil with cheaper alternatives.
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssusche-meng.8b05272.
XRD, TEM, DRIFT, NH3-TPD, H2-TPR, XPS, XAS,
and solid-state NMR of 1%Zn/ZSM-5 at various life stages; solution-state NMR and GC-MS of gas and liquid products; and reaction rate measurement and calculation (PDF)
■
AUTHOR INFORMATION Corresponding Author*Fax: +1 (403) 284-4852. Phone: +1 (403) 220-3792. E-mail: sonh@ucalgary.ca. ORCID Michelle Ha:0000-0002-4447-529X Vladimir K. Michaelis: 0000-0002-6708-7660 Lijia Liu: 0000-0003-2439-0739 Hongbo Zeng: 0000-0002-1432-5979 Hua Song:0000-0002-2791-1723 Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSThe authors gratefully acknowledgefinancial support from the Natural Sciences and Engineering Research Council of Canada
(NSERC, RGPIN/04385-2014). We also acknowledge Uni-versity of Alberta, Alberta/Technical UniUni-versity of Munich International Graduate School for Hybrid Functional Materials (ATUMS) training program supported by NSERC CREATE and NSERC-DG for research support.
■
REFERENCES(1) Resende, F. L. P. Recent advances on fast hydropyrolysis of biomass. Catal. Today 2016, 269, 148−155.
(2) Bridgwater, A. V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68−94.
(3) Stocker, M. Biofuels and biomass-to-liquid fuels in the biorefinery: catalytic conversion of lignocellulosic biomass using porous materials. Angew. Chem., Int. Ed. 2008, 47 (48), 9200−11.
(4) Alonso, D. M.; Wettstein, S. G.; Dumesic, J. A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41 (24), 8075−98.
(5) Serrano-Ruiz, J. C.; Dumesic, J. A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels. Energy Environ. Sci. 2011, 4 (1), 83−99.
(6) Alonso, D. M.; Bond, J. Q.; Dumesic, J. A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12 (9), 1493.
(7) French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91 (1), 25−32.
(8) Lu, Q.; Zhang, Z. F.; Dong, C. Q.; Zhu, X. F. Catalytic upgrading of biomass fast pyrolysis vapors with nano metal oxides: An analytical Py-GC/MS study. Energies 2010, 3 (11), 1805−1820.
(9) Wang, A.; He, P.; Yung, M.; Zeng, H.; Qian, H.; Song, H. Catalytic co-aromatization of ethanol and methane. Appl. Catal., B 2016, 198, 480−492.
(10) Wang, A.; Austin, D.; Karmakar, A.; Bernard, G. M.; Michaelis, V. K.; Yung, M. M.; Zeng, H.; Song, H. Methane upgrading of acetic acid as a model compound for a biomass-derived liquid over a modified zeolite catalyst. ACS Catal. 2017, 7 (5), 3681−3692.
(11) Bu, Q.; Lei, H.; Ren, S.; Wang, L.; Holladay, J.; Zhang, Q.; Tang, J.; Ruan, R. Phenol and phenolics from lignocellulosic biomass by catalytic microwave pyrolysis. Bioresour. Technol. 2011, 102 (13), 7004−7007.
(12) Wang, X.; Rinaldi, R. A route for lignin and bio-oil conversion: dehydroxylation of phenols into arenes by catalytic tandem reactions. Angew. Chem., Int. Ed. 2013, 52 (44), 11499−11503.
(13) Prasomsri, T.; To, A. T.; Crossley, S.; Alvarez, W. E.; Resasco, D. E. Catalytic conversion of anisole over HY and HZSM-5 zeolites in the presence of different hydrocarbon mixtures. Appl. Catal., B 2011, 106 (1−2), 204−211.
(14) Choudhary, T. V.; Phillips, C. B. Renewable fuels via catalytic hydrodeoxygenation. Appl. Catal., A 2011, 397 (1−2), 1−12.
(15) Gilkey, M. J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6 (3), 1420−1436.
(16) Shafaghat, H.; Sirous Rezaei, P.; Daud, W. M. A. W. Catalytic hydrogenation of phenol, cresol and guaiacol over physically mixed catalysts of Pd/C and zeolite solid acids. RSC Adv. 2015, 5 (43), 33990−33998.
(17) Jongerius, A. L.; Jastrzebski, R.; Bruijnincx, P. C. A.; Weckhuysen, B. M. CoMo sulfide-catalyzed hydrodeoxygenation of lignin model compounds: An extended reaction network for the conversion of monomeric and dimeric substrates. J. Catal. 2012, 285 (1), 315−323.
(18) Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An overview on catalytic hydrodeoxygenation of pyrolysis oil and Its model compounds. Catalysts 2017, 7 (6), 169.
(19) Gu, G. H.; Mullen, C. A.; Boateng, A. A.; Vlachos, D. G. Mechanism of dehydration of phenols on noble metals via first-principles microkinetic modeling. ACS Catal. 2016, 6 (5), 3047− 3055.
(20) Mortensen, P. M.; Grunwaldt, J.-D.; Jensen, P. A.; Jensen, A. D. Screening of catalysts for hydrodeoxygenation of phenol as a model compound for bio-oil. ACS Catal. 2013, 3 (8), 1774−1785.
(21) Do, P. T. M.; Foster, A. J.; Chen, J.; Lobo, R. F. Bimetallic effects in the hydrodeoxygenation of meta-cresol on γ-Al2O3 supported Pt−Ni and Pt−Co catalysts. Green Chem. 2012, 14 (5), 1388.
(22) Tan, Q.; Wang, G.; Nie, L.; Dinse, A.; Buda, C.; Shabaker, J.; Resasco, D. E. Different product distributions and mechanistic aspects of the hydrodeoxygenation of m-Cresol over platinum and ruthenium catalysts. ACS Catal. 2015, 5 (11), 6271−6283.
(23) Robinson, A.; Ferguson, G. A.; Gallagher, J. R.; Cheah, S.; Beckham, G. T.; Schaidle, J. A.; Hensley, J. E.; Medlin, J. W. Enhanced hydrodeoxygenation of m-Cresol over bimetallic Pt−Mo catalysts through an oxophilic metal-induced tautomerization path-way. ACS Catal. 2016, 6 (7), 4356−4368.
(24) Liu, G.; Robertson, A. W.; Li, M. M.-J.; Kuo, W. C. H.; Darby, M. T.; Muhieddine, M. H.; Lin, Y.-C.; Suenaga, K.; Stamatakis, M.; Warner, J. H.; Tsang, S. C. E. MoS2monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9 (8), 810.
(25) Griffin, M. B.; Ferguson, G. A.; Ruddy, D. A.; Biddy, M. J.; Beckham, G. T.; Schaidle, J. A. Role of the support and reaction conditions on the vapor-phase deoxygenation of m-Cresol over Pt/C and Pt/TiO2catalysts. ACS Catal. 2016, 6 (4), 2715−2727.
(26) Hong, Y.; Zhang, H.; Sun, J.; Ayman, K. M.; Hensley, A. J. R.; Gu, M.; Engelhard, M. H.; McEwen, J.-S.; Wang, Y. Synergistic catalysis between Pd and Fe in gas phase hydrodeoxygenation of m-Cresol. ACS Catal. 2014, 4 (10), 3335−3345.
(27) Peters, J. E.; Carpenter, J. R.; Dayton, D. C. Anisole and guaiacol hydrodeoxygenation reaction pathways over selected catalysts. Energy Fuels 2015, 29, 909−916.
(28) Xiao, Y.; Varma, A. Kinetics of guaiacol deoxygenation using methane over the Pt−Bi catalyst. React. Chem. Eng. 2017, 2 (1), 36− 43.
(29) Gao, D.; Xiao, Y.; Varma, A. Guaiacol hydrodeoxygenation over platinum catalyst: reaction pathways and kinetics. Ind. Eng. Chem. Res. 2015, 54 (43), 10638−10644.
(30) Sajiki, H.; Mori, A.; Mizusaki, T.; Ikawa, T.; Maegawa, T.; Hirota, K. Pd/C-catalyzed deoxygenation of phenol derivatives using Mg metal and MeOH in the presence of NH4OAc. Org. Lett. 2006, 8 (5), 987−990.
(31) Mettler, M. S.; Vlachos, D. G.; Dauenhauer, P. J. Top ten fundamental challenges of biomass pyrolysis for biofuels. Energy Environ. Sci. 2012, 5 (7), 7797.
(32) Zhang, H.; Cheng, Y. T.; Vispute, T. P.; Xiao, R.; Huber, G. W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with ZSM-5: the hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4 (6), 2297.
(33) Anunziata, O. Improvement of methane activation using n-hexane as co-reactant over Zn/HZSM-11 zeolite. Catal. Commun. 2004, 5 (8), 401−405.
(34) Luzgin, M. V.; Rogov, V. A.; Arzumanov, S. S.; Toktarev, A. V.; Stepanov, A. G.; Parmon, V. N. Methane aromatization on Zn-modified zeolite in the presence of a co-reactant higher alkane: How does it occur? Catal. Today 2009, 144 (3−4), 265−272.
(35) He, P.; Gatip, R.; Yung, M.; Zeng, H.; Song, H. Co-aromatization of olefin and methane over Ag-Ga/ZSM-5 catalyst at low temperature. Appl. Catal., B 2017, 211, 275−288.
(36) Choudhary, V. R.; Mondal, K. C.; Mulla, S. A. R. Simultaneous conversion of methane and methanol into gasoline over bifunctional Ga-, Zn-, In-, and/or Mo-modified ZSM-5 zeolites. Angew. Chem. 2005, 117 (28), 4455−4459.
(37) He, P.; Shan, W.; Xiao, Y.; Song, H. Performance of Zn/ZSM-5 for In situ catalytic upgrading of pyrolysis bio-oil by methane. Top. Catal. 2016, 59 (1), 86−93.
(38) He, P.; Song, H. Catalytic conversion of biomass by natural gas for oil quality upgrading. Ind. Eng. Chem. Res. 2014, 53 (41), 15862− 15870.
(39) Yang, Z.; Kumar, A.; Apblett, A. Integration of biomass catalytic pyrolysis and methane aromatization over Mo/HZSM-5 catalysts. J. Anal. Appl. Pyrolysis 2016, 120, 484−492.
(40) Hensley, A. J. R.; Wang, Y.; McEwen, J.-S. Phenol deoxygenation mechanisms on Fe(110) and Pd(111). ACS Catal. 2015, 5 (2), 523−536.
(41) Chen, H.Y. T.; Pacchioni, G. Role of oxide reducibility in the deoxygenation of phenol on ruthenium clusters supported on the anatase titania (101) surface. ChemCatChem 2016, 8 (15), 2492− 2499.
(42) Zanuttini, M. S.; Dalla Costa, B. O.; Querini, C. A.; Peralta, M. A. Hydrodeoxygenation of m-cresol with Pt supported over mild acid materials. Appl. Catal., A 2014, 482, 352−361.
(43) Nie, L.; de Souza, P. M.; Noronha, F. B.; An, W.; Sooknoi, T.; Resasco, D. E. Selective conversion of m-cresol to toluene over bimetallic Ni−Fe catalysts. J. Mol. Catal. A: Chem. 2014, 388−389, 47−55.
(44) Nie, L.; Resasco, D. E. Kinetics and mechanism of m-cresol hydrodeoxygenation on a Pt/SiO2 catalyst. J. Catal. 2014, 317, 22− 29.
(45) Xiao, Y.; Varma, A. Catalytic deoxygenation of guaiacol using methane. ACS Sustainable Chem. Eng. 2015, 3 (11), 2606−2610.
(46) Pines, A.; Gibby, M. G.; Waugh, J. S. Proton-enhanced nuclear induction spectroscopy. A method for high resolution NMR of dilute spins in solids. J. Chem. Phys. 1972, 56 (4), 1776−1777.
(47) Bennett, A. E.; Rienstra, C. M.; Auger, M.; Lakshmi, K. V.; Griffin, R. G. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995, 103 (16), 6951−6958.
(48) Earl, W. L.; VanderHart, D. L. Measurement of13C chemical shifts in solids. J. Magn. Reson. (1969-1992) 1982, 48 (1), 35−54.
(49) Thilakaratne, R.; Tessonnier, J. P.; Brown, R. C. Conversion of methoxy and hydroxyl functionalities of phenolic monomers over zeolites. Green Chem. 2016, 18 (7), 2231−2239.
(50) Long, L.; Wang, X.; Ding, Z.; Xie, L.; Zhang, Z.; Dong, J.; Lin, H.; Fu, X. Cyclopentadiene transformation over H-form zeolites: TPD and IR studies of the formation of a monomeric cyclopentenyl carbenium ion intermediate and its role in acid-catalyzed conversions. J. Catal. 2008, 255 (1), 48−58.
(51) Khachatryan, L.; Adounkpe, J.; Maskos, Z.; Dellinger, B. Formation of cyclopentadienyl radical from the gas-phase pyrolysis of hydroquinone, catechol, and phenol. Environ. Sci. Technol. 2006, 40 (16), 5071−5076.
(52) Zhu, L.; Bozzelli, J. W. Kinetics and thermochemistry for the gas-phase keto-enol tautomerism of phenol to 2,4-cyclohexadienone. J. Phys. Chem. A 2003, 107, 3696−3703.
(53) Dejaifve, P.; Gravelle, P. C.; et al. Methanol conversion on acidic ZSM-5, offretite, and mordenite zeolites: A comparative study of the formation and stability of coke deposits. J. Catal. 1981, 70 (1), 123−136.
(54) Topaloǧlu Yazıcı, D.; Bilgiç, C. Determining the surface acidic properties of solid catalysts by amine titration using Hammett indicators and FTIR-pyridine adsorption methods. Surf. Interface Anal. 2010, 42 (6−7), 959−962.
(55) Veses, A.; Puértolas, B.; López, J. M.; Callén, M. S.; Solsona, B.; García, T. Promoting deoxygenation of bio-oil by metal-loaded hierarchical ZSM-5 zeolites. ACS Sustainable Chem. Eng. 2016, 4 (3), 1653−1660.
(56) Ekicibil, A.; Ozkendir, O. M.; Farha, A. H.; Ufuktepe, Y. Study of the electronic properties of Zn0.8−4xHoxOy(0.05≤ x ≤ 0.09) by X-ray absorption and photoemission spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2015, 202, 56−61.
(57) Yuste, M.; Escobar Galindo, R.; Caretti, I.; Torres, R.; Sánchez, O. Influence of the oxygen partial pressure and post-deposition annealing on the structure and optical properties of ZnO films grown by dc magnetron sputtering at room temperature. J. Phys. D: Appl. Phys. 2012, 45 (2), No. 025303.
(58) Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents,
Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29 (9), 2176−2179. (59) Pretsch, E.; Buhlmann, P.; Badertscher, M. Structure Determination ofOrganic Compounds Tables of Spectral Data Fourth; Springer, 2009.
(60) Schmidt, J. E.; Poplawsky, J. D.; Mazumder, B.; Attila, O.; Fu, D.; de Winter, D. A.; Meirer, F.; Bare, S. R.; Weckhuysen, B. M. Coke formation in a zeolite crystal during the methanol-to-hydrocarbons reaction as studied with atom probe tomography. Angew. Chem., Int. Ed. 2016, 55 (37), 11173−7.
(61) Sad, M. E.; Padró, C. L.; Apesteguía, C. R. Study of the phenol methylation mechanism on zeolites HBEA, HZSM5 and HMCM22. J. Mol. Catal. A: Chem. 2010, 327 (1−2), 63−72.
(62) Wang, X.; Xu, J.; Qi, G.; Wang, C.; Wang, Q.; Deng, F. Alkylation of benzene with carbon monoxide over Zn/H-ZSM-5 zeolite studied using in situ solid-state NMR spectroscopy. Chem. Commun. (Cambridge, U. K.) 2014, 50 (77), 11382−4.
(63) Gabrienko, A. A.; Arzumanov, S. S.; Moroz, I. B.; Toktarev, A. V.; Wang, W.; Stepanov, A. G. Methane activation and transformation on Ag/H-ZSM-5 zeolite studied with solid-state NMR. J. Phys. Chem. C 2013, 117 (15), 7690−7702.
(64) Liu, Y.; Li, D.; Wang, T.; Liu, Y.; Xu, T.; Zhang, Y. Efficient conversion of methane to aromatics by coupling methylation reaction. ACS Catal. 2016, 6 (8), 5366−5370.
(65) Luzgin, M. V.; Rogov, V. A.; Arzumanov, S. S.; Toktarev, A. V.; Stepanov, A. G.; Parmon, V. N. Understanding methane aromatiza-tion on a Zn-modified high-silica zeolite. Angew. Chem., Int. Ed. 2008, 47 (24), 4559−62.
(66) Zhao, Z.; Shi, H.; Wan, C.; Hu, M. Y.; Liu, Y.; Mei, D.; Camaioni, D. M.; Hu, J. Z.; Lercher, J. A. Mechanism of phenol alkylation in zeolite H-BEA using in situ solid-state NMR spectros-copy. J. Am. Chem. Soc. 2017, 139 (27), 9178−9185.
(67) Hu, J. Z.; Solum, M. S.; Taylor, C. M. V.; Pugmire, R. J.; Grant, D. M. Structural determination in carbonaceous solids using advanced solid state NMR techniques. Energy Fuels 2001, 15 (1), 14−22.
(68) Gabrienko, A. A.; Arzumanov, S. S.; Toktarev, A. V.; Stepanov, A. G. Solid-state NMR characterization of the structure of intermediates formed from olefins on metal oxides (Al2O3and Ga2O3). J. Phys. Chem. C 2012, 116 (40), 21430−21438.
(69) Gabrienko, A. A.; Stepanov, A. G. Structure of allylic intermediate on zinc oxide, π or σ? J. Phys. Chem. C 2012, 116 (20), 11096−11099.