Study of the HIV-1-mediated induction of BAFF in
primary human monocytes and monocyte-derived
macrophages.
Thèse
Alejandro Martin Gomez
Doctorat en Microbiologie-Immunologie
Philosophiae Doctor (Ph.D)
Québec, Canada
iii
Résumé
L’infection par le VIH-1 (Virus de l’Immunodéficience Humaine de type I) est caractérisée par une réplication virale persistante, une activation chronique du système immunitaire, une déplétion des cellules T CD4+ et de plusieurs dysfonctionnements immunitaires qui sont observés même chez les
cellules qui ne sont pas ciblées par le virus telles que les cellules B. Plusieurs cytokines et facteurs de croissance, qui sont augmentées dans le sérum des individus infectés par le VIH-1, ont été suggérés de déclencher directement et/ou indirectement l'activation des cellules B, et l'un d'eux est BAFF (B cell activating factor). BAFF est un composant essentiel de l'homéostasie des lymphocytes B mais sa surproduction résulte en une hyperplasie des cellules B, lymphoprolifération, une hypergammaglobulinémie et des symptômes d’autoimmunité. Les différentes études présentées dans cette thèse convergent vers l'objectif général de mieux caractériser les mécanismes qui mènent à la surproduction de BAFF par les monocytes primaires humains et les macrophages dérivés des monocytes dans le contexte de l’infection par le VIH-1. Nous démontrons que le VIH-1 cause la sécrétion de BAFF dans les monocytes par un processus dépendant de l’interféron (IFN) de type-I. De plus, nous avons identifié les cellules dendritiques plasmacytoïdes (CDP) comme la source de l’IFN de type-I dans nos cultures de monocytes, ce qui démontre que l’interaction entre ces cellules est nécessaire pour la production de BAFF générée par le VIH-1. En plus, nous avons aussi démontré que le VIH-1 régule la production de BAFF dans les macrophages et ce processus nécessite une infection productive par le virus et est influencé par le statut du phénotype cellulaire mais est indépendant de la transduction de signal par les récepteurs de type Toll, l’IFN de type-I ainsi que l'action de la protéine virale Nef. En résumé, cette thèse offre de nouvelles connaissances dans les mécanismes d'augmentation de BAFF dans le contexte de l’infection par le VIH-1. Celles-ci pourraient être pertinentes dans le développement de thérapies qui pourraient aider à restaurer la fonctionnalité normale du compartiment des cellules B chez les sujets infectés par le VIH-1.
v
Summary
HIV-1 (Human Immunodeficiency Virus I) infection is characterized by persistent viral replication, chronic immune activation, CD4+ T cell depletion and several immune dysfunctions that are observed
even in cells that are not targeted by the virus such as B lymphocytes. Some B-cell abnormalities observed in HIV-1-infected individuals include hypergammaglobulinemia, nonspecific B-cell activation, class switching, increased cell turnover, breakage of tolerance as well as a loss of the capacity to generate and maintain memory, among others. Several cytokines and growth factors that are increased in the serum of HIV-1-infected individuals have been suggested to directly and/or indirectly trigger B-cell activation, and one of these is the B-cell-activating factor (BAFF). BAFF is an essential component of B-cell homeostasis but excess production results in B-cell hyperplasia, lymphoproliferation, hypergammaglobulinemia, and symptoms of autoimmunity. The mechanisms of BAFF upregulation in the context of HIV-1 infection are not fully understood and no previous studies have addressed the ability of fully competent HIV-1 to induce BAFF production by myeloid cells. The different studies presented in this thesis converge to the general objective of better characterizing the mechanisms underlying BAFF upregulation by primary human monocytes and monocyte-derived macrophages in the context of HIV-1 infection. We show here that HIV-1 drives BAFF secretion in monocytes by a type-I interferon (IFN)-dependent process. Moreover, we identified plasmacytoid dendritic cells (pDCs) as the cellular source of this type-I IFN-directed modulatory effect in our monocyte cultures, demonstrating that a pDC/monocyte interplay is required for the HIV-1-induced BAFF production. In addition, we provide evidence that HIV-1 upregulates BAFF production in monocyte-derived macrophages and this process relies on productive virus infection, which is itself influenced by the cell phenotype status, and is independent of Toll-like receptors and type-I IFN signal transduction as well as the action of Nef. Altogether, this doctoral project provides new insights for the increased BAFF levels observed during HIV-1 infection. These findings might be relevant for the design of therapies that could help restore the functionality of the B-cell compartment in HIV-1-infected individuals.
vii
Table of contents
Résumé ... iii
Summary ... v
Table of contents... vii
List of Tables ... xi
List of Figures ... xiii
List of Abbreviations... xv
Acknowledgements... xxi
Foreword ... xxiii
Chapter 1: Introduction ... 1
1 Section 1: The Human Immunodeficiency Virus (HIV) ... 1
1.1 The discovery of HIV as the cause of AIDS ... 1
1.2 Origin, classification and diversity of HIV ... 2
1.3 Epidemiology ... 3
1.4 Transmission ... 4
1.5 Structure ... 5
1.5.1 Genome organization ... 5
1.5.2 Morphology ... 6
1.5.3 Viral proteins and their functions ... 7
1.6 Viral tropism ... 8
1.6.1 CD4 receptor , CXCR4 and CCR5 coreceptors ... 8
1.6.2 Alternative receptors and coreceptors ... 9
1.6.3 Other attachment molecules ... 10
1.7 Replicative cycle ... 10
1.7.1 Early steps ... 10
1.7.1.1 Attachment and entry ... 10
1.7.1.2 Uncoating and reverse transcription ... 12
1.7.1.3 Nuclear import and Integration ... 13
1.7.2 Late steps ... 14
1.7.2.1 Transcription and translation ... 14
viii
1.8 Restriction factors of viral replication ... 16
1.8.1 APOBEC3G ... 16 1.8.2 TRIM5α ... 17 1.8.3 SAMHD1 ... 17 1.8.4 Tetherin ... 18 1.9 Pathogenesis ... 18 1.9.1 Phases of infection ... 18
1.9.1.1 Eclipse and acute phase ... 18
1.9.1.2 Chronic or latent phase ... 20
1.9.1.3 AIDS ... 21
1.10 Prevention and Treatment ... 21
1.10.1 Reverse transcriptase inhibitors ... 22
1.10.2 Protease Inhibitors ... 22
1.10.3 Integrase Inhibitors ... 22
1.10.4 Entry Inhibitors ... 22
2 Section 2: Immunopathogenesis of HIV-1 infection ... 23
2.1 Cells involved in HIV-1 immunopathogenesis. ... 23
2.1.1 Cells of the lymphoid lineage ... 24
2.1.1.1 CD4+ T lymphocytes ... 24
2.1.1.2 CD8+ T lymphocytes ... 26
2.1.1.3 NK cells ... 27
2.1.1.4 B lymphocytes ... 27
2.1.1.5 Plasmacytoid dendritic cells. ... 27
2.1.2 Cells of the lymphoid myeloid lineage ... 29
2.1.2.1 Monocytes ... 29
2.1.2.2 Macrophages ... 32
2.1.2.3 Dendritic cells ... 35
2.1.3 Other cellular types ... 35
2.2 Innate immune recognition of HIV-1. ... 36
2.2.1 Toll-like receptors ... 36
2.2.2 RIG-like receptors ... 38
2.2.3 Other cytosolic sensors ... 38
ix
2.4 Dysfunction of the B-cell compartment. ... 42
2.4.1 Direct effects of viral replication on B cells. ... 43
2.4.2 Indirect effects of viral replication on B cells. ... 43
2.4.3 Effect of ART in B-cell functionality. ... 44
3 Section 3: The B-cell-activating factor (BAFF) ... 46
3.1 Structure ... 46
3.2 Expression ... 46
3.3 BAFF receptors and their functions ... 47
3.4 BAFF-induced signals ... 48
3.5 Physiological roles of BAFF... 49
3.6 The role of BAFF in human diseases ... 51
3.6.1 Autoimmunity ... 51
3.6.2 Cancer ... 51
3.6.3 Infectious diseases ... 51
3.7 BAFF-targeting therapy ... 52
3.8 BAFF and HIV-1 infection ... 53
Chapter 2: Research hypothesis and objectives ... 57
1 Research hypothesis ... 57
2 Research objectives ... 57
Chapter 3: HIV-1-triggered release of type I IFN by plasmacytoid dendritic cells induces BAFF production in monocytes. ... 59
1 Preface ... 59
2 Article ... 60
Chapter 4: HIV-1-mediated BAFF secretion in macrophages does not require TLR, type-I interferon and Nef but depends on the cellular phenotype status. ... 91
1 Preface ... 91
2 Article ... 92
Chapter 5: Discussion ... 129
1 HIV-1-mediated BAFF production by monocytes relies on a type-I IFN response produced by pDCs………….. ... 130
2 HIV-1-mediated BAFF production by monocyte-derived macrophages ... 136
Chapter 6: Conclusions and Perspectives ... 143
Bibliography ... 147
xi
List of Tables
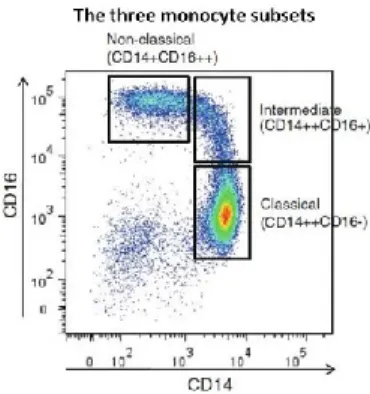
Table 1: Functions of HIV-1 proteins ... 8 Table 2: Phenotypic and functional differences between the three monocyte subsets ... 30
xiii
List of Figures
Figure 1: People living with HIV in the world ... 4
Figure 2: Organization of the HIV-1 genome ... 6
Figure 3: Schematic representation of HIV-1 ... 7
Figure 4: The replicative cycle of HIV-1 ... 11
Figure 5: HIV-1 restriction and the resistance factors ... 16
Figure 6: Time course of a typical HIV-1 infection ... 19
Figure 7: Flow cytometry dot plot showing the gating of the three monocyte subsets ... 29
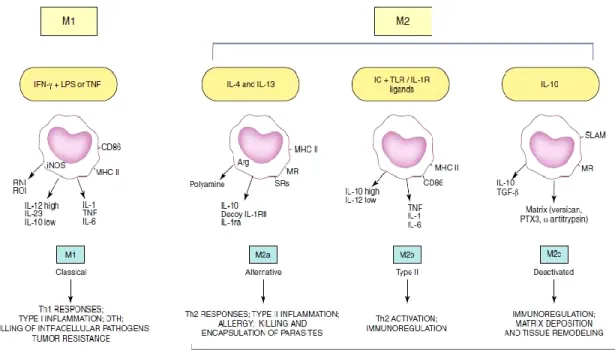
Figure 8: General concepts and properties of polarized macrophages ... 34
Figure 9: Mammalian TLR distribution and their signalization pathways in the cell ... 37
Figure 10: Summary of the differences between non-pathogenic and pathogenic SIV infection ... 40
Figure 11: Factors associated with HIV-induced immune activation ... 41
Figure 12: Direct and indirect effects of HIV-1 replication on B cells ... 42
Figure 13: BAFF, APRIL and their receptors ... 48
xv
List of Abbreviations
Ab Antibody
ADCC Antibody-Dependent Cell-mediated Cytotoxicity
AGM African Green Monkeys
AID Activation-Induced cytidine Deaminase
AIDS Acquired Immune Deficiency Syndrome
APC Antigen-Presenting Cell
APOBEC3G Apolipoprotein B mRNA Editing enzyme, Catalytic polypeptide-like 3G
APRIL A Proliferation-Inducing Ligand
ART Antiretroviral Therapy
ARV AIDS-associated Retrovirus
ATP Adenosin Triphosphate
AZT Zidovudine
BAFF B-cell-Activating Factor
BAFF-R BAFF Receptor
BBB Blood Brain Barrier
BCMA B-Cell Maturation Antigen
BCR B-Cell Receptor
BSA Bovine Serum Albumin
CA Capsid
Cdk Cyclin-dependent kinase
cDNA Complementary Deoxyribonucleic Acid
CRD Cysteine-Rich Domain
cGAMP Cyclic GMP-AMP
CRF Circulating Recombinant Forms
CSR Class Switch Recombination
CTL Cytotoxic T-Cell
xvi
CTRL Control
CVID Common Variable Immunodeficiency
CyA Cyclophilin A
DC Dendritic Cell
DCIR Dendritic Cell Immunoreceptor
DC-SIGN Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing
Non-integrin
DMEM Dulbecco's Modified Eagle Medium
DNA Deoxyribonucleic acid
dNTP Deoxynucleosidetriphosphate
dsRNA Double-stranded Ribonucleic Acid
EAE Experimental Autoimmune Encephalomyelitis
EBV Epstein-Barr Virus
EDTA Ethylenediaminetetraacetic acid
EFV Efavirenz
ELISA Enzyme-Linked Immunosorbent Assay
Env Envelope
ER Endoplasmic Reticulum
FBS Fetal Bovine Serum
FCRL4 Fc Receptor-Like 4
FDC Follicular Dendritic Cell
GALT Gut-Associated Lymphoid Tissue
GC Germinal Center
GSL Glycosphingolipid
GVHD Graft Versus Host Disease
HAART Highly Active Antiretroviral Therapy
HAD HIV-Associated Dementia
HCV Hepatitis C Virus
xvii
HEV High Endothelial Venules
HIV Human Immunodeficiency Virus
HTLV Human T-lymphotropic virus
iDC Immature Dendritic Cell
IDO Indoleamine-pyrrole 2,3-dioxygenase
IDU Injection Drug Use
IFI16 IFN-Inducible Protein 16
IFN Interferon
IL Interleukin
IN Integrase
IRF Interferon Regulatory Factor
ISG Interferon-Stimulated Gene
LAV Lymphadenopathy-Associated Virus
LGP2 Laboratory of Genetics and Physiology 2
LPS Lipopolysaccharide
LTNP Long-Term Non-Progression
LTR Long Terminal Repeat
MA Matrix
M-CSF Macrophage Colony-Stimulating Factor
MDA5 Melanoma Differentiation-Associated gene 5
mDC Myeloid Dendritic Cell
MDDC Monocyte-Derived Dendritic Cell
MDM Monocyte-Derived Macrophage
MSM Men who have Sex with Men
MVC Maraviroc
MZ Marginal Zone
Nab Neutralizing antibody
xviii
Nef Negative regulatory factor
NNRTI Non-Nucleoside Reverse Transcriptase Inhibitor
NOD Nucleotide-binding Oligomerization Domain
NRTI Nucleoside Reverse Transcriptase Inhibitor
O.D Optical Density
ORF Open Reading Frame
PAMP Pathogen-Associated Molecular Pattern
PBMC Peripheral Blood Mononuclear Cell
PBS Phosphate-Buffered Saline
pDC Plasmacytoid Dendritic Cell
PD-1 Programmed Cell Death 1
PI Protease Inhibitor
PIC Pre-Integration Complexes
PR Protease
PRR Pattern Recognition Receptor
RA Rheumatoid Arthritis
Rev Regulator of expression of virion proteins
RIG-I Retinoic acid-Inducible Gene-I
RLR RIG-like Receptor
RNA Ribonucleic Acid
RNAPII Ribonucleic Acid Polymerase II
RPMI Roswell Park Memorial Institute medium
RSV Respiratory Syncytial Virus
RT Reverse Transcriptase
RTC Reverse-Transcription Complexes
SAMHD1 Sterile Alpha Motif and Histidine-aspartic acid Domain containing protein 1
sCD4 Soluble CD4
xix
SEAP Secreted Embryonic Alkaline Phosphatase
siRNA Small interference Ribonucleic Acid
SIV Simian Immunodeficiency Virus
SLE Systemic Lupus Erythematosus
SS Sjögren's Syndrome
ssRNA Single-stranded Ribonucleic Acid
SU Surface
TACI Transmembrane Activator and CAML Interactor
TAR Transactivation Response
Tat Trans-activator of transcription
TBK TANK-Binding Kinase
Tcm T central memory cell
TCR T-Cell Receptor
Te T effector cell
Tem T effector memory cell
Tfh T follicular helper
Th T helper
TIM-3 T cell Immunoglobulin domain and Mucin-3
TLR Toll-like receptor
TM Transmembrane
Tn T naive cell
TNF Tumor necrosis factor
TRAF TNF Receptor Associated Factor
Tregs T regulatory cells
TREX1 Three Prime Repair Exonuclease 1
TRIM5α Tripartite Motif-containing protein 5
UNAIDS United Nations Programme on HIV and AIDS
xx
Vif Viral infectivity factor
xxi
Acknowledgements
First and foremost, I would like to thank Dr. Michel J. Tremblay for giving me the opportunity to join his lab to pursue my doctoral studies. Thanks for trusting in me and giving me the freedom to develop my research projects. Thank you for your generosity, your disposal, your efficacy, your advices, your humanity and your work ethic, which have contributed to my development as a researcher and will remain an example I will aspire to. I want to thank the members of the jury for accepting to be part of my thesis committee. Thank you for your time and contribution. I would also like to thank Dr. Michel Ouellet who gave me the freedom to develop my competences at my own pace, but who was always there if I needed advice or a different perspective. I would like to thank the past and present team members, who contributed to generate a pleasant environment of work, for being patient with my Spanish-French-English in my first days and for the friendship that I have established with some of them.
Being away from home is not easy and my days in Québec would not have been the same without the close friendship I had with people from around the world. Thanks Lupe for being there when I needed, not only as a friend but also as a scientist, thank you for being my Argentinean connection, for helping me to come to Quebec, your advices, discussions and for opening me the doors of your home and family. Thanks so much Laetitia, Réjean, Alma, Adriano, Andres, Julie, Lupe, Pablo and Rafik for making me feel at home and for being my “family” during these last five years.
Je voudrais remercier les Québécois qui ont fait partie de ma vie personnelle pendant mes années au Québec. Merci pour votre soutien pendant mon doctorat, pour votre patience, pour m’avoir montré le Québec avec vos yeux, pour votre générosité, pour m’avoir aidé à me sentir comme un québécois dans votre terre, pour l’échange culturel, pour me comprendre et de me faire vivre des beaux moments qui resteront toujours dans mon cœur.
Finalmente quiero agradecer a mi gran familia y amigos en Argentina. Gracias por estar siempre ahí a pesar de las distancias. Gracias por entenderme y aceptar mis decisiones. Gracias por ayudarme en los momentos en los cuales me sentía solo y perdido. Gracias a mis padres que me dieron
xxii
siempre la libertad de elegir, por los valores que me transmitieron, por la educación que me dieron, por todo el sacrificio que hicieron para que llegue a donde estoy, por su incondicional amor, gracias eternamente. Gracias Patricia, Mariana, Ivana, Alejandra, Paula, Fernanda, Marina, Eugenia y Guillermo por todo el cariño, sin ustedes no sería la persona que soy hoy.
xxiii
Foreword
As part of this thesis, I present a review of the most up-to-date literature on HIV-1 infection. In this regard, general aspects such as the history and discovery of HIV, origin, epidemiology, treatments and clinical aspects of HIV infection are addressed. The structure of the viral particle, the virus replicative cycle and restriction factors of viral replication are also described. Particular attention was paid to the cells studied in this project and involved in HIV-1 immunopathogenesis, the innate immune recognition of HIV-1, the dysfunction of the B-cell compartment and the role of BAFF.
The results of this doctoral project are presented in the form of two scientific articles (chapters 3 and 4). The first article is already published in an international peer-reviewed journal and available through Pubmed. The second article was submitted to an international journal and is currently under revision.
1
Chapter 1: Introduction
1 Section 1: The Human Immunodeficiency Virus (HIV)
1.1 The discovery of HIV as the cause of AIDS
During the early 1980s, the world was confronted with a devastating new epidemic when the first cases of AIDS (Acquired Immunodeficiency Syndrome) were observed [1]. This new and unusual syndrome was characterized by generalized lymphadenopathy, opportunistic infections (typically Pneumocystis carinii pneumonia, Toxoplasma gondii encephalitis, cytomegalovirus-associated retinitis, and cryptococcal meningitis), and a variety of unusual cancers (non-Hodgking’s lymphoma and Karposi’s sarcoma) [2-4]. Affected individuals showed depletion of the CD4+ T lymphocyte subset
in the peripheral blood. These clinical observations were first brought to the attention of the general medical community by June 1981 in the United States, when the Centers for Disease Control described five California men with severe immunodeficiency in the Morbidity and Mortality Weekly Report [2], and then similar clinical reports were quickly detected across the world.
The epidemic, which was initially associated with men who have sex with men (MSM), then with injecting drug users, people who had received blood transfusions and finally the general population, was rapidly spreading across the world, urging the scientific and medical community to react promptly and investigate the origin and causes of this deadly disease [5]. Between late 1981 and early 1983, numerous microorganisms were proposed as possible etiologic agents for the AIDS but the virus was finally identified in 1983 when Barré-Sinoussi et Montagnier at the Pasteur Institute recovered a new human retrovirus at the time coined lymphadenopathy-associated virus (LAV) from the lymph node biopsy sample of a patient with generalized lymphadenopathy of unknown origin [6]. During its replication in cultured cells, this virus released high titers of progeny virions that contained reverse transcriptase (RT) activity and exhibited features typical of retroviruses. However, unlike the commonly studied retroviruses of diverse vertebrate origin, it was highly cytopathic in human peripheral blood mononuclear cells (PBMC), specifically killing the CD4+ T cell subset in the cell
2
Within a year, similar virus had been isolated from patients with AIDS. Gallo and colleagues at the National Institutes of Health in United States reported the isolation of retroviruses from AIDS patients [8], which they named HTLV-III and subsequently, Levy and associates [9] recovered a similar retrovirus from both AIDS patients and healthy individuals from the various risk groups, which they named the AIDS-associated retrovirus (ARV). The new retrovirus, associated with AIDS in the United States, Europe, and central Africa and exhibiting morphologic and genetic characteristics typical of the Lentivirus genus, was named human immunodeficiency virus (HIV) (and subsequently HIV-1) in 1986 [10]. In the same year, a related but immunologically distinct and less pathogenic human retrovirus (now called HIV-2) was recovered from individuals residing in several west African countries [11].
1.2 Origin, classification and diversity of HIV
HIV originated from multiple zoonotic transmissions of simian immunodeficiency virus (SIV) from non-human primates into humans in West and Central Africa. Cross-species transmission probably occurred in the process of hunting and butchering of primates for bush meat and the capture, trade and keeping of monkeys as pets [12-13]. Independent zoonotic transmission events from non-human primates to humans have generated several HIV lineages: HIV-1 groups M (Major), N (non-M, non-O), O (Outlier) and P (“pending the identification of further human cases”) and HIV-2 groups A-H. HIV-1 groups M and N originate directly, but independently, from SIVcpz found in the
chimpanzee Pan troglodytes troglodytes in West-Central Africa [14-15] whereas group O and the newly discovery group P are related to SIVgor identified in wild-living Western lowland gorillas (Gorilla
gorilla gorilla in Cameroon) [16-17]. On the other hand, the different HIV-2 groups are the results of at least nine independent transmissions of SIV from sooty mangabeys in West Africa [18-20]. It is conceivable that further HIV lineages in humans will be discovered in the future, as all HIV lineages may not yet have been identified and new cross-species transmissions may take place in the future [13].
Among the four HIV-1 groups, only HIV-1 group M has spread worldwide, which includes over 90% of the global virus isolates. As shown in a biopsy from 1960 and a serum from 1959, HIV-1 M strains circulated already among humans in Kinshasa (in what is now the Democratic Republic of Congo) 20 years before the first AIDS cases were observed in the USA. Molecular clock analyses showed that HIV-1 group M started to diverge in the human population at the beginning of the 20th
3
century [21-23]. Based on phylogenetics analysis, HIV-1 group M can be further subdivided into nine subtypes (A-D, F-H, J, K), sub-subtypes (A1-A4 and F1 and F2), and numerous circulating forms (CRF) and unique recombinant forms (URF) [20].The epidemic patterns of HIV-1 subtypes are continuously evolving. Subtype B accounts for 12% of infections worldwide (predominant in Europe, Australia and North America), whereas subtype C accounts for approximately 50% of human cases (predominant in South Africa and India) [24].
The other HIV-1 groups are less prevalent and remain mainly restricted to Cameroon. HIV-2 only spread to some extent in West Africa, and similarly as for HIV-1, the different HIV-2 groups have also a different epidemic spread. Today, HIV-2 prevalence is decreasing and HIV-1 is becoming predominant in West Africa, most likely because HIV-2 is less pathogenic and less transmissible [20, 25].
The pronounced variability of HIV isolates could be attributed to several factors; the high mutation rate of reverse transcriptase (RT) enzyme, which lacks a proof-reading mechanism and high recombination rate of proviral DNA, together with high rates of virus replication (109 virions/day),
provides the basis for the continuous emergence of new virus variants [26]. These mechanisms result in the rapid generation of genetically diverse viral populations denominated “quasispecies” in each infected individual. Consequently, HIV can acquire resistance to antiretroviral drugs rapidly and escape mutants to neutralizing antibodies and cytotoxic T cells that may emerge in vivo [27].
1.3 Epidemiology
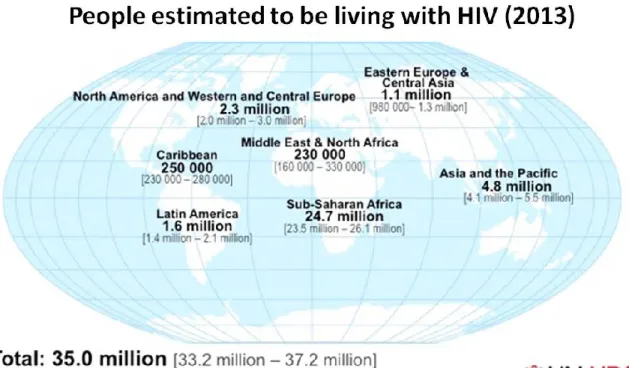
Since the beginning of the epidemic in 1981, around 78 million people have become infected with HIV and 39 million people have died of AIDS-related illness. According to the last epidemiological global report from UNAIDS [28], in 2013 there were 35 million people living with HIV. As shown in Figure 1, the vast majority of infected people are found in low- and middle-income countries; particularly in Sub-Saharan Africa. Despite these data, the report highlights continued progress towards the global vision of reducing new HIV infections, discrimination and AIDS-related deaths. The number of new infections has fallen dramatically by 38% since 2001 whereas the number of AIDS-related deaths has fallen by 35% since its peak in 2005. These achievements were possible due to more individuals having access to antiretroviral therapy (ART) and continuing efforts to prevent new infections.
4
According to the Public Health Agency of Canada [29] in 2011 approximately 71,300 people in Canada were living with HIV infection (including AIDS), an increase of about 11.4% from the 2008 estimate of 64,000. The increase in the number of infected people is mainly due to new infections continuing to occur each year; and fewer HIV-related deaths among people living with HIV because antiretroviral treatment has improved survival. The greatest proportion of prevalent HIV infections in 2011 continued to be attributable to the MSM exposure category (46.7%), followed by the heterosexual exposure category (32.5%) and the injection drug use (IDU) exposure category (16.9%) [29].
1.4 Transmission
HIV is transmitted by human bodily fluids via three major routes: i) non-protected sexual intercourse through vaginal, rectal, or penile tissues; ii) parenteral, through direct injection with HIV-contaminated drugs, needles, syringes, blood or blood products; and iii) vertical transmission from HIV-infected mother to the foetus in utero, through intrapartum inoculation from mother to infant or
Figure 1: People living with HIV in the world (2013). This epidemiologic data
compile information from WHO, UNAIDS, and the Kaiser Family Foundation’s Global Health Policy Division. Adapted from: https://www.aids.gov/
5
during breast-feeding. Most of the new infections are caused by non-protected sexual intercourse and the most important factor that increases the risk of transmission is the number of copies of HIV-1 RNA per ml of plasma (viral load) [30]. Acute HIV infection, which causes very high plasma viral loads in the first months, is an important driver of HIV epidemics [31]. Other factors associated with increased risk of sexual transmission of HIV include sexually transmitted infections (genital ulcers of any cause, herpes simplex type-2 infection and bacterial vaginosis) [32], pregnancy [33], and receptive anal intercourse [34]. Conversely, male circumcision is associated with a reduced risk of sexual transmission of HIV [35].
Behavioural factors that increase transmission include many sexual partners, and concurrent partnerships. Sex inequality is another important driver of the HIV epidemic, especially in Sub-Saharan Africa where woman account for 57% of people living with HIV [36]. In the last global report of AIDS epidemic, UNAIDS has identified stigma against HIV, and discrimination and punitive laws against high-risk groups as barriers for people to undergo HIV testing, access care, and access prevention measures [28].
1.5 Structure
1.5.1 Genome organization
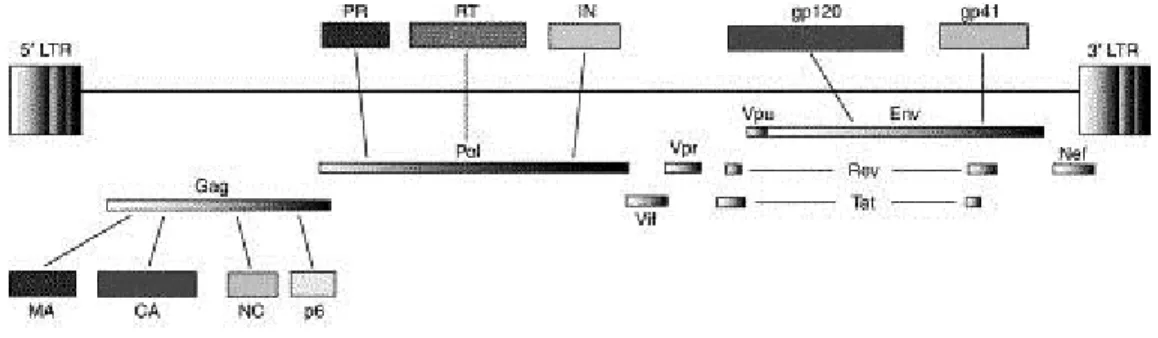
HIV-1 genome is composed of two copies of non-covalently linked, unspliced, positive-sense single-stranded RNA. As with all lentiviruses, HIV possesses a complex genome (approximately 9.7 kb) containing accessory and regulatory genes (Figure 2). An additional, novel open reading frame (ORF), vpu separates the pol and env regions. In total 9 genes are present that can be classified into three functional groups. Gag, pol, and env are structural genes, while tat and rev are regulatory genes and vpu, vpr, vif and nef are accessory genes [37]. In addition to the coding sequences for proteins, the HIV-1 genome is flanked by two non-coding sequences called “Long terminal repeat” (LTR), which drive HIV-1 gene expression and contain many binding sites shown to interact with a vast array of host and viral factors [38].
6
Taken from [39]
1.5.2 Morphology
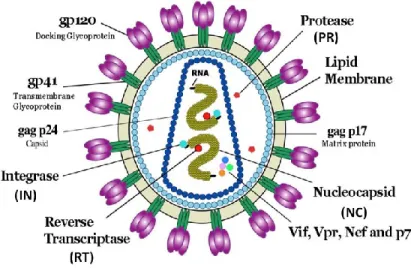
HIV-1 is a member of the lentivirus genus, which includes retroviruses that forms spherical, membrane-enveloped, pleomorphic virions, 1,000–1,500 Å in diameter and contain two copies of its single-stranded, positive-sense RNA genome (Figure 3) [40]. All lentiviruses are enveloped by a lipid bilayer that is derived from the membrane of the host cell. Exposed surface glycoproteins (SU, gp120) are anchored to the virus via interactions with the transmembrane protein (TM, gp41) [41]. The lipid bilayer also contains several cellular membrane proteins derived from the host cell, including major histocompatibility antigens, actin, ubiquitin, ICAM-1 and CD40L, to name a few [42-45].
A matrix shell comprising many copies of the matrix protein (MA, p17) lines the inner surface of the viral membrane, and a conical capsid core particle composed of the capsid protein (CA, p24) is located in the center of the virus. The capsid particle encapsidates two copies of the unspliced viral genome, which is stabilized as a ribonucleoprotein complex composed of the nucleocapsid protein (NC, p7), and also contains three essential virally encoded enzymes called protease (PR), reverse transcriptase (RT) and integrase (IN). Virus particles also package the accessory proteins Nef, Vif and Vpr. The three additional accessory proteins Rev, Tat and Vpu that function in the host cell do not appear to be packaged in the virion [41].
Figure 2: Organization of the HIV-1 genome. The location of the long terminal repeats (LTRs) and the genes encoded
by HIV-1 are showed. Gag, Pol and Env proteins are initially synthesized as polyprotein precursors. The products generated by their cleveage are also shown. Gag proteins: matrix (MA), capsid (CA), nucleocapsid (NC) and p6. Pol enzymes: PR, reverse transcriptase (RT) and integrase (IN). Env proteins: gp120 and gp41. Adapted from [39].
7
1.5.3 Viral proteins and their functions
As is the case for all retrovirus, the three primary HIV-1 translation products are initially synthesized as polyprotein precursors, which are subsequently processed by viral or cellular proteases into mature particles-associated proteins (Figure 2). The 55-kDa Gag precursor Pr55Gag is
cleaved into the matrix (MA), capsid (CA), nucleocapsid (NC) and p6 proteins. Autocatalysis of the 160-kDa Gag-Pol polyprotein, Pr160Gag-Pol, gives rise to the protease (PR), the heterodimeric reverse
transcriptase (RT), and the integrase (IN) proteins, whereas proteolytic digestion by cellular protease furin converts the glycosylated 160-kDa Env precursor gp160 to the gp120 surface (SU) and gp41 transmembrane (TM) cleavage products.
The remaining six HIV-1-encoded proteins (Vif, Vpr, Tat, Rev, Vpu and Nef) are the primary translation products of spliced mRNAs [46]. Tat and Rev are essential regulatory proteins for viral replication, whereas Vif, Vpu, Vpr and Nef are accessory proteins that increase viral infectivity through different mechanisms [41, 47]. The functions of HIV-1 proteins are summarized in Table 1.
Figure 3: Schematic representation of HIV-1. Structural (p17, NC, p24, gp41,
gp120), regulatory (PR, IN, RT) and accessory (Vif, Vpr, Nef) viral proteins are showed in this illustration. Adapted from: http://commons.wikimedia.org/wiki/File:HIV_Virion-en.png
8
Table 1. Functions of HIV-1 proteins (summarized from [41, 46])
Protein Principal functions
p24 Capsid protein. Structural function.
p17 Matrix protein. Structural function.
p7 Nucleocapsid protein. Regulation of the condensation and
encapsidation of viral genome.
p6 Regulation of viral budding.
RT (reverse transcriptase) Synthesis of viral double stranded DNA.
IN (Integrase) Integration of viral cDNA into the cellular genome.
PR (Protease) Cleavage of Pr55Gag and Pr160Gag-Pol polyproteins to generate a mature virion.
gp120 Viral attachment and entry.
gp41 Fusion of viral and cellular membranes. Viral entry.
Tat (Transactivator of transcription) Positive regulator of viral transcription.
Rev (Regulator of viral expression) Export of unspliced and incompletely spliced transcripts from the nucleus to the cytoplasm.
Nef (Negative factor) Augmentation of viral infectivity. Modulation of the cellular activation pathways. Downregulation of CD4, MHC-I and MHC-II, CD3 and CD28 on the surface of infected cell. Determinant of the pathogenesis.
Vpu (Viral protein U) Promotes the release of virus particles. CD4 degradation. Acts against the restriction factor BST- 2 (Tetherin)
Vif (Viral infectivity factor) Inactivation of the restriction factor APOBEC3G
Vpr (Viral protein R) Nuclear import of the pre-integration complex. Transcriptional activator of viral and cellular promoters. Apoptosis
1.6 Viral tropism
1.6.1 CD4 receptor , CXCR4 and CCR5 coreceptors
The main target of HIV-1 is activated CD4+ T lymphocytes. Virus entry occurs via interactions
between the gp120 protein with CD4 receptor and chemokine coreceptors, mainly CCR5 or CXCR4 [48]. Other cells bearing CD4 and chemokine receptors are also infected, including resting CD4+ T
cells, monocytes and macrophages, and dendritic cells. CD4-independent HIV infection of cells can happen, notably in astrocytes [48] and renal epithelial cells [49]. The receptor and coreceptors are the major determinants of HIV tropism.
9
The HIV-1 isolates using CXCR4 are designated X4-tropics, whereas those exclusively utilizing the CCR5 receptor are called R5-tropic. Dual-tropic viruses are designated R5/X4 when they are able to use both coreceptors [50]. The X4 strains infect mainly CD4+ T lymphocytes. For this
reason, they are also called T-tropic and generally induce syncytia formation (multinucleated cells that can result from multiple cell fusions of uninuclear cells due to gp120 expression on their cellular membranes). The R5 strains are also called M-tropic because of their capacity to infect macrophages and are in general non-syncytium-inducing. However, R5-tropic strains can also infect memory CD4+
T cells that express CCR5 [51]
HIV tropism evolves during the course of infection. R5-strains are generally found at the initial stages of HIV infections but X4/R5- (around 50% of patient isolates) and X4-tropic strains can quickly emerge following mutations in the virus [52]. The emergence of X4 virus in 50% of infected people causes the progression to AIDS [53]. Therefore, viral tropism is closely related to the pathogenesis of infection.
The apparent resistance to HIV infection in individuals expressing a non-functional CCR5 (bearing the mutation CCR5-Δ32) confirms that sexually transmitted viruses are mainly R5-tropic [54-56]. The restriction to the transmission of X4 virus is thought to be multifactorial. The vaginal, penile and colorectal mucosa have all the mechanisms that limit the passage of X4-tropic viruses and promote transmission of R5-tropic viruses [57].
1.6.2 Alternative receptors and coreceptors
Although the expression of CD4 and CCR5 or CXCR4 in most of the cases is essential for viral entry, it is possible that the infection occurs in the absence of one or any of these molecules. Indeed, it has been demonstrated that galactosylceramide could be used as an alternative receptor in combination with CXCR4, for example in the infection of oral keratinocytes [58] or epithelial cells of the salivary glands [59] by an X4-tropic virus. There are also alternative coreceptors to CCR5 and CXCR4, such as IBRS, CCR2b, CCR3, CCR8, CCR9 and CXCR2, among others [51].
10
1.6.3 Other attachment molecules
Different molecules promote the attachment of the virus particle to the cell and therefore can be considered participants in the viral tropism. For example the C-type lectins DC-SIGN and DCIR promote virus attachment and infection of dendritic cells (DC) while langerin restricts the infection of Langerhans cells [60-62]. Moreover, glycosphingolipids (GSL) allow infection of neurons [63] and epithelial cells of the colon [64]. Furthermore, the presence of the LFA-1 molecule at the surface of memory CD4+ T lymphocytes seems to be crucial for their preferential infection [65]. It was
also demonstrated that gp120 could bind to integrin α4β7, specifically expressed on T cells with a
tropism for the intestinal mucosa [66]. This would promote the massive depletion of CD4+ T cells in
the gut-associated lymphoid tissue (GALT).
1.7 Replicative cycle
The life cycle of retroviruses is arbitrarily divided into two distinct phases: the early phase refers to the steps of infection from cell binding to the integration of the proviral DNA into the cell genome, whereas the late phase begins with the expression of viral genes and continues through to the release and maturation of progeny virions. The different steps of the viral replicative cycle will be described in this section and are represented in Figure 4.
1.7.1 Early steps
1.7.1.1 Attachment and entry
The initial step of the retroviral replicative cycle is the adsorption of viral particles to the surface of their target cells. It remains unclear whether this binding occurs through specific interactions, but it is thought that such attachment usually involves molecules (see sections 1.6.2 and 1.6.3) which are distinct from the viral receptor responsible for the entry process [67]. As the affinity of HIV envelope glycoproteins for CD4 is relatively low, the existence of other attachment factors may serve to concentrate the virus on the target cell surface prior to specific receptor engagement.
11 Adapted from [68]
Following the initial step of binding, retroviral particles use cell-surface proteins as specific receptors to enter their target cells through interactions with the viral envelope glycoproteins. Firstly, the envelope glycoprotein gp120, present on the surface of viral particles as gp41/gp120 trimers, recognises the primary receptor CD4. This interaction leads to conformational changes in both CD4 and gp120 and to the recruitment of coreceptors CXCR4 and CCR5 [69]. A second interaction then takes place between gp120 and one of these coreceptors, which triggers new conformational shifts in the envelope glycoproteins [70]. These sequential conformational changes finally lead to the dissociation of gp120 from gp41, and to the transition of gp41 to its fusogenic conformation. Entry of virions into the cell is achieved by insertion of the gp41 fusion peptide into the target membrane, resulting in the fusion of viral and cellular membranes and the release of the viral core in the cytoplasm [71].
Figure 4: The replicative cycle of HIV-1. The Env of HIV binds CD4 first, undergoes a conformational change, then
binds one of two chemokine receptors — CCR5 or CXCR4 — and enters cells by fusion of the viral and cellular membranes. Viral RNA is uncoated and reverse transcription begins; yielding double-stranded viral complementary DNA which is transported to the nucleus and, by action of the IN, is integrated into the host genome. Cellular activation increases the level of transcription of the provirus, which is augmented greatly by the viral transcriptional transactivator protein (Tat). Regulator of virion gene expression (Rev) transports singly spliced (ss) and unspliced genomic transcripts from the nucleus to the cytoplasm. Viral structural and enzymatic proteins are synthesized and transported to the plasma membrane, where the viral assembly and budding take place. Adapted from [68].
12
Most retroviruses, including HIV, enter target cells by direct fusion with the plasmatic membrane, as indicated by their resistance to drugs blocking the acidification of endosomes [72]. Interestingly, although HIV entry is pH independent, the majority of viral particles that bind to the cell surface enter by endocytosis [73]. It seems that a balance exists between these two entry pathways of HIV-1 into the target cell, since the inhibition of one route increases entry of particles by the alternative mechanism [74]. Endocytic HIV entry could offer advantages relative to fusion with the plasmatic membrane by ensuring virus delivery to the perinuclear space, protection from cytosolic restriction factors, and by shortening the cell surface exposure of gp41 intermediates targeted by fusion inhibitors and neutralizing antibodies. Current data appear to support the view that the HIV entry route depends on the cell type and their activation status [75].
Finally there are some examples of direct infection from cell to cell. This is the case of dendritic cells which can transmit HIV particles to T-cells by direct contact (virological synapse) without themselves being infected [76-78] The fact that most of the infectious HIV produced by primary macrophages is assembled in late endocytic membranes rather than at the plasma membrane suggests that a direct transmission of virions from infected macrophages to T-cells during antigen presentation could also occur [79-80].
1.7.1.2 Uncoating and reverse transcription
Immediately after its release into the cytoplasm, the viral core undergoes a partial and progressive disassembly, known as uncoating, that leads to the generation of subviral particles called reverse-transcription complexes (RTCs) and pre-integration complexes (PICs). It seems that initiation of reverse transcription is coupled to the onset of uncoating of the viral core [81]. Some viral and cellular proteins appear to influence the uncoating and/or the reverse transcription of HIV-1. This has been exemplified by HIV-1 Nef and Vif and the cellular protein cyclophilin A [82-84]. However, we still do not know all the cellular factors involved in the process of uncoating.
By definition, retroviruses possess the ability to convert their single-stranded RNA genomes into double-stranded DNA during the early stages of the infection process. The enzyme that catalyses this reaction is RT, in conjunction with its associated RNaseH activity. RT has three enzymatic activities: (a) RNA-directed DNA polymerization (for minus-strand DNA synthesis), (b)
13
RNaseH activity (for degradation of the tRNA primer and genomic RNA present in DNA-RNA hybrid intermediates), and (c) DNA-directed DNA polymerization (for second- or plus-strand DNA synthesis). As with other DNA polymerase reactions, reverse transcription is dependent on the 3’-OH group of a primer to initiate polymerization (HIV-1 utilises tRNALys3 for this function). The mechanism of tRNA
selection and placement on the template is complex, involving interaction with RT and NC as well as with the 18-nt primer binding site near the 5’ end of the viral genome [46]. The process of reverse transcription is extremely error-prone, and the resulting mutations may cause drug resistance or allow the virus to evade the host's immune system.
1.7.1.3 Nuclear import and Integration
The retroviral life cycle requires the integration of the viral DNA into the host cell genome to form the so-called provirus. To achieve this, the reverse-transcribed DNA associated with viral proteins to form PICs, must enter the nucleus [85]. HIV PICs are composed of the double-stranded linear DNA associated with the viral proteins MA, RT, IN and Vpr. Then, PICs are transported through the nuclear membrane; this process is complex and involves the interaction of PICs with nuclear pore complexes (NPCs) [86].
IN catalyzes the insertion into the genome of the infected human cell of viral DNA produced by the reverse transcription process. This enzyme mediates three highly specific and coordinated steps which are required for integration. IN initially assembles at specific sequences within the long terminal repeat (LTR) regions at each end of the fully reverse transcribed HIV-1 DNA.
It is only in the context of PICs that the enzyme will then catalyze the subsequent two reactions required for integration: 3' end processing which removes the terminal 3' dinucleotide from each end of the viral DNA and strand transfer which results in the covalent linkage of the viral DNA with the host's DNA. These two enzymatic reactions are displaced in both space and time during HIV-1 infection. The initial 3' processing reaction occurs concomitant with or soon after reverse transcription in the cytoplasm and is critical to generate the 3' OH nucleophile that IN requires for the subsequent DNA-joining reaction which takes place following nuclear import of the PICs [87].
14
In some case however, viral DNA is not integrated and can be found in three forms: circular with two LTR which results from the ligation of the two ends of the viral DNA, circular but with only one LTR which corresponds to homologous recombination of the viral genome or it can remain as linear DNA [88]. Several studies have shown that these non-integrated forms can be transcriptionally active [89-90].
1.7.2 Late steps
1.7.2.1 Transcription and translation
Regulation of HIV gene expression involves a complex interplay between chromatin-associated proviral DNA, cellular transcription factors and the virally encoded Tat protein. The process of viral transcription can be divided into two distinct phases. The first phase occurs early in transcription and is mediated by direct interaction between cellular transcription factors and the 5’ LTR. This first step allows the transcription of tat, rev and nef genes. During the second phase following the accumulation of Tat, long transcripts are generated.
In the host genome, the 5′ LTR functions like other eukaryotic transcriptional units. It contains downstream and upstream promoter elements, which include the initiator (Inr), TATA-box (T) and three Sp1 sites [91]. These regions help position the RNA polymerase II (RNAPII) at the site of initiation of transcription and assemble the pre-initiation complex. Transcription begins but the polymerase fails to elongate efficiently along the viral genome. In the process, short non-polyadenylated transcripts are synthesized, which are stable and persist in cells due to the formation of an RNA stem loop called the transactivation response (TAR) element [92]. A little upstream of the promoter is the transcriptional enhancer, which binds nuclear factor κB (NF-κB), nuclear factor of activated T cells (NFAT), and Ets family members. NF-κB increases rates of initiation as well as elongation of viral transcription [93-94].
Tat protein is responsible for shifting viral gene expression into a higher rate. In association with cyclin T1, Tat binds TAR and recruits the cellular cyclin-dependent kinase 9 (Cdk9) to the HIV LTR. In the positive transcription-elongation factor b (P-TEFb) complex, Cdk9 phosphorylates the C-terminal domain of RNAPII, which marks the transition from initiation to elongation of eukaryotic
15
transcription [95-96]. After the transcription of the viral genome, more than a dozen HIV-specific transcripts are generated. Some are processed co-transcriptionally and, in the absence of inhibitory RNA sequences are rapidly transported into the cytoplasm. These multiply spliced mRNA species encode Nef, Tat and Rev. Various other individually spliced or unspliced viral transcripts remain in the nucleus and are relatively stable. The incompletely spliced viral transcripts encode the structural proteins and enzymes as well as the viral RNA genome that are needed for the assembly of fully infectious virions. Their transport into the cytoplasm depends on the production of a threshold amount of Rev [97-98].
Then, the translation of viral proteins is produced in the cytoplasm with exception of the Env proteins that are translated by endoplasmic reticulum (ER)-associated ribosomes as a single precursor polypeptide, called gp160, which is transported to the plasma membrane via the cellular secretory pathway. During transport, gp160 is extensively glycosylated, associates into trimers, and is cleaved by the cellular protease furin into mature subunits [99].
1.7.2.2 Assembly, viral budding and maturation
All of the components of the virion are assembled at the plasma membrane where budding occurs. This process is directed by Gag-Pol and Gag polyproteins but is also influenced by Env and involves the recruitment of two copies of the viral RNA genome as well as Vpr [100].
Due to the myristylation and palmitoylation of the Gag polyprotein, Gag associates preferentially with cholesterol- and glycolipid-enriched membrane microdomains [101-102]. Virion budding occurs through these specialized regions in the lipid bilayer yielding virions with cholesterol-rich membranes. This lipid composition favors release, stability and fusion of virions with the target cell [103]. Maturation transforms the non-infectious immature virion into a particle that is capable of transmitting infection. This process occurs in conjunction with (or immediately following) budding, and is driven by viral PR cleavage of the Gag and Gag-Pro-Pol polyproteins at ten different sites, ultimately producing the fully processed MA, CA, NC, p6, PR, RT, and IN proteins.Over the course of maturation, these processed proteins rearrange dramatically to create the mature infectious virion, with its characteristic conical core [40, 104]
16
1.8 Restriction factors of viral replication
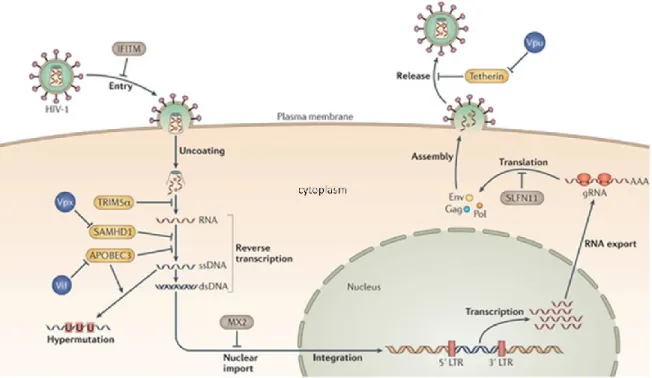
Although HIV -1 is an effective parasite in the host cell, the latter has developed defense mechanisms to control viral replication. The major restriction factors involved in the known intrinsic immunity of the cell will be described thereafter and are represented in Figure 5.
Taken from [105]
1.8.1 APOBEC3G
Apolipoprotein B mRNA-editing enzyme catalytic subunit-like 3G (APOBEC3G), also known as CEM15, was the first identified restriction factor for HIV-1 [106], and is a prominent member of the large family of APOBEC cytidine deaminases that inhibit retroviral infection. APOBEC3 proteins, of which APOBEC3G is the most abundant in vivo, are expressed in hematopoietic cells, including B lymphocytes, T lymphocytes and myeloid cells [107]. APOBEC3G inhibits viral replication by converting cytosines to uracils on newly synthesized minus-strand viral cDNAs, leading to changes in the viral sequences that compromise viral genome integrity due to Figure 5: HIV-1 restriction and resistance factors. TRIM5α promotes accelerated fragmentation of viral cores,
preventing cDNA synthesis. SAMHD1 depletes the cellular levels of dNTPs, which are required for efficient cDNA synthesis. APOBEC3 proteins interfere with the processivity of HIV-1 reverse transcriptase and induce hypermutation of viral cDNA by cytidine deamination. Tetherin prevents the release of budded virions from the infected cell. Several viral proteins (blue) antagonize these cellular restriction factors. Adapted from [105].
17
catastrophic mutations. This restriction factor is highly inducible by IFN, particularly in myeloid cells. Interestingly, APOBEC3 proteins can be incorporated in the virion, but this is prevented by the HIV-1 accessory protein Vif [107-109].
1.8.2 TRIM5α
TRIM5α is a cytoplasmic protein that inhibits replication of HIV-1 and other retroviruses at a step early after entry of the core particle into the target cell [110]. The restriction activity of TRIM5α is determined by recognition of the HIV-1 CA via its C-terminal PRYSPRY domain. TRIM5α is a multi-domain protein with RING, B-box and coiled-coiled multi-domains that all contribute to retroviral restriction activity [111]. This restriction factor also possesses an E3 ubiquitin ligase activity that mediates proteasomal degradation of CA components, IN and RT, resulting in impaired reverse transcription and nuclear import. Cyclophilin A (CypA) is another cellular factor that binds to the HIV-1 CA and counteracts TRIM5α activity. Although CypA can be incorporated in the nascent virion, its expression in the target cell is more important for promoting HIV-1 infectivity [112-113].
1.8.3 SAMHD1
SAMHD1, or sterile alpha motif and histidine-aspartic acid domain containing protein 1, is a host deoxynucleoside triphosphate phosphorohydrolase (dNTPase) that depletes the cellular dNTP pool [114]. SAMHD1 potently inhibits HIV-1 replication in myeloid cells, including monocyte-derived macrophages (MDM) and monocyte-derived dendritic cells (MDDC), either after cell-free infection or cell-associated transmission. In contrast to HIV-1, HIV-2 and certain SIV strains can attenuate restriction to infection of myeloid cells via expression of the virion-contained accessory molecule Vpx [115]. Interestingly, when introduced into cells via virus-like particles, Vpx can also attenuate HIV-1 restriction in macrophages and dendritic cells. SAMHD1 also inhibits HIV-1 replication in resting CD4+ T cells, but not in activated CD4+ T cells. The low efficiency of SAMHD1 restriction in dividing
cells can be explained by the observation that SAMHD1 activity is regulated in a cell-cycle-dependent manner via cyclin-dependent kinase 1 (CDK1) dependent phosphorylation [116-117]. SAMHD1 contains a SAM domain, which is largely dispensable for retroviral restriction, and an HD domain, that contains the enzymatic functions crucial for its function in retroviral restriction, including the triphosphohydrolase activity, RNA binding and nuclease activity. The retroviral restriction activity
18
exerted by SAMHD1 has been attributed to depletion of the cellular dNTP pool below levels required for productive reverse transcriptase [117].
1.8.4 Tetherin
Tetherin, also known as BST-2 (Bone Marrow Stromal Cell Antigen 2) or CD317, is another IFN-inducible restriction factor that anchors HIV-1 particles to the plasma membrane and prevents release of viral particles. As many other host restriction factors, the activity of tetherin is antagonized by the HIV-1 accessory protein Vpu [118]. The mechanism of action of tetherin relies on the conformation of the molecule which infiltrates into the viral envelope with one of its anchor domains, while the other anchor remains associated with the plasma membrane [119].
1.9 Pathogenesis
1.9.1 Phases of infection
The course of HIV-1 infection can be monitored by different means over symptoms, such as fever, wasting, opportunistic infections, neurological symptoms as well as blood levels of CD4+ T
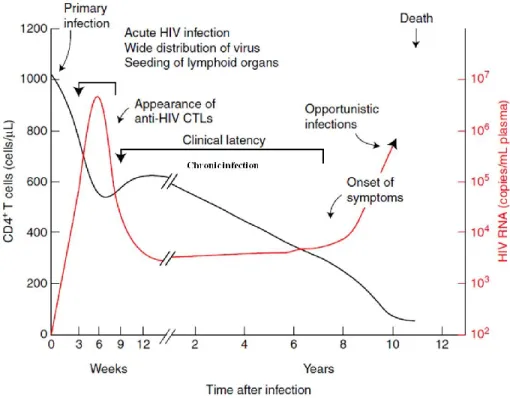
cells, antiviral antibodies and viremia (virus in blood). A typical time course of infection is shown in Figure 6. Although their timing varies considerably from individual to individual, the general outline is essentially the same in almost every infected person who does not receive effective antiviral therapy.
1.9.1.1 Eclipse and acute phase
During the eclipse phase, the virus is freely replicating and spreading from the initial site of infection to many tissues and organs that provide the sites for active replication. Viral load is undetectable in peripheral blood and neither immune response nor symptoms of infection are yet visible [120]
The acute or primary phase of infection (~1-4 weeks) is characterized by relatively high levels of viremia (infected individuals are also more susceptible to transmit the infection) and a large fraction of infected CD4+ T cells in blood and lymph nodes [121]. This phase is often, but not always,
accompanied by “flu-like” symptoms (fever, enlarged lymph nodes, etc.). Eclipse phase and acute infection are difficult to study in humans because the event is usually over before a patient sees a
19
physician. Studies in non-human primate models have provided invaluable information about the early infection events and the interactions between the virus and immune system cells at the entry sites in the mucosal epithelium [122]. Antigen-presenting cells (APC) such as dendritic cells migrates to the lymph nodes and can passively deliver HIV to T cells via their immunological synapses [123]. It is now clearly established that the earliest and greatest damage caused by HIV infection is to the gut-associated lymphoid tissue (GALT) [124], which is characterized by a dramatic depletion of CD4+ T
cells (principally memory cells) and damage of the integrity of the gut epithelium itself, so that commensal gut bacteria leak into the GALT and their lipopolysaccharides (LPS) cause inflammatory responses [125]. This process could be an important contributor to the chronic immune activation that characterizes HIV-1 infection (discussed in further detail in section 2.3).
Taken from [120]
With the explosive replication of HIV-1 in the GALT, the viral load detected in the peripheral blood reaches a peak. However, a fall in viral load occurs soon after seroconversion, and is thought to be due to the appearance of HIV-specific CD8+ cytotoxic T cells [121]. Nevertheless, two additional
factors might contribute to the decrease in viral load. First, although neutralizing antibodies only Figure 6: Time course of a typical HIV-1 infection. CD4+ T cell counts decline and
viremie vary from one patient to another. After several years of infection, people without antiretroviral treatment die as consequence of opportunistic infections. Adapted from [120].
20
appear several weeks or months later, non-neutralizing antibodies appear at this time and in conjunction with innate immune factors that might help opsonisation and clearance of virus particles [126]. Second, the depletion of CD4+ CCR5+ T cells in the gut might be too severe, thus leading to
insufficient new target cells for HIV-1 to maintain a high virus load [127].
1.9.1.2 Chronic or latent phase
After seroconversion and the reduction in peak virus production, viral load in the peripheral blood decreases to a “set point”, which is predictive of the rate of progression to AIDS [128]. The higher the set point, the more rapid the advance towards immune deficiency tends to be. As a rule, patients in this phase (~1-20 years) are asymptomatic and usually unaware that they have been infected. Despite the term “latency”, the viral infection is far from latent, with active HIV-1 replication and destruction of CD4+ T cells in the GALT and lymph nodes [125], but the regenerative power of
CD4+ T cells is significant, with a high daily turnover of both cells and virus [129]. However, due to the
persistent immune activation, this regenerative capacity of CD4+ T cells is eventually impaired [130].
Decreasing CD4+ T cell counts in the peripheral blood is a crucial independent marker from viral load
of disease progression, and is taken into account by patients and their physicians in decisions such as when to begin antiretroviral therapy. When the CD4+ T cell counts fall below 200 cells/µl,
opportunistic infections occur leading to AIDS [120].
According to the rate of progression to AIDS, HIV infection may be divided in: i) rapid progression, where AIDS develops within 3 years of infection, ii) intermediate progression (most of infected people), where AIDS develops slowly between a span of 3 and 10 years after seroconversion, iii) long-term non-progression (LTNP) where HIV infected people maintain high CD4+
and CD8+ T-cell counts for years (>10 years) without therapy representing 2-4% of infected
people[131]. As viral load testing became available, long-term non-progressors were further divided into two groups, one showing low plasma viral load (<5000 HIV-RNA copies/ml), termed long-term non-progressors and a second group showing plasma HIV-RNA values persistently below 50 copies/ml, and termed “elite” or “natural controllers” representing less than 1% of infected individuals [131]. Understanding the mechanisms that allow the control of viral infection in certain individuals is a major challenge of current HIV research.
21
1.9.1.3 AIDS
Finally, the number of CD4+ T cells declines (~200 cells/ml) to the point at which immune
control of infectious agents can no longer be maintained, and opportunistic infections begin to appear such as tuberculosis, Pneumocystis, cytomegalovirus (CMV), cerebral Toxoplasma or Candida [121]. People with AIDS also have an increased risk of developing various cancers such as Kaposi's sarcoma, cervical cancer and lymphomas. The specific opportunistic infections that AIDS patients develop depend in part on the prevalence of these infections in the geographic area in which the patient lives. Control of HIV-1 infection itself is also lost, and the level of viremia rises during the AIDS phase, culminating in death of the infected patient. Indeed, untreated HIV-1 infection is one of the most lethal infectious diseases known, with a mortality rate over 95% [120].
1.10 Prevention and Treatment
To date, there is not a cure or treatment that allows the complete elimination of HIV-1 in an infected person. Only the “Berlin patient” represents a case of functional cure of HIV-1 [132-133]. This patient was infected with HIV and developed acute myelogenous leukemia (AML), for which he was treated with an allogeneic hematopoietic stem cell transplant from a donor who was homozygous for the CCR5Δ32 deletion, which confers resistance to infection with CCR5-tropic virus. The patient interrupted ART soon after the transplant and has had no detectable plasma HIV RNA for over 5 years [133]. Despite the extensive efforts put in the development of a vaccine, major challenges still exist concerning HIV vaccine design. However, the RV144 vaccine trial done in Thailand provided the only evidence up to now that vaccine protection could be achieved with 31% reduction in HIV acquisition [134]. Immune correlates of protection from this vaccine trial together with new vaccine designs that improve the breadth of T-cell responses as well as the identification of targets for broadly neutralizing antibodies can help in the development of more effective vaccines [135].
However, combination antiretroviral therapy regimens, highly active antiretroviral therapy (HAART), that are able to suppress viral replication were developed in middle 1990s and transformed HIV from a progressive illness with a fatal outcome into a chronic manageable disease [30]. HAART can reduce viral replication to undetectable levels in peripheral blood, which result in considerably slower progression to AIDS and reduction of viral transmission. The combination of different inhibitors targeting different steps of the viral replication cycle reduces the development of resistance that were
22
observed rapidly in monotherapy regimens. Standard ART combines two nucleoside reverse transcriptase inhibitors with a non-nucleoside reverse transcriptase inhibitor, protease inhibitor, or integrase inhibitor [30].
1.10.1 Reverse transcriptase inhibitors
There are two types of RT inhibitors: the nucleoside reverse transcriptase inhibitors (NRTIs) were the first agents available for the treatment of HIV infection and include zidovudine (AZT). These are nucleoside analogues that are phosphorylated inside the cell and incorporated into the viral DNA during reverse transcription by the RT. NRTIs are chain terminators; they do not have a 3’ OH group required for polymerisation thus, once incorporated, work by preventing the incorporation of other cellular nucleosides into the DNA chain. On the other hand, the non-nucleoside reverse transcriptase inhibitors (NNRTIs) bind directly to RT, causing a conformational change that inhibits its activity. This class of drugs include efavirenz (EFV) and nevirapine (NVP) [136].
1.10.2 Protease Inhibitors
HIV protease inhibitors (PIs) bind directly to HIV protease and prevent the cleavage of gag and gag/pol precursor proteins thus blocking viral maturation. PIs were first introduced in 1995 and are an integral part of treatment of HIV infection. They exhibit activity against clinical isolates of both HIV-1 and HIV-2 [137].
1.10.3 Integrase Inhibitors
These agents inhibit the transfer of viral DNA strand into the cellular DNA by binding metallic ions in the integrase active site. There are several integrase inhibitors currently under clinical trials, and raltegravir became the first to receive FDA approval in October 2007 [138].
1.10.4 Entry Inhibitors
These drugs interfere with binding, fusion and entry of HIV-1 to the host cell. Maraviroc and enfuvirtide (T-20) are the two currently available agents in this class. Maraviroc is a small molecule that selectively and reversibly binds the CCR5 coreceptor and causes a change in its conformation that prevents any interactions with the V3 loop of gp120. Maraviroc is active against HIV-1 R5-tropic viruses. It has no activity against X4-tropic or dual/mixed tropic virus [139]. (T-20) blocks the second
![Table 1. Functions of HIV-1 proteins (summarized from [41, 46])](https://thumb-eu.123doks.com/thumbv2/123doknet/6575653.178005/32.918.109.785.139.836/table-functions-hiv-proteins-summarized.webp)

![Table 2. Phenotypic and functional differences between the three monocyte subsets (Adapted from [202])](https://thumb-eu.123doks.com/thumbv2/123doknet/6575653.178005/54.918.105.777.354.805/table-phenotypic-functional-differences-monocyte-subsets-adapted.webp)