Chromosomal Instability and Tumorigenesis:
Genetic Analysis of the Murine Spindle Checkpoint gene Mpsl
byStephanie Zhi-Juan Xie B.A. Biology University of Chicago, 1996
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOLOGY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY FEBRUARY 2007
C 2007 Massachusetts Institute of Technology. All rights reserved.
Signature of Author: Certified by: Peter K. Sorger Professor of Biology Thesis Supervisor Accepted by: Stephen P. Bell Professor of Biology Chairman, Biology Graduate Committee OF TECHNOOGY v
FEB 2 2W007
SLIBRARP,•S.__
• .. I _ = v __ _ _
Chromosomal Instability and Tumorigenesis:
Genetic Analysis of the Murine Spindle Checkpoint Gene Mpsl by
Stephanie Zhi-Juan Xie
Submitted to the Department of Biology on January 11, 2007 in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Biology
ABSTRACT
The ubiquity of aneuploidy in human cancers, particularly solid tumors, suggests a fundamental link between errors in chromosome segregation and tumorigenesis. The spindle checkpoint ensures accurate chromosome segregation by delaying anaphase onset until all kinetochores achieve bipolar attachment to the mitotic spindle. Abrogation of the mammalian spindle checkpoint can cause chromosomal instability (CIN), the frequent gain or loss of entire chromosomes during cell divisions. While CIN has been proposed to be sufficient to initiate tumorigenesis, no direct evidence has shown that CIN itself is a
causal agent for human cancer.
To determine if CIN can promote tumor formation, I generated mice containing a
conditional mutant allele of the spindle checkpoint kinase Mpsl (MpslA) and induced its expression in thymocytes. I show that MpslA is a partial loss of function allele for spindle checkpoint activation which results in chromosome missegregation in MEFs and embryonic lethality in mice. Expression of MpslA in thymocytes causes sporadic
lymphomas and a decrease in thymus size. Strikingly, when MpslA was introduced into a p53 heterozygous background, all mice developed thymomas due to a loss of
heterozygosity of the wildtype p53 allele. In contrast, no p53 heterozygous
micedeveloped thymomas in the same time frame when Mpsl is wildtype. Moreover, MpslA also accelerated tumor formation in p 19AF heterozygous thymocytes. I propose that MpslA-induced CIN activates p53, and results in apoptosis of the majority of
SpsI A- expressirn cells. Therefore, there- ;is strong selecptiove pressure- for ;nactivation of the p53 pathway, and in those cells where p53 loss does occur, MpslA-induced CIN
drives tumor development. In summary, I show that CIN caused by a weakened checkpoint is sufficient to facilitate tumorigenesis when the p53 pathway is also impaired.
Thesis Supervisor: Peter K. Sorger
Title: Professor of Biology, Professor of Biological Engineering
STEPHANIE ZHIJUAN XIE
Education
Ph.D. student in Biology at Massachusetts Institute of Biology, 2006 Laboratory of Peter K. Sorger
B.A. degree in Biology, University of Chicago, 2000 Undergraduate thesis in the laboratory of Douglas Bishop Hunter College High School, NYC, 1996
Honors and Awards
Paul and Cleo Schimmel Scholar, 2000-2004 Elected to Sigma Xi, 2000
Honorable Mention, National Science Foundation Graduate Fellowship, 2000 Honorable Mention, Howard Hughes Predoctoral Fellowship in Biological
Sciences, 2000
Student Marshall, University of Chicago, 2000
Publications
A partial loss of function mutation in the mitotic checkpoint causes CIN and drives tumorigenesis in mice. Xie, S. and Sorger, P.K.
Intended for submission to Nature, Spring 2007
Chromosome segregation and genomic stability. Draviam, V.M., S., Sorger, P.K. Curr Opin Genet Dev. 2004 Apr;14(2):120-5.
Poster Presentations
AACR: Cancer Susceptibility and Cancer Susceptibility Syndromes 2006 ASCB Annual Meeting, San Francisco 2005
ISREC: Cell and Molecular Biology of Cancer, Lausanne 2005 ASCB Annual Meeting, Washington, D.C. 2004
ACKNOWLEDGEMENTS
I would like to thank Peter for taking me into his lab and giving me the freedom to find my own way. I am grateful to you for your enthusiasm and support and for creating a wonderful research environment to go to these last five years.
I would like to thank the members of my thesis committee, Tyler Jacks and Steve Bell, for your invaluable advice over the years. I would like to thank Jianzhu Chen and Randall King for joining my defense committee.
I would like to thank all the members of the Sorger lab, past and present, especially Ying Yue, Aurora Burds Conner, Annagret Schulze-Lutum, Rob Hagan, John Albeck, Suzanne Gaudet, Viji Draviam, Patrick Meraldi, Irina Shapiro, Jessica Tytell and Alexa Turke. I am would also like to thank Margaret White. I am grateful for all your encouragement, support and intellectual advice. You made going to lab fun!
To all the people that helped this yeast geneticist to successfully make the transition to becoming a mouse geneticist, many thanks! I would like to extend a special thanks to Tyler and Jianzhu for making the expertise in their labs accessible, and to the many members of their labs who took
time out of their busy lives to listen to my questions. I would not have been able to complete much of my research without invaluable advice from Qing Ge and Chris Dillon, who were kind enough to teach a complete novice immunology. Thanks to all those at DCM and the CCR histology core - particularly Stephanie Haskell and Michael Brown.
I would like to give a special thanks to all those kind enough to read the rough drafts of this thesis, particularly Ying Yue and Aurora Burds Conner, who prevented incoherence from late nights to be propagated.
I would like to thank all my classmates from biograd 2000, especially Kevin Lai, Sunny Wong, Mimi Lee, Mandy Tam, David Doroquez and Mark Rosenzweig, for the not so tranquil (but very
fun) first year and all the advice and support through the years.
I would like to extend a special thanks to all my friends, particularly from Hunter, Tang and 69 and 71 Elm St, for all that you are. You know who you are and I won't embarrass you guys.
Well, I'll embarrass some of you anyway, at least all the mahjong and dimsum buddies -Aaron, David, Due, Jenny, Justin, Kev, Lesley, Mandy, Mark, Mimi, Nate, Pat, Sal, Sarah, Shirley, Tony. Thanks Ina and Matthew for listening. Thank you Shirley for periodically reminding me that I'm a girl and girls should have fun.
To Derek, thank you for all the love and encouragement, for sharing all the good and not so good times, and reminding me to "play safe". I love you.
And to my family, mom, dad and bro, thank you for reminding me what is most important. I am so grateful for your love and support through these years.
Table of Contents
Chapter 1 Introduction: The spindle checkpoint, chromosomal instability and cancer 9
I. Chromomsomal Instability and Cancer 10
A. Introduction 10
B. Searching for the molecular basis of CIN 14
C. Spindle checkpoint deficiencies in tumors 14
D. Senescence or tumorigenesis? 20
E. Connection between DNA damage and mitotic progression and cancer? 21
F. Conclusion 22
II. The spindle checkpoint 23
A. Cell cycle and cell cycle checkpoints 26
B. The canonical players of the spindle checkpoint 27 C. Interactions between the spindle checkpoint and the cell cycle 29
D. Spindle checkpoint signaling 30
E. Mpsl: a spindle checkpoint kinase 36
F. Meiosis and the spindle checkpoint 38
III. Tumor suppressor genes and oncogenes 39
A. p53: DNA damage, aneuploidy and cancer 40
B. p53-dependent and p53-independent functions of p 9ARF 43
C. Oncogenic K-ras and lung cancer 45
IV. Conclusion 46
References 48
Chapter 2 A mutation in Mpsl causes chromosomal instability and accelerates
tumorigenesis in mice 67 Abstract 68 Introduction 69 Results 70 Discussion 96 Methods 106
Acknowledgements References
Chapter 3 Loss ofp53, not pl9ARF can rescue MpslA induced lethality in mouse embryonic fibroblasts
Abstract Introduction Results
MpslA decreases viability of MEFs, which is rescued by p53 inactivation 125 MpslA increases chromosome segregation defects, but not centrosome amplification
in p53 null MEFs 129
Inactivating pl 19ARF does not rescue MpsA induced cellular lethality 139
Mpsl"'; pl9 RF+; Lckcre+ mice develop tumors with an increase penetrance over
MpslfEf; Lckcre+ mice 139
Discussion Methods
Acknowledgements References
Chapter 4 siRNA depletion of Mpsl causes chromosome missegregation and accelerates mitotic timing in HeLa cells
Abstract Introduction Results
siRNA depletion of Mps abolishes kinetochore localization of Madl and Mad2 165
Complete siRNA depletion of Mps 1 by Mps 1-4 abolishes the checkpoint 169
Mpsl controls mitotic timing in addition to monitoring chromosome segregation 173 112 113 119 120 121 125 142 150 153 154 160 161 162 165 Discussion Methods 178 184 Methods Acknowledgements References
Acknowledgements 185
References 186
Chapter 5 Conclusions, Discussion and Future Directions 190
Conclusions and Discussion 192
MpslA is a partial loss of function mutation in the spindle checkpoint 192
p53 inhibits CIN 194
The spindle checkpoint kinase Mpsl has multiple functions 197
CIN as a facilitator of tumorigenesis 198
Future Directions 199
MpslA as a universal facilitator of tumorigenesis? 199
What is the mechanism of Mps 1A induced chromosome missegregation? 200 The intersection of p53 and the spindle checkpoint in tumorigenesis 202
Tissue specific effects of spindle checkpoint inactivation and genetic modifiers 204
Mpsl and human cancer 207
Summary 208
References 210
Appendix Determining the effect of MpslA in two post mitotic tissues: a conditional
K-ras lung cancer model and liver-specific deletion by Albumin-Cre 217
Introduction 218
Results and Discussion 220
MpslA causes a modest decrease in mouse mortality when expressed in the liver 220
MpslA has a modest effect on the tumor spectrum of K-rasG1 2 lungs 224
Methods 229
Acknowledgements 230
References 231
List of Figures
Figure 1.1: The canonical spindle checkpoint proteins. ... 24
Figure 2.1: MpslA is a truncation mutation that retains kinetochore localization. ... 71
Figure 2.2: MpslA causes embryonic lethality in mice before embryonic day 10.5... 73
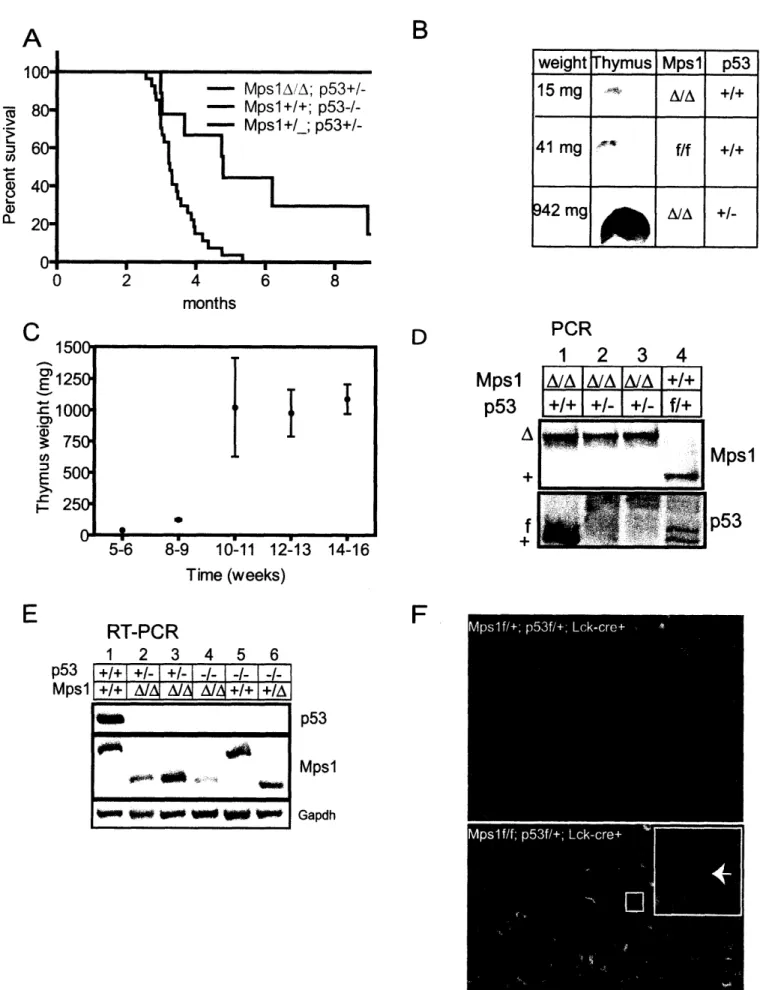
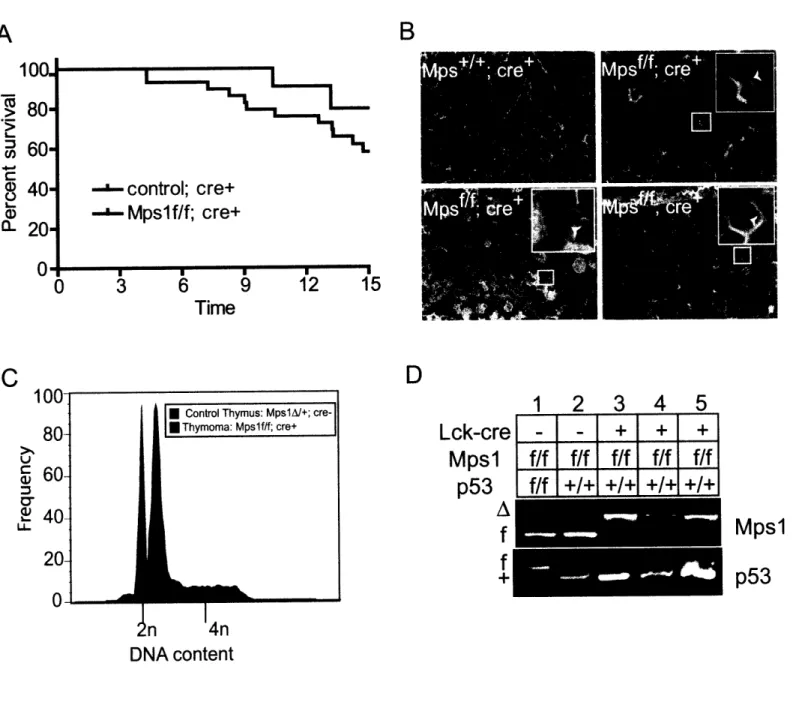
Figure 2.3: MpslA facilitates tumorigenesis in p53-heterozgyous thymocytes by inducing LOH of p53 ... 77
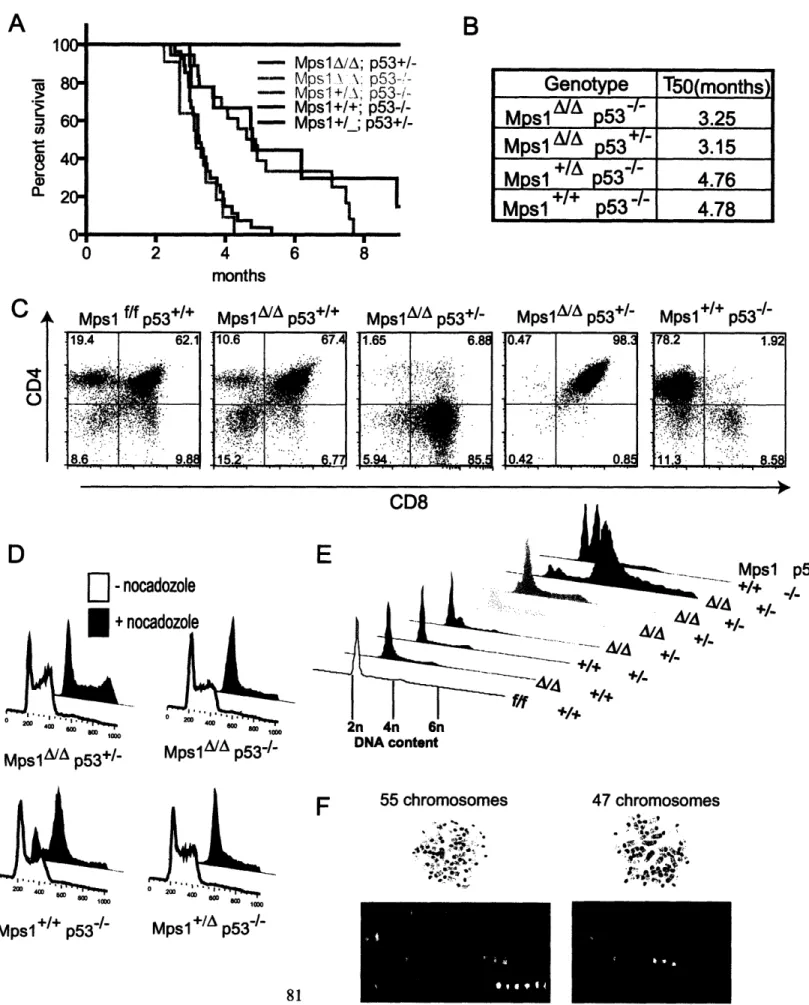
Figure 2.4: MpslA accelerates lymphomagenesis in p53-null knockout thymocytes... 81
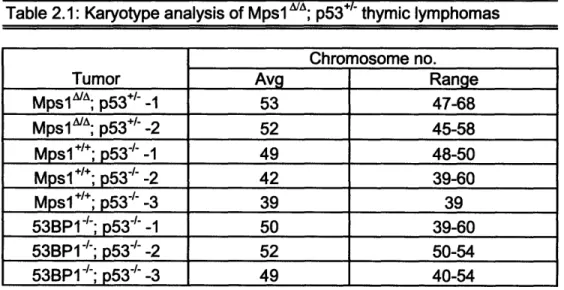
Table 2.1: Karyotype analysis of MpslA/A; p53+/- thymic lymphomas... 85
Figure 2.5: MpslA decreases thymus size and increases heterogeneity in thymocytes. . 86 Figure 2.6: MpslA is a partial loss of function mutation in the spindle checkpoint that induces CIN and sporadic lymphomagenesis ... ... 88
Table 2.2: Number of cells in immune organs of mice at 6 weeks of age ... 92
Table 2.3: T cell development in thymi of mice at 6 weeks of age. ... 93
Table 2.4: Percentage circulating peripheral T cells of mice at 3-4 weeks of age ... 94
Figure 2.7: MpslA accelerates tumorigenesis in p53-heterozygous conventional knockout m ice ... 99
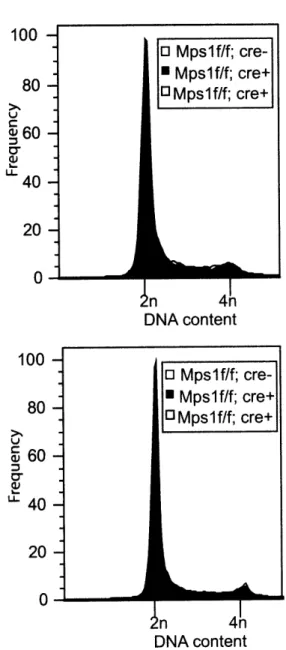
Table 2.5: MpslA does not induce centrosome duplication in thymocytes ... 101
Figure 3.1: MpslA decreases MEF viability which is rescued by inactivating p53... 126
Figure 3.2: MpslA is a partial loss of function mutation that increases chromosome missegregation in p53-null MEFs ... 130
Table 3.1: Analysis of mitotic timing in MEFs ... 132
Table 3.2: MpslA does not induce centrosome duplication in MEFs ... 133
Figure 3.3: MpslA increases CIN in p53-null MEFs through multiple passages... 137
Figure 3.4: MpslA decreases viability of pl9ARF knockout MEFs, but increases tumorigenesis in pl 9ARF+/- mice... 140
Table 4.1: Mpsl siRNA sequences ... 166
Figure 4.1: siRNA depletion of Mps abolishes kinetochore localization of Mad2. ... 167
Figure 4.2: Complete siRNA depletion of Mpsl by Mpsl-4 abolishes the checkpoint. 170 Table 4.2: Analysis of mitotic timing in cells depleted of Mps 1 ... 172
Figure 4.3: Mps1 controls mitotic timing ... 174
Figure 4.4: Mpsl depletion causes chromosome missegregation... 176
Figure 4.5: A model for the dual functions of Mpsl during mitosis in the spindle checkpoint and mitotic timing. ... 180
Figure Al: Mps lA causes a modest increase in mortality when expressed in the liver 221 Table Al: Mice from Alb-cre survival study that were found dead or found with liver tum ors. ... 223
Figure A2: MpslA causes a modest shift in K-rasGl2D lungs from adenomas to papillom as... 225
Chapter 1
Introduction:
The spindle checkpoint, chromosomal instability and
cancer
Note:
Sections of this chapter have been adapted, with permission, from the following review:
Draviam, V. M., Xie, S., and Sorger, P. K. Chromosome segregation and genomic stability. Curr Opin Genet Dev. 2004. Apr; 14 (2):120-5.
I. Chromomsomal Instability and Cancer
The acquisition of genomic instability, an abnormal cell state associated with an increased rate of heritable alterations, is a crucial step in the development of human cancer. Genomic instability has multiple causes of which chromosomal instability (CIN), the frequent gain or loss of entire chromosomes during cell divisions, and microsatellite
instability (MIN), associated with defects in DNA mismatch repair, have received the most attention. CIN has been observed in many human tumors and increased CIN is often
correlated to the severity of the cancer (Loeb et al., 2003; Wilkens et al., 2004). Whereas the connection between a MIN phenotype and cancer is well established, the argument
that CIN causes cancer remains circumstantial (Lengauer et al., 1997; Shibata et al., 1994). Nonetheless, the ubiquity of aneuploidy in human cancers, particularly solid tumors, suggests a fundamental link between errors in chromosome segregation and tumorigenesis. Current research in the field is focused on elucidating the molecular basis
of CIN, including the possible roles of defects in the spindle checkpoint and defects in other regulators of mitosis.
A. Introduction
Since the late-19th century, abnormal chromosome number has been recognized as a nearly ubiquitous feature of human cancers (Hansemann, 1890). Careful study of colorectal cancers has shown that about 85% are aneuploid, and contain cells with an average of 60 to 90 chromosomes (reviewed in (Pellman, 2001). Moreover, tumors with higher clinical grades and poorer prognosis are typically associated with greater degrees of aneuploidy. Despite its long history and clinical relevance, the study of aneuploidy has yet to prove Boveri's postulate that abnormal chromosome number is a cause rather than
a consequence of the cancerous state (Boveri, 1914). More precisely, it remains unclear whether aneuploidy arises early in tumorigenesis and plays a role in tumor development or whether it arises late and reflects a general breakdown of cell cycle control.
Genomic instability is thought to be critical in the multi-step process by which cells accumulate the mutations characteristic of a cancerous state. For example, human colorectal cancers, which usually exhibit significant genomic instability, have been found to have 11,000 or more genetic alterations, and presumably, an ability to overcome a wide variety of negative controls on proliferation (Stoler et al., 1999). The mutator hypothesis posits that genomic instability arises early in tumorigenesis to increase subsequent occurrence of tumor-promoting mutations and genetic lesions, some of which will be tumor-promoting (Loeb et al., 2003; Nowell, 1976). Mutator genes, unlike oncogenes or tumor suppressor genes (TSGs), do not directly affect rate of cell growth or death, but instead increase the chance that oncogene and TSG mutations will appear (Baranovskaya et al., 2001).
An important advance in the study of aneuploidy was the discovery by Vogelstein and colleagues that many cancer cell lines exhibit chromosome instability (CIN), a phenotype in which cell division is accompanied by an abnormally high rate of chromosome loss and gain (Lengauer et al., 1997). Cell fusion studies involving these cell lines also suggest that specific recessive mutations are responsible for CIN. These findings argue that aneuploidy is a consequence of GCIN-promoting mutations rather than of sporadic errors in mitosis. Thus, CIN can be considered as one form of genomic instability, along with elevated rates of mutation, errors in DNA repair and somatic hyper-recombination (reviewed in (Masuda and Takahashi, 2002), see Table 1.1). The
potential consequences of CIN are best understood within the context of the mutator hypothesis, and modeling studies have shown that CIN is sufficiently powerful as a mutagen to drive tumor progression (Nowak et al., 2002). Indeed, the extreme view has been put forth that CIN alone is sufficient for cancer - in the absence of either oncogene or TSG mutation (Duesberg and Li, 2003).
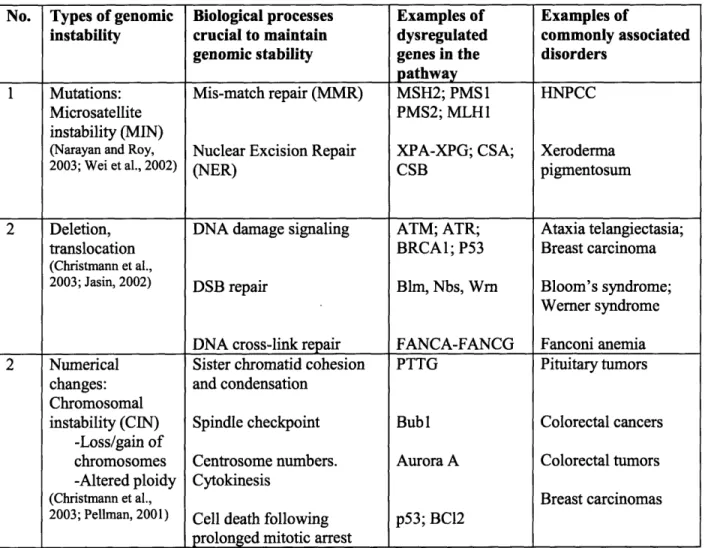
Table 1.1: Ways to acquire genomic instability
No. Types of genomic Biological processes Examples of Examples of
instability crucial to maintain dysregulated commonly associated genomic stability genes in the disorders
pathway
1 Mutations: Mis-match repair (MMR) MSH2; PMS1 HNPCC
Microsatellite PMS2; MLH1
instability (MIN)
(Narayan and Roy, Nuclear Excision Repair XPA-XPG; CSA; Xeroderma
2003; Wei et al., 2002) (NER) CSB pigmentosum
2 Deletion, DNA damage signaling ATM; ATR; Ataxia telangiectasia;
translocation BRCAl; P53 Breast carcinoma
(Christmann et al.,
2003; Jasin, 2002) DSB repair Blm, Nbs, Wrn Bloom's syndrome;
Werner syndrome DNA cross-link repair FANCA-FANCG Fanconi anemia 2 Numerical Sister chromatid cohesion PTTG Pituitary tumors
changes: and condensation Chromosomal
instability (CIN) Spindle checkpoint Bub 1 Colorectal cancers -Loss/gain of
chromosomes Centrosome numbers. Aurora A Colorectal tumors -Altered ploidy Cytokinesis
(Christmann et al., Breast carcinomas
2003; Pellman, 2001) Cell death following p53; BCl2 prolonged mitotic arrest
Other experiments challenge the idea that CIN is important for tumorigenesis. Studies in mice, for example, have shown that adenomas can develop without changes in karyotype or other obvious genomic instability (Haigis et al., 2002). The difficulty in proving a connection between CIN and cancer is that the molecular mechanisms of chromosome segregation, and the specific lesions that give rise to CIN, remain largely unknown. This situation contrasts with our current understanding of microsatellite instability (MIN), a type of genomic instability associated with errors in DNA mismatch repair (MMR) and a consequent 1000-fold increase in the rate of DNA mutation
(reviewed in (Christmann et al., 2003)). Interestingly, CIN and MIN appear to be mutually exclusive in many tumor cells examined suggesting the mutations that lead to either form of instability are in different sets of genes (Lengauer et al., 1997; Loeb,
1991). Prior to the discovery that the cancer-susceptibility syndrome HNPCC results from mutations in the MMR system, attempts to show that tumor cells have a higher intrinsic mutation rate than normal cells were unsuccessful. The clinical significance of links between cancer and MMR is emphasized by data showing that mutations in MSH1 and MSH2 (MMR genes) are found not only in HNPCC, but also in sporadic human colorectal cancers (Narayan and Roy, 2003). Moreover, knockout studies with the murine MMR genes demonstrate that MMR defects directly promote tumorigenesis (Wei et al., 2002). Finally, biochemical and cell biological studies show that MMR defects directly increase the mutation rate. The argument that MIN plays a direct role in the development of cancer is therefore supported by three strong pieces of evidence: a connection to hereditary and sporadic tumors in humans, a cancer model in mice, and compelling
biochemical data on the mechanism. As yet, none of these types of data are available for
CIN.
B. Searching for the molecular basis of CIN
Aneuploidy is thought to arise from errors in chromosome segregation such as non-disjunction and loss. Accurate segregation is maintained both by the intrinsic properties of the mitotic machinery and by a spindle checkpoint (reviewed in (Yu, 2002)
and described later in this chapter). Thus, in eukaryotes ranging from yeast to mice to humans, mutations in cell cycle regulators, checkpoint proteins and structural
components of the mitotic spindle can cause CIN (Masuda and Takahashi, 2002). However, two potential causes of CIN have received particular attention: mutations in
spindle checkpoint proteins and defects in the regulation of centrosome numbers
(reviewed in (Nigg, 2002)). Centrosome amplification, observed in many tumor cells, can often cause multipolar mitosis and lead to aberrant chromosome segregation (Marx, 2001; Sato et al., 1999). By decoupling the structural events of the chromosome cycle from cell cycle progression, defects in the spindle checkpoint lead to non-disjunction and the gain or loss of chromosomes.
C. Spindle checkpoint deficiencies in tumors
The spindle checkpoint proteins form a signal transduction system containing at least three key functional entities: a sensor that monitors the state of kinetochore-microtubule attachment, an amplifier that makes cell cycle progression sensitive to the mis-alignment of a single chromosome, and one or more regulators that control the activity of the anaphase promoting complex responsible for degrading securin and mitotic
cyclins (Yu, 2002). The spindle checkpoint acts to delay the onset of anaphase until all pairs of duplicated (sister) chromatids have achieved bipolar attachment to the
microtubules of the mitotic spindle. Bipolar attachment involves the binding of one kinetochore in a pair of sisters to microtubules emanating from one spindle pole and the binding of the other kinetochore to microtubules emanating from the opposite pole. The majority of checkpoint genes (Madl, Mad2, BubR1, Bubl, Bub3, Mpsl, and Aurora-B) have been highly conserved among higher and lower eukaryotes; although two genes (Rod and Zwl 0) are restricted to metazoans and one gene (Rae 1) appears to regulate the checkpoint in mice but not in yeast (Babu et al., 2003; Carmena and Earnshaw, 2003; Yu, 2002). Commonly used anti-neoplastic agents, such as paclitaxel and vincristine, affect microtubule dynamics and trigger the spindle checkpoint. These drugs cause mitotic arrest and cell death in checkpoint-proficient cancer cells. In checkpoint-impaired tumors, anti-microtubule drugs may increase chromosome missegregation and aggravate CIN (reviewed in (Mollinedo and Gajate, 2003)). Alternatively, a lack of checkpoint control may sensitize cells to chemotherapeutics because severe failure of cell division gives rise to inviable daughter cells. Thus, the spindle checkpoint status of a tumor is likely to be an important consideration for chemotherapy, but no systematic clinical studies
investigating this possibility have been reported to date.
A high percentage of solid tumors fail to arrest in response to microtubule poisons such as nocodazole, suggesting an impaired spindle checkpoint, and prompting a search in human tumors for mutations in checkpoint genes. Such mutations have been found in some CIN human colorectal cancer lines (Cahill et al., 1998) and the expression of a truncated form of the Bub kinase similar to that found in tumors has been shown to
cause defects in chromosome segregation (Taylor et al., 1998). However, only a small fraction of CIN cancer cells appear to carry mutations in the Mad and Bub checkpoint genes (Tighe et al., 2001). To date, the strongest evidence for a direct role for aneuploidy as a direct cause in human cancer is the discovery of BubR1 mutations in several
individuals with mosaic variegated aneuploidy (MVA), a rare disorder where individuals are prone to cancer (Hanks et al., 2004; Matsuura et al., 2006). Although one study was unsuccessful in detecting Mad2 mutations in 32 human primary gastric cancers, a recent report found Mad2 was mutated in 45% of the tested gastric cancer tissues (Kim et al.,
2005b; Tanaka et al., 2001). Moreover, two Mad2 mutant alleles containing amino acid substitutions were identified which when overexpressed in HeLa cells and then
challenged with nocadazole caused aneuploidy (Kim et al., 2005b). However, none of the checkpoint mutations found in human cells have been shown to be tumorigenic when reintroduced into mice. Sequestration of checkpoint proteins has also been proposed as a way in which to impair the spindle checkpoint (see below) in tumorigenesis. In adult T-cell leukemias, the Tax viral oncoprotein appears to bind and mislocalize Madl thereby inactivating the checkpoint and presumably aiding tumor progression (Kasai et al., 2002). Breast cancer-specific gene 1 (BCSG1) may also exert its oncogenic effect by binding to and reducing BubRl protein levels (Gupta et al., 2003). Furthermore, Breast cancer gene 1 (BRCA1) appears to regulate Mad2 through binding the transcription factor Octl and mutating Brcal in mice decreases Mad2 expression levels (Wang et al., 2004b). Future study of these phenomena may help to reconcile the observed high rate of spindle checkpoint inactivation with the infrequent mutation of the canonical checkpoint genes.
The connection between inactivation of the spindle checkpoint and tumorigenesis has also been studied in mice. One complication of these studies is that the deletion of Mad and Bub genes in mice causes widespread apoptosis during early development and embryonic lethality (Babu et al., 2003; Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). Thus, it has not been possible to compare directly rates of tumor formation in wild-type and checkpoint-deficient animals. However, the loss of one copy of BubR1 or the conditional loss of CENP-E in mice cause abnormal chromosome number in both
splenic and liver cells, respectively (Putkey et al., 2002; Wang et al., 2004a). Moreover, heterozygous Mad2 (Mad2 /') has been shown to promote the formation of non-lethal lung tumors (Michel et al., 2001). Similarly, Bub3 /- Rael+- /compound mice are characterized by spindle checkpoint dysfunction, chromosome missegregation, elevated rates of aneuploidy, and increased tumor formation following treatment with a carcinogen (Babu et al., 2003). BubR1+ - mice also exhibit enhanced carcinogen-induced tumor development (Dai et al., 2004). Furthermore, BubR1 haploinsufficiency increases the incidence of colonic tumors in ApcMirn+ mice (Rao et al., 2005). Overall, these data suggest a real but weak connection between tumor incidence and mutation of spindle checkpoint genes.
Recent studies suggest that mitotic regulators other than Mads and Bubs may be promising candidates for genes whose mutation cause CIN. Aurora A has been
implicated in mitotic entry and centrosome duplication (Giet et al., 2002; Hirota et al., 2003; Meraldi et al., 2002). The Aurora-A gene lies in a chromosomal region commonly amplified in human epithelial cancers, and Aurora-A mRNA is overexpressed in a wide variety of human cancers (Miyoshi et al., 2001). Overproduction of Aurora-A appears to
disrupt the binding of BubR1 to Cdc20 (an activator of the anaphase promoting complex) and to abrogate the spindle checkpoint, thereby generating a CIN phenotype (Jiang et al., 2003). Adenomatous Polyposis Coli (APC), a microtubule associated protein localized to kinetochores, is another gene frequently mutated in human colorectal cancers. Dominant mutations in APC cause chromosome segregation errors, although the connection
between APC and CIN in human tumors remains controversial (Green and Kaplan, 2003; Haigis et al., 2002; Kaplan et al., 2001; Sieber et al., 2002). Several other structural proteins with a role in spindle assembly are overexpressed in tumors (see Table 1.2), although their role in CIN is not yet known.
To date, the study of human cancers and of mouse models has failed to identify a checkpoint gene whose mutation, like that of p53 in the DNA damage response, has a direct and widespread role in tumorigenesis. Why is this? One possibility is that the spindle checkpoint is simply not involved in CIN in human tumors. Alternatively, a wide variety of different genes -including Mads and Bubs- may play a role in CIN but each mutation may be found in only a small percentage of tumors (i.e. BubR1 and MVA). Another possibility is that we are not searching for mutations in the right spindle checkpoint gene or that alterations in the checkpoint need not be dramatic to exert an effect on tumor development. The mitotic delay established by the spindle checkpoint is seldom permanent and in the presence of an activated checkpoint, many cells eventually bypass the delay and eventually progress to G1, often as tetraploid cells (Rieder and Maiato, 2004; Shi and King, 2005). Both the biochemical signal from unattached kinetochores and the subsequent response by checkpoint protens are not all-or-none events, and can be weakened, particularly in partial depletion studies of spindle
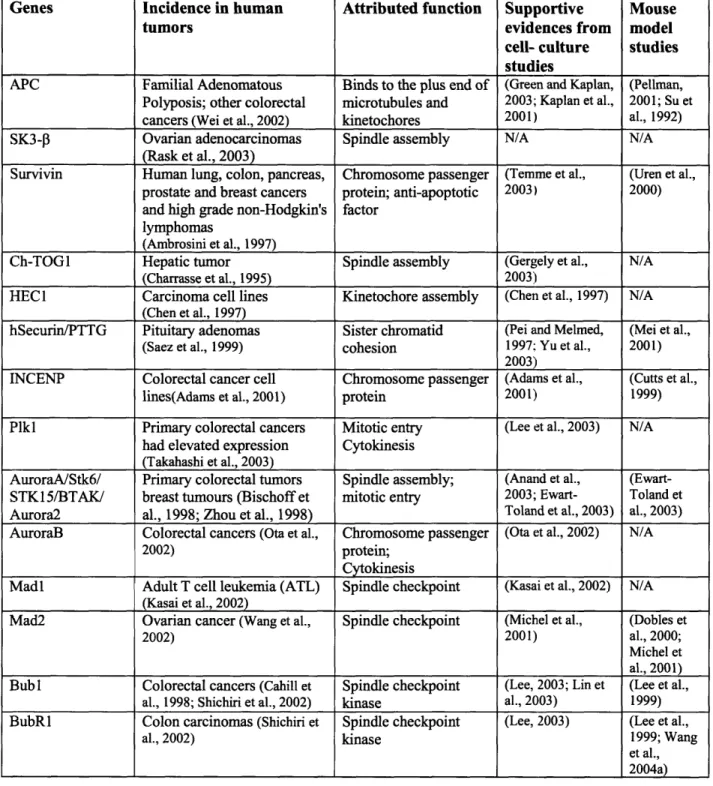
Table 1.2: Chromosome segregation genes mutated in human cancers
Genes Incidence in human Attributed function Supportive Mouse
tumors evidences from model
cell- culture studies
studies
APC Familial Adenomatous Binds to the plus end of (Green and Kaplan, (Pellman, Polyposis; other colorectal microtubules and 2003; Kaplan et al., 2001; Su et cancers (Wei et al., 2002) kinetochores 2001) al., 1992) SK3-P Ovarian adenocarcinomas Spindle assembly N/A N/A
(Rask et al., 2003)
Survivin Human lung, colon, pancreas, Chromosome passenger (Temme et al., (Uren et al., prostate and breast cancers protein; anti-apoptotic 2003) 2000) and high grade non-Hodgkin's factor
lymphomas
(Ambrosini et al., 1997)
Ch-TOGI Hepatic tumor Spindle assembly (Gergely et al., N/A (Charrasse et al., 1995) 2003)
HEC 1 Carcinoma cell lines Kinetochore assembly (Chen et al., 1997) N/A (Chen et al., 1997)
hSecurin/PTTG Pituitary adenomas Sister chromatid (Pei and Melmed, (Mei et al., (Saez et al., 1999) cohesion 1997; Yu et al., 2001)
2003)
INCENP Colorectal cancer cell Chromosome passenger (Adams et al., (Cutts et al., lines(Adams et al., 2001) protein 2001) 1999) Plkl Primary colorectal cancers Mitotic entry (Lee et al., 2003) N/A
had elevated expression Cytokinesis (Takahashi et al., 2003)
AuroraA/Stk6/ Primary colorectal tumors Spindle assembly; (Anand et al., (Ewart-STK15/BTAK/ breast tumours (Bischoff et mitotic entry 2003; Ewart- Toland et Aurora2 al., 1998; Zhou et al., 1998) Toland et al., 2003) al., 2003) AuroraB Colorectal cancers (Ota et al., Chromosome passenger (Ota et al., 2002) N/A
2002) protein;
Cytokinesis
Madl Adult T cell leukemia (ATL) Spindle checkpoint (Kasai et al., 2002) N/A (Kasai et al., 2002)
Mad2 Ovarian cancer (Wang et al., Spindle checkpoint (Michel et al., (Dobles et
2002) 2001) al., 2000;
Michel et al., 2001) Bubl Colorectal cancers (Cahill et Spindle checkpoint (Lee, 2003; Lin et (Lee et al.,
al., 1998; Shichiri et al., 2002) kinase al., 2003) 1999) BubR1 Colon carcinomas (Shichiri et Spindle checkpoint (Lee, 2003) (Lee et al.,
al., 2002) kinase 1999; Wang
et al., 2004a) checkpoint components by RNA interference (Martin-Lluesma et al., 2002; Meraldi et
al., 2004; Rieder and Maiato, 2004). Thus, a weakened checkpoint would still cause CIN
and possibly promote tumorigenesis, but not necessarily inactivate checkpoint signaling ((Baker et al., 2006) and Chapter 2).
D. Senescence or tumorigenesis?
Recent studies of mice with mutated spindle checkpoint genes suggest that although all spindle checkpoint mutations induce aneuploidy, CIN is as likely to cause cellular senescence and early aging as tumorigenesis (reviewed in (Baker et al., 2005)). BubR +/- mice, as indicated earlier, exhibit aneuploidy, but have little elevated
predisposition to cancer (Wang et al., 2004a). Instead, decreased expression of BubR1 causes early onset aging phenotypes and decreased lifespan in mice as demonstrated with a series of BubR1 genetic mutants combining hypomorphic and knockout alleles (Baker et al., 2004). This effect can be directly attributed to increased CIN. In fact, wildtype mice show declining levels of BubR1 with age in several tissues, particularly in the testes. BubR1 mutant mice are also infertile due to meiotic defects. (Meiosis and the
spindle checkpoint will be discussed further later in this chapter.) How do we reconcile this with the human BubR1 mutations in MVA? First, since MVA is quite rare, the primary phenotype for BubR1 inactivation may be early aging. Second, patients with MVA may exhibit early aging since they are prone to developing cataracts ((Furukawa et al., 2003; Matsuura et al., 2006). A link between early onset aging and checkpoint induced aneuploidy is further strengthened from a study of Bub3+/-; Rae+/- compound mice (Baker et al., 2006). Like BubR1 haploinsufficient mice, Bub3+/-; Rae+/- mice have a low propensity for spontaneous tumor formation (Babu et al., 2003). However, Bub3+'; Rae+/- compound mutant mice develop signs of aging earlier than either single mutant.
Furthermore, Bub3 /-; Rae+/- mouse embryonic fibroblasts (MEFs) undergo premature senescence which appears to be dependent on the p53-regulated growth arrest pathway since several proteins in the pathway including pl 9ARF, p53, p21, and p16 are upregulated. In contrast, Mad2'/- MEFs do not display premature cellular senescence (Baker et al., 2006). Instead, Mad2 inactivation predisposes mice to tumor development ((Michel et al., 2001); Ying Yue, personal communication). These data suggest that inactivation of spindle checkpoint components can be assigned to at least two groups: 1) those that cause premature senescence and early aging and 2) those that cause cancer. The DNA damage and repair field is rampant with examples of mutations that can either cause diseases displaying either early aging or cancer (Dimri, 2005; Dolle et al., 2006; Fuss and Cooper, 2006). Evidence is accruing that links spindle checkpoint components to protective effects against both cancer and aging, but clear aetiological evidence to an extent similar to the DNA repair field remains to be shown.
E. Connection between DNA damage and mitotic progression and cancer?
MIN and CIN are often presented as alternative routes to genomic instability, but considerable evidence is accumulating that DNA damage pathways interact strongly with spindle checkpoint mutations. For example, whereas Mad2/-1 MEFs are inviable, Mad2/
p53-/- double knockout MEFs are viable (Burds et al., 2005). This suggests an important
link between the spindle checkpoint and the DNA damage pathway. Mpsl, a spindle checkpoint kinase, may link mitosis to the G2 DNA damage checkpoint. Mpsl has been
shown to interact with Chk2 and phosphorylate Chk2 on threonine 68 (Wei et al., 2005). Recently, Mpsl has also been shown to phosphorylate the Bloom syndrome gene BLM during mitosis (Leng et al., 2006). Upon phosphorylation, BLM, a member of the RecQ
family of helicases, can interact with the mitotic kinase Plkl (Leng et al., 2006). Rad5 1, another component of the DNA damage pathway, has also been shown to interact with the breast-cancer associated protein Brca2 (reviewed in (Jasin, 2002)). Mice harboring homozygous Brca2 truncations (Brca2TrrTr) develop thymic lymphomas with an impaired spindle checkpoint and mutations in p53, Bubl or BubR1 (Lee et al., 1999). Intriguingly, expression of a truncated Bub 1 protein also appears to induce transformation of Brca2Trr/T MEFs (Lee et al., 1999). Moreover, BRCA2 has been reported to interact with PLK1 and the spindle checkpoint kinase hBubR1 in vitro (Futamura et al., 2000; Lee et al., 2003). These data offer the possibility that CIN may arise from a synergy between errors in mitosis and lesions in DNA repair pathways.
F. Conclusion
The evidence that genomic instability plays a critical role in tumorigenesis is strong, and extensive circumstantial data points to CIN as an important source of genomic instability. Chromosome missegregation resulting from the deregulation of the spindle checkpoint is thought to be one cause of CIN. However, evidence for this connection remains weak, and the molecular basis of CIN remains largely unknown. To better elucidate the connections between aneuploidy and tumorigenesis, it will be
necessary to gain a better understanding of chromosome segregation events in normal cells and to identify target genes whose mutations might cause CIN in tumors. Sensitive assays for CIN in live animal cells will be necessary for this analysis (Meraldi et al., 2004; Rieder and Khodjakov, 2003). Such assays have already been shown in yeast to be essential for the identification and analysis of CIN-promoting mutations (Nigg, 2002). Finally, the effects of CIN on tumor formation must be investigated in mice. Only then
will we be able to establish the role that errors in chromosome segregation play in genomic instability and the development of human cancer.
II. The spindle checkpoint
Chromosomal instability (CIN) refers to an abnormal cellular state associated with an increased rate of chromosome loss or gain (Draviam et al., 2004; Kops et al., 2005). During mitosis, a cell must properly attach all its chromosomes spindle microtubules and align them such that one daughter cell receives one complete set of chromosomes and the other daughter cells receives the second identical set. The spindle checkpoint, also known as the spindle assembly checkpoint or the mitotic checkpoint, monitors genomic stability by ensuring faithful transmission of chromosomes to the daughter cells during mitosis (Amon, 1999; Gardner and Burke, 2000; Shah and Cleveland, 2000; Wassmann and Benezra, 2001). The spindle checkpoint delays anaphase until all sister chromatids have made bipolar attachments to microtubules emanating from opposite poles of the mitotic spindle (Fig. 1.1). Chromosomes have specialized multi-protein complexes called kinetochores, that bind to their centromeres and attach the DNA to microtubules. It is crucial that the mitotic spindle only begin to pull sister chromatids apart once every kinetochore has properly bound microtubules. Strikingly, a single misattached kinetochore is sufficient to activate the spindle checkpoint and delay mitotic progression to allow additional time for chromosomes to become properly attached to the spindle (Li and Nicklas, 1995; Rieder et al., 1995). Abrogation of the spindle checkpoint would be detrimental to genomic stability as chromosome missegregation leads to CIN. In this section, I explain the paradigm of a cell
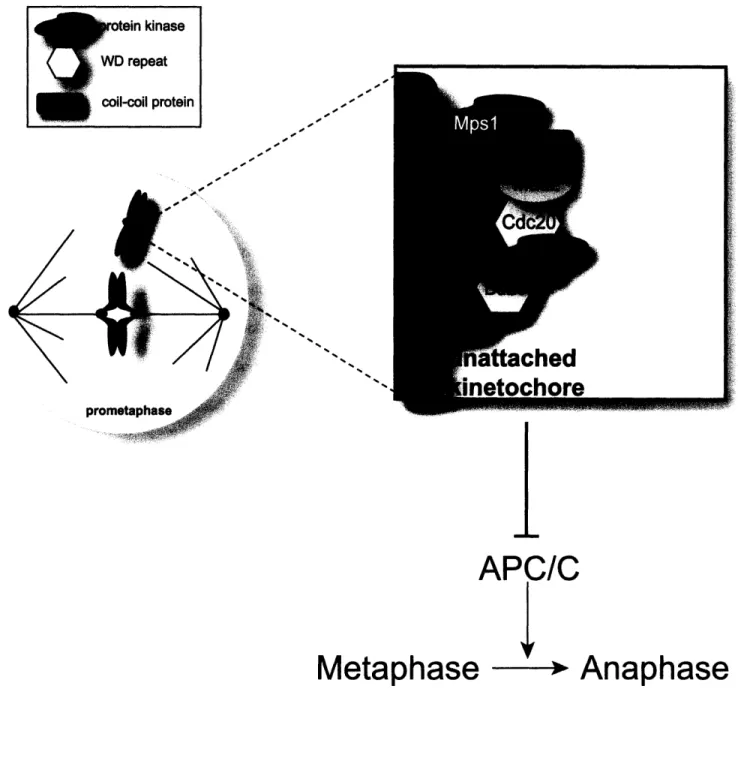
Figure 1.1
APC/C
Y
Metaphase
Anaphase
kinase >eat I protein rFigure 1.1: The canonical spindle checkpoint proteins.
Spindle checkpoint proteins localize to unattached kinetochores and prevent the metaphase to anaphase transition by inhibiting the activation of the APC/C. Mpsl is required for kinetochore localization of Madl and Mad2. Mad2 and BubR1 can directly as well as in combination with Bub3 and BubR1 inhibit Cdc20 from activating the APC/C, thus preventing ubiquitination of securin and mitotic progression.
cycle checkpoint, describe the discovery of spindle checkpoint components and briefly examine checkpoint signaling and its importance in meiosis.
A. Cell cycle and cell cycle checkpoints
Cell duplication proceeds through a series of events called the cell cycle that occurs in a tightly regulated and temporally ordered fashion (Dash and El-Deiry, 2004; Kastan and Bartek, 2004). The cell must first accumulate the necessary nutrients for growth during Gl before it can replicate its DNA in S phase. In G2, a cell undergoes a further period of growth before it is ready to enter mitosis when the cell must first segregate its chromosomes before it undergoes cytokinesis. To ensure the cell cycle proceeds in the correct manner, checkpoints act to delay downstream events until upstream events have been properly transpired. For example, DNA damage surveillance mechanisms operate prior to DNA replication and mitosis in G1 and G2 to maintain genome fidelity by arresting cell cycle progression until the damage has been repaired (Hartwell and Weinert, 1989). The original checkpoint paradigm, RAD9, was identified in Saccharomyces cervisiae in a genetic screen for mutations that allow budding yeast to
continue to undergo cell division despite radiation induced DNA damage (Weinert and Hartwell, 1988). Whereas wildtype yeast arrest in G2 upon exposure to low dosages of X-ray irradiation, rad9 cells fail to arrest and proceed to mitosis with DNA damage and may die. However, when rad9 cells are treated simultaneously with X-rays and
nocodazole to arrest cells in mitosis, cells have sufficient time to repair DNA lesions and viability is restored. Furthermore, rad9 cells exhibit normal growth and viability in the absence of exposure to DNA damaging agents (Weinert and Hartwell, 1988). Thus, cell cycle checkpoints were proposed to be non-essential surveillance systems that monitor
essential cell processes and respond in the event of a rare problem by inducing cell cycle arrest (Hartwell and Weinert, 1989; Weinert and Hartwell, 1988).
B. The canonical players of the spindle checkpoint
The spindle checkpoint delays the onset of anaphase by inhibiting the anaphase promoting complex/ cyclosome (APC/C), a multi-subunit E3 ubiquitin ligase, until all chromosomes have achieved stable microtubule attachments and congressed to the metaphase plate (Amon, 1999). Yeast with a wildtype spindle checkpoint undergo very accurate chromosome segregation as less than one missegregation event can be detected in 105 cell divisions (Hartwell et al., 1982). Like the DNA damage checkpoint, genetic screens in S. cervisiae identified the canonical components of the spindle checkpoint (Hoyt et al., 1991; Li and Murray, 1991; Winey et al., 1991). Two genetic screens searching for mutations that would allow cells to bypass spindle damage induced by the microtubule destabilizing drug benomyl identified six genes: MADI, M4D2, and MAD3 for Mitotic Arrest Deficient, and BUB], BUB2, and BUB3 fo Budding Uninhibited by Benzimidazole (Hoyt et al., 1991; Li and Murray, 1991). In budding yeast, a mutation in any one of the six genes disables the ability of a cell to arrest in response to spindle damage suggesting these genes were part of a spindle assembly checkpoint (Hoyt et al., 1991; Li and Murray, 1991). BUB2 was subsequently shown to be required for mitotic exit, not the spindle checkpoint. Mutations in any of the MAD and BUB genes do not decrease viability in the absence of spindle damage, although mad mutations increase the chromosome loss rate 10-fold under normal growth conditions and up to 1000-fold in the presence of benomyl (Li and Murray, 1991). Two additional components were
Spindle 1) and Cdc20. The Mads and Bubs were shown to target an activator of the APC/C: Cdc20 (Hwang et al., 1998). MPS1 was initially identified in a screen for mutants defective in spindle pole body (SPB) duplication in S. cerevisae, but was later
shown to be essential for the spindle checkpoint (Weiss and Winey, 1996; Winey et al., 1991). The SPB duplication and spindle checkpoint functions of Mps lp are genetically separable; and it is the inactivation of Mpslp in SPB duplication that causes budding yeast inviability (Castillo et al., 2002; Schutz and Winey, 1998). Thus, the original definition of a cell cycle checkpoint, as an auxiliary system dispensable for normal growth, appears to hold true for the yeast spindle assembly checkpoint.
The spindle assembly checkpoint has been functionally conserved in all eukaryotes (Wassmann and Benezra, 2001). All six budding yeast checkpoint
components - MAD1-3, BUB1, BUB3, and MPSI -have metazoan homologues.
However, BubR1, the metazoan homologue of Mad3p, is actually a hybrid of sequences from Mad3p and Bublp, with a Mad3p-like N-terminal domain and a C-terminal Bubl-like kinase domain (Taylor et al., 1998). Like Bub and BubR1, Mps is also a kinase (Winey and Huneycutt, 2002). Madl is a coiled-coil protein (Sironi et al., 2002). Bub3 contains WD repeats and is also very similar to Rae 1, which functions in nucelo-cytoplasmic transport (Wang et al., 2001). Additional genes have been found to be required for the spindle checkpoint including Ipll/Aurora B kinase, Birlp/Survivin and Slil5p/INCENP; CENP-E (centromere-associated protein E); Rael; CMT2 (Caught by MAD Two); and also Rod and ZwlO which are unique to vertebrates (Adams et al., 2001; Babu et al., 2003; Chan et al., 2000; Cutts et al., 1999; Habu et al., 2002; Kallio et al., 2001; Putkey et al., 2002; Uren et al., 2000; Wang et al., 2001; Weaver et al., 2003). The
canonical checkpoint components - Bubl, BubR1, Bub3, Madl, Mad2, and Mpsl - are
part of a bona fide mammalian spindle checkpoint as inactivation of these proteins abrogates the checkpoint and results in chromosome missegregation, aneuploidy and failure to arrest in mitosis in response to microtubule destabilizing agents (Meraldi et al., 2004; Stucke et al., 2002; Taylor et al., 1998; Yu, 2002).
C. Interactions between the spindle checkpoint and the cell cycle
The importance of the spindle checkpoint for viability seems to increase with an organism's complexity. The loss of the checkpoint in S. cerevisiae leads to increased rate of chromosome loss, but is only lethal in the pressure of anti-microtubule drugs (Li and Murray, 1991). However, the checkpoint is essential in metazoans under normal growth conditions and inactivation of spindle checkpoint genes leads to embryonic lethality in mice (Dobles et al., 2000; Kalitsis et al., 2000; Wang et al., 2004a). In a multicellular organism like the mouse, where viability is dependent on the interaction of all of the cells, the apoptosis of aneuploid cells in G1 or mitotic catastrophe caused by impaired checkpoint function is disastrous for the embryo (Dobles et al., 2000). Furthermore, the spindle checkpoint is not required in mouse embryonic fibroblasts lacking p53,
presumably because apoptosis is impaired (Burds et al., 2005). In unicellular organisms like S. cerevisiae, failure to undergo cell cycle arrest and transmission of genetic alterations either leads to death or adaptation to the mutations accumulated in the cell. Though these outcomes are tolerated in yeast, metazoans have evolved a more stringent set of cell cycle checkpoints that can induce apoptosis as well as cell cycle arrest to protect genome fidelity and prevent the propagation of cancerous cells.
Expression levels of several spindle checkpoint components are regulated in a cell cycle dependent manner by E2F. The E2F family of transcription factors is crucial to the control of cell cycle progression by both tranactivating and repressing expression of genes integral to DNA replication, DNA repair and mitosis (Hansemann, 1890; Polager et al., 2002). The tumor suppressor pRB regulates the E2F proteins. Mad2 is a direct target of E2F 1 and cells with mutations in the Rb pathway have misregulated expression of Mad2 protein (Hernando et al., 2004). E2F1 and E2F3 were found to up-regulate the expression of several mitotic genes including BUBR1 (BUB B) and Aurora B (AIM1) by DNA microarray analysis in Rat cells (Polager et al., 2002). Another study utilizing a combination of chromatin immunoprecipitation and DNA microarray analysis in human cell lines found the repressive E2F4 efficiently binds to the promoters of hMAD2 (MAD2L1), hBUB3 and hMPS1 (TTK) (Ren et al., 2002). Moreover, the promoter of HEC 1, whose gene product Ndc80 is required for the kinetochore localization of both Mad2 and Mps 1, is regulated by both repressing and activating E2Fs (E2F4 and E2F 1) (Ren et al., 2002). Thus, one way the spindle checkpoint is integrated with the rest of the cell cycle is via regulation by the E2F family of transcription factors.
D. Spindle checkpoint signaling
To understand the mechanism of spindle checkpoint signaling, we must understand the signal for checkpoint activation, the signal transmission system, the mechanism of mitotic arrest and the cessation of checkpoint signaling that allows mitosis to continue. The underlying biochemistry of how the checkpoint is activated and then turned off is incomplete, but it is believed the "prevent anaphase" signal must originate from the kinetochores. The kinetochore is a complex structure built onto the centromere
of each chromosome that mediates attachments to microtubules and encompasses as many as 60 proteins (De Wulf et al., 2003; McAinsh et al., 2003). The canonical
mammalian spindle checkpoint proteins -Mpsl, Madl, Mad2, Bubl, BubR1, and Bub3
-all localize to kinetochores during mitosis (Fisk and Winey, 2001; Jablonski et al., 1998; Kallio et al., 1998; Li and Benezra, 1996; Martinez-Exposito et al., 1999; Meraldi et al., 2004; Taylor et al., 1998; Taylor and McKeon, 1997). The timing of their arrival at kinetochores varies in mitosis, but all are present by nuclear envelope breakdown in late prophase and their abundance decreases following chromosome attachment, suggesting checkpoint components are monitoring kinetochore-microtubule interactions.
Two models summarize an ongoing debate over the exact nature of the "prevent anaphase" signal sensed by spindle checkpoint components: 1) the spindle checkpoint components monitor the microtubule occupancy status on kinetochores; or 2) the checkpoint components monitor the tension generated across sister kinetochores by the bipolar attachment of sister chromatid pairs to microtubules emanating from opposite poles of the mitotic spindle. Many attempts have been made to uncouple kinetochore-microtubule attachment from the tension generated across kinetochore pairs resulting from that attachment (Kapoor et al., 2000; Rieder et al., 1995; Stem and Murray, 2001). Support for the kinetochore-microtubule attachment model comes from experiments in PtK cells where anaphase initiates about 23 minutes after the last kinetochore captures microtubules (Rieder et al., 1994). Moreover, when the last unattached kinetochore is destroyed by laser ablation, cells are able to proceed to anaphase and undergo
chromosome segregation (Rieder et al., 1995). Mad2 specifically localizes to unattached kinetochores in PtK cells, but those kinetochores that have lost tension due to
microtubule stabilization by taxol treatment fail to recruit Mad2 (Waters et al., 1998). However, when DNA replication is prevented by a mutation in CDC6, an initiator of DNA replication in budding yeast, then chromatids can bind microtubules, but are not under tension since there is no sister and the spindle checkpoint is activated (Stem and Murray, 2001). Furthermore, when cells are exposed to monastrol, which inhibits the mitotic motor Eg5, synthelic (microtubules from the same pole) kinetochores attached to microtubules still retain Mad2 suggesting microtubule attachment alone is insufficient to satisfy the checkpoint (Kapoor et al., 2000). These experiments have shown that either the lack of microtubule attachment or tension can be sufficient for activation of the spindle checkpoint. For further complexity, the Iplil/Aurora B kinase has been
implicated in spindle checkpoint function, specifically for the sensing and correction of synthelic attachments which do not produce tension (Hauf et al., 2003; Tanaka et al., 2002). However, the debate may never be resolved due to the interdependence of
microtubule attachment and tension at kinetochores; kinetochore-microtubule attachment is stabilized by tension and both attachment and tension appear to be monitor by the spindle checkpoint (Nicklas et al., 2001). Nonetheless, the spindle checkpoint can monitor the status of sister chromatid bi-orientation and respond to any perturbations by arresting cells at the metaphase to anaphase transition.
Checkpoint signaling is presumably transmitted by the three kinases in the canonical spindle checkpoint -Bubl, BubR1, and Mpsl. Yet, little is known about spindle checkpoint signal transmission since few targets have been described for these three checkpoint kinases. The kinase function of Mpsl is necessary for checkpoint activation, but whether the kinase domain of Bubl is required for the checkpoint is
unclear (Abrieu et al., 2001; Chen, 2004; Warren et al., 2002). A truncated form of Bub1
lacking its C-terminal kinase domain is sufficient for checkpoint function in budding
yeast, but inhibition of Bub 1 kinase activity in Xenopus extracts causes partially
compromises the checkpoint (Chen, 2004; Warren et al., 2002). Mps1 and Bub 1 have
been shown to phosphorylate yeast Madl in vitro, but this has not been observed in vivo
(Hardwick et al., 1996; Seeley et al., 1999). Surprisingly, the kinase domain of BubR1 is
not required for inhibition of the APC/C in vitro (Tang et al., 2001). However, BubR1
kinase activity, which is activated by binding to CENP-E, has been shown to be essential
for checkpoint signaling in MEFs (Putkey et al., 2002; Weaver et al., 2003).
Checkpoint signal transmission does require multiple complexes of checkpoint
proteins. Bub3 must be in a complex with Bubl or BubR1 to bind to kinetochores (Taylor
et al., 1998;). Interaction with Bub3 is mediated by a GLEBs motif located in Bubl and
BubR1/Mad3 (Wang et al., 2001). Madl and Mad2 require Mpsl for kinetochore
localization in mammals (Liu et al., 2003; Martin-Lluesma et al., 2002). Mad2 also
requires the Rod/ZwlO0 complex to localize to unattached kinetochores (Buffin et al.,
2005). Mad 1 forms a homodimer via its coiled-coiled domain and recruits Mad2 to
unattached kinetochores. Once there, Mad2 can bind and inhibit Cdc20, a WD domain
protein that localizes to the kinetochore, by binding to an N-terminal 12-residue stretch of
Cdc20 (Luo et al., 2002; Sironi et al., 2001). Mad2 may only bind Madl and be recruited
to the kinetochore in an unphosphorylated form (Wassmann et al., 2003a). Crystal
structures of the Madl-Mad2 complex reveal a tetramer of two Madl -Mad2
subcomplexes with the Mad2 C-terminal tails as flexible elements that undergo
et al., 2002). Both Mad2 and BubR1 can independently interact directly with Cdc20 and inhibit the APC/C in vitro (Chan et al., 1999; Fang, 2002; Kallio et al., 1998; Sudakin et al., 2001; Tang et al., 2001). BubR1 and Mad2 inhibition of Cdc20 may also be
cooperative. A mitotic checkpoint complex (MCC) purified from HeLa cells that contained BubR1, Bub3, Cdc20 and Mad2 was found to be - 3000-fold more potent at inhibiting the APC/C than recombinant Mad2 alone (Sudakin et al., 2001). The isolation of these complexes suggests that the spindle checkpoint functions as a complex multi-protein machine in contrast to a standard signal transduction cascade, but conflicting data suggests the individual molecular interactions require additional elucidation. What has been well characterized is the ultimate target of the checkpoint: Cdc20 and the APC/C (reviewed in Zachariae and Nasmyth, 1999).
The APC/C is a highly regulated multi-subunit E3 ubiquitan ligase whose ubiquitination activity controls the timing of sister chromatid separation and mitotic exit via its regulators Cdc20 and Cdhl (Morgan, 1999; Visintin et al., 1997). Newly
replicated sister chromatids are physically held together by a protein complex called cohesin composed of Smcl, Smc3, Sccl and Scc3 (Uhlmann, 2003). Conserved throughout evolution, cohesins localize to the centromeres and along the arms of sister chromatids following DNA replication. However, chromosomes cannot segregate until sister chromatid cohesion is lost following cleavage of the cohesin subunit Scc by Esplp/separase. Separase is bound to and inhibited by Pdslp/securin until the APC/C is activated by Cdc20 to ubiquitinate securin, targeting it for degradation by the 26S proteosome (Cohen-Fix et al., 1996; Coux et al., 1996). Mad2 and BubR1 physically interact with Cdc20 and prevent activation of the APC/C to prevent premature
chromosome segregation (Chan et al., 1999; Fang, 2002; Kallio et al., 1998; Sudakin et al., 2001; Tang et al., 2001). Once all chromosomes have achieved bipolar attachment to the mitotic spindle, Cdc20 is released by the spindle checkpoint to activate the APC/C to dissolve sister chromatid cohesion (Kallio et al., 1998; Tavormina and Burke, 1998). Thus, when chromosomes are not properly attached or under proper tension during mitosis, a signal is generated to activate the spindle checkpoint machinery to prevent the degradation of securin, maintaining chromosome cohesion and preventing mitotic progression.
Silencing of the spindle checkpoint to allow anaphase entry appears to be an active process. Cdc20 must be released by Mad2 to activate the APC/C for chromosome
segregation to occur. Recently, a model for Mad2 activation has been proposed that suggests Mad2 exists in two conformations: an unbound open state that does not bind Cdc20 and a closed conformation when bound to Madl or Cdc20 (De Antoni et al., 2005; Nezi et al., 2006). Checkpoint activation promotes the conversion of cytosolic open Mad2 to the Madl/kinetochore bound closed conformation facilitating binding to and inhibiting Cdc20. When all kinetochores have properly bound to microtubules, Mad2 also interacts with an additional protein - CMT2/p31comet (Habu et al., 2002; Mapelli et al., 2006). CMT2 binds to Cdc20/Mad2 complexes and promotes the dissociation of Mad2 from Cdc20 (Habu et al., 2002). Robert Hagan and colleagues in the Sorger group have shown that CMT2 is a kinetochore-associated protein critical for exit from mitosis. RNAi depletion of CMT2 arrests cells in mitosis even though spindle checkpoint components have left kinetochores and this mitotic arrest is dependent on Mad2 (Robert Hagan,
personal communication). Thus, mammalian cells require the active inhibition of a Mad2-dependent spindle checkpoint by CMT2 for normal mitotic progression.
E. Mpsl: a spindle checkpoint kinase
Mps1 is a dual-specificity kinase that is unique among the checkpoint proteins in S. cerevisiae because it is required for the spindle assembly checkpoint and SPB
duplication, equivalent to the mammalian centrosome (Winey and Huneycutt, 2002). MPS 1 is the only essential checkpoint gene in budding yeast, but this is due to its role in SPB duplication (Castillo et al., 2002; Schutz and Winey, 1998). Mps lp interacts with yeast Damlp, an essential component of the DASH microtubule ring complex, which links kinetochores to microtubules thereby facilitating chromosome segregation (Jones et al., 1999; Li et al., 2002; Miranda et al., 2005). In S. pombe however, spMpslp/Mphlp acts exclusively in the spindle checkpoint (He et al., 1998). Epistatic analysis of the budding yeast checkpoint genes has placed scMPS 1 as the most upstream component of the checkpoint pathway (Weiss and Winey, 1996). Overexpression of Mps lp
constitutively activates the spindle checkpoint, in the absence of spindle damage, which is dependent on the Mads and Bubs (Hardwick et al., 1996; He et al., 1998). Although
scMpslp has been reported to phosphorylate scMadlp, this interaction has not been recapitulated in vertebrates (Hardwick et al., 1996). Nonetheless, Mps 1 appears to be upstream of other checkpoint components in metazoans as well since Mad2 and Madl localization to the kinetochore requires Mpsl (Abrieu et al., 2001; Liu et al., 2003; Martin-Lluesma et al., 2002). Homology between scMpslp and the vertebrate orthologs is confined to the C-terminal kinase region (Winey and Huneycutt, 2002). The mouse and human homologs of Mps 1, ESK and TTK, were first described in the literature as dual
specificity kinases able to phosphorylate both tyrosine and serine/threonine residues and highly expressed in tissues with a high proliferation rate such as the thymus and the testis (Douville et al., 1992; Hogg et al., 1994; Mills et al., 1992). Subsequently, ESK and TTK have definitely been shown to be the Mpsl functional homolog in mouse and human; homologs have also been found in the fly, frog, zebrafish (Abrieu et al., 2001; Fischer et al., 2004; Fisk and Winey, 2001; Poss et al., 2002; Stucke et al., 2002). Four findings hold true for Mps 1 throughout evolution: 1) Mps1 localizes to kinetochores, 2) the kinase function of Mps I is required for the checkpoint, 3) Mps likely acts early in the
checkpoint pathway and 4) Mpsl protein and mRNA levels are regulated during the cell cycle.
Although the checkpoint function of Mps 1 has been conserved in vertebrates, there is conflicting data on the role of Mps1 in centrosome duplication. Although mouse Mps 1 has been described to be required for centrosome duplication, three reports have both argued for and against human Mpsl localizing to centrosomes and regulating their duplication (Fisk et al., 2003; Fisk and Winey, 2001; Liu et al., 2003; Stucke et al., 2004; Stucke et al., 2002; Winey and Huneycutt, 2002). Moreover, drosophila Mpsl does localize to centrosomes, but Mps 1 mutants do not have any centrosome defects (Fischer et al., 2004). Although the existence of a role for mammalian Mps 1 in centrosome duplication remains controversial, recent reports describing interactions with Chk2 and Blm suggest hMps may have other functions outside of the spindle checkpoint in the DNA damage response (see section IE) (Leng et al., 2006; Wei et al., 2005). Thus, Mps is a mitotic kinase essential for the spindle checkpoint from yeast to man and may have
acquired additional functions that link the DNA damage response to accurate chromosome segregation.
F. Meiosis and the spindle checkpoint
In humans, aneuploidy has been linked to spontaneous abortions and
developmental defects as well to many cancers (Hassold and Hunt, 2001). A trisomy or monosomy event has been identified in at least 5% of human pregnancies (Hassold and Hunt, 2001). Although aneuploidy almost always causes embryonic lethality and results in spontaneous abortions, 1 in 300 infants are born with aneuploidy, usually in a sex chromosome or with an additional chromosome 13, 18, or 21. The latter event causes Down syndrome, a developmental disorder in which individuals show decreased mental capacitity and physical defects (reviewed in (Scarbrough et al., 1982)). The strongest causal basis for aneuploidy in humans is increasing maternal age. Women over 40 have at least a one in three probability of a pregnancy complicated by abnormal chromosome numbers (Hassold and Hunt, 2001; Thomas et al., 2001). The basis of this age effect is unclear, although it is intriguing to speculate that a decrease in spindle checkpoint function plays a role (Baker et al., 2005; Ma et al., 2005). BubR1 hypomorphic mice undergo meiotic chromosome missegregation and have decreased fertility (Baker et al., 2004). A Mad2-dependent spindle checkpoint has been shown to exist during the first meiotic division in mouse oocytes, and overexpression of Mad2 causes a metaphase I arrest (Wassmann et al., 2003b). Moreover, Mad2÷/ - mouse oocytes undergo meiosis with abnormal mitotic timing and chromosome segregation defects (Katja Wassmann,
personal communication). Mad2 downregulation by siRNA interference in mouse oocytes also decreased the length of meiosis I (Wang et al., 2006). Mpsl mutations in
zebrafish and fruit flies cause aneuploidy associated meiotic defects as well (Gilliland et al., 2005; Poss et al., 2004). Interestingly, while gaining or losing a single chromosome is a lethal event, gaining an entire set of chromosomes is less detrimental. Although
extremely rare, triploidy babies, those that have a 3N complement of the genome instead of the normal 2N, can survive to birth and possibly live for an additional few hours after birth (Leisti et al., 1974). Moreover, a cell often chooses to forgo cytokinesis and become polyploid after a chromosome missegregation event instead of gaining or losing a single
chromosome (Shi and King, 2005). This suggests that balancing gene expression is vital to a cell and therefore, understanding those cell cycle checkpoints that monitor genome
stability and maintain proper gene dosage such as the spindle checkpoint is critical to preventing and curing human disease.
III. Tumor suppressor genes and oncogenes
It was Theodore Boveri who first drew attention to the abnormal number of chromosomes in human tumors and hypothesized that the act of losing or gaining chromosomes may be sufficient to cause a cancerous state (Boveri, 1914). Although the role of aneuploidy in tumorigenesis is still debated, the gain or loss of a few genes called tumor suppressor genes (TSPs) or oncogenes are firmly established as causes of human tumorigenesis (Balmain, 2001; Bertwistle et al., 2004; Duesberg and Li, 2003).
Disregulation of TSPs and oncogenes desensitizes tumor cells to both proliferative signals and cell cycle checkpoints. Although current research is still trying to establish a clear relationship between components of the spindle assembly checkpoint and
tumorigenesis, more than two decades of scientific literature clearly link the DNA