HAL Id: hal-02375679
https://hal-normandie-univ.archives-ouvertes.fr/hal-02375679
Submitted on 9 Dec 2019
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Distributed under a Creative Commons Attribution| 4.0 International License
Comparative Genome Analysis of Staphylococcus
lugdunensis Shows Clonal Complex-Dependent Diversity
of the Putative Virulence Factor, ess/Type VII Locus
Jérémie Lebeurre, Sandrine Dahyot, Seydina Diene, Amandine Paulay, Marion
Aubourg, Xavier Argemi, Jean-Christophe Giard, Isabelle Tournier, Patrice
François, Martine Pestel-Caron
To cite this version:
Jérémie Lebeurre, Sandrine Dahyot, Seydina Diene, Amandine Paulay, Marion Aubourg, et al..
Com-parative Genome Analysis of Staphylococcus lugdunensis Shows Clonal Complex-Dependent Diversity
of the Putative Virulence Factor, ess/Type VII Locus. Frontiers in Microbiology, Frontiers Media,
2019, 10, pp.2479. �10.3389/fmicb.2019.02479�. �hal-02375679�
fmicb-10-02479 October 29, 2019 Time: 15:58 # 1 ORIGINAL RESEARCH published: 31 October 2019 doi: 10.3389/fmicb.2019.02479 Edited by: Frank T. Robb, University of Maryland, Baltimore, United States Reviewed by: Timothy J. Foster, Trinity College Dublin, Ireland Ben Pascoe, University of Bath, United Kingdom Simon Heilbronner, University of Tübingen, Germany *Correspondence: Jérémie Lebeurre jeremie.lebeurre@hotmail.fr
Specialty section: This article was submitted to Evolutionary and Genomic Microbiology, a section of the journal Frontiers in Microbiology Received: 22 July 2019 Accepted: 15 October 2019 Published: 31 October 2019 Citation: Lebeurre J, Dahyot S, Diene S, Paulay A, Aubourg M, Argemi X, Giard J-C, Tournier I, François P and Pestel-Caron M (2019) Comparative Genome Analysis of Staphylococcus lugdunensis Shows Clonal Complex-Dependent Diversity of the Putative Virulence Factor, ess/Type VII Locus. Front. Microbiol. 10:2479. doi: 10.3389/fmicb.2019.02479
Comparative Genome Analysis of
Staphylococcus lugdunensis Shows
Clonal Complex-Dependent Diversity
of the Putative Virulence Factor,
ess/Type VII Locus
Jérémie Lebeurre1* , Sandrine Dahyot2, Seydina Diene3, Amandine Paulay1,
Marion Aubourg4, Xavier Argemi5, Jean-Christophe Giard4, Isabelle Tournier6,
Patrice François3and Martine Pestel-Caron2
1UNIROUEN, GRAM EA2656, Normandie Université, Rouen, France,2UNIROUEN, GRAM EA2656, Rouen University Hospital, Normandie Université, Rouen, France,3Genomic Research Laboratory, Service of Infectious Diseases, Geneva University Hospitals, Geneva, Switzerland,4EA4655 U2RM (Équipe Antibio-Résistance), Université de Caen
Basse-Normandie, Caen, France,5CHRU de Strasbourg, VBP EA7290, Fédération de Médecine Translationnelle
de Strasbourg (FMTS), Institut de Bactériologie, Université de Strasbourg, Strasbourg, France,6UNIROUEN, Inserm U1245, Normandy Centre for Genomic and Personalized Medicine, Rouen University Hospital, Normandie Université, Rouen, France
Staphylococcus lugdunensis is a commensal bacterium of human skin that has emerged as a virulent Coagulase-Negative Staphylococcus in both community-acquired and healthcare associated infections. Genotyping methods have shown a clonal population structure of this pathogen but failed to identify hypervirulent lineages. Here, complete genomes of three pathogenic and three carriage S. lugdunensis strains were obtained by Single-Molecule sequencing (PacBio) and compared to 15 complete genomes available in GenBank database. The aim was to identify (i) genetic determinants specific to pathogenic or carriage strains or specific to clonal complexes (CCs) defined by MultiLocus Sequence Typing, and (ii) antibiotic resistance genes and new putative virulence factors encoded or not by mobile genetic elements (MGE). Comparative genomic analysis did not show a strict correlation between gene content and the ability of the six strains to cause infections in humans and in a Galleria mellonella infection model. However, this study identified new MGEs (five prophages, two genomic islands and one plasmid) and genetic variations of some putative virulence-associated loci, especially in CC3 strains. For a clonal population, high variability and eight CC-dependent genetic organizations were observed for the ess locus, which encodes a putative type VII secretion system (T7SS) homologous to that of S. aureus. Further phenotypic and functional studies are needed to characterize this particular CC3 and to evaluate the role of T7SS in the virulence of S. lugdunensis.
Keywords: Staphylococcus lugdunensis, genomic comparative analysis, virulence, ess/type VII locus, clonal complexes
fmicb-10-02479 October 29, 2019 Time: 15:58 # 2
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
INTRODUCTION
Staphylococcus lugdunensis is a Coagulase-Negative
Staphylococcal species (CoNS) that is a part of the normal human skin flora. However, unlike other CoNS,S. lugdunensis exhibits a high degree of virulence similar in some respects to Staphylococcus aureus (Frank et al., 2008).
Staphylococcus lugdunensis is responsible for infections ranging from minor skin and soft tissue infections (Bocher et al., 2009; Arias et al., 2010) to invasive diseases such as infective endocarditis (Non and Santos, 2017), bone and joint infections (Douiri et al., 2016;Lourtet-Hascoët et al., 2016;Argemi et al., 2017a), prosthetic device-infections (Anguera, 2005;Shah et al., 2010) and vascular catheter-related infections (Ebright et al., 2004), both in healthy host and immune-compromised patients. One of the characteristics of S. lugdunensis infections is the development of deep-seated abscess lesions that cannot be cleared without surgical intervention associated with effective antibiotic therapy (Arias et al., 2010). Interestingly, few virulence factors have been described to date to explain the high ability of S. lugdunensis to cause acute and suppurative infections, and particularly tissue damage and elevated mortality observed for infections such as infective endocarditis (Anguera, 2005;
Sabe et al., 2014).
Several molecular typing methods have been described to search for a potential link between within-species genetic variations and pathogenic potential or virulence. Multi-Locus Sequence Typing (MLST) has shown a clonal population structure of this pathogen (Chassain et al., 2012), and has allowed the description of seven Clonal Complexes (CCs)1 but failed
to identify hypervirulent lineages or clusters specific to carriage strains. Multi-Virulence Locus Sequence Typing (Didi et al., 2014) as well as Multiple Locus Variable Number Tandem Repeat Analysis (MLVA) and Tandem Repeat Sequence Typing (TRST) recently developed, have confirmed this absence of link between clustering and clinical settings (Dahyot et al., 2018).
Whole-genome sequencing (WGS) has become a powerful tool in microbiology allowing the sequencing of hundreds of S. aureus genomes but to date only 15 complete genomes of S. lugdunensis are available at the National Center for Biotechnology Information (NCBI). The only two studies
comparing S. lugdunensis whole genomes revealed that the
genome (i) is closer to that of S. aureus than other CoNS, (ii) harbors mobile genetic elements (MGE) such as prophages and plasmids, but (iii) displays a closed pan-genome and several systems (restriction-modification, toxin/antitoxin and CRISPR/Cas systems) to prevent horizontal gene transfer (Argemi et al., 2017b, 2018).
In this context, we first sequenced the whole genome of three pathogenic and three carriage strains, previously characterized by MLST (Didi et al., 2014) and collected from four geographic origins in France and Sweden. These whole-genome sequences were compared with data from 15 genomes available in the GenBank database to identify genetic variations specific to
1http://bigsdb.web.pasteur.fr/staphlugdunensis
pathogenic or carriage strains or to CCs, antibiotic resistance genes and new putative virulence factors carried or not by MGE.
Comparative genomic analysis did not show correlation between gene content and the ability of the six strains to
cause infections in humans and in a Galleria mellonella
infection model. It showed CC-dependent genetic variations of some putative virulence-associated loci among 21 genomes analyzed. Surprisingly for a highly clonal population, a great diversity was observed for the ess locus, which putatively
encodes a T7SS homologous to that of S. aureus in which
this multiprotein complex plays a key role in pathogenesis. Indeed, it promotes chronically long-term persistence infections with abscesses, lesions and lethal outcomes in a mouse model (Burts et al., 2005, 2008).
MATERIALS AND METHODS
G. mellonella Infection Experiments
Infection of G. mellonella larvae with S. lugdunensis was
performed as previously described (Michaux et al., 2011). Briefly, using a syringe pump (KD Scientific, Holliston, MA, United States), larvae (about 0.3 g and 3 cm in length) were infected subcutaneously with washed cells ofS. lugdunensis from an overnight culture in Brain Heart Infusion, with 3(±2) × 106 CFU per larva administered in 10µl of sterile saline buffer. In each test, 10 insects were infected, and experiments were repeated at least four times. Larval killing was then monitored at 1-, 2-and 3-days post-infection. Sterile saline buffer was also tested under the same conditions as control. Results were analyzed using one-way analysis of variance with a Bonferroni correction with free “R” software. For all comparisons, ap value<0.05 was considered significant.
Pacific Biosciences Sequencing
The complete genome of three pathogenic and three carriage strains of S. lugdunensis previously characterized by MLST and Multi-Virulence Locus Sequence Typing (Chassain et al., 2012; Didi et al., 2014) was sequenced using Single Molecule sequencing Pacific Biosciences technology (Gupta, 2008). Strains SL13, SL29 and SL55 were, respectively, isolated from infective endocarditis, vascular prosthesis infection and skin and soft tissue infection. Carriage strains were isolated from groin (SL118 and 122) and toe (SL117) (van der Mee-Marquet et al., 2003;
Bieber and Kahlmeter, 2010). Four strains belonged to CC1 (SL13, SL29, SL117, SL122) and 2 to CC6, as defined by MLST (Chassain et al., 2012; Table 1).
High molecular weight genomic DNA was purified from 20-ml exponential-phase cultures by using genomic-tip-500g kit (Qiagen) following manufacturer’s instructions. DNA was precipitated in isopropanol, extensively washed with cold 75% ethanol and in 1 × Tris-EDTA buffer. Purity and quantity were assessed by using NanoDrop and Qubit assays, respectively, before size evaluation with a Tape Station (Agilent). DNA was sequenced on a Pacific Biosciences RS system at the Lausanne Genomic Technologies Facility, Center for Integrative Genomics. Using P6-C4 chemistry, libraries of approximately 8–20 kb were
fmic b-10-02479 October 29, 2019 T ime: 15:58 # 3 Lebeurr e et al. Staphylococcus lugdunensis CC-Dependent Diversity
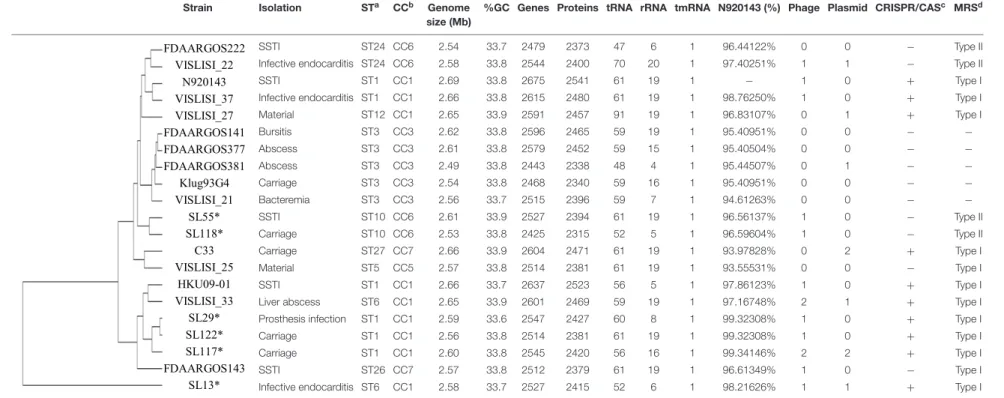
TABLE 1 | Whole genome sequence content of the 6 S. lugdunensis strains sequenced with Pacific Biosciences technology and the 15 S. lugdunensis genomes available on NCBI.
Strain Isolation STa CCb Genome
size (Mb)
%GC Genes Proteins tRNA rRNA tmRNA N920143 (%) Phage Plasmid CRISPR/CASc MRSd
FDAARGOS222 VISLISI_22 N920143 VISLISI_37 VISLISI_27 FDAARGOS141 FDAARGOS377 FDAARGOS381 Klug93G4 VISLISI_21 SL55* SL118* C33 VISLISI_25 HKU09-01 VISLISI_33 SL29* SL122* SL117* FDAARGOS143 SL13* SSTI ST24 CC6 2.54 33.7 2479 2373 47 6 1 96.44122% 0 0 − Type II
Infective endocarditis ST24 CC6 2.58 33.8 2544 2400 70 20 1 97.40251% 1 1 − Type II
SSTI ST1 CC1 2.69 33.8 2675 2541 61 19 1 − 1 0 + Type I
Infective endocarditis ST1 CC1 2.66 33.8 2615 2480 61 19 1 98.76250% 1 0 + Type I
Material ST12 CC1 2.65 33.9 2591 2457 91 19 1 96.83107% 0 1 + Type I Bursitis ST3 CC3 2.62 33.8 2596 2465 59 19 1 95.40951% 0 0 − − Abscess ST3 CC3 2.61 33.8 2579 2452 59 15 1 95.40504% 0 0 − − Abscess ST3 CC3 2.49 33.8 2443 2338 48 4 1 95.44507% 0 1 − − Carriage ST3 CC3 2.54 33.8 2468 2340 59 16 1 95.40951% 0 0 − − Bacteremia ST3 CC3 2.56 33.7 2515 2396 59 7 1 94.61263% 0 0 − − SSTI ST10 CC6 2.61 33.9 2527 2394 61 19 1 96.56137% 1 0 − Type II Carriage ST10 CC6 2.53 33.8 2425 2315 52 5 1 96.59604% 1 0 − Type II Carriage ST27 CC7 2.66 33.9 2604 2471 61 19 1 93.97828% 0 2 + Type I Material ST5 CC5 2.57 33.8 2514 2381 61 19 1 93.55531% 0 0 − Type I SSTI ST1 CC1 2.66 33.7 2637 2523 56 5 1 97.86123% 1 0 + Type I
Liver abscess ST6 CC1 2.65 33.9 2601 2469 59 19 1 97.16748% 2 1 + Type I
Prosthesis infection ST1 CC1 2.59 33.6 2547 2427 60 8 1 99.32308% 1 0 + Type I
Carriage ST1 CC1 2.56 33.8 2514 2381 61 19 1 99.32308% 1 0 + Type I
Carriage ST1 CC1 2.60 33.8 2545 2420 56 16 1 99.34146% 2 2 + Type I
SSTI ST26 CC7 2.57 33.8 2512 2379 61 19 1 96.61349% 1 0 − Type I
Infective endocarditis ST6 CC1 2.58 33.7 2527 2415 52 6 1 98.21626% 1 1 + Type I
aSequence type.bClonal complex.cClustered, regularly interspaced, short, palindromic repeat-associated (cas) genes.dModification-restriction system.∗
Genomes sequenced in this study. SSTI, skin and soft tissue infection. Fr ontiers in Micr obiology | www .fr ontiersin.org 3 October 2019 | V olume 10 | Article 2479
fmicb-10-02479 October 29, 2019 Time: 15:58 # 4
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
generated. Analyses of reads were carried out by using SMRT pipe version 2.3.0 software (Pacific Bioscience). The quality value (QV) threshold for motif calling was set at 30.
Computational Analysis
In addition to the 6 genomes sequenced by PacBio, 15 complete genomes published or available were retrieved from GenBank for comparative analyses (Supplementary Table S1). All complete genomes were annotated using the software Prokka v1.13 (Seemann, 2014). Genomes were compared using orthologsorter2
to generate ortholog clustering sequences among the overall 21 strains and to define the pan-genome, the core-, and
the accessory-genome of S. lugdunensis. Genomes were also
compared according to the clinical context (infection vs. carriage) and the CC of strains. A similarity matrix of complete genomes and a phylogenetic tree were created based on the neighbor-joining method with the software BioNumerics v7.6. Sequence Types (STs) and CCs defined by MLST were identified by extracting sequences of genes aroE, dat, ddl, gmk, ldh, recA and yqiL (Chassain et al., 2012). STs and CCs were obtained by submission of gene sequences into the international MLST database3. The presence of genes encoding restriction enzymes
was predicted by comparison and Blastn with datasets of the Restriction Enzyme Database (Roberts et al., 2013).
Integrated prophage regions were identified and annotated by using the online PHAge Search Tool Enhanced Release (PHASTER) (Arndt et al., 2016)4 with default parameters for
the six genomes sequenced by PacBio, the complete genome sequences of the 5 FDAARGOS strains and the Klug93-G4 strain, available on NCBI, and for which no phage has been described to date. PHASTER provided the region length and position of the prophages on the chromosome, their GC content and the
most common related phages. Known prophage sequences8SL1
(Heilbronner et al., 2011),8SL2, 8SL3, 8SL4 and 8SL5 (Argemi et al., 2017b) were compared with prophage regions identified and Mauve alignments were used to confirm PHASTER results.
Plasmid identification was done by using Prokka v1.13 for annotation of extra-chromosomic contigs obtained by PacBio sequencing and was compared to plasmid sequences retrieved from the NCBI database (pVISLISI_1, pVISLISI_2, pVISLISI_3, pVISLISI_4 pVISLISI_5 (Argemi et al., 2017b) and to unnamed plasmid from the FDAARGOS381 strain). Nucleotide sequence similarities were investigated by Blastn to identify the closest related plasmid.
Virulence factors were identified using the Virulence Factors database (VFDB)5 (Chen et al., 2016) and tBlastn searches
were performed to compare the presence or absence of known virulence genes of staphylococci. Comparisons of agr and cap hypervariable regions were performed by extracting and aligning agr and cap loci with MUSCLE. Allelic agr type-I (agrSl-type I, accession number: AF173933) and type-II (agrSl-type II, accession number: AF346728) described byDufour et al. (2002)
2https://git.facom.ufms.br/bioinfo/orthologsorter 3https://bigsdb.pasteur.fr/staphlugdunensis/ 4http://phaster.ca
5http://www.mgc.ac.cn/VFs
were used as reference. The hypervariable regions (capH to capK) from S. aureus cap8 and cap5 loci were downloaded from the NCBI database. Phylogenetic trees were constructed using the maximum likelihood method implemented in the PhyML program v3.0.
Resistance genes were identified by using ResFinder v3.16
(Zankari et al., 2012). A cutoff of 80% minimum length and 80% identity were used to search for resistance genes in the 21 genomes and plasmids ofS. lugdunensis.
Comparisons of individual sequences of the ess locus were performed on the 21 genomes ofS. lugdunensis by using Blastn and Blastp against the NCBI database, and were facilitated by using the Artemis Comparison Tool (Carver et al., 2005). T7SS genes and proteins were identified from available genomes of HO 5096 0412 (Accession number HE861097) (Holden et al., 2013) and RJ-ST398 reference strains (Accession number AM990992) (Wang et al., 2016). Pfam database was also used to identify protein domains.
RESULTS
G. mellonella Infection Experiments
In order to evaluate and compare the virulence of 6S. lugdunensis strains (SL13, SL29, SL55, SL117, SL118 and SL122) under
the same conditions in vivo, a G. mellonella model of
infection was used.
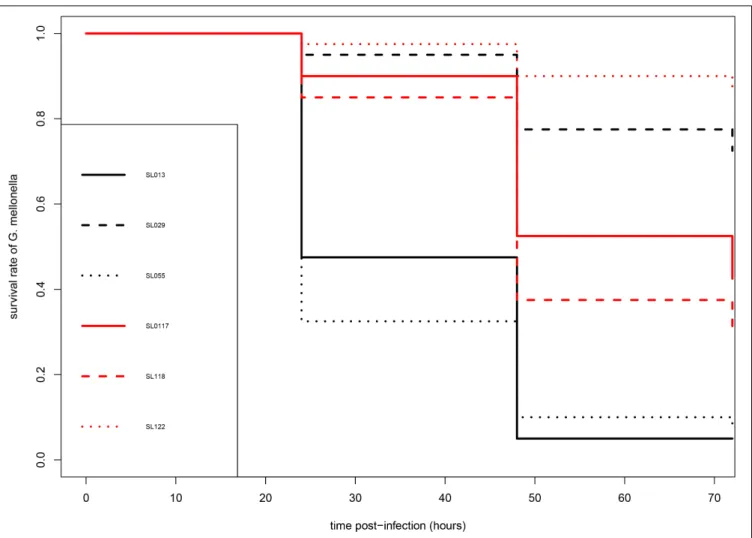
As shown in Figure 1, carriage isolates (SL117, SL118, and SL122) as well as the pathogenic isolate SL29 (from vascular prosthesis infection) revealed moderate virulence in this model of infection. After 72 h, 31–87% of the larvae infected by these four isolates were still alive. However, they could not be clustered because statistical analysis displayed significant differences between them (p values<0.05) (Table 2). In contrast, isolates SL13 (from infective endocarditis) and SL55 (from a skin and soft tissue infection) were significantly more virulent than the others since only 4 and 8% of the infected caterpillars survived 3-days post infection, respectively (Figure 1 and Table 2). No mortality was observed for the larvae into which sterile saline buffer was injected (data not shown). Of note, no differences were observed between strains in terms of colony morphology, hemolysis (β-hemolysis on blood agar) and growth characteristics under standard laboratory conditions.
Pacific Biosciences Genome Sequences
and Functional Genome Annotation
PacBio sequencing of 6 S. lugdunensis strains resulted in full genome recovery on a single contig with sequence length ranging from 2.53 (SL118) to 2.61 Mb (SL55). Between 67457 (SL55) and 79450 (SL118) post-filter polymerase reads were generated with an average read length ranging from 13.7 (SL55) to 14.8 kb (SL13). The N50 read length was comprised between 19.2 and 22.7 kb, and the average coverage between 272× and 367×. In addition to the contigs corresponding to
6https://cge.cbs.dtu.dk/services/ResFinder
fmicb-10-02479 October 29, 2019 Time: 15:58 # 5
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
FIGURE 1 | Kaplan-Meier survival curves of infected larvae of G. mellonella after infection by S. lugdunensis strains. Pathogenic (SL13, SL29, SL55) and carriage (SL117, SL118, SL122) are shown in black and red, respectively.
genomes, 0 (SL29 and SL118) to 3 (SL117) extra-chromosomic contigs were generated.
The complete genome sequences of 15 differentS. lugdunensis strains available in the NCBI database (Supplementary Table
S1) were compared to the six genomes sequenced by PacBio. The size of these 21 complete genomes ranged between 2.53 (SL118) and 2.69 Mb (N920143). All strains presented similar %GC-content (33.6–33.9%). The number of genes automatically predicted by Prokka software ranged from 2443 (FDAARGOS381) to 2675 (N920143) genes encoding between 2338 and 2541 proteins. Genomic data for the 21 strains are shown in Table 1.
Pan-, Core- and Accessory-Genome
Analysis
Pan-genome analysis led to identification of the total gene repertoire of the species and sets of core and accessory genes available in each S. lugdunensis genome. The pan-genome of the 21 S. lugdunensis strains was found to be composed of a total repertoire of 3077 genes. Of these, 2185 genes (71.01%) were part of the core-genome, and 892 genes (28.99%) were
part of the accessory genome. Based on this analysis, no gene retrieved from the accessory genome belonged only to pathogenic or only to carriage strains. In the same way, no gene was specifically associated with strains isolated from a deep infection such as infectious endocarditis, deep abscess, or device-associated infection.
However, CC-dependent genetic variations were observed. Accessory-genome analysis of CC1 strains revealed that 18 genes were unique to these nine strains. Among them, nine genes were clustered, regularly interspaced, short, palindromic
TABLE 2 | Pair test statistical comparison (p values) of the virulence of 6 strains of S. lugdunensis in a Galleria mellonella infection model.
SL13 SL29 SL55 SL117 SL118
SL29 2.14e-10 NA NA NA NA
SL55 1.00 1.71e-10 NA NA NA
SL117 5.72e-06 0.11 4.33e-06 NA NA
SL118 0.0006 0.002 0.0003 1.00 NA
SL122 2.12e-13 1.00 1.52e-13 0.0004 2.68e-06
fmicb-10-02479 October 29, 2019 Time: 15:58 # 6
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
repeat (CRISPR) andloci-associated corresponding to type III-A proteins: Cas1, Cas2, Cas10, Csm2, Csm3, Csm4, Csm5,
Csm6, and Cas6. The cas locus from CC1 strains showed
a sequence homology of 76% with that of S. epidermidis
RP62a strain. In all CC1 strains, except strains SL29 and SL122, a potential frameshift is located in the gene encoding the Csm6 protein and probably results in a non-functional CRISPR/Cas system. The remaining nine genes unique to CC1 strains were carried by a transposon and encode arsenical resistance proteins.
Eight genes were unique to CC6 strains. Six of them encode hypothetical proteins but two encode a type II restriction-modification system (RMS). Indeed, we identified a C-5 cytosine-specific DNA methylase and a Type-2 restriction
enzyme Sau3AI sharing 82% identity with RMS type II from
S. haemolyticus JCSJ1435 strain. Only 2 genes were found specific to CC7 strains: one encoding a DUF600 protein and one encoding a hypothetical protein. No gene was identified as specific to CC3 strains.
Mobile Genetic Elements
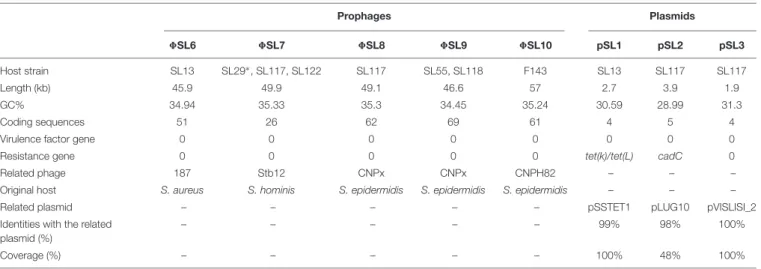
Five new intact prophage regions named 8SL6 to 8SL10
were identified by PHASTER analysis for 12 of the 21 strains
(Table 3). Prophage regions 8SL6, 8SL7 and 8SL8 were
identified in CC1 strains (SL13, SL29, SL117, and SL122)
while prophage regions 8SL9 and 8SL10 were found in
CC6 (SL55 and SL118) and CC7 (FDAARGOS143) strains, respectively. The total length of these prophages ranged from
46.6 kb (8SL9) to 49.9 kb (8SL7); they encoded 26–69
proteins. Among these proteins, no virulence factors or antibiotic resistance-associated proteins were identified. However, we identified a putative bifunctional autolysin, a cell wall hydrolase lytN precursor, a collagen triple-helix protein and a LexA repressor which might be involved in the remodeling of the bacterial cell wall.
Functional annotation by using Prokka on additional contigs led to identification of three plasmids in two of the
six newly sequenced genomes: pSL1 in strain SL13, and pSL2 and pSL3 in strain SL117 (Table 3). The total length of the plasmids identified ranged from 1.9 (pSL3) to 3.9 kb (pSL1); they encode between 4 and 5 proteins. Plasmids pSL2 and pSL3 respectively correspond to cadmium
resistance-associated plasmid pLUG10 (Poitevin-Later et al., 1992)
and plasmid pVISLISI_2 (Argemi et al., 2017b) previously described. Of note, no virulence factor was carried on these plasmids but tetracycline and cadmium resistance genes were found.
In addition, we identified a region of 13 kb length restricted to CC1 strains SL13 and VISLISI_33. Around 5 kb of this region shared a sequence homology of 94% with the pathogenicity
islands SaPITokyo11212 and SaPIivm60 of S. aureus strains
Tokyo11212 and IVM60, respectively. This region encodes an integrase, an excisionase, a primase and a terminase. Other interesting genes were carried by this putative pathogenicity island as those encoding Ear penicillin binding protein, FhuD ferrichrome ABC transporter and a protein homologous to part of theS. aureus Pathogenicity Island (SAPI) domain.
Resistome
Several antibiotic resistance-associated genes were identified by using the web tool ResFinder v3.0 (Table 4). A gene homologous (83% identity) to the fosB gene of Bacillus cereus AS4-12 was identified in the chromosome of allS. lugdunensis strains. A penicillin resistance-associated gene, blaZ was also identified. This gene was carried by a transposon and found in 7 of the 21 S. lugdunensis strains. The blaZ gene showed
a homology ranging from 92 to 100% with the blaZ gene
carried by the plasmid pSAP106A ofS. epidermidis. Otherwise, annotation of the extra-chromosomal contig of SL13 strain led to identification of a tetK tetracycline resistance gene
which shared 99% identity with tetK gene from the plasmid
pT181 of S. aureus. Of note, no gene of methicillin resistance,
within a composite staphylococcal chromosome cassette mec
element, was found.
TABLE 3 | Prophage regions and plasmids identified among 21 genomes of S. lugdunensis.
Prophages Plasmids 8SL6 8SL7 8SL8 8SL9 8SL10 pSL1 pSL2 pSL3 Host strain SL13 SL29∗, SL117, SL122 SL117 SL55, SL118 F143 SL13 SL117 SL117 Length (kb) 45.9 49.9 49.1 46.6 57 2.7 3.9 1.9 GC% 34.94 35.33 35.3 34.45 35.24 30.59 28.99 31.3 Coding sequences 51 26 62 69 61 4 5 4
Virulence factor gene 0 0 0 0 0 0 0 0
Resistance gene 0 0 0 0 0 tet(k)/tet(L) cadC 0
Related phage 187 Stb12 CNPx CNPx CNPH82 – – –
Original host S. aureus S. hominis S. epidermidis S. epidermidis S. epidermidis – – –
Related plasmid – – – – – pSSTET1 pLUG10 pVISLISI_2
Identities with the related plasmid (%)
– – – – – 99% 98% 100%
Coverage (%) – – – – – 100% 48% 100%
∗
duplication.
fmicb-10-02479 October 29, 2019 Time: 15:58 # 7
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
T ABLE 4 | Antibiotic resistance-associated genes identified among 21 genomes of S. lugdunensis and their genomic location. Antibiotic Gene Strains Genomic location SL13 SL29 SL55 SL117 SL118 SL122 HKU09-01 N920143 V21 V22 V25 V27 V33 V37 C33 F141 F143 F222 F377 F381 Fosfomycin fosB + + + + + + + + + + + + + + + + + + + + Chr omosome Penicillin blaZ + − − − − − + − + − + + − − + − − + − T ransposon (chr omosome) Fusidic acid fusD − − − − − − − − − − + − − − − − − − − Genomic island (chr omosome) Tetracyclin tetK + − − − − − − − − − − − − − − − − − − − Plasmid
Finally, we also identified a fusidic acid resistance gene located in a genomic island of 16 kb, only in strain VISLISI_27. This gene had 91% homology with thefusB gene carried by a genomic island (SaRIfusB) of theS. aureus CS6-EEFIC strain. In addition, in this genomic island, we identified a gene encoding a putative virulence factor,vapE (for virulence-associated protein E). vapE gene was also found in a genomic island of 40 kb in the genome of strain HKU09-01 with an identity of 85%. However, Blastn comparison of these two genomic islands showed 80% identity for only 3 kb, suggesting that strains VISLISI_27 and HKU09-01 acquired these regions independently and from different sources.
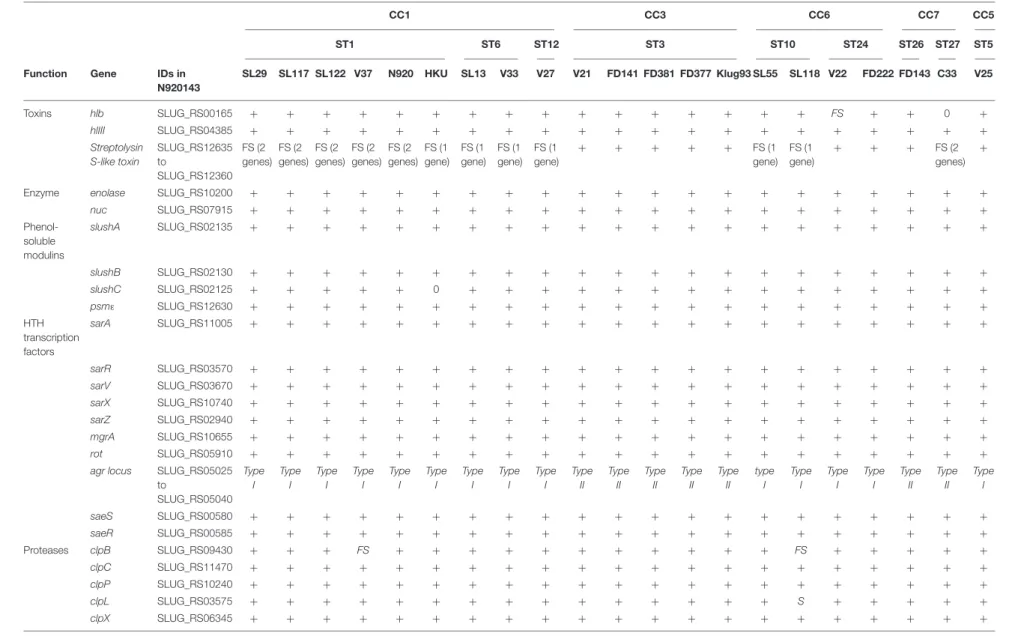
Virulome
Analysis of virulence gene composition based on the Virulence Factor Database (VFDB) showed a number of putative virulence factors that were detected in most of the S. lugdunensis isolates. To improve the identification of notable features, tblastn searches of well-characterized staphylococcal virulence factors and transcriptional regulators were performed (Table 5). No gene duplication was detected, except inisd locus for one strain (HKU09-01) and in sst locus for 20 of 21 strains.
The accessory gene regulator (agr) locus (agrA, agrB, agrC andagrD) was found in all S. lugdunensis strains. Phylogenetic analysis based on alignment of the hypervariableagr region (agrB toagrC) led to identification of 2 genetic groups, agrSl-types I and II (Supplementary Figure S1A). Of note, all CC3 isolates were clustered with alleleagrSl-type II whereas all other strains were closer to alleleagrSl-type I.
In a region of 16 kb in the genome of the 21S. lugdunensis strains, 16 genes homologous to those encoding polysaccharidic capsule type 5 or 8 inS. aureus were detected (Supplementary
Figure S1B). InS. lugdunensis, only the central region composed ofcapH, capI, capJ and capK genes, was found different between strains; the 12 other genes of thecap locus were conserved. Based on phylogenetic analysis, the caplocus of CC3 and VISLISI_25 strains (CC5) was closer to thecap8 locus of S. aureus while other strains were related to thecap5 locus.
In the genome of S. lugdunensis, up to seven sls genes
(slsA to slsG) were identified. Comparative analysis of the predictedS. lugdunensis surface (Sls) proteins revealed important diversity between the 21S. lugdunensis strains. The size of SlsA, varying from 1634 to 5228 amino acids, correlated with the number of IgG-like domain ranging from 8 to 45. Sequence comparison of SlsA proteins using Blastp showed 51–52% identity with the Bhp protein ofS. epidermidis RP62a and 63– 64% with a cell wall associated biofilm Bhp/SesD protein from Staphylococcus caprae JMUB590. Pfam analysis of these two staphylococcal proteins also showed the presence of a similar IgG-like domain, with 18 (Bhp) and 50 (Bhp/SesD) repeats. Interestingly, slsA gene was absent from the genomes of all CC3 isolates and from FDAARGOS377 isolates (CC5), and
a nonsense mutation was detected in the slsA gene of the
isolate FDAARGOS222 (CC6). Furthermore, in the slsD gene,
a nonsense mutation located upstream of the region coding
the LPXTG motif was found in 14 of the 21 S. lugdunensis
fmic b-10-02479 October 29, 2019 T ime: 15:58 # 8 Lebeurr e et al. Staphylococcus lugdunensis CC-Dependent Diversity
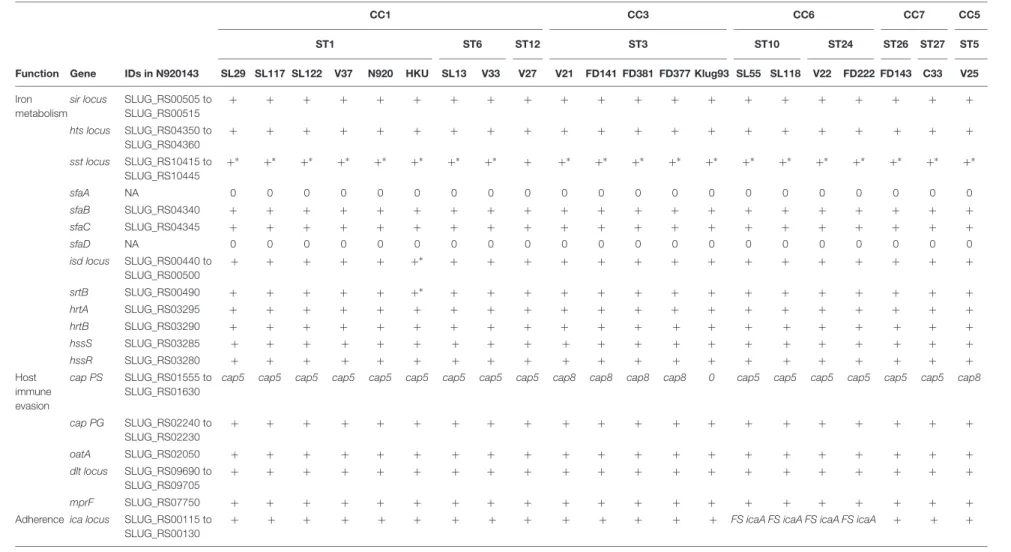
TABLE 5 | Comparison of virulence factors among 21 S. lugdunensis strains according to clonal complexes (CCs).
CC1 CC3 CC6 CC7 CC5
ST1 ST6 ST12 ST3 ST10 ST24 ST26 ST27 ST5
Function Gene IDs in
N920143 SL29 SL117 SL122 V37 N920 HKU SL13 V33 V27 V21 FD141 FD381 FD377 Klug93 SL55 SL118 V22 FD222 FD143 C33 V25 Toxins hlb SLUG_RS00165 + + + + + + + + + + + + + + + + FS + + 0 + hlIII SLUG_RS04385 + + + + + + + + + + + + + + + + + + + + + Streptolysin S-like toxin SLUG_RS12635 to SLUG_RS12360 FS (2 genes) FS (2 genes) FS (2 genes) FS (2 genes) FS (2 genes) FS (1 gene) FS (1 gene) FS (1 gene) FS (1 gene) + + + + + FS (1 gene) FS (1 gene) + + + FS (2 genes) +
Enzyme enolase SLUG_RS10200 + + + + + + + + + + + + + + + + + + + + +
nuc SLUG_RS07915 + + + + + + + + + + + + + + + + + + + + + Phenol-soluble modulins slushA SLUG_RS02135 + + + + + + + + + + + + + + + + + + + + + slushB SLUG_RS02130 + + + + + + + + + + + + + + + + + + + + + slushC SLUG_RS02125 + + + + + 0 + + + + + + + + + + + + + + + psmε SLUG_RS12630 + + + + + + + + + + + + + + + + + + + + + HTH transcription factors sarA SLUG_RS11005 + + + + + + + + + + + + + + + + + + + + + sarR SLUG_RS03570 + + + + + + + + + + + + + + + + + + + + + sarV SLUG_RS03670 + + + + + + + + + + + + + + + + + + + + + sarX SLUG_RS10740 + + + + + + + + + + + + + + + + + + + + + sarZ SLUG_RS02940 + + + + + + + + + + + + + + + + + + + + + mgrA SLUG_RS10655 + + + + + + + + + + + + + + + + + + + + + rot SLUG_RS05910 + + + + + + + + + + + + + + + + + + + + +
agr locus SLUG_RS05025
to SLUG_RS05040 Type I Type I Type I Type I Type I Type I Type I Type I Type I Type II Type II Type II Type II Type II type I Type I Type I Type I Type II Type II Type I saeS SLUG_RS00580 + + + + + + + + + + + + + + + + + + + + + saeR SLUG_RS00585 + + + + + + + + + + + + + + + + + + + + + Proteases clpB SLUG_RS09430 + + + FS + + + + + + + + + + + FS + + + + + clpC SLUG_RS11470 + + + + + + + + + + + + + + + + + + + + + clpP SLUG_RS10240 + + + + + + + + + + + + + + + + + + + + + clpL SLUG_RS03575 + + + + + + + + + + + + + + + S + + + + + clpX SLUG_RS06345 + + + + + + + + + + + + + + + + + + + + + (Continued) Fr ontiers in Micr obiology | www .fr ontiersin.org 8 October 2019 | V olume 10 | Article 2479
fmic b-10-02479 October 29, 2019 T ime: 15:58 # 9 Lebeurr e et al. Staphylococcus lugdunensis CC-Dependent Diversity TABLE 5 | Continued CC1 CC3 CC6 CC7 CC5 ST1 ST6 ST12 ST3 ST10 ST24 ST26 ST27 ST5
Function Gene IDs in N920143 SL29 SL117 SL122 V37 N920 HKU SL13 V33 V27 V21 FD141 FD381 FD377 Klug93 SL55 SL118 V22 FD222 FD143 C33 V25
Iron metabolism
sir locus SLUG_RS00505 to SLUG_RS00515 + + + + + + + + + + + + + + + + + + + + + hts locus SLUG_RS04350 to SLUG_RS04360 + + + + + + + + + + + + + + + + + + + + + sst locus SLUG_RS10415 to SLUG_RS10445 +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ + +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ +∗ sfaA NA 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 sfaB SLUG_RS04340 + + + + + + + + + + + + + + + + + + + + + sfaC SLUG_RS04345 + + + + + + + + + + + + + + + + + + + + + sfaD NA 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
isd locus SLUG_RS00440 to SLUG_RS00500 + + + + + +∗ + + + + + + + + + + + + + + + srtB SLUG_RS00490 + + + + + +∗ + + + + + + + + + + + + + + + hrtA SLUG_RS03295 + + + + + + + + + + + + + + + + + + + + + hrtB SLUG_RS03290 + + + + + + + + + + + + + + + + + + + + + hssS SLUG_RS03285 + + + + + + + + + + + + + + + + + + + + + hssR SLUG_RS03280 + + + + + + + + + + + + + + + + + + + + + Host immune evasion cap PS SLUG_RS01555 to SLUG_RS01630
cap5 cap5 cap5 cap5 cap5 cap5 cap5 cap5 cap5 cap8 cap8 cap8 cap8 0 cap5 cap5 cap5 cap5 cap5 cap5 cap8
cap PG SLUG_RS02240 to SLUG_RS02230 + + + + + + + + + + + + + + + + + + + + + oatA SLUG_RS02050 + + + + + + + + + + + + + + + + + + + + + dlt locus SLUG_RS09690 to SLUG_RS09705 + + + + + + + + + + + + + + + + + + + + + mprF SLUG_RS07750 + + + + + + + + + + + + + + + + + + + + +
Adherence ica locus SLUG_RS00115 to SLUG_RS00130
+ + + + + + + + + + + + + + FS icaA FS icaA FS icaA FS icaA + + +
(Continued) Fr ontiers in Micr obiology | www .fr ontiersin.org 9 October 2019 | V olume 10 | Article 2479
fmic b-10-02479 October 29, 2019 T ime: 15:58 # 10 Lebeurr e et al. Staphylococcus lugdunensis CC-Dependent Diversity TABLE 5 | Continued CC1 CC3 CC6 CC7 CC5 ST1 ST6 ST12 ST3 ST10 ST24 ST26 ST27 ST5
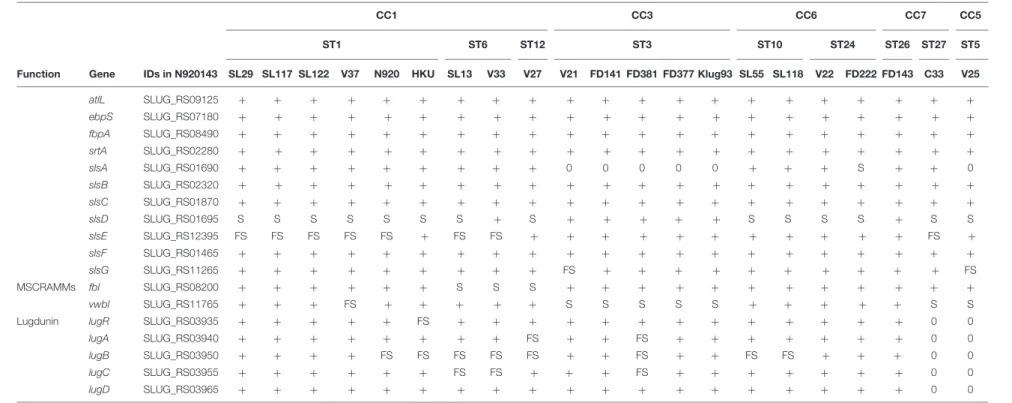
Function Gene IDs in N920143 SL29 SL117 SL122 V37 N920 HKU SL13 V33 V27 V21 FD141 FD381 FD377 Klug93 SL55 SL118 V22 FD222 FD143 C33 V25
atlL SLUG_RS09125 + + + + + + + + + + + + + + + + + + + + + ebpS SLUG_RS07180 + + + + + + + + + + + + + + + + + + + + + fbpA SLUG_RS08490 + + + + + + + + + + + + + + + + + + + + + srtA SLUG_RS02280 + + + + + + + + + + + + + + + + + + + + + slsA SLUG_RS01690 + + + + + + + + + 0 0 0 0 0 + + + S + + 0 slsB SLUG_RS02320 + + + + + + + + + + + + + + + + + + + + + slsC SLUG_RS01870 + + + + + + + + + + + + + + + + + + + + + slsD SLUG_RS01695 S S S S S S S + S + + + + + S S S S + S S slsE SLUG_RS12395 FS FS FS FS FS + FS FS + + + + + + + + + + + FS + slsF SLUG_RS01465 + + + + + + + + + + + + + + + + + + + + + slsG SLUG_RS11265 + + + + + + + + + FS + + + + + + + + + + FS MSCRAMMs fbl SLUG_RS08200 + + + + + + S S S + + + + + + + + + + + + vwbl SLUG_RS11765 + + + FS + + + + + S S S S S + + + + + S S
Lugdunin lugR SLUG_RS03935 + + + + + FS + + + + + + + + + + + + + 0 0
lugA SLUG_RS03940 + + + + + + + + FS + + FS + + + + + + + 0 0
lugB SLUG_RS03950 + + + + FS FS FS FS FS + + FS + + FS FS + + + 0 0
lugC SLUG_RS03955 + + + + + + FS FS + + + FS + + + + + + + 0 0
lugD SLUG_RS03965 + + + + + + + + + + + + + + + + + + + 0 0
The presence of a putative virulence gene is indicated by “+” while absence is indicated by “0.” PS, polysaccharidic; PG, polyglutamic; FS, frameshift; S, non-sens mutation.∗
Duplication; NA, not available. Systematic IDs of the genes are those of the S. lugdunensis N920143 strain.
Fr ontiers in Micr obiology | www .fr ontiersin.org 10 October 2019 | V olume 10 | Article 2479
fmicb-10-02479 October 29, 2019 Time: 15:58 # 11
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
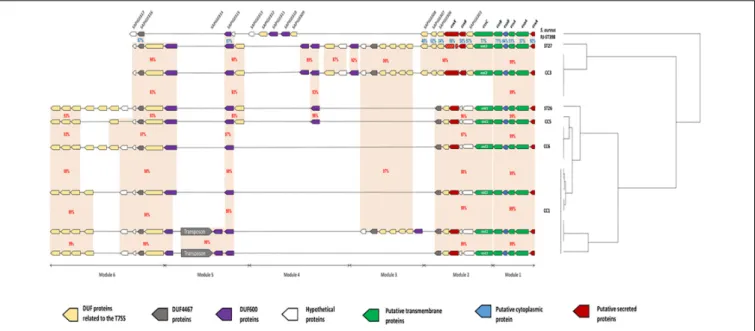
FIGURE 2 | Schematic representation of 8 genetic organizations of the ess locus from genomes of 21 strains of S. lugdunensis. For each strain, clonal complexes (CC) are indicated to the right. The ess locus was divided into 6 modules depending on gene-based content and relative conservation defined with the Artemis comparison tool. Module 1 contained 5 genes homologous of the core components (esxA to essB) of S. aureus T7SS. Module 2 included the essC gene, of which there are 2 variants, each associated with a distinct cluster of 5–6 genes. Modules 3, 4 and 6 contained a complex arrangement of coding sequences that encode a variable number of uncharacterized proteins. Module 5 is composed of 2 distinct genetic organizations: one of them is composed of a transposon associated with 3 coding sequences encoding hypothetical DUF600 proteins whereas the other one is comprised of only 2 genes. Orange areas highlight conserved coding sequences between strains with the percentage of amino acid identity in red between S. lugdunensis strains and in blue with S. aureus strain RJ-ST398.
strains but was not present in CC3 strains or the VISLISI_33 strain (CC1). Sequence comparison of SlsD proteins using Blastp showed an identity of 40 and 32% with SdrF and SdrG adhesins from S. epidermidis, respectively. The size of the SlsE predicted protein sequences varied from 2818 to 4166 amino acids among the 21 S. lugdunensis strains. Interestingly, a frameshift was located in theslsE gene, upstream of the region encoding the LPXTG motif and was found in all CC1 strains except the strain HKU09-01. The size of the SlsG predicted protein sequences, varying from 1604 to 2286 amino acids, correlated with the Rib repeat number ranging from 7 to 21. A frameshift was located within Rib repeats for VISLISI_21 and VISLISI_25 isolates. On the other hand, SlsB, SlsC and SlsF proteins were highly conserved (99–100% of identity) in the 21 S. lugdunensis strains.
Of note, notable diversity was observed inlug locus encoding a peptide antibiotic (lugdunin). Of the 21 strains, 8 contained a frameshift in one to 2 genes of the locus while the latter was absent in 2 strains.
Description of the CC-Dependent
Genetic Diversity of the S. lugdunensis
ess Locus
Comparative genomic analysis showed variations in the size of the ess locus of the 21 S. lugdunensis isolates. Indeed, ess locus size ranged from 22134 bp (HKU09-01) to 38834 bp (SL29 and SL122). This difference was the result of an important polymorphism identified in CC1 strains. For a
better understanding of these variations, we divided the ess locus into 6 distinct modules, each based on gene content and relative conservation (Figure 2). Eight genetic organizations were observed.
Module 1 contained five genes present in all sequences analyzed and homologous to genes encoding proteins of T7SS in S. aureus: EsxA, EsaA, EssA, EsaB and EssB. This module was highly conserved among all S. lugdunensis strains, with slight modifications in the amino acid sequences of these five proteins (99–100% identity).
Module 2 was variable betweenS. lugdunensis CCs. Indeed, 2 distinct organizations of the ess locus were identified: one shared by all CC3 strains and by one CC7 strain (C33), and the other one exhibited by other CC strains (CC1, CC5 and CC6). The first important variation concerned the essC gene
for which 2 essC sequence variants (essC1 and essC2) were
observed. The 50
region of theessC gene was conserved within all S. lugdunensis isolates (3762 bp) whereas the 30
region (around 681 bp) differed according to CCs. The essC1 variant, identified in 15 strains, encodes a putative protein of 1476 residues whereas the essC2 variant encodes a putative protein of 1481 amino acids. This latter was only identified in 5 CC3 strains and in one CC7 strain (C33). Of note, S. lugdunensis N920143 contained a frameshift in the gene encoding the membrane-associated protein EssC. Otherwise, each essC gene variant was associated with its own cluster of genes. essC1 gene was associated with five genes highly conserved among 15 S. lugdunensis isolates while essC2 gene variant was associated with six genes conserved for six isolates. Conversely, these six
fmicb-10-02479 October 29, 2019 Time: 15:58 # 12
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
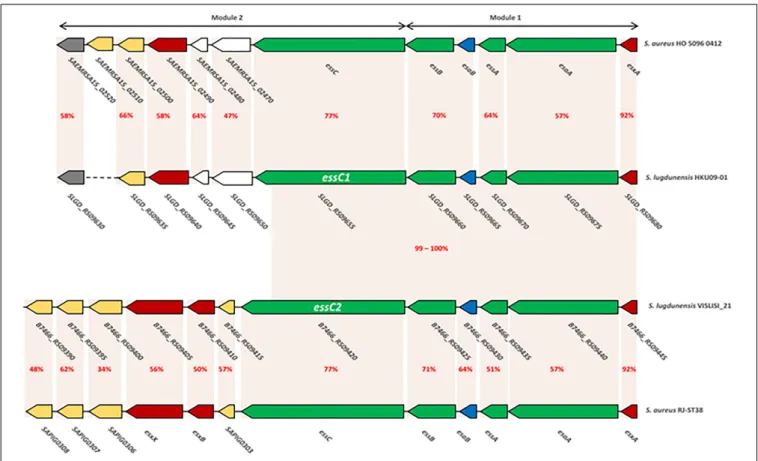
FIGURE 3 | Comparison of ess modules 1 and 2 of S. aureus HO 5096 0412 and RJ-ST398 strains with HKU09-01 and VISLISI_21 S. lugdunensis strains. Gene-based content and relative conservation defined with the Artemis comparison tool. Orange areas highlight conserved coding sequences between strains with the percentage of amino acid identity in red.
genes shared no identity with the five genes associated with the essC1 variant (Figure 3).
In modules 3, 4 and 6, a complex arrangement of sequences encoding a variable number of uncharacterized proteins was observed. In particular, an important number of genes which encode proteins containing Domains of Unknown Function (DUFs) were identified. Among them, DUF600 and some others (DUF5079, DUF5080, DUF5020, DUF5084 and DUF5085) are suspected to be involved in T7SS on the basis of homology with characterized T7SS systems. The number of genes encoding DUF600 proteins varied from 2 in CC6 strains to 5 in CC3 strains while the number of genes encoding the other DUF proteins ranged from 6 to 11 among all strains (Figure 2).
In module 5, a transposon was found in 4 of the 9 CC1 isolates, associated with genes encoding hypothetical DUF600 proteins (Figure 2).
Here, the comparison focuses on modules 1 and 2 which are the only ones to have a strong homology with those ofS. aureus (48–92% identity). This was performed on the one hand between
the genomes of S. aureus HO 5096 0412 and S. lugdunensis
HKU09-01 strains, and on the other hand between the genomes of S. aureus RJ-ST398 and S. lugdunensis VISLISI_21 strains. These 4 strains were chosen to be representative of the diversity of thelocus in both species (Figure 3).
Comparative analysis of module 1 showed thatS. lugdunensis
genes SLGD_RS09680 (HKU09-01) and B7466_RS09445
(VISLISI_21) which encode a putative secreted protein shared
92% identity with the EsxA protein of S. aureus strains.
This putative protein of 97 amino acids contains a WXG100 domain and a conserved C-terminal hydrophobic pattern for the formation of specific α-helical surface. Moreover, several putative transmembrane proteins in S. lugdunensis HKU09-01 and VISLISI_21 isolates showed homologies (51–64% identity) with the transmembrane proteins EsaA, EssA and EssB of S. aureus strains (Tables 6, 7). The EsaB cytoplasmic protein
of S. aureus strains exhibited 64% identity with proteins
SLGD_RS09065 (HKU09-01) and B7466_RS09420 (VISLISI_21) which contained a YukD family domain. Thus, functional analysis of T7SS core components using pfam database allowed the prediction and identification of the same protein domains for S. aureus and S. lugdunensis.
Then, pairwise comparison of module 2 showed homologous genes and comparable organization (Figure 3 and Tables 6, 7). First, theessC1 and essC2 variants of S. lugdunensis strains encode proteins sharing 77% identity with the EssC proteins ofS. aureus strains. The putative EssC proteins ofS. lugdunensis contain a N-terminal FtsK-SpoIIIE domain for DNA transportation and 2 domains of the SpoIIIE-FtsK-like ATPase family, like those of S. aureus. Second, in both S. aureus and S. lugdunensis, variants
fmicb-10-02479 October 29, 2019 Time: 15:58 # 13
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
TABLE 6 | Comparison of proteins encoded by ess modules 1 and 2 of S. aureus HO 5096 0412 with HKU09-01 S. lugdunensis strains.
S. aureus HO 5096 0412 strain S. lugdunensis HKU09-01 strain
Proteins Length (amino acids) pfam family
domain
Proteins Length (amino acids) pfam family
domain
% identity
EsxA 97 WXG100 SLGD_RS09680 97 WXG100 92%
EsaA 1009 N/A SLGD_RS09675 1006 N/A 57%
EssA 153 EssA SLGD_RS09670 144 EssA 64%
EsaB 80 YukD SLGD_RS09665 80 YukD 70%
EssB 444 YukC SLGD_RS09660 437 YukC 70%
EssC 1476 FtsK SpoIIIE N two
FtsK SpoIIIE
SLGD_RS09655 1476 FtsK SpoIIIE N two
FtsK SpoIIIE
77%
SAEMRSA15_02470 359 N/A SLGD_RS09650 360 N/A 47%
SAEMRSA15_02480 96 N/A SLGD_RS09645 96 N/A 64%
SAEMRSA15_02490 420 LXG SLGD_RS09640 420 LXG 58%
SAEMRSA15_02500 137 DUF5082 SLGD_RS09635 137 DUF5082 66%
SAEMRSA15_02510 157 DUF5083 N/A N/A N/A N/A
SAEMRSA15_02520 124 DUF4467 SLGD_RS09630 127 DUF4467 58%
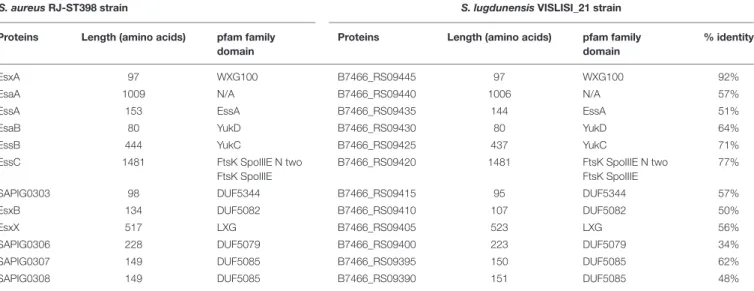
TABLE 7 | Comparison of proteins encoded by ess modules 1 and 2 of S. aureus RJ-ST398 strains with VISLISI_21 S. lugdunensis strains.
S. aureus RJ-ST398 strain S. lugdunensis VISLISI_21 strain
Proteins Length (amino acids) pfam family
domain
Proteins Length (amino acids) pfam family
domain
% identity
EsxA 97 WXG100 B7466_RS09445 97 WXG100 92%
EsaA 1009 N/A B7466_RS09440 1006 N/A 57%
EssA 153 EssA B7466_RS09435 144 EssA 51%
EsaB 80 YukD B7466_RS09430 80 YukD 64%
EssB 444 YukC B7466_RS09425 437 YukC 71%
EssC 1481 FtsK SpoIIIE N two
FtsK SpoIIIE
B7466_RS09420 1481 FtsK SpoIIIE N two
FtsK SpoIIIE
77%
SAPIG0303 98 DUF5344 B7466_RS09415 95 DUF5344 57%
EsxB 134 DUF5082 B7466_RS09410 107 DUF5082 50%
EsxX 517 LXG B7466_RS09405 523 LXG 56%
SAPIG0306 228 DUF5079 B7466_RS09400 223 DUF5079 34%
SAPIG0307 149 DUF5085 B7466_RS09395 150 DUF5085 62%
SAPIG0308 149 DUF5085 B7466_RS09390 151 DUF5085 48%
of the essC gene are associated with their own gene cluster. Among putative encoded proteins, there are proteins with a N-terminal LXG domain or a central WXG motif (Tables 6,
7). For example, the putative proteins B7466_RS09410 and
B7466_RS09405 of S. lugdunensis VISLISI 21 have 50% and
56% identity with the EsxB and EsxX proteins of S. aureus
RJ-ST398, respectively.
DISCUSSION
Staphylococcus lugdunensis is associated with a wide range of diseases including skin and soft tissue infections, infective endocarditis and bone and joint infections (Frank et al., 2008). This organism is considered as an opportunistic pathogen which can cause infections with unusual severity compared to other CoNS. This particular virulence is not currently explained by the rare virulence factors described so far, and no typing method
(Chassain et al., 2012; Didi et al., 2014) has yet been able to identify invasive clonal complexes as described for Neisseria meningitidis (Bratcher et al., 2012) orStreptococcus pneumoniae (Dagerhamn et al., 2008).
The advent of high throughput sequencing technology offers new insights into the identification of determinants associated with bacterial virulence. Before this study, only 15 complete genomes of S. lugdunensis were available in GenBank and no comparison between pathogenic and carriage strains had been conducted. In this context, we sequenced the genome of 6 new strains (3 pathogenic and 3 carriage strains) whose virulence was characterized in aG. mellonella infection model.
This model has been applied to characterize the virulence and pathogenesis of a wide range of microorganisms (Kavanagh and Reeves, 2004; Ramarao et al., 2012) but has never been implemented for S. lugdunensis. Interestingly, our results suggest that this model could be useful for assessing the virulence of S. lugdunensis in vivo as the SSTI-associated
fmicb-10-02479 October 29, 2019 Time: 15:58 # 14
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
strain (SL55) and the infective endocarditis strain (SL13) were significantly more lethal than the four other isolates. Although insects lack an adaptative immune response, their innate immune response still retains remarkable similarities with the immune response in vertebrates; this allowed to obtain relevant information about the infection process (Kavanagh and Reeves, 2004). In addition, infected larvae can be incubated at 37◦
C, which favors the study of human pathogenic bacteria (Kavanagh and Reeves, 2004). Of course, these preliminary results must be confirmed in a larger collection of isolates. Although it will not replace mammalian models, the larvae of G. mellonella provide a fast and cost-effective alternative to collect initial data.
With the addition of six new S. lugdunensis genomes, this study has raised the total number of complete genomes available for comparative purposes from 15 to 21. This result has increased the genetic diversity of the genomic data for this species as these strains were collected from one Swedish and 3 different French cities, whilst 7 of the 15 genomes in GenBank originated from a unique French location (Argemi et al., 2018).
Here, we have characterized the pan-, core- and accessory-genome of the species, particularly according to CCs defined by MLST. Furthermore, we have described the first genomic comparison of carriage and pathogenic strains
representative of the major lineages of S. lugdunensis to
explore whether virulence can be linked to specific genes or genetic variations.
Presence of MGE Despite a Closed
Pan-Genome
Comparative genomic analysis favored a closed pan-genome, as suggested by Heaps’ law of extrapolation applied byArgemi et al. (2018). The S. lugdunensis pan-genome appeared in fact to be poorly expanding with the inclusion of six new complete genomes reaching a total of only 3077 genes.
On the contrary, S. aureus and S. epidermidis pan-genomes
reached more than 3800 genes, with a constant increase
when adding new genomes (Argemi et al., 2018). This
should be confirmed by the analysis of human S. lugdunensis strains from other continents but also of environmental and animal origin.
In this context, it is not surprising that a large number of S. lugdunensis strains have barriers to horizontal gene transfer like CRISPR/Cas system or RMS (Argemi et al., 2018). Our study shows that 48% of strains possess a CRISPR/Cas system. These findings confirm the particular status of this species among CoNS. Indeed, Rossi et al. (2017) showed that only 9% (i.e., 3/34) and 3% (i.e., 1/32) of S. epidermidis and Staphylococcus haemolyticus strains respectively presented characteristic CRISPR/Cas systems. Of note, the CRISPR/Cas system was only found in CC1 strains. This high prevalence may be explained by the fact that most of the sequenced genomes belonged to CC1 strains (9/21, i.e., 43%). This CC is currently over-represented in NCBI in comparison with other CCs (CC3 [5/21, i.e., 24%], CC5 [1/21, i.e., 4%], CC6 [4/21, i.e., 19%] and CC7 [2/21, i.e., 10%]) in
relation to its global distribution (Yeh et al., 2016; Dahyot et al., 2019). It would be interesting to sequence the whole genome of CC2 and CC4 strains, as none are available in the GenBank database.
Despite the presence of CRISPR/Cas immunity systems and several type I and type II RMS, most (15/21, i.e., 71%) of the S. lugdunensis strains have acquired MGE probably from other staphylococcal species by horizontal gene transfer. This was particularly observed for CC1 strains. This latter could be explained by a frameshift within the csm6 gene that might impact the initial role of the CRISPR/Cas system. Indeed, in S. epidermidis, the protein Csm6 was found essential for preventing plasmid transformation (Hatoum-Aslan et al., 2014;
Jiang et al., 2016). Further investigations in a larger collection of S. lugdunensis strains and functional characterization are needed to understand the ability of CRISPR/Cas to prevent MGE acquisition. Another hypothesis would be that MGE may have been present before acquisition of the CRISPR/Cas region. Enriching the database with genomes of ancestral strains would help to resolve this issue.
On the contrary, while CC3 strains do not exhibit CRISPR/Cas systems or RMS, only one plasmid was detected within their genome. This can be related to the great difficulties encountered in genetically manipulating S. lugdunensis. The rare mutant strains published were only constructed after the introduction of plasmids by protoplast transformation (Szabados et al., 2011; Marlinghaus et al., 2012) or by electroporation of Escherichia coli strains expressing hsdM/hsdS RMS genes of S. lugdunensis. Interestingly, despite a new electroporation protocol, Heilbronner et al. (2013) highlighted that CC3 and CC4 isolates were harder to transform than CC1, CC2 and CC5 strains. This suggests that other mechanisms may play the role of barrier against the acquisition of MGE in S. lugdunensis. Bacteria can display physical barriers such as extracellular polysaccharidic substances, including capsules to prevent phage infection (Wilkinson and Holmes, 1979; Labrie et al., 2010). Thus, cap8 could be involved in the synthesis of capsule in S. lugdunensis CC3 strains and act as a protective barrier.
S. lugdunensis exhibits a highly conserved antibiotic susceptibility to most anti-staphylococcal antibiotics, which may be related to these different barriers against the integration of foreign DNA (Higaki et al., 1999; McHardy et al., 2017). Indeed, only one plasmid carrying an antibiotic resistance gene (tetK) was identified in a newly sequenced genome. Fosfomycin, penicillin and fusidic acid resistance-associated genes were identified with a chromosomic location. Fosfomycin resistance-associated gene was highly conserved among the 21 strains while the distribution of penicillin and fusidic acid resistance genes was variable between isolates. This finding is in accordance with the results of McHardy et al. (2017). who showed that the blaZ gene was detected in 39.1% of oxacillin-susceptible S. lugdunensis isolates. The presence of the blaZ gene was not correlated with CCs.
In this study, no specific virulence determinant was identified in S. lugdunensis pathogenic isolates compared to carriage strains. Nevertheless, two genomic islands were only identified in three pathogenic strains. One genomic island of 16 kb carries
fmicb-10-02479 October 29, 2019 Time: 15:58 # 15
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
fusidic acid resistance fusB gene and vapE gene which might contribute to bacterial adaptation and virulence (Novick et al., 2010; Chen et al., 2013). A second one of 13 kb, partially homologous to the pathogenicity islands of S. aureus (SaPI) (Novick and Subedi, 2007), was found inS. lugdunensis strains SL13 (causing infective endocarditis) and VISLISI_33 (causing liver abscess). In S. aureus, SaPIs play an important role in genomic evolution, leading to an increase in its ability to cause severe infections by acquisition of numerous virulence determinants such as pore-forming toxins and superantigens (Malachowa and DeLeo, 2010; Novick and Ram, 2017). In S. lugdunensis, no enterotoxin, superantigen or toxic shock syndrome toxin-1 was found in this region. However, an Ear penicillin binding protein and a FhuD ferrichrome ABC transporter were carried by this potential pathogenicity island. S. aureus Ear protein may act as a superantigenic exoprotein which could play a role in virulence (Singh et al., 2017). Otherwise, the ferric hydroxamate uptake D (FhuD) protein could be involved in iron acquisition by S. lugdunensis as it can promote ferrisiderophore import in S. aureus (Beasley et al., 2009) and facilitate heme uptake in blood and serum (Malachowa et al., 2011). Therefore, identification of these genes in the genomes of S. lugdunensis suggests that genomic island, other than MGE as bacteriophage or plasmids, could also contribute to the acquisition of antibiotic resistance and virulence genes in this species even if the phenomenon is probably rare (Argemi et al., 2018).
CC-Dependent Genetic Variations of
Putative Virulence-Associated loci
Based on genomic comparison using VFDB, we identified CC-dependent polymorphisms in several loci (agr, cap, sls and ess) putatively associated withS. lugdunensis virulence.
Our study shows that all CC3 strains belonged to the agrSl-type II whereas all other strains were associated with
the allele agrSl-type I. However, no link has been shown
between S. lugdunensis agr polymorphisms (agrSl-types) and specific clinical infection entities. Many studies have attempted
to associate agr types with one or more of the biological
characteristics of staphylococcal species.Tseng et al. (2015)thus showed that 7 of 11 isolates collected from bacteremia belonging toagrSl-type II presented higher hemolytic and protease activities than 4 agrSl-type I isolates. In S. aureus, agr groups have been shown to vary by clonal lineage distribution, antibiotic resistance profile, biofilm formation, expression of virulence factors, and autoinducing peptide structures (Holtfreter et al., 2007; Tan et al., 2018). In addition, Jarraud et al. (2000)
have shown that S. aureus endocarditis strains were mainly associated with phylogenetic group AF2 (agr groups I or II) while phylogenetic group AF1 (agr group IV) strains are mainly responsible for generalized exfoliative syndromes and bullous impetigo. Similarly, agr-specificity groups in S. epidermidis are also associated with specific clinical infections (Hellmark et al., 2013; Harris et al., 2017). The potential impact of this CC-agrSl-type association on the regulation of virulence
could be explored by a functional analysis of the agr locus inS. lugdunensis.
Interestingly, CC3 strains are also distinguished from all other strains by the allele of the cap gene (cap8). Except for the study of Lambe et al. (1994) that showed that S. lugdunensis possessed glycocalyx, no work has yet described the prevalence, composition, diversity and role of the capsule in this species. In S. aureus, several studies have shown that serotype 5 or 8 capsular isolates account for approximately 80% of isolates recovered from humans, the remaining isolates being not encapsulated or non-typeable (Arbeit et al., 1984; Roghmann et al., 2005; Cocchiaro et al., 2006). Like S. aureus, the genes specifying capsular polysaccharides in S. lugdunensis are encoded by a highly conserved cap locus including 4 genes, capHIJK, which may specify chemical diversity among serotypes (O’Riordan and Lee, 2004).
Heilbronner et al. (2011) described 7 genes encoding “S. lugdunensis surface proteins” named SlsA to SlsG within the genome of S. lugdunensis N920143 strain. Here, we have shown that several Sls proteins were absent or potentially non-functional in some S. lugdunensis strains according to CC. These CC-dependent differences could result in variations in the biofilm-forming and adhesion capacities between isolates. Indeed, SlsA contains putative protein domains and structural organization homologous to a known cell wall-associated biofilm Bhp protein (Post et al., 2017), which has been shown to promote primary attachment to abiotic surfaces as well as intercellular adhesion during biofilm formation (Cucarella et al., 2001; Bowden et al., 2005). In addition, the SlsD protein has putative structural similarity with members of the Sdr adhesin family such as SdrF and SdrG, crucial for the attachment of S. epidermidis to surfaces coated with human fibrinogen or type I collagen (Bowden et al., 2005; Arrecubieta et al., 2007). A functional characterization of these Sls proteins could be helpful to explore whether they are involved in the ability of S. lugdunensis to bind medical devices or biotic surfaces, such as host tissues.
On the other hand, significant genetic variations independent of CCs were detected in thelug locus. The impact of this diversity on the production and activity of lugdunine as well as on the structure of the microbiota of ecological niches ofS. lugdunensis remains to be demonstrated (Zipperer et al., 2016).
High Genetic Diversity of the ess/Type VII
Locus, a Potential Key Virulence
Determinant
This study provides, to the best of our knowledge, the first description of the CC-dependent genetic organization of the ess locus from S. lugdunensis. Initially mentioned for one strain of S. lugdunensis (Warne et al., 2016), this locus has shown considerable diversity for a clonal species (eight genetic CC-dependent organizations among only 21 strains). Homologous genes of those encoding the six core components of S. aureus T7SS were identified among the 21 genomes: the four membrane-associated proteins (EsaA, EssA, EssB, and EssC), a soluble cytosolic protein (EsaB) and the Early Secreted Antigenic
fmicb-10-02479 October 29, 2019 Time: 15:58 # 16
Lebeurre et al. Staphylococcus lugdunensis CC-Dependent Diversity
Target-6 kDa (ESAT-6, also termed EsxA) (Warne et al., 2016;
Unnikrishnan et al., 2017). Membrane and cytoplasmic proteins have been demonstrated to be necessary to export effector proteins across the bacterial membrane during host infection (Unnikrishnan et al., 2017), as proteins from the superfamily WXG100 (Burts et al., 2005) or LXG domain-containing proteins (Dai et al., 2017).
Whereas S. aureus T7SS can secrete two WXG100 proteins
(EsxA, EsxB), only one member of the WXG100 protein superfamily, EsxA, has been identified inS. lugdunensis. As for S. aureus EsxA, the putative S. lugdunensis EsxA protein contains conserved C-terminal hydrophobic patterns HxxxD/ExxhxxxH (where H stands for highly conserved hydrophobic, h for less conserved hydrophobic residues, x for any amino acid and D/E for either aspartic or glutamic acids, respectively). This domain could constitute a key component of T7SS-substrate recognition (Poulsen et al., 2014).
In addition, S. lugdunensis possesses a gene encoding a
LXG domain-containing protein which is found in a group of polymorphic toxins predicted to use the T7SS pathway (Zhang et al., 2011;Whitney et al., 2017). Thereby, these data highlight the possible extracellular translocation of these proteins. In S. aureus, proteins with domain LXG are involved in immune evasion and virulence as the protein EsxX (Dai et al., 2017) or in bacterial competition as the EsaD nuclease (Anderson et al., 2013; Cao et al., 2016). Of note, no gene homologous
to the esaD gene encoding the anti-bacterial toxin with a
C-terminal nuclease domain was identified within the genome of S. lugdunensis strains.
A great diversity in the genetic organization of the ess locus was described by Warne et al. (2016) in 153 S. aureus isolates belonging to different CCs. They showed that the S. aureus essC gene exhibited 4 sequence variants, each associated with a specific cluster of 4–6 different genes. In S. aureus,
the essC4 variant (homologous to the S. lugdunensis essC1
variant) was only found in CC22 isolates whereas the essC2 variant (homologous to the S. lugdunensis essC2 variant) was identified in CC15 and ST398 strains. In S. lugdunensis, a
strong association between CCs and essC variants was also
observed. These results suggest that S. lugdunensis strains may have acquired these regions independently and from difference sources.
Moreover, genetic diversity was observed downstream of modules 1 and 2, based on (i) a variable number of genes encoding uncharacterized proteins, (ii) the presence of a cluster of 7 genes specific to CC1 and CC3, (iii) the insertion of a transposon in CC1 strains, and (iv) the variable copy number of genes encoding DUF600 proteins. The mechanisms of occurrence of these inter- and intra-CC variations are unclear and need further investigations in a larger collection of S. lugdunensis strains, and especially in strains belonging to CC2 and CC4 which could present different organizations of thislocus.
Thus, we describe here homologs of a large number of genes encoding T7SS in S. lugdunensis which shares a high number of clinical features with S. aureus, including ability to form
abscesses. S. lugdunensis is the sole CoNS to possess all the components required for functional Ess machinery. T7SS could play a key role in abscess development and hence in the currently poorly understood virulence of this bacterial species.
Taken together, these results support the “closed” state of the pan-genome of S. lugdunensis, but identification of MGE carrying putative virulence and resistance genes suggests some possible horizontal gene transfers between S. lugdunensis and other staphylococcal species. No specific variation or virulence determinant was associated with the pathogenic strains, but the distribution of potential genomic islands constitutes an interesting option to explore in a larger collection of strains. Here, we identified several CC-dependent variations, especially for CC3 strains which showed specific variations in severalloci potentially associated with virulence. Further phenotypic and functional studies are needed to fully characterize this particular CC and to evaluate the role of T7SS in the virulence of S. lugdunensis. Finally, whole genome sequencing of CC2 and CC4 strains is needed for a complete picture of the genetic diversity ofS. lugdunensis.
DATA AVAILABILITY STATEMENT
The datasets generated for this study can be found in the GenBank database, accession: CP041722, CP041723, CP041724, CP041725, CP041726, CP041727.
AUTHOR CONTRIBUTIONS
JL, SDa, PF, and MP-C designed the study. JL, SDi, and
AP analyzed the data. J-CG and MA performed the Galleria
mellonella infection experiments. JL, SDa, and MP-C wrote the manuscript. All authors have read and approved the final version of the manuscript.
FUNDING
This study was supported by an educational grant for JL from the Charles Nicolle Foundation.
ACKNOWLEDGMENTS
The authors are grateful to Nikki Sabourin-Gibbs (Rouen University Hospital) for her help in editing the manuscript.
SUPPLEMENTARY MATERIAL
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb. 2019.02479/full#supplementary-material