Catalytically active peptides affected by self-assembly and residues order
Texte intégral
Figure

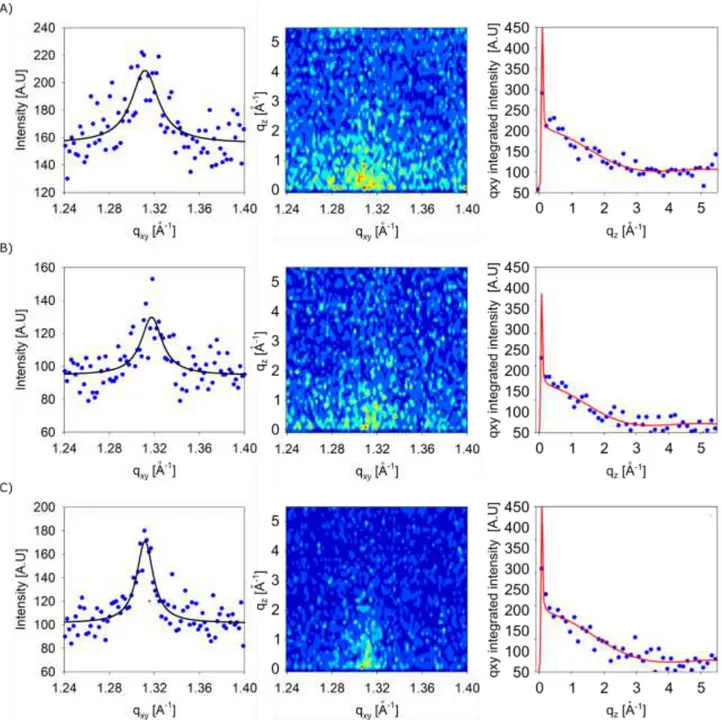


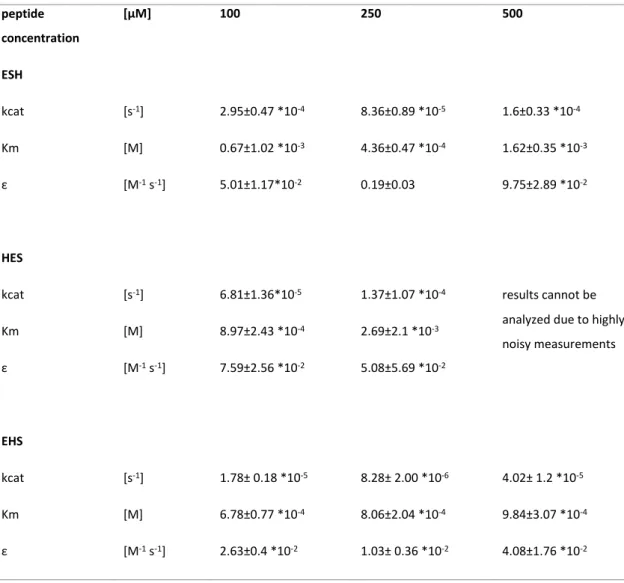


Documents relatifs
Based on our recent progress in the utilization of compounds 1 to access masked chiral b 2,2 -AAs 2 under chiral phase-transfer catalysis, 14 we now became interested in
de cristallites est manifeste. Cela indique qu’il reste des germes cristallins.. indétectables par DRX qui se manifestent au réchauffage par une recristallisation effective de
pour former tout d’abord le plexus pelvien et le nerf de Remak 5
considered as an attractive method since it can start with waste materials, generating an inexpensive energy and providing an alternative waste treatment. It can be divided in
In all families, LCLs from carriers of an RP11-associated isoallele (asymptomatics and AG261) express more PRPF31 pre-mRNA than those from affected patients, but all cell lines
abiotic process of random peptide formation via the polymerization of a-amino acid mixed anhydrides with phosphate (aa-PMAs) or phosphate esters (aa-PEMAs) and adenylates (aa-AMPs)
Above a critical concentration P/L*, there is a coexistence region in which a fraction of the peptide molecules are inserted and the rest remain on the surface.. The
